

Abstract
Calcium activates the ATPase activity of tissue-purified myosin V, but not that of shorter expressed constructs. Here, we resolve this discrepancy by comparing an expressed full-length myosin V (dFull) to three shorter constructs. Only dFull has low ATPase activity in EGTA, and significantly higher activity in calcium. Based on hydrodynamic data and electron microscopic images, the inhibited state is due to a compact conformation that is possible only with the whole molecule. The paradoxical finding that dFull moved actin in EGTA suggests that binding of the molecule to the substratum turns it on, perhaps mimicking cargo activation. Calcium slows, but does not stop the rate of actin movement if excess calmodulin (CaM) is present. Without excess CaM, calcium binding to the high affinity sites dissociates CaM and stops motility. We propose that a folded-to-extended conformational change that is controlled by calcium and CaM, and probably by cargo binding itself, regulates myosin V's ability to transport cargo in the cell.
Keywords: myosin V; calmodulin; calcium regulation; molecular motors; motility assay
Introduction
Unconventional myosin V is a member of the well-known actin-based superfamily of motor proteins. Its cellular roles range from mRNA transport and membrane trafficking to the establishment of cell polarity (for review see Reck-Peterson et al., 2000). Each head of dimeric myosin V consists of a motor domain that binds actin and hydrolyzes ATP, followed by a long neck region composed of six IQ motifs to which CaM is bound. A unique feature is that all the IQ motifs bind CaM in the absence of calcium, unlike most targets, which bind CaM only in the presence of calcium. The coiled-coil rod formed by both heavy chains (HCs) ends in a globular cargo-binding domain (Reck-Peterson et al., 2000). Single-molecule analyses have shown that myosin V is processive (Mehta et al., 1999; Rief et al., 2000), a feature that allows a single motor to transport its cargo for long distances without dissociating from its actin track. The two heads and extended neck region of myosin V enable the molecule to stride along the 36-nm pseudo-helical repeat of actin (Purcell et al., 2002; Sakamoto et al., 2003). Because CaM is bound to the neck, there is also the potential for calcium to regulate myosin V's function. Nonetheless, the mechanism by which calcium might regulate myosin V's function in the cell remains elusive.
An unresolved issue is why EGTA decreases the actin-activated ATPase activity of tissue-purified myosin V as well as its affinity for actin (Cheney et al., 1993; Nascimento et al., 1996; Tauhata et al., 2001), as these features were not observed with any of the shorter baculovirus-expressed constructs (Trybus et al., 1999; Wang et al., 2000). Such observations raise the question of whether the expressed constructs lack a feature that is retained by the tissue-purified myosin, e.g., a critical post-translational modification, or if the longer tail length allows the full-length molecule to exhibit unique calcium-dependent properties. Common features are that calcium causes dissociation of some CaM, and that calcium inhibits actin movement in the in vitro motility assay regardless of whether the construct is an expressed monomer (Trybus et al., 1999), an expressed short-tailed dimer (Homma et al., 2000), or the tissue-purified full-length molecule (Cheney et al., 1993). Two of these papers (Cheney et al., 1993; Trybus et al., 1999) showed restoration of motility in the presence of excess Ca2+-CaM, albeit at a slower rate. This observation suggests that calcium-dependent CaM dissociation causes the molecule to be ineffective as a motor, a finding that is confirmed here. An opposing view is that inhibition of motility occurs because of a conformational change in the bound Ca2+-CaM (Homma et al., 2000).
Here, we resolve these disparate results by expressing and characterizing four recombinant murine myosin V constructs, including for the first time an expressed full-length myosin V (dFull). Importantly, the actin-activated ATPase data show that only full-length myosin V has low actin-activated ATPase activity in the absence of calcium, whereas any shorter construct has a high and relatively calcium-insensitive actin-activated ATPase activity. These results suggest that in solution, full-length myosin V can adopt an inhibited structural state that is not possible with any shorter construct. Hydrodynamic data and EM suggest that the inhibited state is a compact conformation of the molecule that can be unfolded to an active state by calcium near physiological ionic strength, as well as by increasing the ionic strength. We propose that a folded-to-extended conformational change, which is regulated by calcium and potentially by cargo binding, is responsible for regulating myosin V's motor activity in the cell.
Results
Myosin V constructs
Murine myosin V (“dilute”) constructs of different lengths were expressed and purified to assess the effect of calcium on function. We focused on four constructs: a monomer with the motor domain and the first two IQ motifs (MD2IQ), a two-headed molecule that contains six IQ motifs followed by a coiled-coil tail (dilute heavy meromyosin, dHMM), a construct lacking only the globular cargo-binding domain (long-dHMM), and the full-length molecule V (dFull) (Fig. 1). All constructs were coexpressed with CaM to ensure full occupancy of the IQ motifs. This is the first report of successful expression of functional full-length myosin V in the baculovirus/insect cell system. An SDS gel of the purified proteins is shown (Fig. 1).
Figure 1.

Myosin V constructs. Schematic representations of MD2IQ, dHMM, long-dHMM, and the full-length construct, dFull. (Bottom right) SDS-PAGE of mol wt standards (kD, lane 1) and the purified expressed proteins: dFull (lane 2), dHMM (lane 3), and MD2IQ (lane 4).
CaM dissociation by calcium inhibits MD2IQ motility
MD2IQ, the smallest construct whose motility is inhibited by calcium (Trybus et al., 1999), was used to determine if inhibition of motility is caused by CaM dissociation or by a calcium-dependent change in bound CaM. The first strategy to distinguish between these possibilities was to generate three mutant CaMs that were deficient in calcium binding. The mutant “CaM-high” retained only the high affinity COOH-terminal pair of sites, “CaM-low” only the low affinity NH2-terminal pair of sites, and “CaMΔall” had no functional calcium-binding sites (Fig. 2, top; Beckingham, 1991). These CaM mutants were coexpressed with MD2IQ in Sf9 cells, resulting in the purified proteins shown in Fig. 2 (bottom). The goal was to establish if any of the mutant CaMs were capable of inhibiting motility without dissociation from the HC, or if dissociation and inhibition are always coupled. Approximately 0.5 mol of wild-type CaM (WT-CaM) per mol of HC dissociated from MD2IQ in the presence of calcium (Fig. 3 A). CaM does not dissociate from a construct containing only the first IQ motif (MD1IQ), thus CaM dissociation is presumably occurring from the second IQ motif of MD2IQ (Trybus et al., 1999). MD2IQ expressed with the CaM-high mutant behaved like WT-CaM, showing that calcium binding to the high affinity sites alone is sufficient to cause CaM dissociation. In contrast, calcium binding to the low affinity sites of CaM-low does not release CaM from the HC, nor does the CaMΔall mutant dissociate from the HC. These same preparations were tested for their ability to move actin in an in vitro motility assay in the presence of calcium (Fig. 3 B). No movement was seen in the constructs from which CaM dissociated from the HC. In contrast, MD2IQ containing the CaM mutants that remained bound to the HC in the presence of calcium (CaM-low and CaMΔall) retained the ability to move actin, in support of the hypothesis that CaM dissociation is the mechanism for inhibition of motility.
Figure 2.
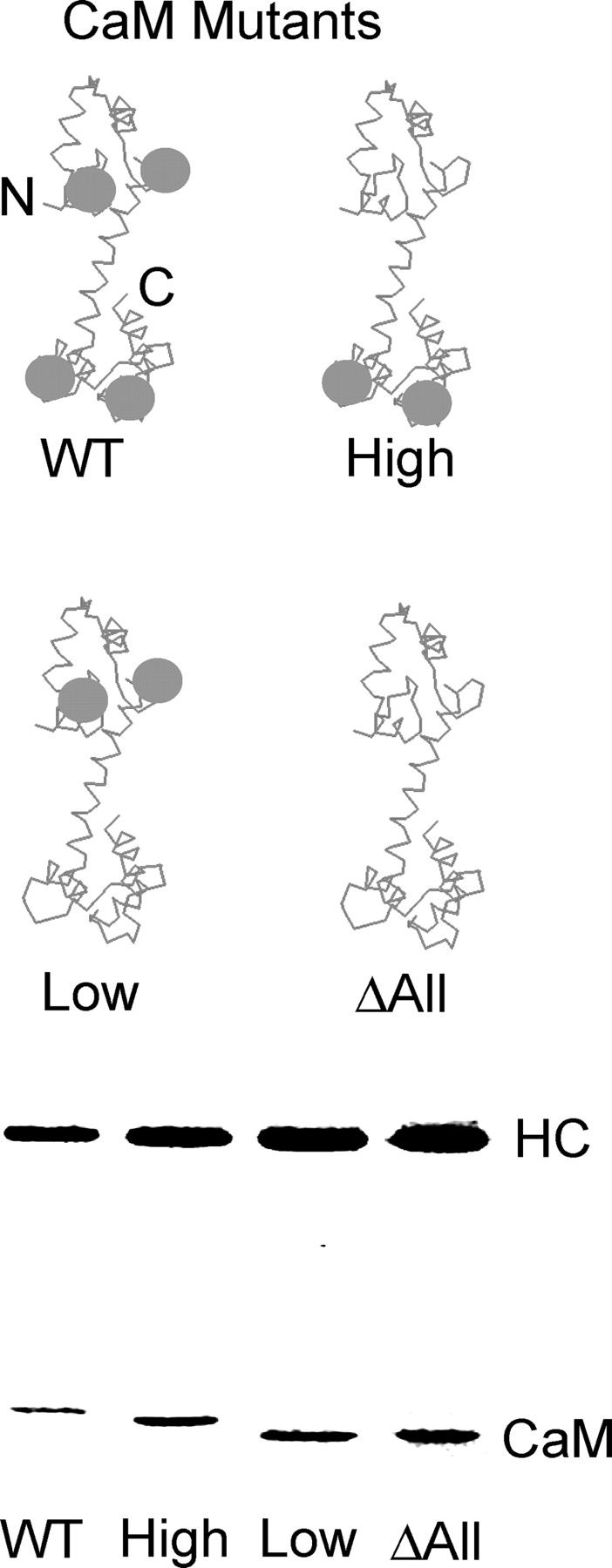
Schematic diagram of CaM mutants and coexpression of CaM mutants with the MD2IQ HC. (Top) WT-CaM has four calcium-binding sites (gray spheres). Mutagenesis was used to render either the NH2-terminal low affinity, the COOH-terminal high affinity, or all the calcium-binding sites nonfunctional. The resulting mutants are called CaM-high (containing only the high affinity sites), CaM-low (containing only the low affinity sites), or CaM Δall, which has no functional calcium-binding sites. (Bottom) SDS gel of MD2IQ coexpressed with WT or mutant CaMs. The protein samples contained EGTA. In the presence of calcium, the mobility of WT-CaM and CaM-high increases, whereas that of CaM-low or CaM Δall does not (not depicted).
Figure 3.

High affinity calcium-binding sites are responsible for calcium-dependent CaM dissociation and motility inhibition of MD2IQ. (A) CaM dissociation from MD2IQ (0.4 mg/ml) coexpressed with various CaM mutants. CaM dissociation was determined using the actin-pelleting assay (see Materials and methods). (B) Actin filament velocity supported by MD2IQ coexpressed with the same CaM mutants. (White bars) 1 mM EGTA; (black bars) 100 μM free Ca2+.
A second approach was to compare the calcium dependence of CaM dissociation and inhibition of motility with MD2IQ containing WT-CaM (Fig. 4 A). No inhibition of motility was observed before CaM dissociation. The fraction of filaments moving dropped sharply as CaM began to dissociate. Motility was completely inhibited as soon as dissociation of CaM began, suggesting that the few CaM-deficient heads act as a substantial load to movement. The CaM-high mutant showed the same calcium dependence of CaM dissociation as WT-CaM (unpublished data). These data confirm the strong correlation between CaM dissociation and inhibition of motility.
Figure 4.

Calcium dependence of CaM dissociation and motility inhibition. (A) MD2IQ. (B) dHMM. Fraction of actin filaments moving in a motility assay (open triangles, normalized to the value in EGTA) and amount of CaM dissociation (solid circles) as a function of calcium concentration. To determine the amount of CaM dissociation, samples were dialyzed into buffers containing the desired amount of free Ca2+ before pelleting with actin (see Materials and methods). The same buffers were used for in vitro motility at that free Ca2+ concentration.
The most compelling experiment to distinguish between the two possible mechanisms is to show if motility can be sustained in calcium in the presence of exogenous CaM. The presence of exogenous CaM along with calcium restored motility to approximately two-thirds the value in EGTA (Fig. 5), whereas addition of the calcium-insensitive CaMΔall mutant fully restored motility. These results suggest that calcium weakens the affinity of some CaMs for the HC, but that as long as CaM is bound to the neck, both the Ca2+- and apo-forms of CaM support movement of actin filaments.
Figure 5.

Rescue of motility in calcium by exogenous CaM for MD2IQ, dHMM, and dFull. Actin filament velocity by three different constructs in EGTA (dark gray bars), in EGTA with 12 μM free WT-CaM (light gray bars). 100 μM free Ca2+ completely inhibited motility (red bars), which was partially restored in 100 μM free Ca2+ and 12 μM free WT-CaM (blue bars), or more fully restored in 100 μM free Ca2+ and 12 μM CaMΔall (pink bars).
Calcium-dependent motility of longer constructs
The motility properties of longer constructs were similar to those of MD2IQ. Dissociation of WT-CaM from dHMM showed the same calcium dependency as inhibition of motility (Fig. 4 B). Three times as much CaM per HC dissociated from the dimeric constructs as from MD2IQ (∼1.5 and ∼1.7 mol CaM per mol HC for dHMM and dFull, respectively), consistent with the release of CaM from two more IQ motifs in addition to the second IQ. Motility of dHMM could be partially rescued by WT-CaM in the presence of calcium, and more completely with the CaMΔall mutant (Fig. 5). Expressed full-length myosin V showed the same pattern of inhibition of motility in calcium, and restoration by exogenous CaM (Fig. 5).
Calcium regulation of actin-activated ATPase activity
The actin-activated ATPase activity was determined in the presence and absence of calcium. A linked assay with a regenerating system was used to prevent buildup of ADP, which inhibits the ATPase rate (De La Cruz et al., 2000; unpublished data). The actin-activated ATPase of MD2IQ was high both in the presence or absence of calcium (∼23 s−1; Fig. 6 A and Table I). The ATPase of both dHMM and long-dHMM was high in calcium, and reduced to 73 and 64% of their respective values in the absence of calcium (Fig. 6, B and C; Table I). Strikingly, the actin-activated ATPase activity of the expressed full-length myosin V was similar to dHMM in the presence of calcium, but Vmax was inhibited over sevenfold in the absence of calcium (Fig. 6 D; Table I). These results show that the full-length molecule, long-dHMM, and dHMM can all achieve the same activated state in the presence of calcium. Although EGTA tends to decrease the activity of both double-headed constructs, only the full-length molecule has a significantly inhibited actin-activated ATPase activity in the absence of calcium.
Figure 6.

Calcium dependence of the actin-activated ATPase activity in the presence of exogenous CaM. (A) MD2IQ, (B) dHMM, (C) long-dHMM, (D) dFull. 1 mM EGTA + 6 μM CaM (open circles), 100 μM free Ca2+ and 6 μM CaM (solid circles). Representative experiments are shown, which gave the following Vmax and Km values, respectively: MD2IQ (22.3 s−1 and 8.1 μM in EGTA, 23.0 s−1 and 11 μM in Ca2+); dHMM (12.8 s−1 and 1.6 μM in EGTA, 18 s−1 and 1.9 μM in Ca2+); long-dHMM (10.3 s−1 and 1.8 μM in EGTA, 16.0 s−1 and 1.0 μM in Ca2+); dFull (2.2 s−1 and 5.1 μM in EGTA, 14.8 s−1 and 1.3 μM in Ca2+). Table I summarizes the Vmax and Km values for multiple preparations.
Table I. Summary of actin-activated ATPase data.
| 1 mM EGTA
|
100 mM Ca2+
|
EGTA + 6 μM CaM
|
Ca2+ + 6 μM CaM
|
|||||
|---|---|---|---|---|---|---|---|---|
| Construct
|
Vmax
|
Km
|
Vmax
|
Km
|
Vmax
|
Km
|
Vmax
|
Km
|
| sec−1 | μM | sec−1 | μM | sec−1 | μM | sec−1 | μM | |
| MD2IQ | 20.8 ± 5.1 (3) | 6.4 ± 2.7 (3) | 19.5 ± 11 (3) | 6.3 ± 1.2 (3) | 23.3 ± 5.7 (3) | 7.6 ± 2.3 (3) | 23.7 ± 6.4 (3) | 9.8 ± 3.3 (3) |
| dHMM | 9.7 ± 0.8 (3) | 2.7 ± 1.4 (3) | 13.6 ± 1.0 (3) | 3.0 ± 2.1 (3) | 12.7 (2) | 1.5 (2) | 17.3 (2) | 1.7 (2) |
| Long-dHMM | 6.5 (2) | 5.8 (2) | 6.5 (2) | 4.7 (2) | 10.6 ± 0.6 (3) | 3.0 ± 1.8 (3) | 16.5 ± 1.2 (3) | 1.5 ± 0.6 (3) |
| dFull | 2.3 ± 0.6 (4) | 7.7 ± 1.0 (4) | 8.7 ± 1.2 (4) | 4.2 ± 1.2 (4) | 2.18 ± 0.1 (3) | 5.1 ± 0.6 (3) | 16.0 ± 1.1 (3) | 1.4 ± 0.1 (3) |
The number in parentheses indicates the number of ATPase assays performed, each done over a range of actin concentrations. Values are means ± SD, or an average when n = 2. Each value was measured on at least two independent preparations.
Actin-activated ATPase rates were in general somewhat lower if additional CaM was not added to the assay, particularly in the presence of calcium, which favors CaM dissociation (Table I). Therefore, the complete inhibition of motility seen in the presence of calcium without added CaM is not due to a complete lack of actin-activated ATPase activity. For full-length myosin V, the presence of extra CaM actually enhances the degree of calcium-dependent regulation of ATPase activity.
Analytical ultracentrifugation shows a calcium- or salt-dependent increase in sedimentation coefficient
Global conformational changes that result in altered activity have been previously observed with other motor proteins, most notably smooth muscle myosin II (Trybus et al., 1982) and kinesin (Stock et al., 1999; Hackney and Stock, 2000). Sedimentation velocity was used to determine if myosin V has the potential to undergo a conformational change that could account for the change in enzymatic activity. Initially, the mutant CaMΔall was expressed with the HC to ensure a more homogeneous preparation with higher yield, which simplifies analysis of the hydrodynamic data. Three independent preparations of the full-length myosin V gave an ∼30% decrease in sedimentation coefficient, as the salt shifted from 0.1 to 0.3 M KCl (Fig. 7 and Table II), consistent with a more compact conformation of the molecule being formed near physiological ionic strength. The mol wt of one of these preparations (Table II, footnote c) was also determined by sedimentation equilibrium, yielding a value of 588 kD (0.1 M KCl) and 643 kD (0.3 M KCl). Thus, the increase in sedimentation coefficient at 0.1 M KCl is consistent with a change in conformation, and not with a change in the state of aggregation of the protein. As controls, the sedimentation velocity of three independent preparations of dHMM changed <8% as the salt shifted from 0.1 to 0.3 M KCl, whereas the values obtained with MD2IQ showed no consistent change with salt.
Figure 7.

A salt- or calcium-induced conformational change in full-length myosin V as shown by sedimentation velocity. (A–C) Absorbance as a function of distance and time; (C–E) the sedimentation coefficients determined from these data by curve fitting to one species, using the dc/dt program (Philo, 2000). In 0.1 M NaCl and 1 mM EGTA (A and D) a folded, inactive myosin V sediments at 13.9 S. With increased salt concentration (B and E), the value decreases to 9.7 S indicating a more extended conformation (0.3 M NaCl). Calcium induces a similar active and extended conformation (10.8 S) near physiological ionic strength (0.1 M NaCl, with added CaM to prevent dissociation). Table II summarizes data from multiple experiments.
Table II. Salt- and calcium-induced conformational changes in myosin V.
| Construct
|
EGTA, 0.1 M NaCla
|
EGTA, 0.3 M NaCla
|
EGTA, 0.1 M NaClb
|
Ca2+, 0.1 M NaClb
|
|---|---|---|---|---|
| S | S | S | S | |
| MD2IQ | 5.8, 5.4 | 5.5, 5.7 | 6.3, 5.8 | 6.2, 5.8 |
| dHMM | 9.6, 8.8, 8.7 | 8.9, 8.0, 9.0 | 9.9, 8.5 | 10.6, 8.7 |
| Long-dHMM | 9.4 | 8.9 | 9.3, 10.3 | 9.7, 10.4 |
| dFull | 14.9, 13.9c, 15.3d | 10.2, 9.7c, 11.4d | 15.3, 13.5, 13.3d | 11.5, 10.9, 10.8d |
Independent protein preparations were used for each pair of comparisons (0.1 M NaCl vs. 0.3 M NaCl, or EGTA vs. calcium), except as noted (footnote d).
These values were obtained with constructs that contained CaMΔall, except where noted (footnote d).
These values were obtained with constructs that contained WT-CaM. 6 μM exogenous CaM was added to prevent CaM dissociation.
The mol wt of this preparation was also determined (see text above).
This preparation, which contained WT-CaM, was analyzed both as a function of salt and calcium.
The more physiologically relevant question is whether calcium induces the same decrease in sedimentation coefficient (increase in asymmetry) at 0.1 M NaCl as was seen by addition of salt to 0.3 M NaCl. Multiple preparations of all four constructs (MD2IQ, dHMM, long-dHMM, and dFull) were expressed with WT-CaM. Sedimentation velocity experiments were performed in the presence of excess CaM to prevent dissociation. Importantly, Fig. 7 and Table II show that calcium induces a decrease in sedimentation coefficient at 0.1 M NaCl similar to that seen when salt was added to 0.3 M. This result is consistent with calcium being the physiological activator of the folded (EGTA, inactive) to extended (calcium, active) conformational transition.
EM of the two conformations of myosin V
Rotary metal-shadowed images of dFull reveal two conformations that are consistent with the hydrodynamic data described above (Fig. 8). The images of the extended form are similar to those previously published (Cheney et al., 1993), which show a tripartite structure with two head and neck domains, connected to the globular cargo-binding domain by a rodlike tail. In this conformation, the tail segment of the “Y” has the longest dimension (Fig. 8 A). At lower ionic strength, the most notable change is that the distance between the head–rod junction and the globular tail is shorter, consistent with a bending of the tail region that would account for the faster sedimentation rate (Fig. 8 B). The molecule looks like a “V” in some cases, with the vertices formed by the two motor domains and the globular tail.
Figure 8.

Metal-shadowed images of full-length myosin V. Myosin V in (A) an extended or (B) more compact conformation. Yellow arrowheads point to the globular tail, and blue arrowheads to the head–rod junction. Note that the length of the tail is shorter in the compact conformation. The molecules shown in A were diluted into high ionic strength for rotary shadowing, whereas those in B were obtained in a lower ionic strength buffer. In rows 1 and 3, the molecules are oriented with the heads up and the tail down. Bar, 30 nm.
Discussion
Our ability to express myosin V constructs of any size, including the full-length molecule, allowed us to show that whole myosin V has an additional degree of regulation not observed with any of the shorter constructs, even one lacking only the globular tail domain. Actin-activated ATPase measurements showed a clear effect of tail length on the degree of calcium-dependent regulation. The activity of monomeric MD2IQ was independent of calcium, whereas dHMM and long-dHMM showed progressively lower activity in the absence of calcium. The expressed full-length molecule was unique in showing a significantly lower actin-activated ATPase in EGTA than any other construct, whereas in calcium, the activity was high and similar to that of dHMM. Thus, the expressed full-length construct mimics the pattern of activity seen with tissue-purified myosin V (Cheney et al., 1993; Nascimento et al., 1996). Our experiments also explain why calpain cleavage of tissue-purified myosin V at the PEST site region, which yields a dHMM-like fragment, results in a calcium-insensitive ATPase activity (Nascimento et al., 1996). As seen with our expressed dHMM, shorter constructs lose calcium sensitivity because they lack a structural element required to obtain inhibited activity in EGTA. Thus, a full-length molecule is required to obtain calcium-dependent regulation of actin-activated ATPase activity.
What accounts for the decrease in activity of the full-length molecule in the absence of calcium? Clearly not CaM dissociation because in the absence of calcium, CaM is more tightly bound to the neck than in its presence. Based on other motor proteins, a theme for regulation has emerged that the full-length molecule can undergo a global conformational change that causes a change in activity. Smooth muscle myosin II with an unphosphorylated regulatory light chain adopts a folded monomeric conformation that has virtually no activity (for review see Trybus, 1991). Full-length kinesin forms a compact conformation with low activity at physiological salt, which is believed to be activated to an extended form upon cargo binding (Coy et al., 1999; Stock et al., 1999; Hackney and Stock, 2000). Based on our data, a similar scenario applies to full-length myosin V. We showed by sedimentation velocity that near physiological ionic strength, full-length myosin V forms a faster-sedimenting species in the absence of calcium (∼14 S compact conformation) than in the presence of calcium (∼11 S extended conformation). Addition of salt to 0.3 M induces the same conformational change as calcium.
Requirements to obtain the inhibited state
The calcium dependence of the conformational change suggests that the CaM-containing neck region is either directly involved in an interaction site that stabilizes the inhibited conformation, or a calcium-dependent change in the neck indirectly places the motor domain in a position that precludes its interaction with actin. Calcium increases the affinity of tissue-purified myosin for actin, suggesting that in EGTA where the compact conformation is formed, the heads are less able to bind to actin (Tauhata et al., 2001). Whether other steps in the ATPase cycle (e.g., ADP release) are also affected by being constrained in the folded conformation remains to be determined.
Likewise, the globular tail domain must be involved in stabilizing the inhibited conformation because its absence precludes full inhibition of activity. Much recent work has focused on this cargo-binding tail domain of myosin V. The picture that emerges is that myosin Va is linked to melanosomes via two adaptor proteins (Nagashima et al., 2002; Strom et al., 2002; Wu et al., 2002). Rab27a binds to the melanosome, and then recruits melanophilin to bind to it. Melanophilin in turn binds directly to myosin V. The binding sites on myosin V are its globular tail and exon F, an alternatively spliced exon present in melanocyte myosin Va but not in brain myosin Va. This observation could explain why different myosin isoforms exhibit different cargo specificities. Another level of regulation of cargo binding is by phosphorylation of a Ser residue in the myosin tail by calcium/CaM-dependent protein kinase II, which results in dissociation of the motor and cargo (Karcher et al., 2001). The fact that the globular tail is required both for stabilizing the compact form and for cargo binding suggests a competition between these two processes whereby cargo binding itself could cause unfolding and activation.
What is apparently not required for adopting the inhibited conformation is the 10-kD dynein light chain that binds to the globular tail (Espindola et al., 2000) because we did not coexpress this subunit with the HC. We do know that our expressed full-length molecule will bind the 10-kD dynein light chain when it is coexpressed with the HC in Sf9 cells (unpublished data). Nucleotide is also not required to adopt the compact state in 0.1 M NaCl and EGTA.
Structure of the inhibited conformation
There are three regions of predicted coiled-coil in the myosin V rod that are separated by two nonhelical regions (Espreafico et al., 1992). After the head–rod junction is ∼30 nm of predicted coiled-coil, a PEST region, ∼13 nm of coiled-coil, a nonhelical segment, and then ∼15 nm more of coiled-coil. This rod region is followed by an ∼45-kD globular cargo-binding tail. Either of the nonhelical regions could be potential hinges for folding of the molecule. Our metal-shadowed images of myosin V are consistent with the idea that the tail domain adopts a more compact conformation in the absence of calcium at physiological ionic strength, suggesting that bending occurs at at least one of these sites. The heads are also likely to adopt a different and constrained conformation relative to the rod by analogy with regulated conformations of smooth muscle myosin II (Wendt et al., 2001). An independent analysis concurrent with ours showed that tissue-purified myosin V also adopts a folded conformation, based on hydrodynamic data that are remarkably similar to our data with the expressed full-length construct, as well as on negatively stained images of myosin V (Wang et al., 2004). Averaging of a small number of these negatively stained images revealed a triangular structure consistent with a shorter tail.
Earlier images of myosin V visualized by quick-freeze, deep-etch EM (Cheney et al., 1993) are described as having a flexible tail domain, with a maximum length of 75 nm. Notably, the authors comment that in some images, the COOH-terminal globular tail superimposed with a smaller globule located ∼30 nm from the head–rod junction, consistent with bending at the second nonhelical hinge region in the rod. These images were the first to show that myosin V has the potential to adopt a folded conformation.
Why does dFull show motility in EGTA?
Given the inhibition of full-length myosin V's actin-activated ATPase activity in the absence of calcium, why does its rate of in vitro motility remain high? There are several possible explanations of this apparent paradox. One possibility is that attachment of myosin V to the nitrocellulose surface activates the motor by preventing formation of the inhibited conformation, as if it were potentially binding “cargo.” Similar activation of full-length kinesin's motility has been observed (Coy et al., 1999). Another possibility is that a small pool of active molecules remains in equilibrium with the inhibited form at low calcium, consistent with the actin-activated ATPase activity not being completely turned off. The high duty cycle of this motor could allow the maximal rate of movement to be maintained in the in vitro motility assay even with a small amount of active protein (Moore et al., 2001). If this scenario holds under cellular conditions, it would still generate a large pool of inactive motors at low calcium that are waiting to be activated by cargo or calcium. Alternatively, it is possible that a small fraction of the molecules in the preparation have been cleaved to generate unregulated molecules that support movement in EGTA. Minor bands can be seen on an SDS gel of the dFull preparation (Fig. 1), and cleavage in the rod by calpain proteases has been shown to occur in vivo (Casaletti et al., 2003).
The effect of calcium on motility depends on CaM concentration
In the presence of calcium without exogenous CaM, motility is inhibited by dissociation of CaM from the IQ motifs. Mutant CaMs were used to show that calcium binding to the high affinity sites in the COOH-terminal lobe of CaM is sufficient to cause dissociation. This result is consistent with crystallographic data of the structure of apo-CaM bound to the first two IQ motifs (unpublished data). In this structure, the N-lobe is in the closed state with few interactions with the HC. In contrast, the C-lobe that contains the high affinity calcium-binding sites is in a gripping semi-open conformation, which provides the major interactions with the HC. Binding of calcium to the C-lobe would thus be expected to cause dissociation provided that the Ca2+-CaM complex has a low affinity for the sequence in that particular IQ motif (e.g., the second IQ motif). It should be noted that each IQ motif has a unique sequence, and only some of the motifs have a lower affinity for CaM in the presence of calcium than in its absence (unpublished data).
A likely mechanism for inhibition of motility in calcium is that dissociation of CaM causes the neck region to become compliant, rendering it unable to act as a lever arm. This mechanism would not require a corresponding change in the actin-activated ATPase activity. In support of this idea, introduction of only two Ala residues between IQ motifs 3 and 4 of myosin V HMM caused greater than a twofold reduction in step size, with no effect on enzymatic activity in solution (Sakamoto et al., 2003).
The effect of calcium on motility is very different in the presence of excess CaM, which prevents CaM dissociation and causes Ca2+-CaM to rebind to the neck by mass action. Ca2+-bound CaM restores motility to approximately two-thirds of the rate seen with apo-CaM. Thus, the neck can act as a lever with either apo-Cam or Ca2+-CaM bound, but the conformation of the bound CaM modifies the observed velocity. CaMΔall restores motility more completely than WT-CaM in calcium because it cannot undergo a conformational change. The observation that calcium completely inhibits motility in the absence of excess CaM, but only partially slows it in the presence of excess CaM, suggests that the degree to which calcium affects transport in vivo will greatly depend on the pool of free CaM in the cell.
Regulation of other CaM-binding myosins
The regulation of myosin 1β by calcium is similar to the regulation of our single-headed myosin V MD2IQ construct. High calcium concentration dissociates some but not all of the bound CaMs (1 of 3 in the case of myosin Iβ) and in this state the molecule shows no motility (Zhu et al., 1998). The high affinity calcium binding sites on CaM were deduced to be the sites responsible for regulation, similar to the results shown here for myosin V.
Myosin VI is a double-headed processive motor that moves toward the pointed end of an actin filament and has a single IQ motif per head (Wells et al., 1999; Rock et al., 2001). Calcium does not dissociate this CaM, but motility of a truncated double-headed construct is slowed threefold. Based on ATPase, motility, and ADP release rates, it was concluded that high calcium uncouples the two heads and causes them to act independently; a feature that would likely effect the degree of processivity (Morris et al., 2003). It is not known if full-length myosin VI would show any further calcium-dependent regulation. At present, it appears that a calcium-dependent global conformational change like we observed for myosin V is not a feature common to all CaM-binding myosins.
Physiological significance
In the absence of calcium at physiological ionic strength, full-length myosin V adopts a state with low actin-activated ATPase, which solution data and microscopy suggest is due to a global conformational change in the whole molecule. Molecules in this state would not be competent to transport cargo, and thus the pool of active motors would be low (see model in Fig. 9). From this state, there are two potential pathways of activation: via calcium and via cargo binding.
Figure 9.

A model for calcium regulation of myosin V in vivo. Two possible pathways of activation of myosin V are shown. Path A: myosin V activation via calcium, by a change from an inactive (compact) to active (extended) conformation. After binding of ATP, actin, and cargo (not necessarily in that order), transport is initiated, provided that CaM is in excess. If CaM is not in excess, motility will probably cease. A subsequent decrease in calcium concentration causes faster myosin V transport. Path B: a hypothetical pathway whereby cargo binding causes unfolding and activation of myosin V at low calcium. Once activated, the velocity of transport varies depending on the calcium and CaM concentrations.
Once calcium levels increase, the enzymatic activity of the full-length molecule is activated due to unfolding from the more compact inhibited conformation. However, the extent to which calcium affects myosin V's mechanical properties highly depends on whether CaM is present in limiting or saturating amounts in the cell. Particularly in the presence of calcium, numerous other CaM-binding proteins will compete with myosin V for the soluble pool of CaM (Tran et al., 2003), and thus this parameter is hard to predict. At limiting CaM, the motor would be inactive as a transporter, whereas in the presence of excess CaM, calcium only slows motility.
At low calcium concentrations, cargo binding to the tail could provide a second pathway of activation. In support of this idea, myosin V has been shown to transport melanosomes along actin filaments in EGTA (Rogers et al., 1999). It is also possible that calcium is required for the cargo to bind, but once the cargo is bound, the tail can no longer fold back, even in the absence of calcium. In this scenario, regions of high intracellular calcium concentration can act as so-called “cargo pick-up or loading zones” for myosin V, whereas those of lower calcium concentration are the “fast transit zones” because myosin V moves along actin faster in the absence of calcium.
A missing piece of the puzzle is whether calcium also affects the processive run length, another important parameter for efficient transport within the cell. Answers to these questions will be needed for a full understanding of calcium regulation of myosin V motor function in the cell. At present, some of the paradoxes have been resolved and the groundwork has been laid for a better understanding of regulation of this molecular motor.
Materials and methods
Myosin V constructs
Murine myosin V constructs of different lengths were cloned into pVL1392 (Invitrogen) for expression in the baculovirus/insect cell system. The constructs were COOH-terminally truncated at aa 820 for MD2IQ, 1098 for dHMM, 1467 for long-dHMM, and 1877 for dFull. The alternatively spliced tail exons in the long-dHMM and dFull constructs were A, C, D, E, and F, which is characteristic of myosin V from skin/spleen (Huang et al., 1998). Each construct contained a COOH-terminal FLAG epitope (DYKDDDDK) for protein purification by affinity chromatography. Before FLAG, the MD2IQ construct also contained the last 15 residues of the chicken neonatal myosin rod sequence (VKSREFHKKIEEERS; Moore et al., 1992). The neonatal sequence provided an epitope tag so that this small construct could be attached via monoclonal 5B4 antibody (Lowey et al., 1991) to the nitrocellulose substratum for in vitro motility experiments.
CaM
WT-CaM and CaM mutants defective in calcium binding were cloned into both bacterial and baculovirus expression plasmids. The CaM clone was derived from Xenopus, but the amino acid sequence is identical to that of mouse and human CaM. CaM has two COOH-terminal high affinity and two NH2-terminal low affinity calcium-binding sites. Calcium binding can be inhibited at any site by mutation of a critical coordinating glutamate to a glutamine residue (Beckingham, 1991). Specifically, the amino acid mutations were E31Q/E67Q (CaM-high, the mutant retaining only the COOH-terminal high affinity sites), E104Q/E140Q (CaM-low, the mutant retaining only the NH2-terminal low affinity sites), and E31Q/E67Q/E104Q/E140Q (CaMΔall, the mutant lacking all calcium-binding sites). The QuikChange® XL site-directed mutagenesis kit (Stratagene) was used to generate these mutants, which were confirmed by DNA sequencing.
Protein expression and purification
Sf9 insect cells were coinfected with viruses encoding for the myosin V HC and for CaM (WT or mutant). 3 d after infection, the cells were harvested by centrifugation and lysed by resuspension in 10 mM sodium phosphate, pH 7.2, 0.6 M NaCl, 5 mM MgCl2, 3 mM NaN3, 7% sucrose, 2 mM EGTA, 2 mM DTT, 1% NP-40, 2 mM MgATP, 25 μg/ml CaM (dHMM and dFull only), and protease inhibitors (0.5 mM AEBSF, 0.78 mg/ml benzamidine, 1 μg/ml leupeptin, and 1 μg/ml calpeptin [dFull only]). The soluble fraction of the cell lysate was fractionated by two successive ammonium sulfate precipitations (25 and 70% cuts, respectively), and then dialyzed into column buffer, 10 mM imidazole pH 7.1, 90 mM NaCl (0.3 M NaCl for dimeric constructs), 1 mM EGTA, 1 mM DTT, and 1 μg/ml leupeptin. MgATP was added to a final concentration of 2 mM, and the protein was clarified by centrifugation. The supernatant was loaded onto a column containing FLAG affinity resin (Sigma-Aldrich), washed with column buffer, and finally eluted using a 0.1-mg/ml solution of Flag peptide in wash buffer. The best fractions were pooled and used within the next 2 d, or concentrated in 50% glycerol for storage at −20°C. Based on gel densitometry, the ratio of CaM/HC in the purified protein ranged from 1.9 to 2 for MD2IQ, and from 4 to 6 for the longer necked constructs. Note that most experiments were performed with and without exogenous CaM.
CaM used for the motility and ATPase rescue experiment was expressed as follows: a T7 polymerase promoter-based bacterial expression plasmid (pNEW) encoding for CaM was transformed into Escherichia coli BL21(DE3) cells, which were grown to saturation overnight in Terrific Broth (Invitrogen), harvested by centrifugation, resuspended in 50 mM Tris-HCl (pH 7.5), 2 mM EDTA, and lysed by sonication. 5 mM CaCl2 was added to the soluble fraction of the lysate, which was then applied to a phenyl Sepharose CL-4B column (15 ml resin/1 l culture, Sigma-Aldrich; Tan et al., 1996). The column was washed with a buffer containing 50 mM Tris-Cl, pH 7.5, 0.1 mM CaCl2, and 1 mM DTT, followed by the same buffer that also contained 0.5 mM NaCl. The protein was eluted with a buffer containing 50 mM Tris-Cl, pH 7.5, 1 mM EGTA, and 1 mM DTT. The best fractions were pooled and concentrated in 50% glycerol.
Actin-pelleting assay
An actin-pelleting assay was used to quantify free and bound CaM. Myosin V constructs and actin were spun for 25 min at 150,000 g at 30°C in the absence of ATP (25 mM imidazole, pH 7.1, 25 mM KCl, 1 mM DTT, and 4 mM MgCl2 with either 1 mM EGTA or various free Ca2+ concentrations). The samples were dialyzed against a 1,000-fold volume of the above buffers overnight before adding actin and pelleting. Either the supernatant or the pellet was analyzed by PAGE and quantified by gel densitometry. Stoichiometries were determined relative to an identically prepared unspun sample. The different calcium concentrations were obtained using varying Ca2+/EGTA ratios (determined using the MaxChelator program; Chris Patton, Stanford University, Pacific Grove, CA) for samples containing 0.1–10 μM free Ca2+ and by directly adding CaCl2 to the buffer without EGTA for higher calcium concentrations.
In vitro motility assay
Myosin V constructs (0.1–0.3 μM) were spun with 0.25 μM actin at 350,000 g for 15 min (25 mM imidazole, pH 7.0, 25 mM KCl for monomers or 0.3 M KCl for dimeric constructs, 4 mM MgCl2, 1 mM MgATP, and 1 mM DTT) in order to remove ATP-insensitive myosin molecules (“rigor heads”). The flow cell (area: 22 × 4 mm; volume: ∼13.5 μl), covered by a nitrocellulose coverslip, was preincubated for 60 s with either 0.01 mg/ml BSA (when assaying dHMM or dFull) or 0.05 mg/ml monoclonal 5B4 antibody (when assaying MD2IQ) in buffer A (25 mM imidazole, pH 7.1, 25 mM KCl, 10 mM DTT, 4 mM MgCl2, 3 mg/ml glucose, 0.1 mg/ml glucose oxidase, 0.018 mg/ml catalase, and 1 mM EGTA). Unbound antibody was removed with two washes of buffer A. 1 vol myosin (spun with actin as described above) was then added for 60 s, and unbound protein was washed out with buffer A. Rhodamine phalloidin–labeled actin filaments (8 nM in buffer A) were added twice for 30 s each. Excess actin was washed out with buffer B (buffer A with 1 mM EGTA or various free calcium concentrations). 1 vol motility buffer C (buffer B with 1 mM MgATP) was added, and the flow cell was immediately placed onto the objective that was temperature controlled at 30°C. Rescue experiments were performed by including 100 μM free Ca2+ and 12 μM exogenous CaM (WT or CaMΔall) in motility buffer C. Filament velocities were quantified using the program described in Work and Warshaw (1992). The surface density of myosin molecules in these experiments was ∼7 × 103 molecules/μm2 (assuming half of the protein was recovered after the actin-MgATP spin, and half of that bound to the nitrocellulose or the antibody).
Gel densitometry
Gel images were captured in digital format using a digital camera (DC290 Zoom; Kodak Digital Science), and the band intensity was quantified using the Kodak Digital Science 1D image analysis software package.
Linked actin-activated ATPase assay
Actin-activated ATPase assays were performed at 37°C in 10 mM imidazole, pH 7.0, 50 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM NaN3, and either 1 mM EGTA or 100 μM free CaCl2. Experiments were done with no added CaM or with 6 μM exogenous CaM, as indicated. The buffers also contained an ATP-regenerating system: 0.2 mM NADH, 0.5 mM phosphoenolpyruvate, 20 U/ml lactate dehydrogenase, and 100 U/ml pyruvate kinase. The rate of the reaction was measured from the decrease in absorbance at 340 nm caused by the oxidation of NADH by lactate dehydrogenase. This assay minimizes the time-dependent ADP-induced inhibition of actin-activated ATPase activity (De La Cruz et al., 2000).
Analytical ultracentrifugation
An analytical ultracentrifuge (Optima XL-I; Beckman Coulter) was used to determine the sedimentation coefficients of the expressed constructs (buffer: 10 mM Hepes, pH 7.0, 1 mM DTT, 1 mM EGTA, 1 mM NaN3, and 0.1 or 0.3 M NaCl). Sedimentation velocity runs were performed in the An60Ti rotor (Beckman Coulter) at 40,000 rpm and 20°C. Sedimentation values were corrected for density and viscosity. Samples of ∼0.5 OD in a 1.2-cm cell were scanned by absorbance at 280 nm. Sedimentation coefficients were determined by curve fitting to one species, using the dc/dt program (Philo, 2000). Sedimentation equilibrium data were collected at 6,000 rpm and 4°C, and the data sets were analyzed with software provided with the Beckman Optima XL-I ultracentrifuge.
EM
Myosin was rotary shadowed with platinum by the Core Electron Microscope Facility at the University of Massachusetts Medical School (Worcester, MA), directed by Dr. Gregory Hendricks. The dFull construct (in 5 mM sodium phosphate, pH 7.0, 0.1 M NaCl, 0.1 mM EGTA, 1 mM DTT, and 50% glycerol) was diluted to ∼10 μg/ml into either 75 mM ammonium acetate (pH 7.2), 66% glycerol or 500 mM ammonium acetate (pH 7.2), 66% glycerol volatile low and high salt buffers.
Acknowledgments
We thank Dr. Gregory Hendricks (director of the Core Electron Microscope Facility at the University of Massachusetts Medical School), for providing the images of metal-shadowed myosin V, Dr. Nancy Jenkins for the myosin V clone, and Dr. Susan Lowey for helpful comments regarding this manuscript.
This work was supported by funds from the National Institutes of Health to K.M. Trybus (HL38113).
D.N. Krementsov and E.B. Krementsova contributed equally to this paper.
Abbreviations used in this paper: dFull, full-length myosin V; dHMM, dilute heavy meromyosin V; HC, heavy chain; long-dHMM, a long heavy meromyosin lacking only the cargo-binding domain; MD2IQ, a monomer with the motor domain and the first two IQ motifs; WT, wild type.
References
- Beckingham, K. 1991. Use of site-directed mutations in the individual Ca2+-binding sites of calmodulin to examine Ca2+-induced conformational changes. J. Biol. Chem. 266:6027–6030. [PubMed] [Google Scholar]
- Casaletti, L., S.B. Tauhata, J.E. Moreira, and R.E. Larson. 2003. Myosin-Va proteolysis by Ca2+/calpain in depolarized nerve endings from rat brain. Biochem. Biophys. Res. Commun. 308:159–164. [DOI] [PubMed] [Google Scholar]
- Cheney, R.E., M.K. O'Shea, J.E. Heuser, M.V. Coelho, J.S. Wolenski, E.M. Espreafico, P. Forscher, R.E. Larson, and M.S. Mooseker. 1993. Brain myosin-V is a two-headed unconventional myosin with motor activity. Cell. 75:13–23. [DOI] [PubMed] [Google Scholar]
- Coy, D.L., W.O. Hancock, M. Wagenbach, and J. Howard. 1999. Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat. Cell Biol. 1:288–292. [DOI] [PubMed] [Google Scholar]
- De La Cruz, E.M., H.L. Sweeney, and E.M. Ostap. 2000. ADP inhibition of myosin V ATPase activity. Biophys. J. 79:1524–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espindola, F.S., D.M. Suter, L.B. Partata, T. Cao, J.S. Wolenski, R.E. Cheney, S.M. King, and M.S. Mooseker. 2000. The light chain composition of chicken brain myosin-Va: calmodulin, myosin-II essential light chains, and 8-kDa dynein light chain. Cell Motil. Cytoskeleton. 47:269–281. [DOI] [PubMed] [Google Scholar]
- Espreafico, E.M., R.E. Cheney, M. Matteoli, A.A. Nascimento, P.V. De Camilli, R.E. Larson, and M.S. Mooseker. 1992. Primary structure and cellular localization of chicken brain myosin-V (p190), an unconventional myosin with calmodulin light chains. J. Cell Biol. 119:1541–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney, D.D., and M.F. Stock. 2000. Kinesin's IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat. Cell Biol. 2:257–260. [DOI] [PubMed] [Google Scholar]
- Homma, K., J. Saito, R. Ikebe, and M. Ikebe. 2000. Ca2+-dependent regulation of the motor activity of myosin V. J. Biol. Chem. 275:34766–34771. [DOI] [PubMed] [Google Scholar]
- Huang, J.D., V. Mermall, M.C. Strobel, L.B. Russell, M.S. Mooseker, N.G. Copeland, and N.A. Jenkins. 1998. Molecular genetic dissection of mouse unconventional myosin-VA: tail region mutations. Genetics. 148:1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karcher, R.L., J.T. Roland, F. Zappacosta, M.J. Huddleston, R.S. Annan, S.A. Carr, and V.I. Gelfand. 2001. Cell cycle regulation of myosin-V by calcium/calmodulin-dependent protein kinase II. Science. 293:1317–1320. [DOI] [PubMed] [Google Scholar]
- Lowey, S., G.S. Waller, and E. Bandman. 1991. Neonatal and adult myosin heavy chains form homodimers during avian skeletal muscle development. J. Cell Biol. 113:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, A.D., R.S. Rock, M. Rief, J.A. Spudich, M.S. Mooseker, and R.E. Cheney. 1999. Myosin-V is a processive actin-based motor. Nature. 400:590–593. [DOI] [PubMed] [Google Scholar]
- Moore, J.R., E.B. Krementsova, K.M. Trybus, and D.M. Warshaw. 2001. Myosin V exhibits a high duty cycle and large unitary displacement. J. Cell Biol. 155:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, L.A., M.J. Arrizubieta, W.E. Tidyman, L.A. Herman, and E. Bandman. 1992. Analysis of the chicken fast myosin heavy chain family. Localization of isoform-specific antibody epitopes and regions of divergence. J. Mol. Biol. 225:1143–1151. [DOI] [PubMed] [Google Scholar]
- Morris, C.A., A.L. Wells, Z. Yang, L.Q. Chen, C.V. Baldacchino, and H.L. Sweeney. 2003. Calcium functionally uncouples the heads of myosin VI. J. Biol. Chem. 278:23324–23330. [DOI] [PubMed] [Google Scholar]
- Nagashima, K., S. Torii, Z. Yi, M. Igarashi, K. Okamoto, T. Takeuchi, and T. Izumi. 2002. Melanophilin directly links Rab27a and myosin Va through its distinct coiled-coil regions. FEBS Lett. 517:233–238. [DOI] [PubMed] [Google Scholar]
- Nascimento, A.A., R.E. Cheney, S.B. Tauhata, R.E. Larson, and M.S. Mooseker. 1996. Enzymatic characterization and functional domain mapping of brain myosin-V. J. Biol. Chem. 271:17561–17569. [DOI] [PubMed] [Google Scholar]
- Philo, J.S. 2000. A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal. Biochem. 279:151–163. [DOI] [PubMed] [Google Scholar]
- Purcell, T.J., C. Morris, J.A. Spudich, and H.L. Sweeney. 2002. Role of the lever arm in the processive stepping of myosin V. Proc. Natl. Acad. Sci. USA. 99:14159–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck-Peterson, S.L., D.W.J. Provance, M.S. Mooseker, and J.A. Mercer. 2000. Class V myosins. Biochim. Biophys. Acta. 1496:36–51. [DOI] [PubMed] [Google Scholar]
- Rief, M., R.S. Rock, A.D. Mehta, M.S. Mooseker, R.E. Cheney, and J.A. Spudich. 2000. Myosin-V stepping kinetics: a molecular model for processivity. Proc. Natl. Acad. Sci. USA. 97:9482–9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock, R.S., S.E. Rice, A.L. Wells, T.J. Purcell, J.A. Spudich, and H.L. Sweeney. 2001. Myosin VI is a processive motor with a large step size. Proc. Natl. Acad. Sci. USA. 98:13655–13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, S.L., R.L. Karcher, J.T. Roland, A.A. Minin, W. Steffen, and V.I. Gelfand. 1999. Regulation of melanosome movement in the cell cycle by reversible association with myosin V. J. Cell Biol. 146:1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto, T., F. Wang, S. Schmitz, Y. Xu, Q. Xu, J.E. Molloy, C. Veigel, and J.R. Sellers. 2003. Neck length and processivity of myosin V. J. Biol. Chem. 278:29201–29207. [DOI] [PubMed] [Google Scholar]
- Stock, M.F., J. Guerrero, B. Cobb, C.T. Eggers, T.G. Huang, X. Li, and D.D. Hackney. 1999. Formation of the compact conformer of kinesin requires a COOH-terminal heavy chain domain and inhibits microtubule-stimulated ATPase activity. J. Biol. Chem. 274:14617–14623. [DOI] [PubMed] [Google Scholar]
- Strom, M., A.N. Hume, A.K. Tarafder, E. Barkagianni, and M.C. Seabra. 2002. A family of Rab27-binding proteins. Melanophilin links Rab27a and myosin Va function in melanosome transport. J. Biol. Chem. 277:25423–25430. [DOI] [PubMed] [Google Scholar]
- Tan, R.Y., Y. Mabuchi, and Z.J. Grabarek. 1996. Blocking the Ca2+-induced conformational transitions in calmodulin with disulfide bonds. J. Biol. Chem. 271:7479–7483. [DOI] [PubMed] [Google Scholar]
- Tauhata, S.B., D.V. dos Santos, E.W. Taylor, M.S. Mooseker, and R.E. Larson. 2001. High affinity binding of brain myosin-Va to F-actin induced by calcium in the presence of ATP. J. Biol. Chem. 276:39812–39818. [DOI] [PubMed] [Google Scholar]
- Tran, Q.K., D.J. Black, and A. Persechini. 2003. Intracellular coupling via limiting calmodulin. J. Biol. Chem. 278:24247–24250. [DOI] [PubMed] [Google Scholar]
- Trybus, K.M. 1991. Regulation of smooth muscle myosin. Cell Motil. Cytoskeleton. 18:81–85. [DOI] [PubMed] [Google Scholar]
- Trybus, K.M., T.W. Huiatt, and S. Lowey. 1982. A bent monomeric conformation of myosin from smooth muscle. Proc. Natl. Acad. Sci. USA. 79:6151–6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trybus, K.M., E. Krementsova, and Y. Freyzon. 1999. Kinetic characterization of a monomeric unconventional myosin V construct. J. Biol. Chem. 274:27448–27456. [DOI] [PubMed] [Google Scholar]
- Wang, F., L. Chen, O. Arcucci, E.V. Harvey, B. Bowers, Y. Xu, J.A. Hammer, and J.R. Sellers. 2000. Effect of ADP and ionic strength on the kinetic and motile properties of recombinant mouse myosin V. J. Biol. Chem. 275:4329–4335. [DOI] [PubMed] [Google Scholar]
- Wang, F., K. Thirumurugan, W.F. Stafford, J.A. Hammer, III, P.J. Knight, and J.R. Sellers. 2004. Regulated conformation of myosin V. J. Biol. Chem. 279:2333–2336. [DOI] [PubMed] [Google Scholar]
- Wendt, T., D. Taylor, K.M. Trybus, and K. Taylor. 2001. Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc. Natl. Acad. Sci. USA. 98:4361–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Work, S.S., and D.M. Warshaw. 1992. Computer-assisted tracking of actin filament motility. Anal. Biochem. 202:275–285. [DOI] [PubMed] [Google Scholar]
- Wells, A.L., A.W. Lin, L.Q. Chen, D. Safer, S.M. Cain, T. Hasson, B.O. Carragher, R.A. Milligan, and H.L. Sweeney. 1999. Myosin VI is an actin-based motor that moves backward. Nature. 401:505–508. [DOI] [PubMed] [Google Scholar]
- Wu, X.S., K. Rao, H. Zhang, F. Wang, J.R. Sellers, L.E. Matesic, N.G. Copeland, N.A. Jenkins, and J.A. Hammer. 2002. Identification of an organelle receptor for myosin-Va. Nat. Cell Biol. 4:271–278. [DOI] [PubMed] [Google Scholar]
- Zhu, T., K. Beckingham, and M. Ikebe. 1998. High affinity Ca2+ binding sites of calmodulin are critical for the regulation of myosin Iβ motor function. J. Biol. Chem. 273:20481–20486. [DOI] [PubMed] [Google Scholar]