

Abstract
Insulin-like growth factors elicit many responses through activation of phosphoinositide 3-OH kinase (PI3K). The tuberous sclerosis complex (TSC1-2) suppresses cell growth by negatively regulating a protein kinase, p70S6K (S6K1), which generally requires PI3K signals for its activation. Here, we show that TSC1-2 is required for insulin signaling to PI3K. TSC1-2 maintains insulin signaling to PI3K by restraining the activity of S6K, which when activated inactivates insulin receptor substrate (IRS) function, via repression of IRS-1 gene expression and via direct phosphorylation of IRS-1. Our results argue that the low malignant potential of tumors arising from TSC1-2 dysfunction may be explained by the failure of TSC mutant cells to activate PI3K and its downstream effectors.
Keywords: TSC1-2; PI3K; IRS proteins; S6K; insulin
Introduction
The signal transduction mechanisms operating downstream of insulin receptor ligation have highlighted the importance both of the receptor tyrosine kinase substrates (insulin receptor substrates, IRS-1 and IRS-2) and the lipid kinase phosphoinositide 3-OH kinase (PI3K; Saltiel, 2001; Saltiel and Kahn, 2001; Cantley, 2002; White, 2002). Activation of PI3K occurs via recruitment to IRS proteins, and is critical in generating many of the cell's responses to insulin (Shepherd et al., 1998). The insulin–PI3K pathway also plays a conserved role in regulating cell and organism size. Mice deficient in effectors of PI3K, PDK1 and S6K1, exhibit growth defects (Shima et al., 1998; Lawlor et al., 2002), whereas activation of PI3K leads to increases in cell and organ growth (Leevers et al., 1996; Crackower et al., 2002; Shioi et al., 2002). The growth response to insulin-like growth factors involves the S6 kinases and leads to increased translation of 5′-terminal oligopyrimidine track mRNAs (Jefferies et al., 1997), many of which encode ribosomal proteins. By increasing ribosome biogenesis, activation of S6K is thought to lead to an increase in protein synthesis (Volarevic and Thomas, 2001). Two S6K genes, S6K1 and S6K2, together encode four kinase isoforms, three of which are present in the nucleus (Reinhard et al., 1992; Gout et al., 1998). Two signaling pathways have been shown to positively regulate S6K: the insulin–PI3K pathway (Reif et al., 1997; Martin et al., 2001) and a nutrient pathway involving the mammalian target of rapamycin (mTOR; Brown et al., 1995; Park et al., 2002). A genetic regulator of S6K1 activity has also been identified—the tumor suppressor tuberous sclerosis complex (TSC1-2), a complex of the TSC1 and TSC2 gene products, hamartin and tuberin (van Slegtenhorst et al., 1998). Inactivation of TSC1-2 leads to constitutive S6K1 activity (Jaeschke et al., 2002; Kwiatkowski et al., 2002) and growth in Drosophila (Ito and Rubin, 1999; Gao and Pan, 2001; Potter et al., 2001; Tapon et al., 2001). An inhibitory target of TSC1-2 action has been identified as Rheb, a small GTPase (Garami et al., 2003; Saucedo et al., 2003; Stocker et al., 2003; Y. Zhang et al., 2003) with Rheb overexpression resulting in activation of S6K (for review see Li et al., 2004).
A key unresolved question is how S6K activation and growth stimulated by insulin-like growth factors is appropriately coordinated with the other responses downstream of PI3K that include cell proliferation and survival. Negative feedback loops relating a downstream response of insulin–PI3K signaling to the overall level of pathway activation would be a potential means of accomplishing this type of coordination. Here, we show that by suppressing such a negative feedback loop, TSC1-2 is a key positive regulator of PI3K signaling. TSC1-2 promotes PI3K signaling by suppressing S6K, thereby preventing S6K-dependent inactivation of IRS-1 and IRS-2. As a result of IRS inactivation, TSC2-deficient cells exhibit a profound defect in PI3K signaling and fail to activate the serine-threonine kinase PKB or respond to the migration and pro-survival activities of IGF-1. We show that for IRS-1, the inactivation is exerted through S6K-dependent suppression of gene transcription and by direct phosphorylation by S6K1 in a region critical for IRS-1 adaptor function. We suggest that although TSC1-2 loss of function promotes hamartomatous growth in TSC, a failure to activate insulin–PI3K signaling may explain the low malignant potential of such tumors.
Results
TSC1-2 specifically regulates insulin signaling to PI3K
Insulin-stimulated activation of the PI3K-regulated serine-threonine kinase PKB (also known as AKT) is defective in cells deficient in TSC2 (Jaeschke et al., 2002) or TSC1 (Kwiatkowski et al., 2002). To determine whether there is a specific impairment of insulin-like growth factor signaling, we assayed PKB activity after stimulation of cells with insulin, IGF-1, or EGF. In control TSC2 +/+ mouse embryo fibroblasts (MEFs) or TSC2 −/− MEFs in which wild-type TSC2 has been reintroduced (Jaeschke et al., 2002), PKB is activated by insulin, IGF-1, and EGF (Fig. 1 A). In contrast, in TSC2 −/− MEFs or TSC2 −/− MEFs in which a pathogenic TSC2 mutant is reintroduced, PKB is activated strongly only by EGF. Similarly, a downstream effector of PKB, GSK3α/β, is robustly phosphorylated in response to stimulation by all three growth factors in control MEFs, but only strongly by EGF in TSC2 −/− MEFs (Fig. 1 B).
Figure 1.

PI3K activation by insulin-like growth factors is dependent on functional TSC2 . (A) PKB activation in cells expressing or lacking functional TSC2. PKB activation in isogenic TSC2 +/+, TSC2 −/− MEFs, and TSC2 −/− MEF lines in which wild-type (−/− (+WT)) or a pathogenic mutant TSC2 (−/− (+N1643K)) has been reintroduced were starved and stimulated with the indicated growth factors for 10 min. The ability of insulin, IGF-1, and EGF to activate PKB is demonstrated by phosphorylation of Ser-473 (S473-P). (B) Phosphorylation of GSK3α/β, at GSK3α Ser-21 (top) and GSK3β Ser-9 (middle) after insulin, IGF-1, or EGF stimulation. Bottom: total GSK3α/β. (C) PI3K activity in TSC2 +/+ or TSC2 −/− MEFs, or TSC2 −/− MEFs reconstituted with wild-type TSC2 (+WT) or a disease-causing mutant TSC2 (+N1643K) after stimulation with IGF-1. Results are expressed as values relative to unstimulated controls. (D) PIP3 levels in TSC2 +/+ or TSC2 −/− MEFs, or TSC2 −/− MEFs reconstituted with wild-type TSC2 (+WT) or a disease-causing mutant TSC2 (+N1643K) after stimulation with IGF-1 or EGF. The results (means and SD of triplicate determinations) are expressed relative to control unstimulated cells.
Because PKB activation generally requires PI3K activation and the production of phosphatidylinositol 3,4,5-trisphosphate (PIP3) at the membrane (Cantley, 2002), we assayed PI3K activity and measured levels of PIP3 after stimulation with IGF-1 or EGF. TSC2 −/− MEFs neither activate PI3K (Fig. 1 C) nor elevate levels of PIP3 in response to IGF-1 stimulation (Fig. 1 D), whereas PIP3 generated after stimulation with EGF is similar to that of control TSC2-expressing MEFs (Fig. 1 D). Thus, the failure to activate PKB after stimulation with insulin or IGF-1 in TSC2-deficient cells is due to a general failure to activate PI3K and generate its lipid product, PIP3.
TSC1-2 regulates IRS-1 mRNA via S6K
The basis of the insulin signaling defect in cells lacking TSC2 could be either at the level of expression of the insulin/IGF-1 receptors or IRS proteins, or at the level of their activation. Therefore, we performed a microarray analysis of gene expression differences between TSC2 +/+ and TSC2 −/− MEFs, which shows that the level of the IRS-1 mRNA is significantly reduced in TSC2 −/− MEFs (Fig. 2 A). To confirm this result, we analyzed IRS-1 mRNA. IRS-1 mRNA abundance is significantly reduced in TSC2 −/− MEFs (Fig. 2 B). Importantly, this reduction is fully reversed by stable retroviral introduction of wild-type TSC2 (Fig. 2 B). Interestingly, the level of IRS-2 mRNA is unaffected by the absence of functional TSC2. A similar result is obtained comparing the levels of IRS-1 protein in these cells by Western blotting, whereas the levels of IRS-2 protein or of the insulin or IGF receptors is unaffected by TSC2 expression (Fig. 2 C). Thus, IRS-1 expression is maintained by the presence of functional TSC2.
Figure 2.
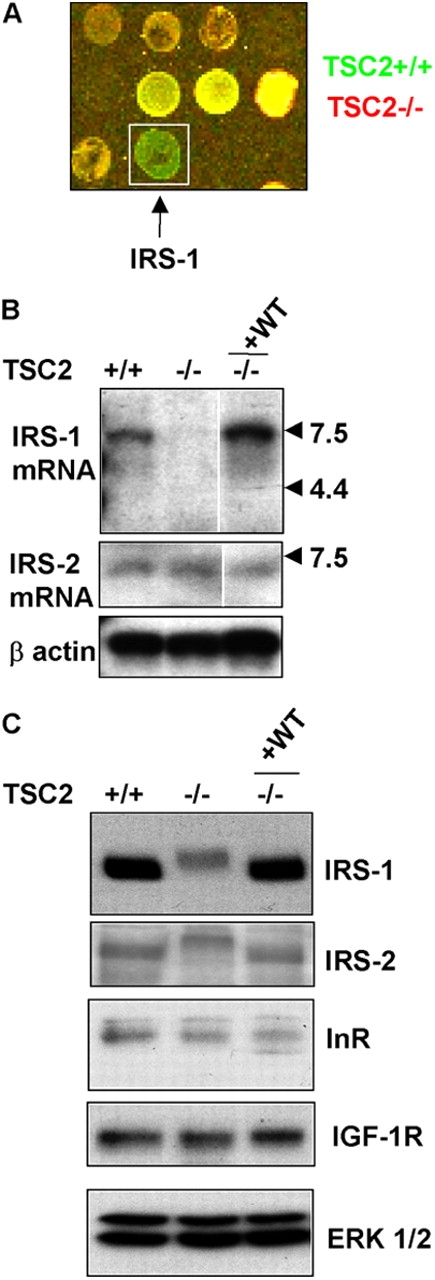
IRS-1 mRNA and protein expression is dependent on TSC2. (A) Microarray analysis of TSC2 +/+ and TSC2 −/− MEFs. IRS-1 mRNA is more abundant in TSC2 +/+ MEFs (green) compared with TSC2 −/− MEFs (red). (B) Northern blotting of IRS-1 (top) or IRS-2 (middle) mRNAs in isogenic TSC2 +/+, TSC2 −/− MEFs, and a TSC2 −/− MEF line in which wild-type TSC2 has been reintroduced (−/− +WT). Aliquots of these samples were run on a separate gel and probed for β-actin as a control for RNA content (bottom). White line indicates that intervening lanes have been removed. (C) IRS-1 (first panel), IRS-2 (second panel), InR (third panel), and IGF1-R (fourth panel) protein levels in TSC2 +/+, TSC2 −/−, and a TSC2 −/− MEF line in which wild-type TSC2 has been reintroduced (−/− +WT). A blot was reprobed for ERK1/2 as a control for protein loading (bottom panel).
IRS-1 protein levels have previously been shown to be rapamycin sensitive (Haruta et al., 2000). Because mTOR/S6K signaling is hyperactivated in TSC2-deficient cells (Jaeschke et al., 2002), we sought to examine the effect of rapamycin on IRS-1 mRNA abundance. Rapamycin treatment of TSC2 −/− MEFs relieves the suppression of IRS-1 mRNA abundance after ∼24 h of treatment (Fig. 3 A). This restoration of IRS-1 mRNA abundance by sustained rapamycin treatment is reversed by actinomycin D (Fig. 3, B and C), indicating that mTOR/S6K1 signaling likely reduces IRS-1 mRNA abundance primarily at the level of gene transcription.
Figure 3.


Inhibition of S6K restores IRS-1 mRNA. (A) IRS-1 mRNA levels (top) in serum-starved TSC2 +/+ or TSC2 −/− MEFs in untreated cells or after the addition of 20 nM rapamycin for various times. The blot was reprobed for β-actin as a control for mRNA loading (bottom). White line indicates that intervening lanes have been removed. (B) IRS-1 mRNA levels (top) in TSC2 −/− MEFs treated with 20 nM rapamycin alone, or in the presence of 10 μg/ml actinomycin D for the final 10 h of a 24-h treatment. The blot was reprobed for β-actin as a control for mRNA loading (bottom). (C) Quantitative RT-PCR analysis of untreated, rapamycin-treated, and rapamycin plus actinomycin D–treated TSC2 −/− MEFs. Left, β-actin; right, IRS-1. A single graph from triplicate determinations showing identical results is shown. (D) RNAi-mediated inhibition of S6K1 and S6K2. Western blotting of extracts from TSC2 +/+ or TSC2 −/− MEFs transfected with a combination of scrambled siRNAs (C) or S6K1, S6K2, or S6K1 plus S6K2 siRNAs. Top, S6K1; middle, S6K2; bottom, anti-pS6 (Ser240/244). Where indicated, cells were stimulated for 10 min with insulin or treated with 20 nM rapamycin for 1 h. (E) IRS-1 mRNA levels in TSC2 +/+ MEFs or TSC2 −/− MEFs transfected with a combination of S6K1 and S6K2 scrambled siRNAs (C), S6K1, or S6K2 siRNAs, or treated for 24 h with 20 nM rapamycin. Top, IRS-1 mRNA; The blot was also reprobed for β-actin as a control for mRNA loading (bottom). (F) Quantitative RT-PCR analysis of untreated TSC2 +/+, TSC2 −/−, or TSC2 −/− treated with 20 nM rapamycin or RNAi to S6K1 or S6K2. Left, β-actin; right, IRS-1. A single graph from triplicate determinations showing identical results is shown.
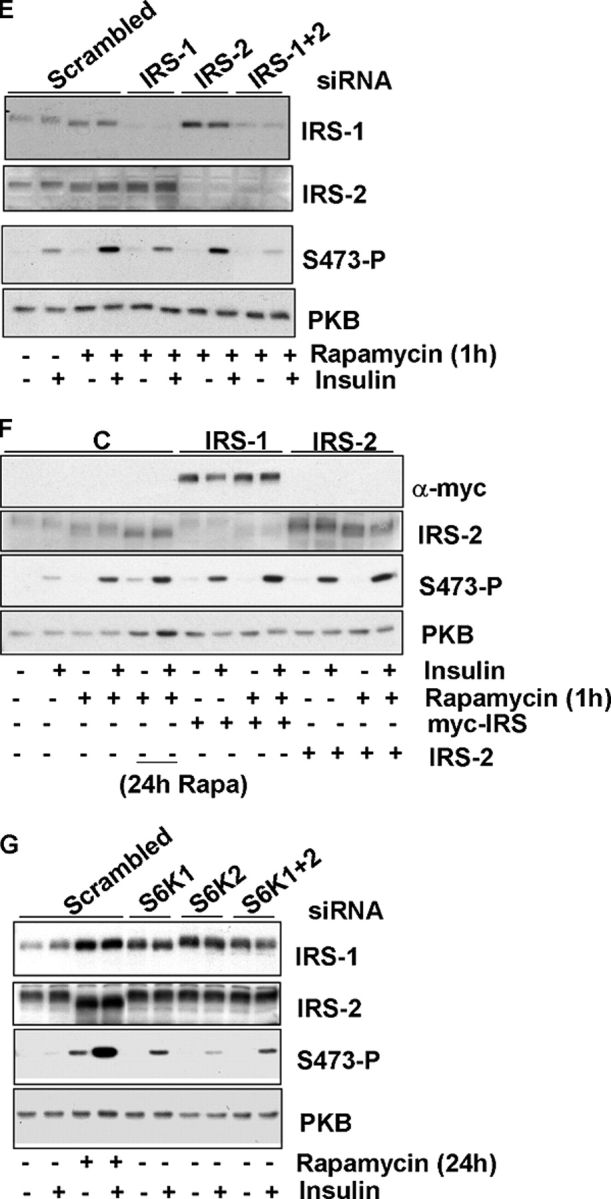
Next, we asked whether the suppression of IRS-1 mRNA was mediated by either S6K1 or S6K2, both of which are rapamycin-sensitive kinases, using a small interfering RNA (siRNA) approach. A near complete knock-down of S6K1 and a 70–80% knock-down of S6K2 is observed (Fig. 3 D). Suppression of either S6K1 or S6K2 mimics the effect of rapamycin treatment and restores IRS-1 mRNA to a level approaching that of TSC2 +/+ MEFs (Fig. 3, E and F). We conclude that the suppression of IRS-1 mRNA we observe in TSC2-deficient cells is mediated by both forms of S6K—S6K1 and S6K2.
TSC1-2 promotes insulin signaling by inhibiting S6K action on IRS-1
IRS-1 and IRS-2 proteins extracted from TSC2 −/− MEFs exhibit reduced migration on SDS-PAGE gels (Fig. 2 C). Treatment of IRS-1 immunoprecipitates with λ-phosphatase indicates that the reduced migration of IRS-1 in TSC2 −/− MEFs is due to differences in phosphorylation, with the protein exhibiting a larger change in mobility than IRS-1 from TSC2 +/+ MEFs after phosphatase treatment (Fig. 4 A). To determine if this change in IRS-1 phosphorylation is caused by mTOR/S6K signaling, we treated TSC2 +/+ or TSC −/− MEFs for various times with rapamycin and examined IRS-1 and IRS-2 protein levels. Both IRS-1 and IRS-2 migrate as slower species in TSC2-deficient cells. After the addition of rapamycin to TSC2 −/− MEFs for 1 h, both IRS-1 and IRS-2 migrate as faster migrating species with no increase in protein levels. However, after 24 h treatment with rapamycin, the migration and level of IRS-1 protein in TSC2 −/− MEFs is similar to that of untreated TSC2 +/+ MEFs (Fig. 4 B). In contrast, the level of IRS-2 is largely unaffected by rapamycin treatment in either TSC2 +/+ or TSC2 −/− MEFs. PKB activation in TSC2 −/− MEFs is partially restored after 1 h of rapamycin treatment, and is fully restored after 24 h of treatment (Fig. 4 C). Consistent with the rescue of PKB activation, the elevation of PIP3 after IGF-1 stimulation of TSC2 −/− MEFs pretreated with rapamycin for 24 h is similar to that produced in stimulated TSC2 +/+ MEFs (Fig. 4 D).
Figure 4.

Inhibition of S6K restores insulin signaling. (A) Treatment of IRS-1 immunoprecipitates with λ-phosphatase indicating that reduced migration of IRS-1 in TSC2 −/− MEFs is due to increased phosphorylation. (B) Treatment of serum-starved TSC2 +/+ or TSC2 −/− MEFs for various times with rapamycin followed by immunoblotting for IRS-1 (top) or IRS-2 (bottom). The slower migrating, more heavily phosphorylated IRS-1 or IRS-2 present in TSC2 −/− MEFs is indicated with an arrow; the faster migrating IRS-1 or IRS-2 present in TSC2 +/+ cells or after rapamycin treatment is indicated with an arrowhead. (C) PKB activation by insulin or IGF-1 after treatment of TSC2 +/+ or TSC2 −/− MEFs with rapamycin for the indicated times. Top, Ser473-phosphorylated PKB; bottom, level of total PKB. (D) PIP3 levels in TSC2 +/+ (gray bars) or TSC2 −/− (white bars) MEFs after IGF-1 stimulation in untreated cells or cells pretreated for 24 h with 20 nM rapamycin. Results from triplicate determinations (means and SDs) are shown normalized to control unstimulated samples and are representative of three independent experiments. (E) PKB activation after RNAi-mediated inhibition of IRS-1, IRS-2, IRS-1 plus IRS-2, or a combination of scrambled IRS-1 and IRS-2 siRNA (Scrambled) in TSC2 −/− MEFs stimulated by insulin for 10 min after treatment with 20 nM rapamycin for 1 h. First panel, IRS-1; second panel, IRS-2; third panel, Ser473-phosphorylated PKB; bottom panel, total PKB. (F) Rescue of PKB activation in TSC2 −/− MEFs after overexpression of IRS-1 or IRS-2. Extracts from mock-transfected TSC2 −/− MEFs (C) or MEFs transfected with expression constructs expressing myc-IRS-1 or IRS-2 were starved for 24 h after transfection and were treated with 20 nM rapamycin for 1 h before stimulation with insulin for 10 min. Where indicated, mock-transfected cells were also treated with 20 nM rapamycin for 24 h before stimulation. First panel, myc-IRS-1 detected with 9E10 monoclonal; second panel, IRS-2; third panel, Ser473-phosphorylated PKB; bottom panel, total PKB. (G) PKB activation in TSC2 −/− MEFs after RNAi-mediated inhibition of S6K1, S6K2, S6K1 plus S6K2, or a mixture of S6K1 and S6K2 scrambled siRNAs (Scrambled) after insulin stimulation for 10 min. Scrambled siRNA-transfected cells were also treated with 20 nM rapamycin for 24 h before insulin stimulation where indicated.
To examine whether the rescuing effect of rapamycin upon insulin signaling is mediated via effects on IRS-1 or IRS-2, we again used siRNAs. The partial rescue of PKB activation we observe with 1 h of rapamycin treatment is diminished by suppression of either IRS-1 or, to a lesser extent, IRS-2, with a more pronounced effect observed when both proteins are suppressed (Fig. 4 E). To determine whether perturbation of IRS function is the critical lesion responsible for diminished insulin signaling in TSC2 −/− MEFs, we then asked whether insulin signaling could be rescued in TSC2 −/− MEFs after overexpression of IRS-1 or IRS-2. We find that overexpression of either IRS-1 or IRS-2 results in an increase in PKB activation, which when combined with short-term inhibition of mTOR/S6K signaling using rapamycin leads to a phenocopy of the effect of sustained (24 h) treatment with rapamycin (Fig. 4 F).
Then, we asked whether the enhanced effect on signaling we observe after rapamycin is due to inhibition of S6K, using siRNAs. The results indicate that suppression of either S6K1 or S6K2 in TSC2 −/− MEFs leads to an increase in PKB activation, with a larger increase elicited by suppression of S6K1 (Fig. 4 G). Suppression of either kinase leads to an equivalent increase in the level of IRS-1 protein, with no effect on IRS-2. Together, these results show that the reduced insulin signaling we observed in TSC2 −/− MEFs is caused by effects on both IRS-1 and IRS-2. In the case of IRS-1, the effects appear to be partially mediated by S6K, with both S6K1 and S6K2 contributing to reduced insulin signaling.
IRS-1 is a novel substrate for S6K
We reasoned that IRS-1 may also be a novel substrate for S6 kinases, and tested the ability of GST-tagged IRS-1 fragments covering the entirety of the protein to be phosphorylated in vitro by S6K, in comparison to S6. A fragment encompassing residues 108–516 (IRS-1108–516) is efficiently phosphorylated by S6K in vitro (Fig. 5 B). Comparison of the S6K phosphorylation site in S6 with IRS-1108–516 reveals three potential phosphorylation sites in which an RxRxxS is followed by distal serines as occurs in S6 (Fig. 5 A). We mutagenized both the proximal (RxRxxS) and distal serines in each of these potential sites and asked whether such mutants could be phosphorylated in vitro. The results show that only in IRS-1108–516 in which serines 302, 307, and 310 are mutated (Fig. 5 A, site 2) is IRS-1 not phosphorylated in vitro by S6K1 (Fig. 5 C, left panels). Individual mutagenesis of serines 307 and 310 has little effect on IRS-1108–516 phosphorylation, indicating that this site is not phosphorylated at multiple serines like that of S6, whereas mutagenesis of serine 302 to alanine ablates phosphorylation (Fig. 5 C, right panels). This result was confirmed using Western blotting with a phospho-Ser302 antibody that only recognizes wild-type IRS-1 after in vitro phosphorylation by S6K (Fig. 5 D). To confirm that this phosphorylation detected in vitro is sensitive to TSC2 in vivo, we probed extracts with this antibody. Although the level of IRS-1 is reduced in TSC2-deficient MEFs compared with TSC2-expressing MEFs (Fig. 2 C and Fig. 5 E), the level of Ser302-phosphorylated IRS-1 is greatly increased (Fig. 5 E) and rapamycin sensitive (Fig. 5 F). We again used S6K siRNAs to determine whether IRS-1 Ser302 phosphorylation is through S6K in vivo. The result indicates that IRS-1 Ser302 phosphorylation is decreased after siRNA-mediated suppression of S6K1, but is unaffected by suppression of S6K2 (Fig. 5 G). Thus, in TSC2 −/− MEFs cells, IRS-1 Ser302 phosphorylation is regulated primarily by S6K1, and not S6K2, although the less effective knock-down of S6K2 expression leaves open the formal possibility of some contribution from this kinase also.
Figure 5.

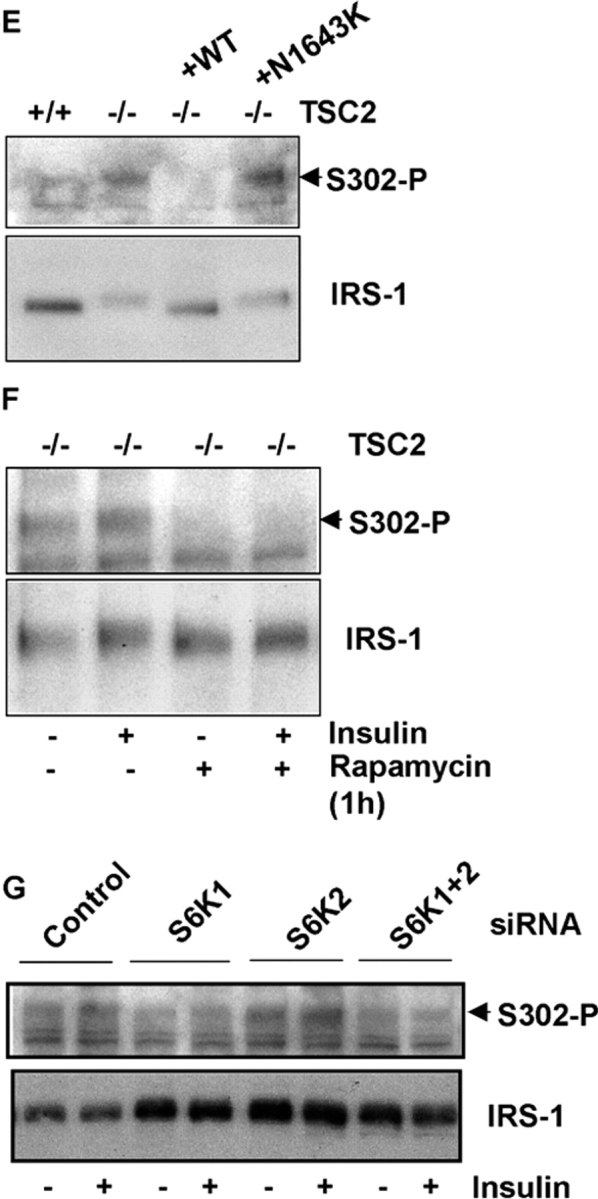
IRS-1 is a novel S6K substrate. (A) Domain structure of IRS-1 indicating regions of GST fusions (aa numbering based on murine IRS-1) used for in vitro kinase assays and comparison of the S6 phosphorylation site with candidate S6-like phosphorylation sites present in IRS-1108–516. Conserved arginines are shown in bold, serines phosphorylated by S6K in S6 and potentially phosphorylated in IRS-1108–516 are shown in red, and serines mutated to alanines in mutants of sites 1–3 are underlined. Also shown is the homologous site to site 2 in murine IRS-2. (B) In vitro kinase assay of GST-IRS-1 fragments with purified S6K2. Full-length GST fusion proteins (1–4, as indicated in A) and 40S ribosomal proteins containing S6 (40S) in these preparations are indicated with asterisks in the Coomassie blue–stained gel (right), and the 32P-labeled proteins after the kinase assay and autoradiography are shown on the left. Two phosphorylated bands are noted in kinase assay with IRS-1108–516, the lower of which represents a poorly resolved doublet of cleaved forms of the protein. An arrow indicates 32P-labeled S6 present in the 40S ribosome preparation. (C) In vitro kinase assay of IRS-1 site-directed mutants by S6K1. Top panels, autoradiographs; bottom panels, Coomassie blue–stained gels; left panels, phosphorylation of IRS-1108–516 (WT) and site 1–3 compound mutants as indicated in Fig. 4 A; right panels, phosphorylation of IRS-1108–516 and individual serine-to-alanine mutants of the serine residues within site 2. (D) Ser302 phosphorylation of IRS-1108–516 wild-type (WT) or IRS-1108–516 with Ser302 mutated to alanine (S302A) detected with a phosphospecific antibody to IRS-1 Ser302 after in vitro phosphorylation by S6K1. (E) IRS-1 Ser302 phosphorylation is elevated in vivo. Top, Ser302-phosphorylated IRS-1 detected by a phosphospecific Ser302 antibody in extracts from serum-starved isogenic TSC2 +/+ and TSC2 −/− MEFs, and TSC2 −/− MEFs lines in which wild-type (−/− (+WT)) or a pathogenic mutant TSC2 (−/− (+N1643K)) has been reintroduced; top, Ser302-phosphorylated IRS-1; bottom, total IRS-1. (F) IRS-1 Ser302 phosphorylation is inhibited by mTOR/S6K inhibition with rapamycin in serum-starved TSC2 −/− MEFs. Top, Ser302-phosphorylated IRS-1; bottom, total IRS-1. Where indicated, MEFs were also stimulated with insulin for 10 min. (G) IRS-1 Ser302 phosphorylation is diminished in TSC2 −/− MEFs after siRNA-mediated inhibition of S6K1. Top, Ser302-phosphorylated IRS-1; bottom, total IRS-1. Where indicated, MEFs were also stimulated with insulin for 10 min.
Phosphorylation by S6K inhibits IRS-1 function
Next, we asked whether IRS-1 could be tyrosine phosphorylated and associate with PI3K in TSC2 −/− MEFs after stimulation with insulin. The results show that in TSC2 −/− MEFs, IRS-1 is both poorly tyrosine phosphorylated and associated with PI3K after stimulation of cells with insulin. Rapamycin treatment for 1 h results in a partial restoration of both IRS-1 tyrosine phosphorylation and association with the p85 subunit of PI3K in TSC2 −/− MEFs (Fig. 6, A and B). Importantly, this short-term treatment does not significantly alter IRS-1 protein levels (Fig. 4, B and E), which argues that the reduction in signaling capacity is attributable to impaired function as well as a reduction in the pool of available IRS-1. Sustained inhibition of S6K signaling by rapamycin treatment of TSC2 −/− MEFs restores both tyrosine phosphorylation and PI3K association to levels approaching that of TSC2 +/+ cells (Fig. 6 A). Then, we asked whether phosphorylation of IRS-1108–516 by S6K directly inhibits its association with activated insulin receptors. We phosphorylated in vitro a GST fusion protein containing the phosphotyrosine-binding domain (PTB) of IRS-1 with S6K, and determined the amount of activated insulin receptor that associated with unphosphorylated or phosphorylated IRS-1 in a pull-down assay. The result shows that phosphorylation of the IRS-1 PTB domain by S6K markedly decreases its association with insulin receptor (Fig. 6, C and D), whereas a Ser302-glutamic acid mutant, which may mimic the effect of phosphorylation at this site, exhibits impaired binding to the insulin receptor (Fig. 6, E and F). Thus, S6 kinases directly phosphorylate IRS-1 at Ser302, and the resulting reduction of binding to insulin receptors is likely to play a major role in the inability of IRS-1 to signal to PI3K.
Figure 6.
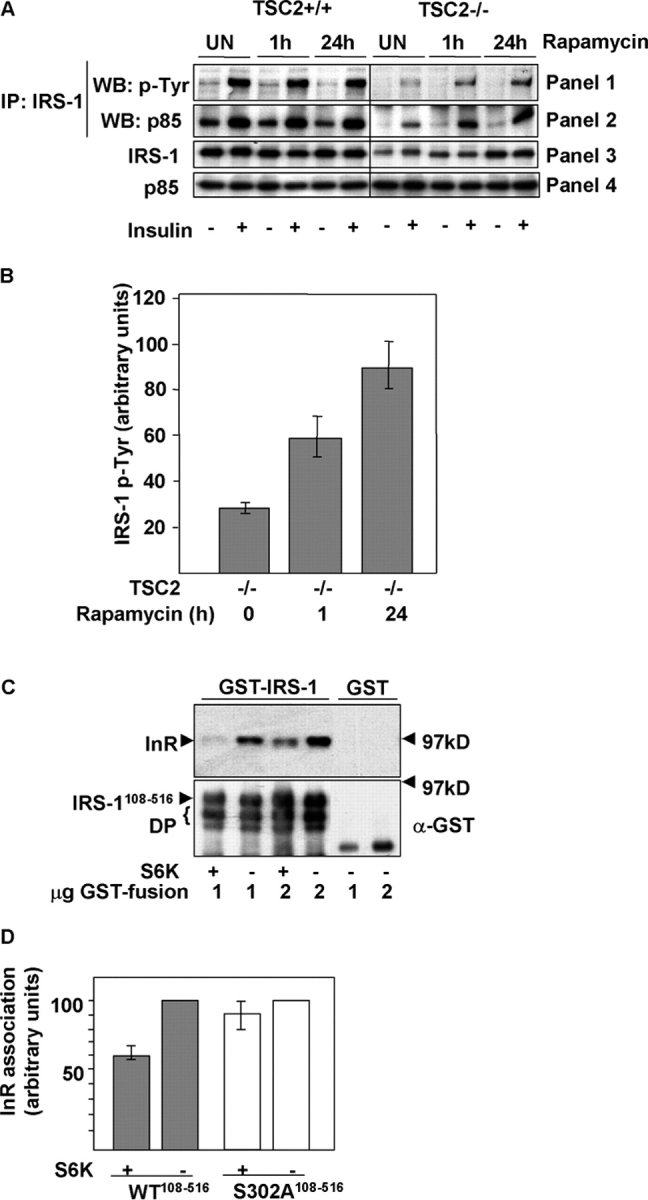
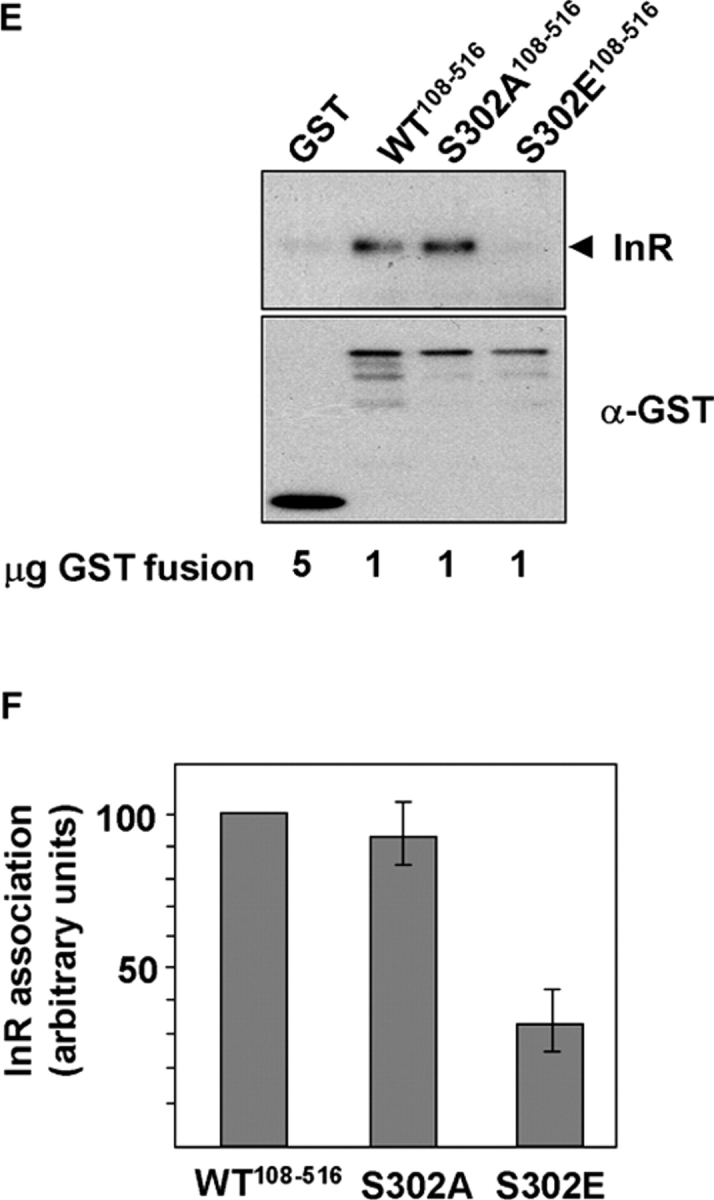
S6K phosphorylation blocks IRS-1 function. (A) IRS-1 tyrosine phosphorylation and association with PI3K is suppressed in TSC2 −/− MEFs and rescued by inhibition of S6K. IRS-1 immunoprecipitates in unstimulated or insulin-stimulated TSC2 +/+ or TSC2 −/− MEFs after rapamycin treatment for the indicated times, immunoblotted for phosphotyrosine (panel 1), or the coimmunoprecipitation of the p85 subunit of PI3K (panel 2). The total levels of IRS-1 and p85 in cell lysates are indicated in panels 3 and 4, respectively. (B) Quantitation of insulin- stimulated IRS-1 tyrosine phosphorylation in untreated TSC2 −/− MEFs or the same cells after rapamycin treatment for the indicated times. An arbitrary value was obtained representing the difference between unstimulated and the corresponding insulin-stimulated level of phosphotyrosine in IRS-1 immunoprecipitations as detected on scanned autoradiographs. The values from three independent experiments were averaged and plotted on a bar chart, with the error bars showing the SEM. (C) Phosphorylation of immobilized GST-IRS-1108–516 PTB domain by S6K2 inhibits insulin receptor (InR) interaction. The interaction with tyrosine-phosphorylated InR (top) was determined in vitro by the addition of glutathione beads containing the indicated amounts of S6K2-phosphorylated or unphosphorylated GST-IRS-1108–516 or GST to lysates prepared from insulin-stimulated CHO-IR cells. An immunoblot of the level of GST-IRS-1 fusion protein or GST is indicated on the bottom. DP indicates degradation products of the GST-IRS-1 protein. (D) Quantitation of InR binding to GST-IRS-1108–516 (WT) or a mutant GST-IRS-1108–516 in which serine 302 is mutated to alanine (S302A) after in vitro phosphorylation by S6K2. Error bars show the SEM from three experiments. (E) InR binding of wild-type GST-IRS-1108–516, or S302A and S302E mutants in a pull-down assay. Top, InR; bottom, anti-GST. (F) Quantitation of InR binding to GST-IRS-1108–516 (WT) or S302A and S302E mutants. An arbitrary value of 100 was assigned for the binding of wild-type GST-IRS-1108–516, and the binding of the mutant IRS-1 proteins was expressed relative to this value. Error bars show the SEM from three experiments.
Impaired PI3K signaling abrogates IGF-1–mediated chemotaxis and survival
PI3K activation by insulin or IGF-1 is required for migration/chemotaxis and survival. Survival is known to involve activation of PKB (Lawlor and Alessi, 2001), whereas chemotaxis requires PI3K activity (Puglianiello et al., 2000). Therefore, we tested the responses of TSC2 +/+ or TSC2 −/− MEFs to IGF-1 in chemotaxis and survival assays. We observe a similar basal and EGF-stimulated rate of migration for both TSC2 genotypes, but a pronounced impairment in IGF-1–stimulated chemotaxis in TSC2 −/− MEFs. Pretreatment with rapamycin for 24 h has little effect on migration of TSC2 +/+ MEFs, but restores IGF-stimulated migration of TSC2 −/− MEFs (Fig. 7, A and B).
Figure 7.


IGF-1–mediated migration and survival are impaired in TSC2 -deficient MEFs. (A) A chemotaxis assay indicating the migration of TSC2 +/+ or TSC2 −/− MEFs in the absence or presence of 50 ng/ml IGF-1 or 20 ng/ml EGF as a chemotactic factor in a Transwell assay. The pictures show fluorescently labeled cells that have traversed a fluorescently inert filter, such that only the cells that have crossed the filter are photographed. Basal migration (left-hand panels) in the presence of 0.5% FCS present in both chambers and IGF-1–stimulated migration (middle panels) or EGF-stimulated migration (rightmost two panels) is shown in untreated cells (top six panels) or cells pretreated with 20 nM rapamycin for 24 h (bottom four panels). (B) Quantitation of IGF-1– or EGF-mediated chemotaxis in the chemotaxis assay of TSC2 +/+ (gray bars) or TSC2 −/− (white bars) MEFs. The results are expressed as a percentage of that of IGF-1–stimulated chemotaxis of TSC2 +/+ MEFs not treated with rapamycin (100%) from four random 10× fields of two duplicate wells. Error bars show the SEM values. (C) Cell survival assay in the absence of serum. A quantitative analysis of the percentage of TUNEL-positive nuclei is shown for TSC2 +/+ (gray bars) and TSC2 −/− (white bars) MEFs after 8 h in serum-free medium alone (−) or supplemented with 200 ng/ml IGF-1 (+) in untreated MEFs or MEFs pretreated for 36 h with 20 nM rapamycin. The results are from four random 10× fields and are typical of experiments performed twice. (D) Relative cell survival of TSC2 +/+ (gray bars) or TSC2 −/− (white bars) MEFs. The results were obtained by dividing the means of the percentages of TUNEL-positive nuclei in the absence of IGF-1 by the means of those in the presence of IGF-1.
Next, we tested the ability of IGF-1 to act as a survival factor for TSC2 +/+ or TSC2 −/− cells. We assessed survival after serum removal, an assay in which IGF-1 is known to act as a survival factor (Tamm and Kikuchi, 1990). IGF-1 significantly protects TSC2 +/+ MEFs from apoptosis, but has little protective effect on TSC2 −/− cells (Fig. 7). TSC2 −/− MEFs exhibit a higher basal rate of survival in the absence of serum, raising the possibility that additional pro-survival pathways may be activated in these cells. Because rapamycin has been shown to lead to increased apoptosis of TSC2-deficient tumor cells in vivo (Kenerson et al., 2002) and S6K1 phosphorylation has been implicated in inactivation of the pro-apoptotic factor Bad (Harada et al., 2001), we assessed basal and IGF-1–mediated survival after treatment of cells with rapamycin. The results show that rapamycin treatment has little effect on the survival of TSC2 +/+ MEFs. However, rapamycin has two effects on TSC2 −/− MEF survival, decreasing basal survival in the absence of IGF-1 while increasing survival in the presence of IGF-1 (Fig. 7, C and D). We conclude that under the conditions of serum withdrawal, IGF-1 does not activate pro-survival signaling in TSC2-deficient cells, and that a weaker pro-survival signal is instead provided by mTOR/S6K signaling.
Discussion
The insulin–PI3K pathway controls cell number (through promoting cell cycle progression and survival via PKB/AKT; Lawlor and Alessi, 2001) and cell growth (via activation of the S6Ks; Dufner and Thomas, 1999). How, then, do cells and tissues coordinate these activities, given that different relative amounts of cells and growth may normally be required to obtain a given organ size, or to modulate this size during development or in the adult? Regulators of coordination may be inferred from human pathologic overgrowth conditions in which such coordination is lost, or in model organisms such as Drosophila where loss-of-function mutations can be analyzed for their effect on organ growth. Both PTEN (Nelen et al., 1997) and TSC1-2 (mutated in TSC; The European Chromosome 16 Tuberous Sclerosis Consortium, 1993; van Slegtenhorst et al., 1997) may play coordinating roles in organ size control (Goberdhan et al., 1999; Tapon et al., 2001). However, PTEN (but not thus far TSC1-2) mutations are known to promote tumorigenesis, with the gene frequently mutated or lost in advanced neoplasms (Li et al., 1997). A connection between PTEN and TSC1-2 has also been proposed via PTEN loss of function leading to hyperactivation of the protein kinase PKB/AKT, which phosphorylates tuberin (Inoki et al., 2002; Manning et al., 2002). Tuberin phosphorylation has been suggested to negatively regulate the TSC1-2 protein complex, allowing activation of S6K and cell growth (Potter et al., 2002).
In this paper, we have explored the failure of insulin to activate PKB in TSC2 −/− cells (Jaeschke et al., 2002). However, the absence of TSC1-2 complex function leads not to a specific failure in PKB activation, but rather to a general failure in insulin's ability to activate PI3K. As a result of TSC1-2 loss of function, insulin-like growth factors do not activate PI3K due to S6K inactivating the adaptor proteins IRS-1 and IRS-2. S6K activation appears to attenuate PI3K signaling through suppressing the adaptor protein IRS-1, both at the level of gene transcription and at the level of its function in coupling activated receptors to PI3K. The former mechanism is unexpected, and is mediated by S6K1 and S6K2, indicating that these protein kinases have a direct role in regulating IRS-1 transcription. A transcription factor, CREM, has already been shown to be a substrate for S6K1 (de Groot et al., 1994).
Although the full range of consequences of activation of the mTOR/S6K pathway remain to be determined, it appears that a reduced responsiveness to growth factors may not be restricted to insulin signaling ( H. Zhang et al., 2003). Future experiments will help clarify whether such “indirect” inhibitory consequences of aberrant mTOR/S6K activation on cell signaling play a role in the distinctive pathology of hamartoma development in TSC.
A second major mechanism of IRS-1 inactivation by S6K is through phosphorylation. We have identified a site (Ser302) proximal to the IRS-1 PTB domain that we show is phosphorylated by S6K in vitro, which shows increased phosphorylation in TSC2-deficient cells and is sensitive to inhibition of mTOR/S6K signaling. Our data indicate that, at least in TSC2 −/− MEFs, the major kinase responsible for phosphorylating IRS-1 Ser302 is S6K1 and that phosphorylation of this site may disrupt the ability of the PTB domain to interact with activated insulin receptors. Reversal of this phosphorylation by short-term treatment with rapamycin, or inhibition of S6K1 by RNAi, partially rescues activation of PKB, arguing that phosphorylation by S6K may be a major mechanism to inactivate IRS-1 function. However, the extent of the band shift we observe in both IRS-1 and IRS-2 extracted from TSC2-deficient cells might suggest that more than one site is phosphorylated by S6K (or perhaps other mTOR-regulated kinases) in vivo; our data support the argument that the direct phosphorylation of the PTB domain site could account for the profound inhibition of IRS-1 function in these cells. Giraud et al. (2004) have also reported that IRS-1 is phosphorylated at Ser302, by an unidentified nutrient- and rapamycin-sensitive kinase. However, using a Ser302-alanine mutant in 32D cells, these workers report that Ser302 phosphorylation may be necessary for association with PI3K p85 and for a restricted set of insulin–PI3K-regulated responses including activation of S6K and DNA synthesis. Surprisingly, their work indicates that activation of PKB is largely unaffected by preventing phosphorylation at this site, suggesting that PIP3 generation is unaffected by preventing Ser302 phosphorylation. However, they did not examine the consequences upon insulin signaling of a phosphomimic of Ser302 (which may reflect the situation occurring upon activation of mTOR/S6K signaling reported here), rendering it difficult to determine what the consequences of increased phosphorylation of this site may be. However, our finding that regulation of IRS by S6K occurs by both long-term (transcriptional) and acute (phosphorylation) mechanisms, and that both these mechanisms act in the same manner (i.e., inhibitory) toward insulin–PI3K activation, is consistent with what we observe in TSC2 −/− cells where S6K is hyperactive and PI3K activation is defective. We suggest that TSC1-2, S6K, and S6K-regulated inactivation of IRS proteins is a negative feedback sensor that may be designed to relate the amount of nutrient/S6K-dependent growth to the magnitude of insulin's activation of PI3K.
The contribution of unregulated S6K activity to tumorigenesis is currently unknown. Some TSC1-2–deficient lesions have increased levels of phosphorylated S6, indicating elevation of S6K activity (Kwiatkowski et al., 2002; Karbowniczek et al., 2003). Treatment of Eker rat tumors, in which TSC2 is inactivated, with the mTOR inhibitor rapamycin leads to increased apoptosis of these cells, indicating that mTOR/S6K signaling may be important here (Kenerson et al., 2002). However, it is also clear that mice rendered TSC-null by genetic means do not develop beyond mid-gestation (Kwiatkowski et al., 2002), whereas in TSC, hamartomatous growths are generally slow or normally growing, and have a low malignant potential. This contrasts with PTEN inactivation in Cowden disease, in which predisposition to malignancy is evident (Starink et al., 1986). Our model (Fig. 8) suggests that an explanation for the low level of malignancy observed in TSC1-2–deficient tumors may be that insufficient pro-tumorigenic PIP3-dependent signals (such as PKB activation) are generated in TSC1-2–deficient tumor cells, and that such signals may be required for malignant conversion. Our data also suggest a paradigm of how appropriate organ size control may be achieved: cell growth through S6 kinases may be either coordinated with PI3K-regulated cell number control or uncoupled from it, depending on the presence or absence of a functional TSC1-2 complex.
Figure 8.

A model for TSC1-2 regulation of insulin signaling to PI3K. The elevated production of the inositol phospholipid PIP3 by PI3K and its antagonism by PTEN (and 5-phosphatases; not depicted) determines the activation of downstream responses such as activation of PKB. A further downstream response to PKB activation may be the inactivation of TSC1-2 via phosphorylation of tuberin, resulting in S6K activation. In our model, TSC1-2 promotes insulin-like growth factor signaling to PI3K by repressing a negative feedback loop from mTOR/S6K to the adaptor molecule IRS-1. This negative regulation by TSC1-2 may be on mTOR effectors (Jaeschke et al., 2002) or on mTOR directly (Tee et al., 2002). A similar negative feedback involving S6K repression of PKB activation has also been described in Drosophila (Radimerski et al., 2002), although the target of dS6K's inhibitory action here is not known. Repression of IRS-1 by mTOR/S6K occurs via both a transcriptional repression of IRS-1 gene expression mediated by both S6K1 and S6K2 and by direct phosphorylation of IRS-1 protein close to the PTB domain by S6K1. Therefore, failure to activate PI3K may restrict the malignant potential of TSC-defective cells.
Materials and methods
Antibodies
Commercial antibodies used were as follows: rabbit anti-GST, anti-p85, anti-IRS-1, and mouse anti-phosphotyrosine (clone 4G10) were from Upstate Biotechnology. Anti-PKB, IGF-1R, pS6 (Ser240/244), PKB Ser473, and total GSK3α/β and GSK3α/β Ser21/9 were from Cell Signaling. Antibodies to ERK1/2 and S6K1 were from Santa Cruz Biotechnology, Inc., and Transduction Laboratories, respectively. An anti-InR mAb CT3 was from K. Siddle (University of Cambridge, Cambridge, UK), and an antibody to S6K2 was from D. Alessi (University of Dundee, Dundee, UK). Antibodies to IRS-2 and IRS-1 phosphorylated at Ser302 were gifts from M. White (Joslin Diabetes Center, Boston, MA).
Cell culture, treatments with growth factors, and siRNA transfections
Starved MEFs (Jaeschke et al., 2002) were stimulated with 1 μg/ml insulin, 50 ng/ml IGF-I, or 20 ng/ml EGF for 10 min. CHO-IR cells were maintained in Ham's F12 medium with 10% FCS and were starved in medium lacking serum for 24 h. To activate insulin receptors, cells were then stimulated for 10 min with 1 μM insulin. siRNAs (Dharmacon) for S6K were: S6K1, 5′-GGACATGGCAGGAGTGTTT-3′; and S6K2, 5′-GAACCAAGAAGTCCAAGAA-3′.
For IRS-1 and IRS-1/IRS-2 double transfections, the sequences used were as described previously (Pirola et al., 2003). For IRS-2 single transfection, the following sequence was used: IRS-2, 5′-CCAAGCACAAGTACCTGAT-3′.
SiRNAs were transfected using LipofectAMINE™ 2000 and cells were harvested after 24 h (for IRS-1 and IRS-2) or 5 d (S6K1 and S6K2).
PI3K assay and PIP3 determination
Assays were performed as described previously (Herbert et al., 2000) and were imaged using a PhosphorImager (Fuji); bands were quantified using AIDA analysis software. Results were expressed as histograms in Microsoft Excel. Estimation of PIP3 was by a TRFRET displacement assay (Gray et al., 2003). The estimates of PIP3 were normalized to total lipid extracted as described previously (Raheja et al., 1973).
RNA analysis
Murine gene oligo microarray slides (∼7.5k genes/slide) were obtained from the Human Mapping Project (Cambridge, UK). Fluorescently labeled cDNA probes were prepared using the Atlas™ Glass Fluorescent Labeling Kit (CLONTECH Laboratories, Inc.) and Cy3 and Cy5 reactive dyes (Amersham Biosciences), and were processed according to the manufacturer's instructions. For Northern blotting, RNA was run on agarose-formaldehyde gels, blotted onto Hybond™-N, and hybridized and washed using standard procedures. Two-step quantitative RT-PCR was performed using cDNA synthesized using the Omniscript™ RT Kit (QIAGEN) using total RNA purified from appropriately treated MEFs using the RNeasy™ Mini Kit (QIAGEN). For the actinomycin D experiment, TSC2 −/− cells were starved for 24 h in the absence or presence of 20 nM rapamycin or for 24 h with rapamycin with addition of 10 μg/ml actinomycin D for the last 10 h. Quantitative PCR was performed using the JOE-labeled mouse β-actin LUX™ Primer set (Invitrogen) and an IRS-1–specific primer pair with a 6-FAM–labeled probe synthesized by QIAGEN. Reactions were performed using the PCR Master Mix for probe assays (Eurogentech) with β-actin LUX™ primers at a final concentration of 200 nM, the IRS-1 primers at a final concentration of 300 nM, and the probe at 100 nM. Reactions were performed in triplicate using a sequence detector (ABI Prism™ 7700; Applied Biosystems). Results were analyzed using Sequence Detector v1.7 software.
Immunoprecipitations
Cells were lysed in buffer (50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 1% NP-40, 1 mM EDTA, 50 mM NaF, 40 mM β-glycerolphosphate, 5 mM Na3VO4, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 mM benzamidine). 300 μg protein was added to 2 μg anti-IRS-1 antibody and immunoprecipitations were performed for 12 h at 4°C, followed by a further 2-h rotation after addition of 10 μl protein G–agarose and washing. For λ-phosphatase experiments, washed immunoprecipitates were washed with λ-phosphatase reaction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM MnCl2, 0.1 mM EDTA, and 5 mM DTT) and resuspended in 15 μl of this buffer with or without 400 U purified λ-phosphatase, and reactions were incubated at 30°C for 1 h with regular agitation.
Protein methods
GST-IRS-1 (aa 21–400, 108–516, 516–895, and 895–1235) were eluted from glutathione-agarose beads in 50 mM Tris-HCl, pH 8.0, and 10 mM glutathione, dialyzed against 50 mM Tris-HCl, pH 8.5, 150 mM NaCl, 10% glycerol, 1 mM PMSF, 1 mM benzamidine, and 1 mM DTT, and were concentrated. Recombinant EE-S6K1 and EE-S6K2 were purified from insect cells infected with generated viruses using mouse EE mAb immobilized to protein A–Sepharose CL-4B (Amersham Biosciences). In vitro kinase assays were performed in kinase assay buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 50 μM cold ATP, 5 μCi γ[32P]-labeled ATP) using either 1 μg GST-IRS-1 fragments or 0.4 μl 40S ribosomal preparation as a substrate (Gout et al., 1998). Reactions were at 30°C for 20 min.
For the InR-binding assay, phosphorylation of GST-IRS-1 was performed for 30 min at 37°C in 15 μl kinase reaction buffer with 200 μM cold ATP and 0.6 μg S6K2. Proteins were bound to glutathione-agarose and washed twice with kinase buffer and once with lysis buffer A (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 50 mM NaF, 1 mM benzamidine, 1 mM EDTA, 1% NP-40, 40 mM β-glycerophosphate, 5 mM sodium orthovanadate, 2 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 10% glycerol). CHO cells overexpressing IR were lysed in buffer A, and 0.5 mg protein was used for the binding assay performed at 4°C for 1 h, followed by three washes with A buffer, addition of SDS-PAGE sample buffer, and SDS-PAGE. Mutants of GST-IRS-1 (aa 108–516) were produced using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene). For the quantitation and statistical analysis of InR pull-down, autoradiographs were scanned and the pixel intensity of bands was determined using Adobe Photoshop® and corrected for background pixel intensity. Insulin receptor associated with GST-IRS-1 wild-type and GST-IRS-1 S302A in each control experiment (i.e., without kinase) was assigned a value of 100%. Insulin receptor associated with GST-IRS-1 wild type and GST-IRS-1 S302A after phosphorylation by S6K was normalized relative to the control value of 100% in each experiment. Values from three independent experiments were analyzed, and the average value was plotted on the graph.
Cell chemotaxis assay
MEFs were starved for 24 h in the presence or absence of 20 nM rapamycin and were labeled 1 h further with 2 μg/ml CellTracker™ Green (Molecular Probes, Inc.) before trypsinization. 20,000 cells were plated in duplicate on Fluoro-block Transwell filters (Becton Dickinson) in DME containing 0.5% FCS, and were placed in wells containing the same media or media containing 50 ng/ml recombinant IGF-1 or 20 ng/ml recombinant EGF (Sigma-Aldrich). Chemotaxis was for 6 h, after which the wells were fixed in PBS/4% PFA. Fluorescence was visualized in 10× fields using FITC excitation/emission on an upright microscope (DM-IRB; Leica) equipped with a digital camera (CoolSnap) and Darkroom image acquisition software (Improvision). For quantitation, three random fields were imaged from each duplicate assay and the number of spread fluorescent cells that had crossed the filter was counted manually. Cell numbers were expressed as a percentage of that of TSC2 +/+ in the presence of IGF-1, and a histogram was generated in Microsoft Excel.
Cell survival assay
20,000 cells were seeded in duplicate onto coverslips in the absence or presence of 20 nM rapamycin. After 36 h, coverslips were washed twice in PBS and placed in wells containing serum-free DME in the presence or absence of 200 ng/ml IGF-1 for 8 h. Cells were fixed in 4% PFA, permeabilized, and subjected to FITC-TUNEL assay using a commercial kit (Apoptosis Detection System; Promega) with the addition of a 30-min labeling stage with 1 μg/ml Hoechst bisbenzamide to label cell nuclei. In each duplicate assay, two 10× fields containing 300–600 nuclei were imaged with DAPI and FITC excitation/emission on a microscope (Axioplan 2; Carl Zeiss MicroImaging, Inc.) with an Axioplan camera and software. In each field, the number of TUNEL-positive nuclei and nuclei for each condition was counted manually and expressed as a percentage and a histogram generated in Microsoft Excel.
Acknowledgments
We wish to thank Alan Ashworth for help with microarray experiments, Tony Ford for advice on quantitative RT-PCR, and Paula Muir for data analysis and DNA sequencing. We are indebted to Stuart Frank for GST-IRS-1 constructs, Morris White for antibodies, Derek LeRoith for expression vectors, and to Chris Marshall for discussion, critical reading of the manuscript, and support.
L.S. Harrington is supported by the Tuberous Sclerosis Association (UK), C.P. Downes by a program grant from the Medical Research Council (UK) and by the Division of Signal Transduction Therapy, and J. Barnett by the Biotechnology and Biological Sciences Research Council via a CASE studentship supported by GlaxoSmithKline. T. Tolkacheva, S. Wigfield, and R.F. Lamb are supported by grants from Cancer Research UK, and G.M. Findlay by a studentship from the Institute of Cancer Research. We gratefully acknowledge the help and support of the Tuberous Sclerosis Association (UK) and LAM Foundation (US).
L.S. Harrington and G.M. Findlay contributed equally to this paper.
Abbreviations used in this paper: IRS, insulin receptor substrate; MEF, mouse embryo fibroblast; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-OH kinase; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PTB, phosphotyrosine-binding domain; siRNA, small interfering RNA; TSC, tuberous sclerosis complex.
References
- Brown, E.J., P.A. Beal, C.T. Keith, J. Chen, T.B. Shin, and S.L. Schreiber. 1995. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature. 377:441–446. [DOI] [PubMed] [Google Scholar]
- Cantley, L.C. 2002. The phosphoinositide 3-kinase pathway. Science. 296:1655–1657. [DOI] [PubMed] [Google Scholar]
- Crackower, M.A., G.Y. Oudit, I. Kozieradzki, R. Sarao, H. Sun, T. Sasaki, E. Hirsch, A. Suzuki, T. Shioi, J. Irie-Sasaki, et al. 2002. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 110:737–749. [DOI] [PubMed] [Google Scholar]
- de Groot, R.P., L.M. Ballou, and P. Sassone-Corsi. 1994. Positive regulation of the cAMP-responsive activator CREM by the p70 S6 kinase: an alternative route to mitogen-induced gene expression. Cell. 79:81–91. [DOI] [PubMed] [Google Scholar]
- Dufner, A., and G. Thomas. 1999. Ribosomal S6 kinase signaling and the control of translation. Exp. Cell Res. 253:100–109. [DOI] [PubMed] [Google Scholar]
- The European Chromosome 16 Tuberous Sclerosis Consortium. 1993. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 75:1305–1315. [DOI] [PubMed]
- Gao, X., and D. Pan. 2001. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 15:1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garami, A., F.J. Zwartkruis, T. Nobukuni, M. Joaquin, M. Roccio, H. Stocker, S.C. Kozma, E. Hafen, J.L. Bos, and G. Thomas. 2003. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell. 11:1457–1466. [DOI] [PubMed] [Google Scholar]
- Giraud, J., R. Leshan, Y.H. Lee, and M.F. White. 2004. Nutrient-dependent and insulin-stimulated phosphorylation of insulin receptor substrate-1 on serine 302 correlates with increased insulin signaling. J. Biol. Chem. 279:3447–3454. [DOI] [PubMed] [Google Scholar]
- Goberdhan, D.C., N. Paricio, E.C. Goodman, M. Mlodzik, and C. Wilson. 1999. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. 13:3244–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout, I., T. Minami, K. Hara, Y. Tsujishita, V. Filonenko, M.D. Waterfield, and K. Yonezawa. 1998. Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase β containing a proline-rich region. J. Biol. Chem. 273:30061–30064. [DOI] [PubMed] [Google Scholar]
- Gray, A., H. Olsson, I.H. Batty, L. Priganica, and C. Peter Downes. 2003. Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal. Biochem. 313:234–245. [DOI] [PubMed] [Google Scholar]
- Harada, H., J.S. Andersen, M. Mann, N. Terada, and S.J. Korsmeyer. 2001. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA. 98:9666–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruta, T., T. Uno, J. Kawahara, A. Takano, K. Egawa, P.M. Sharma, J.M. Olefsky, and M. Kobayashi. 2000. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol. 14:783–794. [DOI] [PubMed] [Google Scholar]
- Herbert, T.P., G.R. Kilhams, I.H. Batty, and C.G. Proud. 2000. Distinct signalling pathways mediate insulin and phorbol ester-stimulated eukaryotic initiation factor 4F assembly and protein synthesis in HEK 293 cells. J. Biol. Chem. 275:11249–11256. [DOI] [PubMed] [Google Scholar]
- Inoki, K., Y. Li, T. Zhu, J. Wu, and K.L. Guan. 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4:648–657. [DOI] [PubMed] [Google Scholar]
- Ito, N., and G.M. Rubin. 1999. gigas, a Drosophila homolog of tuberous sclerosis gene product-2, regulates the cell cycle. Cell. 96:529–539. [DOI] [PubMed] [Google Scholar]
- Jaeschke, A., J. Hartkamp, M. Saitoh, W. Roworth, T. Nobukuni, A. Hodges, J. Sampson, G. Thomas, and R. Lamb. 2002. Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J. Cell Biol. 159:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies, H.B., S. Fumagalli, P.B. Dennis, C. Reinhard, R.B. Pearson, and G. Thomas. 1997. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 16:3693–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowniczek, M., J. Yu, and E.P. Henske. 2003. Renal angiomyolipomas from patients with sporadic lymphangiomyomatosis contain both neoplastic and non-neoplastic vascular structures. Am. J. Pathol. 162:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenerson, H.L., L.D. Aicher, L.D. True, and R.S. Yeung. 2002. Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res. 62:5645–5650. [PubMed] [Google Scholar]
- Kwiatkowski, D.J., H. Zhang, J.L. Bandura, K.M. Heiberger, M. Glogauer, N. el-Hashemite, and H. Onda. 2002. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 11:525–534. [DOI] [PubMed] [Google Scholar]
- Lawlor, M.A., and D.R. Alessi. 2001. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 114:2903–2910. [DOI] [PubMed] [Google Scholar]
- Lawlor, M.A., A. Mora, P.R. Ashby, M.R. Williams, V. Murray-Tait, L. Malone, A.R. Prescott, J.M. Lucocq, and D.R. Alessi. 2002. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 21:3728–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevers, S.J., D. Weinkove, L.K. MacDougall, E. Hafen, and M.D. Waterfield. 1996. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 15:6584–6594. [PMC free article] [PubMed] [Google Scholar]
- Li, J., C. Yen, D. Liaw, K. Podsypanina, S. Bose, S.I. Wang, J. Puc, C. Miliaresis, L. Rodgers, R. McCombie, et al. 1997. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 275:1943–1947. [DOI] [PubMed] [Google Scholar]
- Li, Y., M.N. Corradetti, K. Inoki, and K.L. Guan. 2004. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem. Sci. 29:32–38. [DOI] [PubMed] [Google Scholar]
- Manning, B.D., A.R. Tee, M.N. Logsdon, J. Blenis, and L.C. Cantley. 2002. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell. 10:151–162. [DOI] [PubMed] [Google Scholar]
- Martin, K.A., S.S. Schalm, C. Richardson, A. Romanelli, K.L. Keon, and J. Blenis. 2001. Regulation of ribosomal S6 kinase 2 by effectors of the phosphoinositide 3-kinase pathway. J. Biol. Chem. 276:7884–7891. [DOI] [PubMed] [Google Scholar]
- Nelen, M.R., W.C. van Staveren, E.A. Peeters, M.B. Hassel, R.J. Gorlin, H. Hamm, C.F. Lindboe, J.P. Fryns, R.H. Sijmons, D.G. Woods, et al. 1997. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum. Mol. Genet. 6:1383–1387. [DOI] [PubMed] [Google Scholar]
- Park, I.H., R. Bachmann, H. Shirazi, and J. Chen. 2002. Regulation of ribosomal S6 kinase 2 by mammalian target of rapamycin. J. Biol. Chem. 277:31423–31429. [DOI] [PubMed] [Google Scholar]
- Pirola, L., S. Bonnafous, A.M. Johnston, C. Chaussade, F. Portis, and E. Van Obberghen. 2003. Phosphoinositide 3-kinase-mediated reduction of insulin receptor substrate-1/2 protein expression via different mechanisms contributes to the insulin-induced desensitization of its signaling pathways in L6 muscle cells. J. Biol. Chem. 278:15641–15651. [DOI] [PubMed] [Google Scholar]
- Potter, C.J., H. Huang, and T. Xu. 2001. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 105:357–368. [DOI] [PubMed] [Google Scholar]
- Potter, C.J., L.G. Pedraza, and T. Xu. 2002. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 4:658–665. [DOI] [PubMed] [Google Scholar]
- Puglianiello, A., D. Germani, P. Rossi, and S. Cianfarani. 2000. IGF-I stimulates chemotaxis of human neuroblasts. Involvement of type 1 IGF receptor, IGF binding proteins, phosphatidylinositol-3 kinase pathway and plasmin system. J. Endocrinol. 165:123–131. [DOI] [PubMed] [Google Scholar]
- Radimerski, T., J. Montagne, M. Hemmings-Mieszczak, and G. Thomas. 2002. Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signaling. Genes Dev. 16:2627–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raheja, R.K., C. Kaur, A. Singh, and I.S. Bhatia. 1973. New colorimetric method for the quantitative estimation of phospholipids without acid digestion. J. Lipid Res. 14:695–697. [PubMed] [Google Scholar]
- Reif, K., B.M. Burgering, and D.A. Cantrell. 1997. Phosphatidylinositol 3-kinase links the interleukin-2 receptor to protein kinase B and p70 S6 kinase. J. Biol. Chem. 272:14426–14433. [DOI] [PubMed] [Google Scholar]
- Reinhard, C., G. Thomas, and S.C. Kozma. 1992. A single gene encodes two isoforms of the p70 S6 kinase: activation upon mitogenic stimulation. Proc. Natl. Acad. Sci. USA. 89:4052–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel, A.R. 2001. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell. 104:517–529. [DOI] [PubMed] [Google Scholar]
- Saltiel, A.R., and C.R. Kahn. 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 414:799–806. [DOI] [PubMed] [Google Scholar]
- Saucedo, L.J., X. Gao, D.A. Chiarelli, L. Li, D. Pan, and B.A. Edgar. 2003. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 5:566–571. [DOI] [PubMed] [Google Scholar]
- Shepherd, P.R., D.J. Withers, and K. Siddle. 1998. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem. J. 333:471–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima, H., M. Pende, Y. Chen, S. Fumagalli, G. Thomas, and S.C. Kozma. 1998. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 17:6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioi, T., J.R. McMullen, P.M. Kang, P.S. Douglas, T. Obata, T.F. Franke, L.C. Cantley, and S. Izumo. 2002. Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell. Biol. 22:2799–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starink, T.M., J.P. van der Veen, F. Arwert, L.P. de Waal, G.G. de Lange, J.J. Gille, and A.W. Eriksson. 1986. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin. Genet. 29:222–233. [DOI] [PubMed] [Google Scholar]
- Stocker, H., T. Radimerski, B. Schindelholz, F. Wittwer, P. Belawat, P. Daram, S. Breuer, G. Thomas, and E. Hafen. 2003. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat. Cell Biol. 5:559–565. [DOI] [PubMed] [Google Scholar]
- Tamm, I., and T. Kikuchi. 1990. Insulin-like growth factor-1 (IGF-1), insulin, and epidermal growth factor (EGF) are survival factors for density-inhibited, quiescent Balb/c-3T3 murine fibroblasts. J. Cell. Physiol. 143:494–500. [DOI] [PubMed] [Google Scholar]
- Tapon, N., N. Ito, B.J. Dickson, J.E. Treisman, and I.K. Hariharan. 2001. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 105:345–355. [DOI] [PubMed] [Google Scholar]
- Tee, A.R., D.C. Fingar, B.D. Manning, D.J. Kwiatkowski, L.C. Cantley, and J. Blenis. 2002. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA. 99:13571–13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Slegtenhorst, M., R. de Hoogt, C. Hermans, M. Nellist, B. Janssen, S. Verhoef, D. Lindhout, A. van den Ouweland, D. Halley, J. Young, et al. 1997. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 277:805–808. [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst, M., M. Nellist, B. Nagelkerken, J. Cheadle, R. Snell, A. van den Ouweland, A. Reuser, J. Sampson, D. Halley, and P. van der Sluijs. 1998. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 7:1053–1057. [DOI] [PubMed] [Google Scholar]
- Volarevic, S., and G. Thomas. 2001. Role of S6 phosphorylation and S6 kinase in cell growth. Prog. Nucleic Acid Res. Mol. Biol. 65:101–127. [DOI] [PubMed] [Google Scholar]
- White, M.F. 2002. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283:E413–E422. [DOI] [PubMed] [Google Scholar]
- Zhang, H., G. Cicchetti, H. Onda, H.B. Koon, K. Asrican, N. Bajraszewski, F. Vazquez, C.L. Carpenter, and D.J. Kwiatkowski. 2003. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J. Clin. Invest. 112:1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., X. Gao, L.J. Saucedo, B. Ru, B.A. Edgar, and D. Pan. 2003. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 5:578–581. [DOI] [PubMed] [Google Scholar]