

Abstract
During heart development endocardial cells within the atrio-ventricular (AV) region undergo TGFβ-dependent epithelial-mesenchymal transformation (EMT) and invade the underlying cardiac jelly. This process gives rise to the endocardial cushions from which AV valves and part of the septum originate. In this paper we show that in mouse embryos and in AV explants TGFβ induction of endocardial EMT is strongly inhibited in mice deficient for endothelial β-catenin, leading to a lack of heart cushion formation. Using a Wnt-signaling reporter mouse strain, we demonstrated in vivo and ex vivo that EMT in heart cushion is accompanied by activation of β-catenin/TCF/Lef transcriptional activity. In cultured endothelial cells, TGFβ2 induces α-smooth muscle actin (αSMA) expression. This process was strongly reduced in β-catenin null cells, although TGFβ2 induced smad phosphorylation was unchanged. These data demonstrate an involvement of β-catenin/TCF/Lef transcriptional activity in heart cushion formation, and suggest an interaction between TGFβ and Wnt-signaling pathways in the induction of endothelial-mesenchymal transformation.
Keywords: heart cushion formation; β-catenin; endothelial cells; Wnt-signaling; transforming growth factor β
Introduction
The heart is the first organ to be formed in the developing vertebrate embryo and originates from the splachnic lateral mesoderm. A specific subpopulation of cells, the cardiogenic mesoderm, gives rise to the first two types of heart cells, which are the myocardial and endocardial cells. The latter acquire endothelial markers, like VE-cadherin, Tie-1, Tie-2, VEGFRI/II, and PECAM/CD31. Furthermore, it is known that endothelial/endocardial cells in the atrio-ventricular (AV) canal give rise to the mesenchymal heart cushion cells, which form the mesenchymal portion of cardiac septa and valves (Markwald et al., 1977; Potts et al., 1991). To accomplish this, endocardial cells undergo an epithelial-mesenchymal transformation (EMT), a process, which has been shown in mammals to be largely dependent on TGFβ signaling (Camenisch et al., 2002). The classical TGFβ-signaling pathway is characterized by a heteromeric receptor complex, consisting of two type II and two type I receptors (Derynck and Feng, 1997; Massague, 2000). In endothelial cells two type I receptors, Alk-1 and Alk-5, which interact with the common type II receptor, confer specificity of TGFβ signaling (Goumans et al., 2002). Upon TGFβ-binding, the receptor complex undergoes a series of phosphorylations, activating the serin-threonine kinase function of the type I receptor. This, in turn, phosphorylates proteins of the receptor smad (R-smad) family, which translocate to the nucleus and, together with accessory transcription factors, initiate target-gene transcription (Itoh et al., 2000; Massague, 2000; Moustakas et al., 2001). Recently, evidence has been presented that smads can also interact with β-catenin and TCF/Lef transcription factors, which are downstream effectors of the Wnt-signaling pathway (Labbe et al., 2000; Nishita et al., 2000; Tian and Phillips, 2002).
Many members of the Wnt family of growth and differentiation factors act by stabilizing β-catenin in the cytosol and promoting its accumulation in the nucleus, where it binds and activates TCF/Lef transcription factors (Gumbiner, 1995; Behrens et al., 1996; Miller and Moon, 1996; Zhurinsky et al., 2000; Hurlstone and Clevers, 2002). In the vascular system, the role of Wnt signaling and of β-catenin transcriptional activity has not been fully elucidated yet. To investigate this aspect we specifically inactivated the β-catenin gene in endothelial cells, using the Cre-loxP system. This resulted in embryonic lethality around mid-gestation due to vascular fragility and defects in placentation (Cattelino et al., 2003).
Here, we show that also heart development is severely affected in mutant mice and that endothelial deficiency of β-catenin leads to a lack of heart cushion formation by 10.5 d post coitum (dpc). In vivo and ex vivo we found that TGFβ2 induction of EMT of endocardial cells is accompanied by activation of β-catenin transcriptional activity and is strongly inhibited in mice deficient for endothelial β-catenin. In addition, the impairment of β-catenin null cells to transform into αSMA-positive cells upon TGFβ2 stimulation has been demonstrated also in vitro. The data presented here suggest a cross-talk between TGFβ and Wnt-signaling pathways during EMT in heart cushion formation.
Results
β-Catenin deficiency in endothelial cells leads to defective septum formation in the heart
To specifically inactivate the β-catenin gene in endothelial cells, we crossed transgenic mice carrying a β-catenin allele flanked by two loxP sites (Brault et al., 2001), with mice expressing the Cre recombinase under the endothelial-specific Tie2 promoter (Schlaeger et al., 1997; Kisanuki et al., 2001). These animals die between 11.5 and 13.0 dpc due to an altered development of the vascular system (Cattelino et al., 2003).
In this paper, we focused on the alterations in heart development due to β-catenin deficiency in the endocardium. The lack of endocardial β-catenin was confirmed previously by immunofluorescence (Cattelino et al., 2003). Embryos at 9.5–10.5 dpc were fixed, stained in whole mount for the endothelial marker PECAM/CD31, embedded in paraffin, sectioned and counterstained with nuclear fast red. At 9.5 dpc, before endocardial-mesenchymal transformation initiates, knockout (KO) and wild-type (WT) littermates were indistinguishable concerning the development of the heart (unpublished data). From 9.5 to 10.5 dpc, in all embryos analyzed (30WT; 15 KO), we observed a divergent formation of the heart cushion. At 10.5 dpc, the cardiac cushion of KO embryos showed a dramatically decreased cellularity as compared with WT littermates (Fig. 1, compare A and C with B and D), which was confirmed quantifying the cell numbers in heart cushions (Fig. 1 E). This indicates a reduced or largely missing ability of KO endocardial cells to undergo EMT and to invade the cardiac jelly. Although the endocardium was stained positive for PECAM/CD31, it is noteworthy that the cells inside the heart cushion at 10.5 dpc were negative for this endothelial marker. This confirms previous reports that endocardial cells lose their endothelial markers during EMT (Nakajima et al., 1997; Camenisch et al., 2002; Frid et al., 2002).
Figure 1.
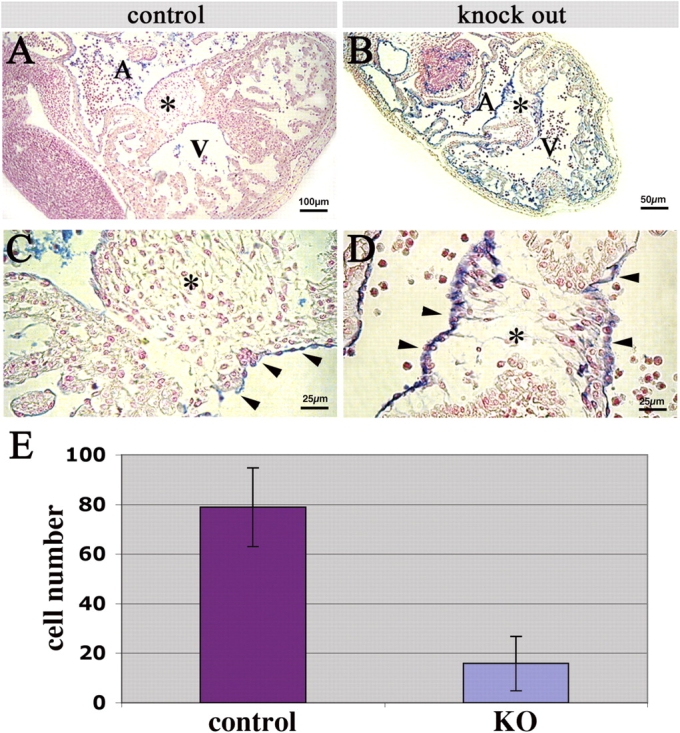
Heart cushion defect in endothelial β-catenin null embryos. (A–D) Paraffin sections of mouse embryos at 10.5 dpc stained in whole mount for PECAM/CD31 (blue) and counterstained with nuclear fast red. (A and C) Sections of a control embryo showing the heart region in low and high magnification, respectively. Note the heart cushion (asterisk) filled with mesenchymal cells. Arrowheads in C point to the endocardium. (B and D) Sections of a representative KO embryo in the same plane of the control. The heart cushion (asterisk) is almost completely devoid of mesenchymal cells. (E) Number of cells in the heart cushion of control and KO embryos, as counted in six serial 6-μm sections of three embryos, respectively. Columns represent means ± SD; P < 0.0001 by t test.
Endocardial cells from β-catenin mutant embryos fail to transform in an ex vivo AV explant assay
To directly characterize the defect of endocardial cells to invade the cardiac jelly and to exclude prelethal, secondary effects at 10.5 dpc, we dissected the AV regions of WT and KO embryos at E9.5 dpc and subjected them to an ex vivo assay for EMT in the AV heart cushion (Runyan and Markwald, 1983; Potts et al., 1991). This assay mimics endocardial EMT in vitro, as the TGFβ2 and other stimuli for the endocardial cells to undergo transformation are provided by the myocardium, which is included. The explants were placed on top of a collagen I gel with the endocardial layer upside down and incubated under standard cell culture conditions for up to 48 h. In total, 64 AV explants were generated of which 46 were β-catenin positive (either homo- or heterozygous) and 18 were deficient for endocardial β-catenin. About 70% of both types of explants, with and without endocardial β-catenin, attached firmly to the collagen gel and spontaneously started contracting in vitro. Only these explants were considered for consecutive investigations. We did not observe any differences between WT and KO explants in attachment and survival. Neither could we observe a difference in the number of apoptotic cells in explants with or without endothelial β-catenin (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200403050/DC1).
In phase contrast, endothelial cells of WT AV explants were widely spread on the gel, showing a largely scattered, mesenchymal morphology (Fig. 2 A). Instead, endothelial cells of KO AV explants grew out predominantly in a monolayer, bearing cobblestone morphology (Fig. 2 D). As the invasion of the collagen gel is considered the hallmark for occurrence of EMT (Potts et al., 1991), we characterized the distribution of the outgrowing endothelial cells both on the surface and inside the collagen lattice. The explants were fixed and stained for F-actin together with a nuclear counterstaining (DAPI) and subjected to confocal microscopy (Fig. 2, B, C, E, and F). In contrast to the long, spindle-like endothelial cells observed in WT explants, those lacking β-catenin maintained epithelioid characteristics, which was visualized by F-actin staining (Fig. 2, B and E, respectively).
Figure 2.

Endocardial cells from β-catenin mutant embryos fail to transform in an ex vivo AV explant assay. (A and D) Phase-contrast micrographs of the AV explant on top of the collagen I gel after 48 h in culture, from control and endothelial β-catenin KO embryos (9.5 dpc), respectively. Note the scattered and widespread appearance of the outgrowing endothelial cells in the control (A), compared with the KO, where endothelial cells grew out in a monolayer on top of the gel, showing an compact, epithelial-like morphology (D). (B and E) AV explants from control and KO embryos, respectively, have been labeled for F-actin (green) and nuclei with DAPI (blue). Confocal stack of images presented as a maximal projection in the z axes. (C and F, left) Nuclear fluorescent staining with DAPI presented as an overlay of confocal xy-images, showing all cell nuclei in a maximal projection in the z axes, of control and KO explants, respectively. (C and F, right) Three different z-scans. The corresponding horizontal section level in the xy-image on the left, and individual cells of interest are indicated by the colored arrowheads. Dashed lines indicate the border of the AV explants in the xy-images on the left panels and the surface of the collagen gel in the z-images on the right panels, respectively. Note that KO cells remain mainly organized in a monolayer on top of the gel (F, arrowheads), whereas some WT cells are regularly found in the gel (C, arrowheads). (G) quantification of cells inside the collagen gel. Confocal sections in focal planes below the gel surface were counted for the presence of cells, indicated by nuclear staining for DAPI. (AV explants, n = 7 for WT and KO, respectively). Columns represent means ± SD; P < 0.0001 by t test.
Performing a confocal scanning of the explants in z axis, we could demonstrate that cells from the WT AV explants invaded the gel (Fig. 2 C), whereas in KO explants cells largely failed to invade the collagen gel and remained on the gel surface (Fig. 2 F). The cells, which have undergone EMT have been quantified and their number per gel volume has been plotted (Fig. 2 G). These data confirm the observation in vivo in heart cushion formation.
TCF/Lef/β-catenin signaling occurs during endothelial-mesenchymal transformation in the developing heart and in AV explants
To directly investigate whether β-catenin transcriptional activity is involved in endocardial EMT, we made use of a transgenic mouse line containing a β-catenin/TCF/Lef–specific reporter construct (BAT-gal), which, upon activation, results in a nuclear accumulation of β-galactosidase (Maretto et al., 2003).
At 10.5 dpc, single cells in the heart cushion stained positive for nuclear β-galactosidase, indicating activation of β-catenin /TCF/Lef transcriptional activity (Fig. 3, A and B). In ex vivo EMT assays described above, AV explants from BAT-gal mice behaved like WTs concerning their ability to grow out and to invade the gel. Interestingly, single cells in the outgrowing endothelial cell layer regularly stained positive for nuclear β-galactosidase, confirming the in vivo data (Fig. 3, C and D). Some, but not all, of these cells could be labeled for both PECAM/CD31 and β-galactosidase indicating their endothelial origin (Fig. 3 D). The fact that several invading cells could not be labeled with PECAM can be explained by the loss of endothelial markers during EMT (Fig. 1 C; Frid et al., 2002). Therefore, to unequivocally demonstrate the endothelial origin of the invading cells in the ex vivo AV explant assay, we crossed mice transgenic for the Tie2-Cre construct (Schlaeger et al., 1997; Kisanuki et al., 2001) with ROSA26 reporter mice. The double-transgenic offspring bears an irreversible expression of the lacZ gene in endothelial cells after Cre-mediated recombination (Soriano, 1999). As reported in Fig. 3 (E–G), essentially all the cells outgrowing from the explant and invading the gel were positive for β-galactosidase indicating their endothelial origin.
Figure 3.

β-Catenin/TCF/Lef transcriptional activity occurs during EMT in the heart cushion in vivo and ex vivo. (A) Paraffin section of a representative BAT-gal embryo (10.5 dpc) stained in whole mount for β-galactosidase and counterstained with Eosin, showing the region of the heart with the heart cushion (asterisk). Single cells in the heart cushion show a nuclear staining in blue. (B) Higher magnification of A, endocardium is indicated by arrowheads. A, Atrium; V, ventricle. (C) Typical AV explant from BAT-gal embryo (9.5 dpc) after 48 h in culture showing single outgrowing cells stained positive for nuclear β-galactosidase (arrowheads). (D) Confocal image of a double staining for β-galactosidase (red) and PECAM (green) confirming the endothelial nature of the cells showing reporter activation (arrowhead). (E) Phase-contrast image of a representative AV explant derived from an embryo positive for the inherited expression of β-galactosidase (blue, staining) in endothelial cells. Note the mesenchymal-like outgrowth of the cells on and into the collagen gel. (F) Bright field image of the same explant described in E, showing clearly that virtually all cells grown out from the explant are of endothelial origin, as indicated by the blue staining for β-galactosidase. Note that also the original endocardial lining of the AV explant, which is still present underneath the myocardium, is positive for β-galactosidase. (G) Higher magnification of F.
β-Catenin–deficient endothelial cells show impaired TGFβ2-induced transformation in vitro
To understand the mechanism of action of β-catenin in promoting TGFβ-induced endothelial EMT we have set up an in vitro system. As reported previously, we isolated, cultured, and characterized endothelial cells from KO animals (Cattelino et al., 2003). The β-catenin–deficient cells were infected with an empty retroviral vector (KOvc), or with the same vector containing the full-length β-catenin gene (KOfl-βcat). The ability of the exogenous β-catenin protein to localize correctly at the membrane and to accumulate in the nucleus, was confirmed by immunofluorescence staining (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200403050/DC1). It has been reported that under long-term stimulation by TGFβ, cultured endothelial cells could transform into mesenchymal cells expressing αSMA in a similar way to what has been described for endocardial EMT (Paranya et al., 2001; Frid et al., 2002). Because TGFβ2 seems to be the most relevant TGFβ-family member for EMT in heart cushion formation (Camenisch et al., 2002), the majority of the experiments were performed using TGFβ2. It is noteworthy that TGFβ1 was also used with comparable results (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200403050/DC1).
Cells were seeded in sparse conditions and cultured in low serum with and without addition of TGFβ2 (1 ng/ml) for 5–7 d. Under these conditions, 44 ± 5.7% of KOfl-βcat cells transformed into cells expressing αSMA, whereas only 20 ± 1.4% of the β-catenin–deficient cells were positive for αSMA (Fig. 4, A, B, and D). The significance of the difference was calculated by a t test analysis and gave a P < 0.0001, whereas in the absence of TGFβ2 the number of αSMA-expressing endothelial cells was equally low for both cell lines (Fig. 4 D). Furthermore, we were able to double stain the majority (>80%) of αSMA-positive cells with the endothelial marker VE-cadherin confirming the endothelial origin of transforming cells (Fig. 4 C). The growth rate of the two cell lines under TGFβ2 stimulation did not differ significantly, as determined by BrdU incorporation (Fig. 4 E).
Figure 4.

β-Catenin is required for TGFβ2-induced endothelial cells transformation into αSMA-positive cells in vitro. Endothelial β-catenin null cells infected with the full-length β-catenin gene (KOfl-βcat) (B and C) or with the empty vector (KOvc) (A), were treated for 6 d with TGFβ2, and fixed and stained for αSMA (green). (C) KOfl-βcat cells, which have receive the same treatment as in A and B, were double stained for VE-cadherin (red) and αSMA (green). The nuclear dye Hoechst (blue) was used to counterstain cells. (D) The number of αSMA-positive cells was expressed as a percentage of total cells (1–2 × 103 cells were counted for each cell line, and columns represents the mean out of four individual experiments ± SD; P < 0. 0001 by t test). (E) BrdU incorporation in KOvc and KOfl-βcat cell after TGFβ2 stimulation. Cells were treated as described above and proliferation was measured for the two cell lines indicated, as described in Materials and methods. The number of cells, which have incorporated BrdU in the nuclei, is presented as the mean percentage of total cells ± SD.
TGFβ2 signaling is not altered in endothelial cells deficient for β-catenin
We tested whether the absence of β-catenin could influence TGFβ2 signaling and in particular the activation of R-smads. In endothelial cells two different TGFβ type I receptors, namely Alk-1 and Alk-5, signal through different R-smads, smad1/5 and smad2/3, respectively (Goumans et al., 2002). Therefore, phosphorylation and nuclear accumulation of smad1/5 and smad2 as indicators of specific receptor activation were evaluated in KOfl-βcat and KOvc cell lines. We found that phospho-smad1/5 (P-smad1/5) and phospho-smad2 (P-smad2) were highly increased upon TGFβ2 stimulation in both cell lines (Fig. 5 A). In particular, the amount of P-smad1/5 in the nuclear fraction of KOfl-βcat and KOvc cells did not differ in resting conditions, and was only slightly lower in the KOvc as compared with the KOfl-βcat after stimulation with TGFβ2 (Fig. 5 A). On the other hand, in KOvc cells, nuclear P-smad2 was not decreased in the absence of β-catenin, both in resting conditions and after TGFβ2 stimulation (Fig. 5 A). Also the total amount of smads, evaluated by antibodies against the nonphosphorylated forms of the proteins, did not differ significantly between KOfl-βcat and KOvc, with and without TGFβ2 stimulation (Fig. 5 B). These data suggest that the overall ability of endothelial cells to respond to TGFβ in terms of activation of R-smads is not significantly altered in the absence of β-catenin.
Figure 5.

The absence of β-catenin does not modify smads and their phosphorylation. (A) Western blots for phospho-smad1 and 5 (P-smad1/5, arrowheads) and phospho-smad2 (P-smad2, arrowhead) from nuclear lysates prepared from KOfl-βcat and KOvc cell lines. Cells were treated with or without TGFβ2 (5 ng/ml) for 2 h before extraction. Both nuclear P-smad1/5 and 2 were increased after TGFβ2 stimulation, with no significant change between KOvc and KOfl-βcat cell lines. (B) Western blots of total lysates of smads1/5 and smads2/3. Note that the total amounts of smads were not altered. α-Tubulin was used as a control for protein loads.
Notch1, delta-like 4, snail1, and VE-cadherin expression is not altered in endothelial cells deficient for β-catenin
It has been reported that Notch activity is required for endocardial EMT. In particular, Notch1 was found to be increased in zebrafish when β-catenin transcriptional activity was increased (Hurlstone et al., 2003). Therefore, we investigated whether inhibition of endothelial EMT in β-catenin–deficient cells could be an indirect effect, due to an alteration of Notch1 expression. As reported in Fig. 6 A, Notch1 was not modified in heart extracts of WT and KO embryos at 10.5 dpc. In cultured endothelial cells with or without β-catenin, Notch1 was not significantly modified under unstimulated conditions and slightly reduced (∼30%) in both cell lines after activation with TGFβ2 (Fig. 6 B). The transcriptional expression level for the Notch ligand delta-like 4 has been determined by quantitative real time PCR and showed no significant differences between cells with or without β-catenin, neither under basal, nor under TGFβ2-stimulated conditions (Fig. 6 D).
Figure 6.

The absence of β-catenin does not modify VE-cadherin, Notch1, delta-like 4, and snail1 expression. (A) Total protein extracts from WT and KO hearts at 10.5 dpc were separated on 7% SDS-PAGE. Immunoblotting for VE-cadherin and Notch1 revealed no significant differences in expression between WT and KO hearts. α-Tubulin was used as a control for protein loads. (B) KOfl-βcat and KOvc endothelial cells, with or without stimulation of TGFβ2, were analyzed by Western blotting for their expression of VE-cadherin and Notch1. In unstimulated conditions, KOfl-βcat and KOvc cells show equal levels of expression for both proteins. After stimulation with TGFβ2, both cell lines show ∼20% reduction in expression for both proteins. α-Tubulin was used as a control for protein loads. (C and D) Total RNA from KOfl-βcat and KOvc endothelial cells, with or without stimulation with TGFβ2, was retro-transcribed into cDNA and the relative level of gene expression (RQ) was analyzed by quantitative real time PCR for snail1 (C) and delta-like 4 (D). In both cell lines snail1 shows no different expression in basal conditions and was comparably up-regulated after TGFβ2 stimulation. Delta-like 4 did not show any significant regulation between the two cell lines, neither with nor without TGFβ2. Error bars (RQmin/RQmax) are based on confidential level of 95%.
The transcription factor snail1 has been reported to play a key role in epithelial and endothelial EMT (Batlle et al., 2000; Cano et al., 2000; Savagner, 2001; Timmerman et al., 2004). In cultured endothelial cells snail1 expression was increased by TGFβ2 as reported previously (Valdes et al., 2002) but this effect was comparable in the presence or absence of β-catenin (Fig. 6 C).
Finally, because cadherins are targets of snail and are known to be down-regulated during EMT, we studied VE-cadherin expression in the embryonic heart and in cells in vitro. We found that in total extract taken from hearts undergoing AV endocardial EMT, VE-cadherin protein levels were not significantly different between WT and KO samples (Fig. 6 A). The same was true for endothelial cells in culture, with or without β-catenin (Fig. 6 B). It is noteworthy that under stimulation with TGFβ2, VE-cadherin protein levels were slightly down-regulated (∼20%) in both cell lines.
Together, these data indicate that the absence of β-catenin does not alter the level of Notch1 receptor and delta-like 4 ligand. Moreover, it does not alter the ability of TGFβ2 to influence the level of snail1 and of VE-cadherin, suggesting that β-catenin is not upstream of snail1.
Discussion
In embryonic development, EMT occurs when epithelial cells become migratory, invade new microenvironments and acquire mesenchymal characteristics such as elongated morphology, rearranged actin cytoskeleton, increased production of lytic enzymes, loss of apical/basal polarity, and junction organization (Thiery, 2002).
During heart development a subset of endocardial cells, located in the AV and cono-ventricular regions, detaches from the endocardial sheet and invades the underlying cardiac jelly to form the endocardial cushions, which give rise to valves and septa. This appears to be a typical EMT of a subset of endocardial cells. TGFβ plays an important role in this phenomenon but the molecular mechanisms controlling the process have only been partially clarified.
In this paper, we provide evidence that induction of EMT in the endocardial cells requires β-catenin transcriptional activity. The requirement for β-catenin was demonstrated by the observation that in mice, in which the β-catenin gene was selectively inactivated in endothelial/endocardial cells, the heart cushion fails to develop. In these embryos, endocardial cells do not undergo EMT. This was demonstrated in vivo by a strong reduction of cellularity in the cardiac jelly and ex vivo in AV explants by the failure of endocardial cells to acquire mesenchymal characteristics and to invade the collagen gel. In addition, cultured endothelial cells deficient for β-catenin exhibit an impaired ability to undergo transformation and to express αSMA upon stimulation with TGFβ2.
These defects are most likely due to a lack of β-catenin transcriptional activity because in reporter mice, cells invading the cardiac jelly or growing out into the collagen gel in AV explants, express the reporter gene LacZ.
These results are in agreement with a recent publication by Hurlstone et al. (2003) in zebrafish, showing that a lack of function mutation in the adenomatous polyposis coli gene, leads to increased β-catenin signaling and results in excessive and misplaced endocardial cushion formation. Conversely, overexpression of adenomatous polyposis coli or inhibition of Wnt by Dickkopf 1 inhibited this phenomenon. These findings support the idea of a prominent role of the Wnt/β-catenin pathway in determining endocardial cell fate.
Interestingly, in vivo and in the AV explants only a limited number of cells were β-galactosidase positive suggesting that activation of β-catenin transcriptional activity is a transient phenomenon, which likely follows a specific temporal pattern. We demonstrated that single cells expressed both endothelial markers and β-galactosidase in explants from BAT-gal reporter mice. This suggests that β-catenin and/or its transcriptional activity likely acts at early/intermediate steps of cell transformation when cells have not yet acquired full mesenchymal characteristics, while losing endothelial characteristics.
Using explants from ROSA26 mice crossed with Tie2-Cre transgenics, in which Tie2-Cre induces LacZ expression in endothelial cells in an irreversible way, we demonstrated that virtually all cells in AV explants, undergoing EMT are of endothelial origin. These data fully support previous work in vivo on mouse heart development (Kisanuki et al., 2001).
In cultured cells, TGFβ2 induced αSMA expression in the endothelium in a β-catenin–dependent way. αSMA is the most frequently used marker of EMT in the endocardium (Paranya et al., 2001) and, with the limitations of a culture system, these results support the data in vivo and in organ culture. In addition, the observations on cultured cells suggest that the effect of TGFβ2 is cell-autonomous, not requiring the interaction with other contiguous cells types present in vivo. Our data support a model in which both β-catenin/TCF/Lef transcriptional activity and TGFβ signaling are required for EMT, but whether these two pathways directly interact, or whether one or the other is upstream, remains to be elucidated. Interestingly, from the data reported here, TGFβ signaling through smads is not significantly altered by the absence of β-catenin, arguing in favor of a role of β-catenin in parallel or downstream of smads. It has been reported that members of the Wnt-signaling pathway like β-catenin and TCF/Lef can independently interact with smad2, smad3, and smad4 (Labbe et al., 2000; Nishita et al., 2000; Tian and Phillips, 2002). Nishita et al. (2000) and Labbe et al. (2000) also show that during development of Xenopus laevis a number of target genes exhibit responsiveness to both Wnt and TGFβ signals.
A recent paper demonstrates that TGFβ3 can up-regulate Lef-1 transcription during EMT in mouse palatal development (Nawshad and Hay, 2003). When smad2 and smad4 are present in the nucleus, Lef-1 is activated without β-catenin. Smad2/4 would therefore interact preferentially with Lef-1 if β-catenin is absent. However, Nawshad and Hay (2003) found that palatal EMT would be promoted in the absence of β-catenin, likely by a Lef–smad2 complex, which is neither the case in the present work in mouse, nor in a zebrafish model (Hurlstone et al., 2003). Therefore, it is conceivable that the mechanism through which TGFβ induces EMT in endocardial cells is not identical to those in the palatal epithelium. Most likely, this might depend on the endothelial-specific expression of TGFβ and/or Wnt receptors.
Other signaling pathways, like EGF, VEGF, and BMP, have been implicated in heart cushion/valve formation (Brown et al., 1999; Dor et al., 2001; Kim et al., 2001; Iwamoto et al., 2003). It was recently reported that also Notch activity promotes EMT during cardiac development via transcriptional induction of the snail1 repressor, which facilitates VE-cadherin down-regulation (Noseda et al., 2004; Timmerman et al., 2004). The authors, using endothelial cells induced to transform by activated Notch, could not find evidence for TGFβ activation. This suggests that Notch may act independently from TGFβ. To understand if in endothelial cells genes of the Notch signaling pathway are altered in the absence of β-catenin, we analyzed the expression of Notch1, delta-like 4, but we could not find any significant regulation neither in vivo, nor in vitro (Fig. 6). Interestingly, upon stimulation with TGFβ2, snail1 is equally up-regulated in cells with and without β-catenin, suggesting on the one hand that β-catenin is not upstream of snail1, on the other hand that snail1 expression is not sufficient to promote control levels of EMT in the absence of β-catenin. As a late target of EMT, protein expression levels of VE-cadherin were analyzed, but we could not find any significant regulation neither in vivo, nor in vitro, between WT and KO (Fig. 6). However, upon TGFβ2 stimulation of cells in vitro, only a slight decrease of protein levels for VE-cadherin could be detected in both cell lines (Fig. 6 B). This observation is supported by immunostaining, in which the majority of cells positive for αSMA also express VE-cadherin (Fig. 4 C). Overall these data suggest that β-catenin may act to a large extent at early stages of EMT, before VE-cadherin down-regulation.
However, keeping in mind that cadherins may indirectly regulate the free pool of β-catenin (Gumbiner, 1995; Nelson and Nusse, 2004), we cannot exclude that the slight down-regulation of VE-cadherin observed upon TGFβ2 might contribute to β-catenin signaling level, thus providing a possible link between Wnt and TGFβ signaling.
In conclusion, endocardial EMT leading to cell invasion of the cardiac jelly is a complex phenomenon, in which different signaling pathways may interplay. There is growing evidence from different in vivo and in vitro systems that vascular endothelial cells may acquire smooth muscle cell markers and can contribute to vessel wall formation during development as well as during pathological conditions, such as artherosclerosis and restenosis (DeRuiter et al., 1997; Frid et al., 2002; Iurlaro et al., 2003; Yurugi-Kobayashi et al., 2003). Therefore, the comprehension of the mechanisms underlying endothelial/endocardial transformation will be crucial not only for the understanding of diseases like congenital heart defects, but also of artherosclerotic degenerations of the vessel wall.
Materials and methods
Transgenic mice and crossings
The generation of β-catenin flox and flox del mice has been described previously (Brault et al., 2001). The Tie2-Cre transgenic mice were a gift of M. Yanagisawa (Schlaeger et al., 1997; Kisanuki et al., 2001). The mating scheme and the genotyping of the animals have been described previously (Cattelino et al., 2003).
Transgenic Wnt-signaling reporter animals have been described previously (Maretto et al., 2003). In brief, BAT-gal was constructed by fusing seven TCF/Lef-binding sites upstream of a 0.13-kb fragment containing the minimal promoter-TATA box of the gene siamois upstream of the β-galactosidase gene (Brannon et al., 1997). Transgenics were crossed into a CD1 outbred mouse background and used for embryo analysis and heart explants.
Antibodies
Mouse mAbs were as follows: anti-αSMA (clone 1A4) was purchased from Sigma-Aldrich and anti–α-tubulin was purchased from Molecular Probes. The rat anti-PECAM/CD31 clone MEC7.46 mAb has been described previously (Vecchi et al., 1994).
pAbs were as follows: anti–P-smad1/5 and anti–P-smad2, as well as anti-smad1/5 and anti-smad2/3 were purchased from Upstate Biotechnology; goat anti–β-galactosidase was purchased from Biogenesis; goat anti–VE-cadherin was purchased from Research Diagnostics Inc.; and goat anti-Notch1 was purchased from Santa Cruz Biotechnology.
Embryological techniques and immunostaining
Whole mount staining with anti-PECAM antibodies has been described previously (Cattelino et al., 2003). For histological examination, fixed embryos were dehydrated, embedded in paraffin, and sectioned at 6 μm according to standard procedures. Sections were dewaxed, rehydrated, and counter stained with Eosin or Nuclear Fast red (Vector Laboratories). Immunostaining on PFA fixed specimens was performed as described previously (Lampugnani et al., 2002; Cattelino et al., 2003). For αSMA, cultured cells were fixed for 5 min at −20°C in methanol and primary ascite was diluted 1:1,000.
Immunostaining of heart explants and of endothelial cells, was performed as described previously (Liebner et al., 2000; Cattelino et al., 2003). For visualizing BrdU incorporation, cells were stained with mouse monoclonal anti-BrdU antibodies (Amersham Biosciences), followed by TRITC-conjugated antibody to mouse immunoglobulin (DakoCytomation). Specimens were conterstained with the nuclear dye DAPI or with Hoechst 33258 (Sigma-Aldrich) and mounted in 90% glycerol containing paraphenylenediamine as an antibleaching agent. Fluorescence was observed either with an epifluorescence microscope (Leica) and digitally documented with a camera (model SenSys; Roper Scientific) or confocal microscope (model TCS2; Leica). Images were computer processed using Adobe Photoshop 7 for Macintosh.
Heart explant assay
All AV tissue was harvested from timed matings at 9.5 dpc of development. The AV canal region was dissected from embryos in sterile PBS and placed on top of drained rat type I collagen (Collaborative Biomedical Products) gel as described previously (Runyan and Markwald, 1983; Potts et al., 1991). The AV explants were maintained in M199 medium supplemented with 1% FBS (Hyclone), 100 U/ml penicillin, 100 mg/ml streptomycin, and 0.1% insulin, transferrin, and selenium (GIBCO BRL) in four-well microculture dishes (Nalge-Nunc). AV explants were allowed to firmly attach overnight (∼14 h) before addition of 100 ml of M199 explant media. AV explants cultures were grown at 37°C with 5% CO2.
Explant cultures were fixed with 4% PFA, rinsed twice with PBS, and stained with Oregon green phalloidin and DAPI (Molecular Probes) to observe cellular and nuclear morphology. Images were acquired according to the procedure described in Embryological techniques and immunostaining.
Culture of endothelial cells and retroviral infection
Endothelial cells with homozygous null mutation of the β-catenin gene (KO) were isolated from 9.5 dpc embryos (Carmeliet et al., 1999; Balconi et al., 2000; Cattelino et al., 2003). Cells were routinely cultured in DME with 15% FCS (Hyclone), endothelial cell growth supplement (5 μg/ml; home-made from calf brain), and heparin (100 μg/ml; Sigma-Aldrich) maintenance medium (Balconi et al., 2000) on gelatin-coated tissue culture vessels.
For the restoration of β-catenin, KO cells were infected with murine full-length β-catenin in pINCO-GFP retroviral vector (KOfl-βcat), or with pINCO-GFP retroviral vector alone, as a control (KOvc). Infected cells were sorted by FACS® for GFP expression.
In vitro transdifferentiation and proliferation assays
To induce differentiation of endothelial cells into αSMA expressing mesenchymal cells, 1–2 × 103 endothelial cells were seeded in a 35-mm-diam Petri dish and cultured in DME medium with 2% FCS in the presence or absence of 1 ng/ml TFGβ2 for 5–7 d with a daily medium change. For BrdU incorporation into nuclear structures, 50 μM BrdU (Roche) was added to cells 5h before fixation. Cells were fixed and analyzed according to the procedure described in Embryological techniques and immunostaining.
Nuclear extraction and Western blot analysis
Cells (105/cm2) were cultured for 4 d in maintenance medium, serum deprived for 48 h and, if desired, stimulated with TGFβ2 for 2 h. Cells were scraped in lysis buffer (20 mM Tris and 150 mM NaCl, pH 7.4, containing 0.5% Triton X-100, 10% glycerol, 1 mM PMSF, 2 mM Ca2+, 15 μg/ml leupeptin, 71 μg/ml phenanthrolyne, and 20 U/μl aprotine [Sigma-Aldrich], 300 μM vanadate, and 600 mM hydrogen peroxide). Extraction of DNA-binding proteins was performed according to the method described by Andrews and Faller (1991). The protein content was measured using the BCA method (Pierce Chemical Co.). Total cell extracts were obtained by lysing the cells in boiling 2× Laemmli buffer. Total and nuclear extracts were separated by 7 or 10% SDS-PAGE under reducing conditions and analyzed in immunoblot with specific antibodies according to standard procedures.
Quantitative real time PCR
2 μg of total RNA from different samples were reverse transcribed in a final volume of 50 μl using Taqman reverse transcriptions reagents. To avoid amplification of contaminating genomic DNA, samples were previously treated with RQ1 RNase-free DNase (Promega). Primers and probe sequences for mouse snail1and for the ribosomal 18S rRNA, which served as a reference gene, were purchased from Applied Biosystems. The probes Taqman predeveloped assay reagent was labeled with the reporter dye VIC. All PCR reactions were performed using an ABI Prism 7700 sequence detection system. For any sample, the expression levels for snail1 and delta-like 4, normalized to the housekeeping gene 18S rRNA, were determined using the comparative threshold cycle method as described previously (Lu et al., 2001; Spagnuolo et al., 2004).
Online supplemental material
Antibody.
Anti–β-catenin mAb was purchased from BD Biosciences.
Apoptosis assay.
AV heart explants were prepared as described in Heart explant assays. After 48 h of incubation ex vivo, explants were fixed and apoptosis was quantified by measuring DNA fragmentation (TUNEL detection method; Roche).
Nuclear localization of β-catenin.
Cells were serum starved for 24 h and treated for 3 h with 6 mM LiCl and 5 ng/ml Leptomycin B (Sigma-Aldrich) in the presence of 15% FCS. Cells were fixed and stained for β-catenin as described in the Materials and methods.
Fig. S1 shows apoptosis in AV heart explants from WT and KO embryos. AV heart explants from WT (A) and KO (B) embryos were stained by TUNEL (red) and a nuclear counterstaining (DAPI, blue) and analyzed by confocal microscopy. To quantify TUNEL-positive cells, stacks of images (n = 4 for WT and KO, respectively) were flattened on one layer, converted into binary colors and automatically analyzed for stained particles using ImageJ 1.32. Columns represent means ± SD. Bar in B is valid for both micrographs.
Fig. S2 shows nuclear localization of β-catenin in KOvc and KOfl-βcat endothelial cell lines. KOvc (A) and KOfl-βcat (B) cell lines were treated with LiCl and Leptomycin B, fixed, and immunostained for β-catenin. Under these conditions, cells reinfected with full-length β-catenin show a pronounced staining at the membrane (arrowheads) as well as an accumulation of β-catenin in the nucleus (arrows), whereas KOvc cells show no specific staining. Bar in A is valid for both micrographs.
Fig. S3 shows TGFβ1-induced endothelial cells transformation into αSMA-positive cells in vitro is impaired in the absence of β-catenin. The experimental setup was as described for Fig. 4, with the exception that KOvc and KOfl-βcat cell lines were treated with TGFβ1 instead of TGFβ2. The number of αSMA-positive cells was expressed as a percentage of total cells (1–2 × 103 cells were counted for each cell line, and columns represents the mean out of three individual experiments ± SD. P < 0.0001 by t test.
Acknowledgments
This work was supported by Associazione Italiana per la Ricerca sul Cancro; the European Community (QLRT-2001-02059; Integrated Project Contract No LSHG-CT-2004-503573;NoE MAIN 502935; NoE EVGN 503254); Associazione Duchenne Parent Project; Italian Ministry of Health and Ministry of University and Scientific and Technological Research; Telethon Italy (grant E.1254), CNR/MIUR (CNR.02.731.DEJA), MIUR/FIRB (RBNE01MAWA_009, RBNE01F8LT_007) and Cofin 2003 (2003058397_04); AIDS Special Program of Istituto Superiore Sanità, Rome, contract 30D.83 and Special Project Stem Cells (CS36 and CS39), Human Frontier Science Foundation.
Abbreviations used in this paper: αSMA, α-smooth muscle actin; AV, atrio-ventricular; dpc, d post coitum; EMT, epithelial-mesenchymal transformation; KO, knockout; P-smad1/5, phospho-smad1/5; P-smad2, phospho-smad2; R-smad, receptor smad; WT, wild-type.
References
- Andrews, N.C., and D.V. Faller. 1991. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 19:2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balconi, G., R. Spagnuolo, and E. Dejana. 2000. Development of endothelial cell lines from embryonic stem cells: a tool for studying genetically manipulated endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 20:1443–1451. [DOI] [PubMed] [Google Scholar]
- Batlle, E., E. Sancho, C. Franci, D. Dominguez, M. Monfar, J. Baulida, and A. Garcia De Herreros. 2000. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2:84–89. [DOI] [PubMed] [Google Scholar]
- Behrens, J., J.P. von Kries, M. Kühl, L. Bruhn, D. Wedlich, R. Grosschedl, and W. Birchmeier. 1996. Functional interaction of β-catenin with the transcription factor LEF-1. Nature. 382:638–642. [DOI] [PubMed] [Google Scholar]
- Brannon, M., M. Gomperts, L. Sumoy, R.T. Moon, and D. Kimelman. 1997. A beta-catenin/XTcf-3 complex binds to the siamois promoter to regulate dorsal axis specification in Xenopus. Genes Dev. 11:2359–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault, V., R. Moore, S. Kutsch, M. Ishibashi, D.H. Rowitch, A.P. McMahon, L. Sommer, O. Boussadia, and R. Kemler. 2001. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 128:1253–1264. [DOI] [PubMed] [Google Scholar]
- Brown, C.B., A.S. Boyer, R.B. Runyan, and J.V. Barnett. 1999. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science. 283:2080–2082. [DOI] [PubMed] [Google Scholar]
- Camenisch, T.D., D.G. Molin, A. Person, R.B. Runyan, A.C. Gittenberger-de Groot, J.A. McDonald, and S.E. Klewer. 2002. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev. Biol. 248:170–181. [DOI] [PubMed] [Google Scholar]
- Cano, A., M.A. Perez-Moreno, I. Rodrigo, A. Locascio, M.J. Blanco, M.G. del Barrio, F. Portillo, and M.A. Nieto. 2000. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2:76–83. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., M.G. Lampugnani, L. Moons, F. Breviario, V. Compernolle, F. Bono, G. Balconi, R. Spagnuolo, B. Oostuyse, M. Dewerchin, et al. 1999. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 98:147–157. [DOI] [PubMed] [Google Scholar]
- Cattelino, A., S. Liebner, R. Gallini, A. Zanetti, G. Balconi, A. Corsi, P. Bianco, H. Wolburg, R. Moore, B. Oreda, et al. 2003. The conditional inactivation of the β-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J. Cell Biol. 162:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRuiter, M.C., R.E. Poelmann, J.C. VanMunsteren, V. Mironov, R.R. Markwald, and A.C. Gittenberger-de Groot. 1997. Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro. Circ. Res. 80:444–451. [DOI] [PubMed] [Google Scholar]
- Derynck, R., and X.H. Feng. 1997. TGF-beta receptor signaling. Biochim. Biophys. Acta. 1333:F105–F150. [DOI] [PubMed] [Google Scholar]
- Dor, Y., T.D. Camenisch, A. Itin, G.I. Fishman, J.A. McDonald, P. Carmeliet, and E. Keshet. 2001. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 128:1531–1538. [DOI] [PubMed] [Google Scholar]
- Frid, M.G., V.A. Kale, and K.R. Stenmark. 2002. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ. Res. 90:1189–1196. [DOI] [PubMed] [Google Scholar]
- Goumans, M.J., G. Valdimarsdottir, S. Itoh, A. Rosendahl, P. Sideras, and P. ten Dijke. 2002. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 21:1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner, B.M. 1995. Signal transduction by β-catenin. Curr. Opin. Cell Biol. 7:634–640. [DOI] [PubMed] [Google Scholar]
- Hurlstone, A., and H. Clevers. 2002. T-cell factors: turn-ons and turn-offs. EMBO J. 21:2303–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlstone, A.F., A.P. Haramis, E. Wienholds, H. Begthel, J. Korving, F. Van Eeden, E. Cuppen, D. Zivkovic, R.H. Plasterk, and H. Clevers. 2003. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 425:633–637. [DOI] [PubMed] [Google Scholar]
- Itoh, S., F. Itoh, M.J. Goumans, and P. Ten Dijke. 2000. Signaling of transforming growth factor-beta family members through Smad proteins. Eur. J. Biochem. 267:6954–6967. [DOI] [PubMed] [Google Scholar]
- Iurlaro, M., M. Scatena, W.H. Zhu, E. Fogel, S.L. Wieting, and R.F. Nicosia. 2003. Rat aorta-derived mural precursor cells express the Tie2 receptor and respond directly to stimulation by angiopoietins. J. Cell Sci. 116:3635–3643. [DOI] [PubMed] [Google Scholar]
- Iwamoto, R., S. Yamazaki, M. Asakura, S. Takashima, H. Hasuwa, K. Miyado, S. Adachi, M. Kitakaze, K. Hashimoto, G. Raab, et al. 2003. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc. Natl. Acad. Sci. USA. 100:3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, R.Y., E.J. Robertson, and M.J. Solloway. 2001. Bmp6 and Bmp7 are required for cushion formation and septation in the developing mouse heart. Dev. Biol. 235:449–466. [DOI] [PubMed] [Google Scholar]
- Kisanuki, Y.Y., R.E. Hammer, J. Miyazaki, S.C. Williams, J.A. Richardson, and M. Yanagisawa. 2001. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230:230–242. [DOI] [PubMed] [Google Scholar]
- Labbe, E., A. Letamendia, and L. Attisano. 2000. Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-beta and wnt pathways. Proc. Natl. Acad. Sci. USA. 97:8358–8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani, M.G., A. Zanetti, F. Breviario, G. Balconi, F. Orsenigo, M. Corada, R. Spagnuolo, M. Betson, V. Braga, and E. Dejana. 2002. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol. Biol. Cell. 13:1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner, S., A. Fischmann, G. Rascher, F. Duffner, E.H. Grote, H. Kalbacher, and H. Wolburg. 2000. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. (Berl.). 100:323–331. [DOI] [PubMed] [Google Scholar]
- Lu, C., G. Schwartzbauer, M.A. Sperling, S.U. Devaskar, S. Thamotharan, P.D. Robbins, C.F. McTiernan, J.L. Liu, J. Jiang, S.J. Frank, and R.K. Menon. 2001. Demonstration of direct effects of growth hormone on neonatal cardiomyocytes. J. Biol. Chem. 276:22892–22900. [DOI] [PubMed] [Google Scholar]
- Maretto, S., M. Cordenonsi, S. Dupont, P. Braghetta, V. Broccoli, A.B. Hassan, D. Volpin, G.M. Bressan, and S. Piccolo. 2003. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA. 100:3299–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwald, R.R., T.P. Fitzharris, and F.J. Manasek. 1977. Structural development of endocardial cushions. Am. J. Anat. 148:85–119. [DOI] [PubMed] [Google Scholar]
- Massague, J. 2000. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 1:169–178. [DOI] [PubMed] [Google Scholar]
- Miller, J.R., and R.T. Moon. 1996. Signal transduction through β-catenin and specification of cell fate during embryogenesis. Genes Dev. 10:2527–2539. [DOI] [PubMed] [Google Scholar]
- Moustakas, A., S. Souchelnytskyi, and C.H. Heldin. 2001. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 114:4359–4369. [DOI] [PubMed] [Google Scholar]
- Nakajima, Y., V. Mironov, T. Yamagishi, H. Nakamura, and R.R. Markwald. 1997. Expression of smooth muscle alpha-actin in mesenchymal cells during formation of avian endocardial cushion tissue: a role for transforming growth factor beta3. Dev. Dyn. 209:296–309. [DOI] [PubMed] [Google Scholar]
- Nawshad, A., and E.D. Hay. 2003. TGFβ3 signaling activates transcription of the LEF1 gene to induce epithelial mesenchymal transformation during mouse palate development. J. Cell Biol. 163:1291–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, W.J., and R. Nusse. 2004. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 303:1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishita, M., M.K. Hashimoto, S. Ogata, M.N. Laurent, N. Ueno, H. Shibuya, and K.W. Cho. 2000. Interaction between Wnt and TGF-beta signalling pathways during formation of Spemann's organizer. Nature. 403:781–785. [DOI] [PubMed] [Google Scholar]
- Noseda, M., G. McLean, K. Niessen, L. Chang, I. Pollet, R. Montpetit, R. Shahidi, K. Dorovini-Zis, L. Li, B. Beckstead, et al. 2004. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 94:910–917. [DOI] [PubMed] [Google Scholar]
- Paranya, G., S. Vineberg, E. Dvorin, S. Kaushal, S.J. Roth, E. Rabkin, F.J. Schoen, and J. Bischoff. 2001. Aortic valve endothelial cells undergo transforming growth factor-beta-mediated and non-transforming growth factor-beta-mediated transdifferentiation in vitro. Am. J. Pathol. 159:1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts, J.D., J.M. Dagle, J.A. Walder, D.L. Weeks, and R.B. Runyan. 1991. Epithelial-mesenchymal transformation of embryonic cardiac endothelial cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth factor beta 3. Proc. Natl. Acad. Sci. USA. 88:1516–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan, R.B., and R.R. Markwald. 1983. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev. Biol. 95:108–114. [DOI] [PubMed] [Google Scholar]
- Savagner, P. 2001. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 23:912–923. [DOI] [PubMed] [Google Scholar]
- Schlaeger, T.M., S. Bartunkova, J.A. Lawitts, G. Teichmann, W. Risau, U. Deutsch, and T.N. Sato. 1997. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc. Natl. Acad. Sci. USA. 94:3058–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano, P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21:70–71. [DOI] [PubMed] [Google Scholar]
- Spagnuolo, R., M. Corada, F. Orsenigo, L. Zanetta, U. Deuschle, P. Sandy, C. Schneider, C.J. Drake, F. Breviario, and E. Dejana. 2004. Gas1 is induced by VE-cadherin and vascular endothelial growth factor and inhibits endothelial cell apoptosis. Blood. 103:3005–3012. [DOI] [PubMed] [Google Scholar]
- Thiery, J.P. 2002. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2:442–454. [DOI] [PubMed] [Google Scholar]
- Tian, Y.C., and A.O. Phillips. 2002. Interaction between the transforming growth factor-beta type II receptor/Smad pathway and beta-catenin during transforming growth factor-beta1-mediated adherens junction disassembly. Am. J. Pathol. 160:1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman, L.A., J. Grego-Bessa, A. Raya, E. Bertran, J.M. Perez-Pomares, J. Diez, S. Aranda, S. Palomo, F. McCormick, J.C. Izpisua-Belmonte, and J.L. De La Pompa. 2004. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 18:99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes, F., A.M. Alvarez, A. Locascio, S. Vega, B. Herrera, M. Fernandez, M. Benito, M.A. Nieto, and I. Fabregat. 2002. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor Beta in fetal rat hepatocytes. Mol. Cancer Res. 1:68–78. [PubMed] [Google Scholar]
- Vecchi, A., C. Garlanda, M.G. Lampugnani, M. Resnati, C. Matteucci, A. Stoppacciaro, H. Schnurch, W. Risau, L. Ruco, A. Mantovani, et al. 1994. Monoclonal antibodies specific for endothelial cells of mouse blood vessels. Their application in the identification of adult and embryonic endothelium. Eur. J. Cell Biol. 63:247–254. [PubMed] [Google Scholar]
- Yurugi-Kobayashi, T., H. Itoh, J. Yamashita, K. Yamahara, H. Hirai, T. Kobayashi, M. Ogawa, S. Nishikawa, and K. Nakao. 2003. Effective contribution of transplanted vascular progenitor cells derived from embryonic stem cells to adult neovascularization in proper differentiation stage. Blood. 101:2675–2678. [DOI] [PubMed] [Google Scholar]
- Zhurinsky, J., M. Shtutman, and A. Ben-Ze'ev. 2000. Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J. Cell Sci. 113:3127–3139. [DOI] [PubMed] [Google Scholar]