

Abstract
Erythropoiesis requires tight control of expansion, maturation, and survival of erythroid progenitors. Because activation of phosphatidylinositol-3-kinase (PI3K) is required for erythropoietin/stem cell factor–induced expansion of erythroid progenitors, we examined the role of the PI3K-controlled Forkhead box, class O (FoxO) subfamily of Forkhead transcription factors. FoxO3a expression and nuclear accumulation increased during erythroid differentiation, whereas untimely induction of FoxO3a activity accelerated differentiation of erythroid progenitors to erythrocytes. We identified B cell translocation gene 1 (BTG1)/antiproliferative protein 2 as a FoxO3a target gene in erythroid progenitors. Promoter studies indicated BTG1 as a direct target of FoxO3a. Expression of BTG1 in primary mouse bone marrow cells blocked the outgrowth of erythroid colonies, which required a domain of BTG1 that binds protein arginine methyl transferase 1. During erythroid differentiation, increased arginine methylation coincided with BTG1 expression. Concordantly, inhibition of methyl transferase activity blocked erythroid maturation without affecting expansion of progenitor cells. We propose FoxO3a-controlled expression of BTG1 and subsequent regulation of protein arginine methyl transferase activity as a novel mechanism controlling erythroid expansion and differentiation.
Keywords: erythropoiesis; Forkhead transcription factors; protein arginine methylation; cDNA microarray; PRMT1
Introduction
Homeostasis of the hematopoietic system requires tight control of expansion, differentiation, and survival of progenitor cells, which is exerted by numerous cytokines and growth factors acting on specific cell types, such as erythropoietin (Epo), or on multiple types of progenitors, such as stem cell factor (SCF). The role of SCF in hematopoiesis is mainly to stimulate expansion and delay differentiation of hematopoietic progenitors in cooperation with more cell type–specific factors (for review see Broudy, 1997).
Erythroid progenitors can be expanded in the presence of Epo, SCF, and dexamethasone (Dex); whereas they differentiate into enucleated, hemoglobinized erythrocytes in the presence of Epo alone (Dolznig et al., 2001; von Lindern et al., 2001). Differentiation involves four differentiation-specific cell divisions with altered cell cycle regulation until a terminal G1 arrest is reached. SCF considerably delays differentiation, eventually yielding ∼20-fold increased numbers of mature erythrocytes. Erythroid differentiation is thought to be an autonomously regulated cascade of events in which Epo signaling is mainly required for survival and constitutive BCL-XL expression is sufficient to allow erythroid differentiation in defined medium lacking any factors (Dolznig et al., 2002). SCF, a potent activator of phosphatidylinositol-3-kinase (PI3K) in erythroid progenitors, is unable to induce cell survival in the absence of Epo (Dolznig et al., 2001; von Lindern et al., 2001). Instead, SCF signaling is required to delay differentiation, which is abrogated by PI3K inhibitor LY294002 (von Lindern et al., 2001). Both Epo and SCF activate PI3K and its target PKB (c-AKT; Bao et al., 1999). However, compared with Epo, SCF is much more potent in inducing PKB phosphorylation (von Lindern et al., 2001).
Phosphorylation and activation of the serine/threonine kinase PKB controls fundamental processes such as cell cycle progression, apoptosis, and mRNA translation (Datta et al., 1999; Raught and Gingras, 1999; Brazil and Hemmings, 2001). The Forkhead box, class O (FoxO) subfamily of Forkhead transcription factors is an important effector of PKB in regulating apoptosis and cell cycle progression. Members of this subfamily, FoxO4 (AFX), FoxO1a (FKHR), and FoxO3a (FKHR-L1), are directly phosphorylated by PKB, leading to cytoplasmic retention and inhibition of their transcriptional activity (Brunet et al., 1999; Kops et al., 1999; Tang et al., 1999). In the absence of phosphorylation, these transcription factors induce expression of genes encoding proteins that inhibit the cell cycle like p27Kip (Dijkers et al., 2000b; Medema et al., 2000), p130-Rb2 (Kops et al., 2002), and cyclin G2 (Ramaswamy et al., 2002), or proapoptotic proteins like Bim (Dijkers et al., 2000a) and the transcriptional repressor BCL-6, which inhibits expression of the antiapoptotic factor BCL-XL (Tang et al., 2002). Whether FoxO activation results in cell cycle arrest, induction of apoptosis, or other cell fates will depend on the cellular context.
We investigated the potential role of FoxO family members during expansion and differentiation of erythroid progenitors. FoxO3a expression increases during differentiation, resulting in nuclear accumulation 48 h after induction of differentiation. Activation of a phosphorylation-insensitive FoxO3a mutant accelerated differentiation under conditions favoring renewal. DNA microarray screens identified B cell translocation gene 1 (BTG1)/antiproliferative protein 2 as a novel FoxO3a target. Ectopic expression of BTG1 inhibited the expansion of mouse erythroid progenitor cells, which was dependent on the protein arginine methyl transferase 1 (PRMT1)–binding domain of BTG. We present data that suggest that modulation of protein arginine methylation activity by BTG1 may present a new FoxO-dependent mechanism regulating erythroid differentiation.
Results
FoxO3a expression and activity increase during erythroid differentiation
In search of PI3K-dependent pathways involved in the maintenance of erythroid progenitor renewal, we analyzed expression and function of the PI3K/PKB-regulated Forkhead family members FoxO4, FoxO1a, and FoxO3a during expansion and differentiation of the p53-deficient I/11 erythroid cell line. After induction of differentiation, expression of FoxO1a and FoxO4 increased for 24 h, but then rapidly declined to levels lower than those observed under renewal conditions (Fig. 1 A). In contrast, FoxO3a expression sharply increased 12–24 h after differentiation induction (Fig. 1 B), reaching maximal expression at 48 h and remaining high until completion of differentiation at 72 h (Fig. 1, A and B). Essentially the same results were obtained with primary erythroid progenitors expanded from E12.5 fetal livers, except that these cells express maximal levels of FoxO3a at 24 h and complete differentiation in 48 h (unpublished data).
Figure 1.
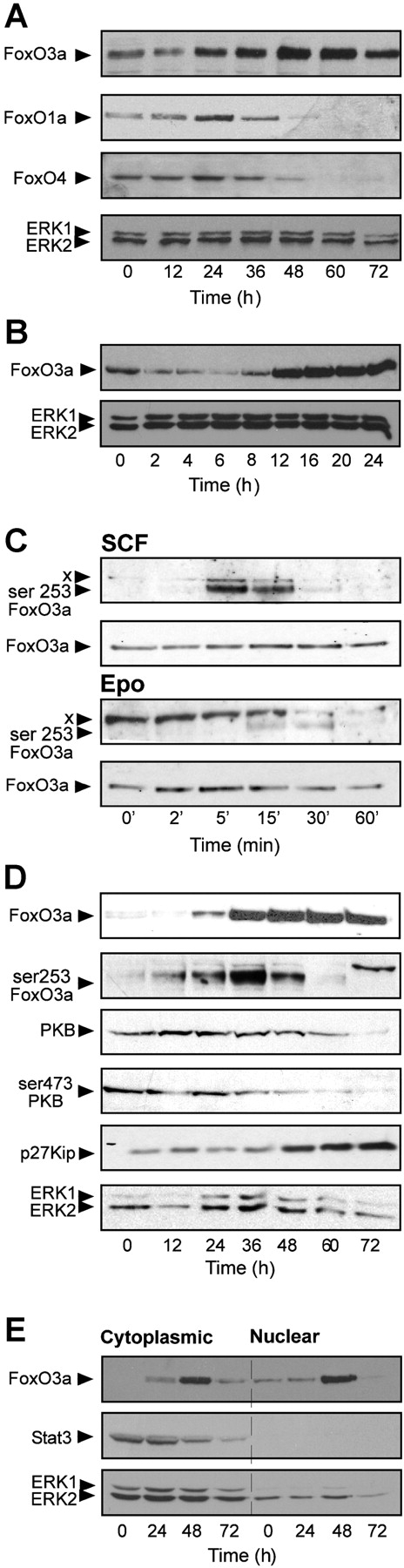
FoxO3a expression and activity increases during erythroid differentiation. (A) Western blots of differentiating I/11 cells (samples taken every 12 h) were analyzed with antibodies recognizing FoxO1a, FoxO3a, and FoxO4. ERK protein levels do not change during differentiation (loading control). (B) Western blot analysis of FoxO3a total protein levels during the first 24 h of differentiation (ERK expression serves as loading control). (C) I/11 cells were factor deprived and stimulated with 5 U/ml Epo and 1 μg/ml SCF for increasing time periods as indicated. Blots were analyzed with phosphospecific FoxO3a antibodies (S253) and antibodies recognizing total FoxO3a. An unidentified, cross-hybridizing protein (X) is dephosphorylated in response to Epo and SCF. Its signal is comparable at time 0 in the Epo and SCF panel when equally exposed. (D) Western blots of differentiating I/11 cells (samples taken every 12 h) were analyzed with antibodies recognizing FoxO3a and S253-phosphorylated FoxO3a, PKB and S473- phosphorylated PKB, and p27Kip and Erk. The protein recognized by phosphoSer253-FOXO3a antibodies at 72 h is unknown and may be the same background band X detected by this antibody in C. (E) Cytoplasmic and nuclear protein extracts of differentiating I/11 cells were analyzed with antibodies recognizing FoxO3a, STAT3, and ERK. Under these conditions, STAT3 is only present in the cytoplasm, indicating minimal contamination of nuclear extracts with cytoplasmic proteins. ERK expression serves as a loading control.
FoxO3a function is regulated by phosphorylation. In factor-deprived and restimulated erythroblasts, SCF induced a strong transient phosphorylation of FoxO3a serine residue 253, whereas Epo only weakly induced phosphorylation of FoxO3a (Fig. 1 C). In Fig. 1 C, the unidentified protein X is phosphorylated in factor-deprived cells and dephosphorylated after Epo or SCF stimulation. Phosphorylation of protein X is equally strong at time 0 in both panels, the much more pronounced signal in the Epo stimulation experiment is due to an accordantly longer exposure of the blot. Upon induction of differentiation, phosphorylation of FoxO3a reached maximum levels after 36 h and decreased thereafter, whereas total FoxO3a expression still increased (Fig. 1 D, the band detected at 72 h by the phosphospecific antibody may be the same protein X detected in Fig. 1 C). FoxO3a phosphorylation coincided with reduced expression and phosphorylation of PKB (Fig. 1 D, third and fourth panels). Consequently, unphosphorylated FoxO3a- and thus FoxO3a-mediated transcription increased from 36 h onwards as monitored by expression of the FoxO3a target p27Kip (Fig. 1 D, third panel). This is accompanied by increased nuclear localization of FoxO3a (Fig. 1 E; 72 h after induction of differentiation the cells are enucleated, hence no nuclear localization can be detected). Together, FoxO3a is up-regulated early in differentiation and is transcriptionally active ∼36–48 h after induction of differentiation, as indicated by its phosphorylation status and the expression of the target p27Kip.
Physiological effects of FoxO3a in erythroid differentiation
Although FoxO1a, FoxO3a, and FoxO4 may control cell cycle progression and cell survival of renewing cells, the expression pattern of Foxo3a suggests an additional role in erythroid differentiation. To investigate the role of FoxO3a, we used an inducible, phosphorylation-insensitive FoxO3a(A3):estrogen receptor (ER) fusion construct in which three serine phosphorylation sites are mutated to alanines. Fusion of this mutant to an estrogen receptor domain allows induction of nuclear translocation of FoxO3a(A3):ER by 4-hydroxytamoxifen (4OHT; Dijkers et al., 2000a). The FoxO3a(A3):ER protein was expressed in I/11 erythroid progenitors by retroviral transduction. Exposure of FoxO3a(A3):ER-expressing cells to 50 nM 4OHT induced p27Kip protein levels in 8 out of 10 clones, which did not occur in empty vector-transduced cells (unpublished data). To test whether activation of FoxO3a(A3):ER could counteract SCF-induced delay of Epo-dependent differentiation, FoxO3a(A3):ER clones and empty vector control clones were cultured in Epo or in Epo plus SCF, both in the presence or absence of 50 nM 4OHT. Cell numbers, cell size, hemoglobin levels, and cell morphology were monitored daily. 4OHT did not affect proliferation or differentiation of control clones, whereas it slightly decreased proliferation and enhanced hemoglobinization of FoxO3a(A3):ER clones in the presence of Epo (Fig. 2 A). In the presence of Epo and SCF, proliferation was impeded, which was accompanied with enhanced differentiation as analyzed by hemoglobinization (Fig. 2 B) and cell morphology (Fig. 2 C). 4OHT increased the number of partially mature and mature hemoglobinized cells, but cytospins showed no evidence for increased cell death (Fig. 2 C). We did not observe an increase in apoptotic cells on FoxO3a(A3):ER activation, using a TUNEL assay (Fig. 2 D). This observation indicated that exogenous, active FoxO3a accelerates differentiation of erythroid progenitors. Also, in the presence of Epo, SCF, and Dex, activation of FoxO3a(A3):ER abrogated renewal and induced differentiation. However, the presence of Dex interferes with hemoglobin synthesis and precludes the use of this parameter to monitor differentiation accurately. Using siRNA, we examined whether underexpression of FoxO3a impaired or delayed erythroid differentiation. An siRNA that fully blocked FoxO3a in a transient assay (Fig. 2 E, FOXi2) resulted in a considerable, but not complete, reduction of FoxO3a expression upon stable expression in erythroid progenitors (Fig. 2 F). After induction of differentiation, reduced FoxO3a expression attenuated differentiation as measured by hemoglobinization (Fig. 2 G). Thus, activation of FoxO3a accelerated differentiation, and reduced levels of Fox3a suppress maturation during differentiation.
Figure 2.
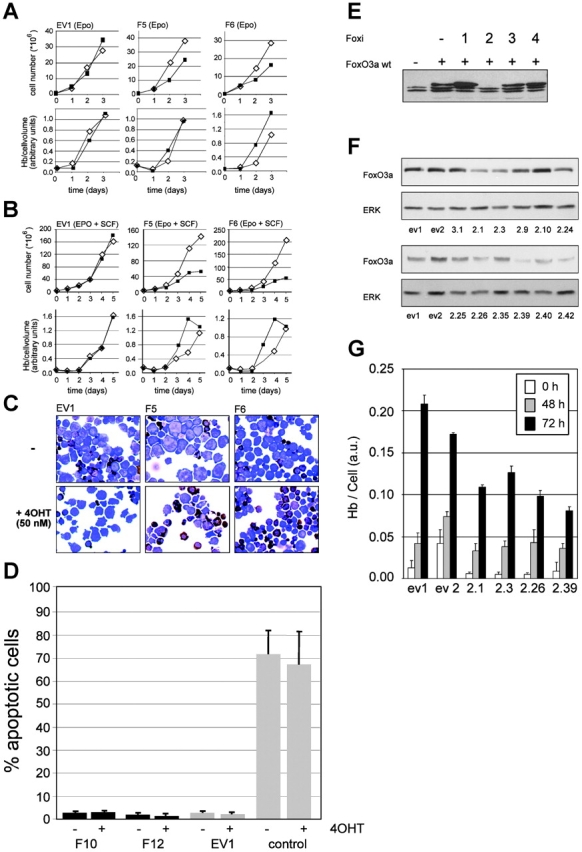
FoxO3a induces erythroid differentiation. All FoxO3a(A3):ER clones used had similar expression levels of the mutant FoxO3a protein, and treatment of all clones induced rapid up-regulation of p27Kip (not depicted). (A and B) A vector-transduced clone (EV1) and FoxO3a(A3):ER-expressing clones F5 and F6 were seeded in differentiation medium containing Epo (A) or Epo plus SCF (B), in the absence (open diamonds) or presence (closed squares) of 50 nM 4OHT. Cumulative cell numbers (top) and hemoglobin content per cell volume (bottom) were determined at daily intervals. (C) At day 4 of the experiments, cell morphology and hemoglobin content was analyzed in cytospins. Hemoglobinized cells can be discriminated by their orange-brown staining. (D) The vector control clone (EV1) and two FoxO3a(A3):ER clones (F10 and F12) were cultured in the presence of Epo, SCF, and Dex in the presence or absence of 4OHT. As a control, parental I/11 cells were seeded in medium lacking factors. After 24 h, the percentage of apoptotic cells was determined by a TUNEL assay. Values represent mean ± SD of apoptotic cells counted in five fields of a cytospin preparation (100 cells/field) in two independent experiments. (E) Phoenix E cells were transfected with FoxO3a wild type alone or in combination with RNAi constructs FOXi1–4 (see Materials and methods). Transient expression of FoxO3a was determined by Western blot (Erk serves as a loading control). (F) I/11 clones, transduced with pSuper-retro vector as a control (ev1 and ev2) or pSuper-retro FOXi2 (clone numbers indicated) were tested for FoxO3a on Western blot. (G) Two empty vector (ev1 and ev2) and four FOXi2 clones (clone numbers indicated) were differentiated in the presence of Epo. The hemoglobin content of the cells was measured at 0, 48, and 72 h in differentiation. Values represent mean ± SD of three experiments.
FoxO3a target genes in erythroid cells
Although the FoxO3a target p27Kip can induce cell cycle arrest and differentiation in some cell types, overexpression of p27Kip induced apoptosis in erythroblasts, whereas erythroid differentiation was normal in p27Kip-deficient cells (Hofmann et al., 2001; unpublished data). In addition to known targets that cause apoptosis or cell cycle arrest, FoxO3a could induce unknown targets that function in erythroid differentiation. To identify such target genes, cDNA derived from a FoxO3a(A3):ER-expressing clone and a control clone exposed to 50 nM 4OHT for 0, 2, or 6 h were hybridized to custom-made “hematopoietic” DNA microarrays containing ∼9,000 cDNAs derived from SSH libraries enriched for transcripts of expanding erythroblasts (I/11 cells) and mature T cells (Kolbus et al., 2003). These microarrays contained multiple copies of abundant erythroid-specific cDNAs (up to a few hundred for β-globin). The complete array results are available as supplemental data (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200307056/DC1). Upon 4OHT treatment of FoxO3a(A3):ER-expressing cells for 2 h, seven transcripts in the array were up-regulated >1.75-fold compared with no treatment, whereas no up-regulated genes were detected in similarly treated control cells. Five out of those seven transcripts represented BTG1, with an average up-regulation of 1.9 ± 0.14 (Fig. 3 A). Upon 6 h of 4OHT treatment, 98 transcripts indicated a >1.75-fold increase compared with no treatment. 11 of these represented BTG1. Their average fold up-regulation was 2.9 ± 0.33 (Fig. 3, A and B). None of these transcripts was detected in the control experiment.
Figure 3.
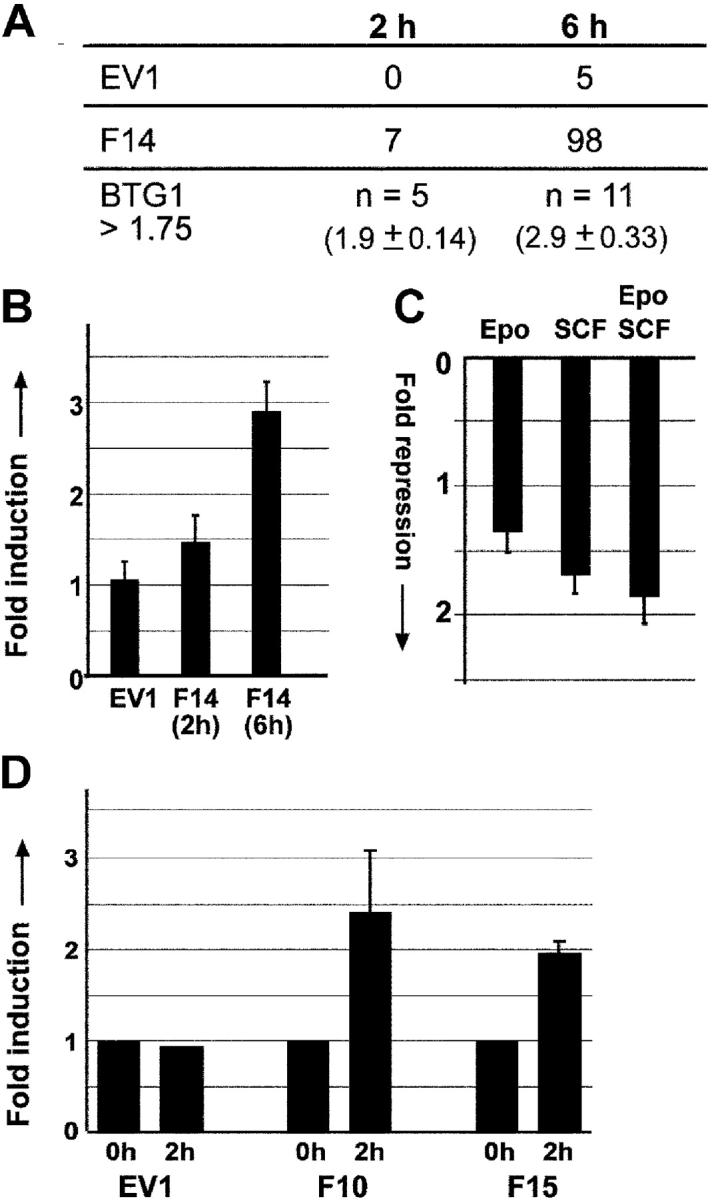
BTG1, a FoxO3a target gene. Labeled cDNA from FoxO3a(A3):ER clone F14 and vector clone EV1, exposed to 50 nM 4OHT for 0, 2, and 6 h in the presence of Epo, SCF, and Dex, were hybridized to a 9,000 cDNA microarray enriched for hematopoietic transcripts. (A) The number of spots that detected a >1.75-fold increase in signal upon treatment with 4OHT compared with nontreated cells is indicated for both clones after 2 and 6 h of treatment. BTG1 was represented at least 11 times on these arrays, 5 of these BTG1 spots showed >1.75-fold up-regulation after 2-h induction with 4OHT. (B and C) The average regulation on the 11 BTG1 spots is calculated after 2 and 6 h of 4OHT treatment (B) and after 2 h of Epo, SCF, or Epo plus SCF induction of factor-deprived cells (C), 1 meaning no regulation. Error bars indicate SD. (D) Control clone EV1 and FoxO3a(A3):ER clones F10 and F15 were treated with 50 nM 4OHT for 2 h, and relative BTG1 expression was determined by real-time PCR, using expression of RNase inhibitor to normalize the values. Values represent mean ± SD of three independent experiments.
The same arrays were also screened for genes up- or down-regulated in I/11 cells that were factor depleted and subsequently restimulated with Epo and/or SCF (Kolbus et al., 2003). Among the FoxO3a targets up-regulated after 6 h, only the transcripts representing BTG1 were down-regulated by Epo and SCF signaling as expected for genes primarily regulated by FoxO3a (Fig. 3 C and not depicted). Therefore, we concentrated on BTG1 as a novel putative FoxO3a target. Two additional FoxO3a(A3):ER clones were treated for 2 h with 50 nM 4OHT, and BTG1 transcript levels were determined by real-time PCR. In both clones, BTG1 expression was twofold up-regulated, whereas control clones showed no change in BTG1 expression (Fig. 3 D). Cyclohexamide treatment of I/11 cells strongly up-regulated BTG1, which precluded its use to determine whether protein synthesis is required for BTG1 up-regulation.
Together, we identified BTG1 as a prominent FoxO3a target in this screen. We also show that mitogenic signaling suppresses BTG1 expression, which is in accordance with the observed Epo- and SCF-induced phosphorylation of FoxO3a.
BTG1 is a direct FoxO3a target
To examine whether BTG1 is a direct target of FoxO3a, we analyzed the BTG1 promoter region for putative FoxO-binding sites. Among the sequences submitted to the public database, mouse cDNA clone L16846 contained the longest 5′UTR sequence. A comparison with genomic sequences in the CELERA database showed that the start of cDNA L16846 (designated +1) is located 40 nt downstream of a conserved TATA-box sequence (Fig. 4 A). We found four potential Forkhead-binding sites (Daf-16 binding element [DBE]) at position −219, −454, −826, and −922 (Fig. 4 B), allowing a 1-bp mismatch compared with the consensus sequence TTGTTTAC (Furuyama et al., 2000) outside the TGTT core sequence. Only DBE1 (position −219) completely matched the consensus sequence, and an alignment between the mouse and human BTG1 promoter sequence (BAC AC025164) revealed that only DBE1 was 100% identical between mouse and human (Fig. 4 A).
Figure 4.
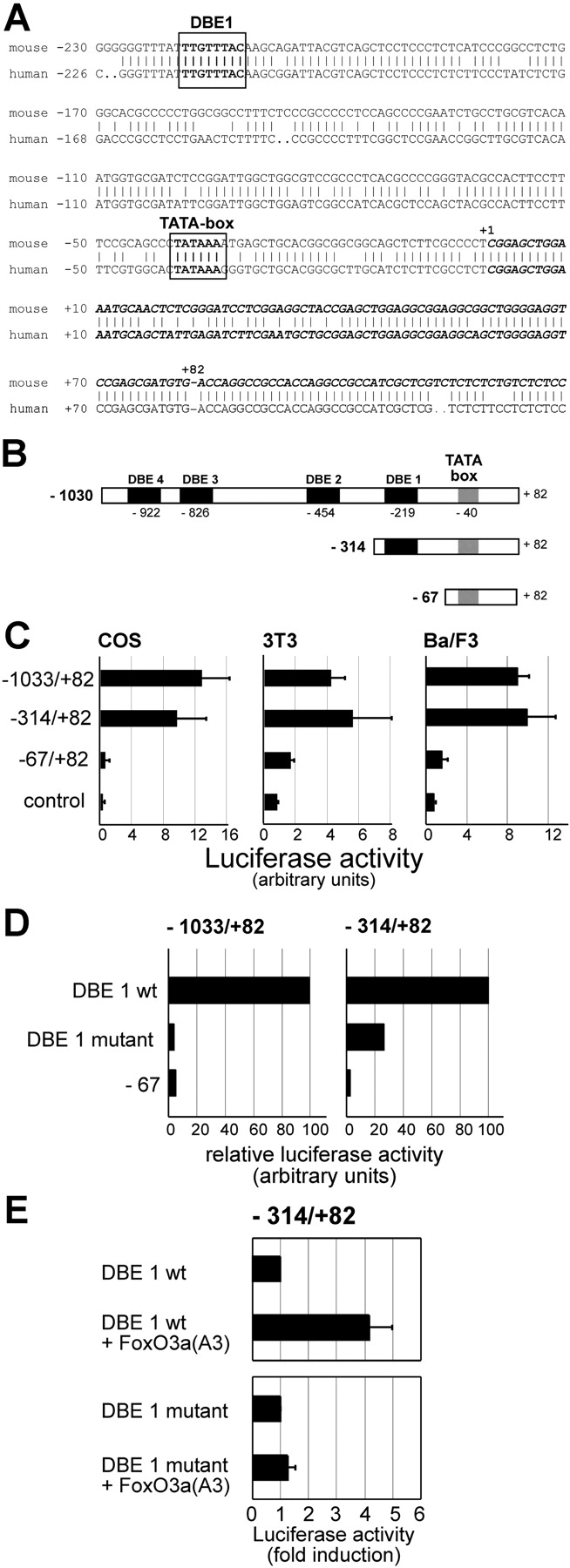
Regulation of BTG1 promoter activity by FoxO3a. (A) Sequence of the promoter region (plain) and part of the first exon (bold italics) of BTG1. Top sequence is derived from the mouse CELERA database and cDNA clone L16846, the bottom sequence is derived from the human BAC AC025164. The start of mouse cDNA L16846 was assigned as position+1. A potential TATA-box and FoxO-binding site (DBE1) are indicated. (B) Schematic drawing of the BTG1 promoter fragments used in reporter assays. Four potential FoxO-binding sites, DBE1–4, were found in the −1033/+82 promoter fragment. (C) Basal BTG1 promoter activity of these fragments was tested in COS, NIH3T3, and Ba/F3 cells and compared with a vector control (pGL3). Luciferase activity is represented as arbitrary units. Values represent mean ± SD of three measurements. (D) Both −1033/+82 and −314/+82 fragments with either a wild-type or a mutated DBE1 were tested for basal promoter activity in COS cells. The −67/+82 fragment serves as a negative control. (E) COS cells were cotransfected with FoxO3a(A3) and the −314/+82 BTG1 promoter with either a wild-type or a mutated DBE1. Luciferase activity is presented as fold induction on the horizontal axis. Values represent mean ± SD of three measurements.
To determine which part of the BTG1 promoter mediates FoxO3a-induced expression, genomic fragments containing all four DBEs (−1033/+82), DBE1 (−314/+82), or no DBE sites (−67/+82) were tested for basal promoter activity in Ba/F3 (hematopoietic, pro-B cells), COS (monkey kidney cells), and NIH3T3 (fibroblasts; Fig. 4, B and C). The activity of the different reporter constructs was similar in all cell types. The promoter activity of the −1033/+82 and the −314/+82 fragments was equally high, whereas the promoter activity of the −67/+82 BTG1 promoter fragment was almost reduced to background levels, indicating the presence of a crucial regulatory element between −314 and −67.
To investigate the role of DBE1, the TGTT core was mutated to AAAT in the −1033/+82 and −314/+82 constructs. This mutation caused a significant loss of basal promoter activity in both fragments (Fig. 4 D), indicating that DBE1 is a critical element in the BTG1 promoter. Cotransfection of FoxO3a(A3) with the wild-type and mutant −314/+82 promoter induced wild-type but not mutant −314/+82 BTG1 promoter activity (Fig. 4 E). Together, these data suggest that FoxO3a is able to activate transcription of BTG1 via the DBE1 element in the BTG1 promoter.
BTG1 is up-regulated in erythroid differentiation
If BTG1 is a FoxO3a target, its mRNA expression should follow FoxO3a activity during differentiation of erythroid progenitors. We determined BTG1 transcript levels during erythroid differentiation by Northern blot and real-time PCR (Fig. 5), using mRNA prepared from I/11 cells harvested at 12-h intervals after differentiation induction. BTG1 mRNA expression was low until 36 h after induction of differentiation when cells still proliferate. BTG1 transcript levels strongly increased 48 h after differentiation induction when cells become postmitotic and remained high until the final stages of erythroid differentiation (Fig. 5). Thus, BTG1 is expressed upon appearance of active FoxO3a during differentiation, suggesting a role of BTG1 in late erythroid differentiation.
Figure 5.
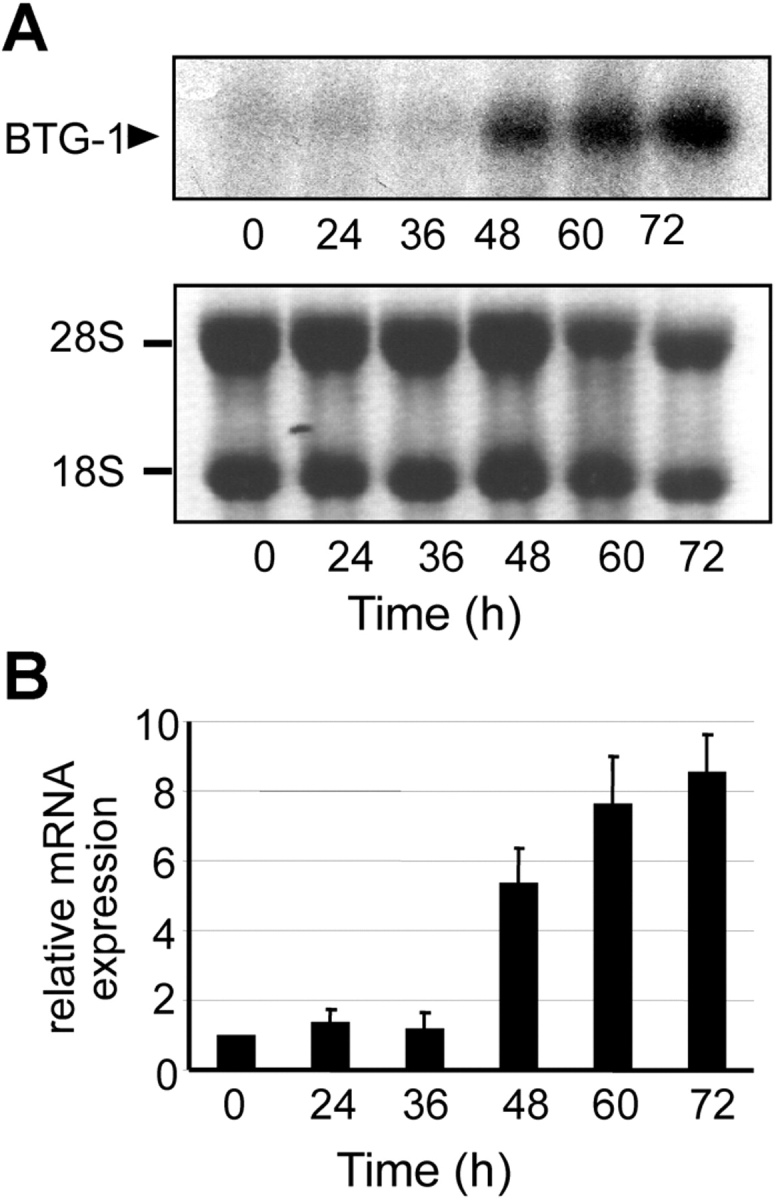
BTG1 is transcriptionally up-regulated during erythroid differentiation. (A) Total RNA was isolated from differentiating erythroid progenitors at 12-h intervals. BTG1 transcript levels were detected using a SmaI–BamHI 211-bp fragment as a probe (top). Ethidium bromide staining (bottom) indicated equal loading. (B) Real-time PCR on the RNA samples confirmed the kinetics in a quantitative way, using SYBR green in Taqman analysis and normalizing to the expression of the RNase inhibitor RI. Values represent mean ± SD of three measurements.
BTG1 may act via protein arginine methylation during differentiation
BTG1 is one of six family members, all sharing two highly homologous domains (BoxA and B; Fig. 6 A). It shares a third region of homology with BTG2 (BoxC), which associates with PRMT1 (Lin et al., 1996; Berthet et al., 2002). Overexpression of the BTG1 BoxC domain has been shown to inhibit differentiation of PC12 cells (Berthet et al., 2002). We expressed BTG1 in the I/11 erythroid progenitors, but only obtained small differentiated clones that could not be expanded (unpublished data). BTG1:ER fusion constructs as used for FoxO3a were not regulated tight enough. Because bone marrow is transduced with much higher efficiencies than I/11 cells, we transduced murine primary bone marrow to examine proliferation and differentiation in suspension cultures and in colony assays, using retroviral expression vectors containing BTG1 wild type or a BTG1 construct lacking boxC (BTG1-ΔBoxC). Colony formation of transduced cells was determined in serum-free semisolid medium supplemented with granulocyte–macrophage colony-stimulating factor (GM-CSF; myeloid colonies) or with a combination of Epo, SCF, and Dex (producing exclusively erythroid colonies as Dex inhibits outgrowth of SCF-dependent, nonerythroid colonies; controlled by cytospins [unpublished data]). Addition of puromycin selected for the outgrowth of transduced cells only. Bone marrow was infected with supernatants containing equal numbers of virus as controlled by dot-blot experiments (Erkeland et al., 2003; unpublished data). Colonies were counted 7 d after plating. Ectopic expression of BTG1 drastically reduced both the size and the number of erythroid colonies (Fig. 6 B, smallest, average, and largest colonies), whereas it only slightly reduced the number of myeloid colonies (Fig. 6 C). In contrast, erythroid colonies expressing BTG1-ΔBoxC were normal in size and only slightly reduced in number compared with the vector–control colonies (Fig. 6, B and C). Though the total number of colonies obtained differed between experiments (between 95 and 266 colonies in the vector control), the relative number of colonies obtained upon transduction of the various constructs was constant. Therefore, colonies are given as a percentage of control. Essentially the same results were obtained in suspension cultures (Fig. 6 D). This result indicates that BTG1 abrogates proliferation and that this effect depends on the presence of the PRMT1 interaction domain in boxC.
Figure 6.
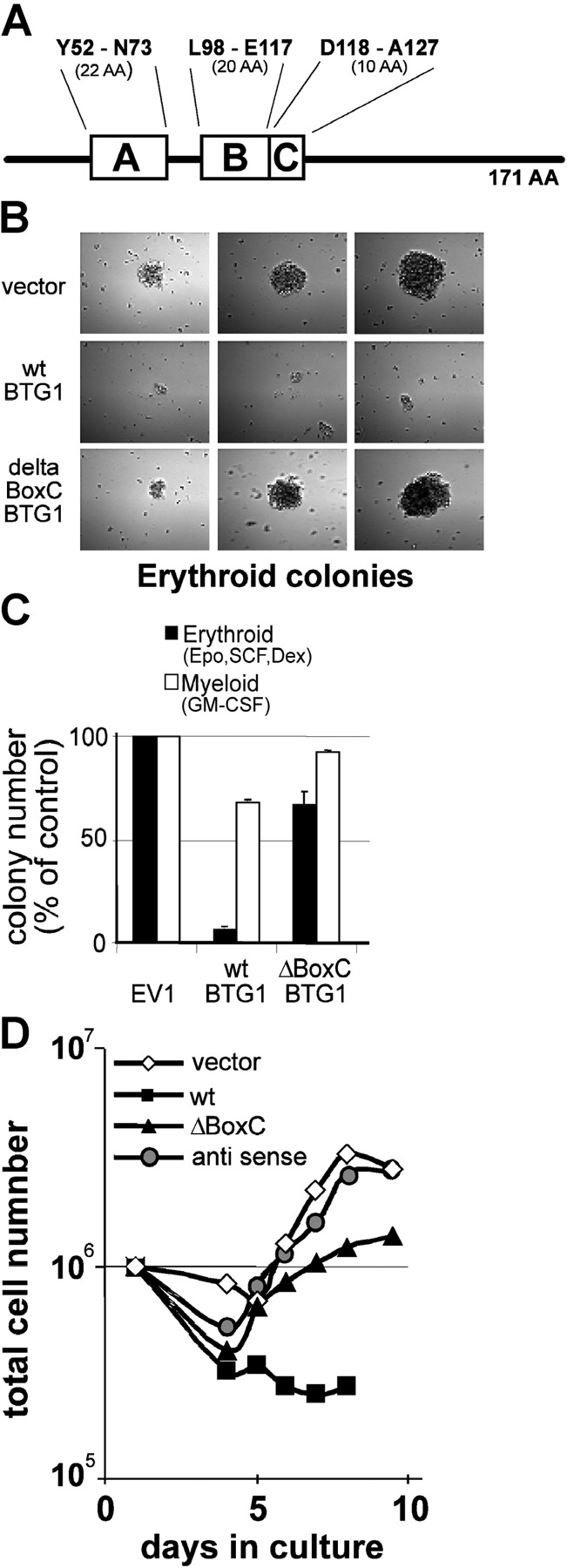
BTG1 inhibits proliferation of erythroid progenitors. (A) Schematic drawing of the 171–amino acid BTG1 protein, indicating the conserved domains A–C. (B–D) Density-purified murine bone marrow progenitors were transduced with an empty vector and retroviral expression vectors encoding BTG1 or BTG1 (ΔBoxC). Transduced cells were selected with puromycin in serum-free semisolid medium supplemented with 2 U/ml Epo, 100 ng/ml SCF, and 10−6 M Dex or with 10 ng/ml GM-CSF. Cytospins showed that all colonies grown with Epo, SCF, and Dex contain erythroid cells, whereas GM-CSF allows colony formation by various myeloid progenitors (not depicted). 7 d after plating, the morphology of erythroid colonies was photographed with a CCD camera (B, left to right: smallest, average, and largest colony) and colonies were counted (C). Values represent mean ± SD of three independent experiments each counted in triplo. Transduced cells were also grown in suspension cultures under conditions favoring expansion of erythroid progenitors, and total cell numbers were determined daily. An antisense BTG1 construct is added as a control (D).
Because boxC of BTG1 interacts with PRMT1, we investigated whether protein arginine methylation is associated with BTG1 expression. Two different antibodies (7E6, recognizing mono- and dimethyl-arginine, and ASYM24, recognizing only asymmetrical dimethyl-arginine) were used to immunoprecipitate arginine-methylated proteins from lysates of cells at different stages of differentiation. Concurrent with the activation of FOXO3a and the up-regulation of BTG1, the 7E6 antibody detected a prominent protein of 100 kD (Fig. 7 A, top panel). The ASYM24 antibody detected proteins of 45, 40, and 37 kD, which appeared after up-regulation of BTG1 (Fig. 7 A, second panel). ASYM24 also detects abundant proteins that are methylated at all stages of differentiation. In contrast, the overall PRMT1 protein levels in the same lysates remained constant during differentiation (Fig. 7 A, bottom panel).
Figure 7.
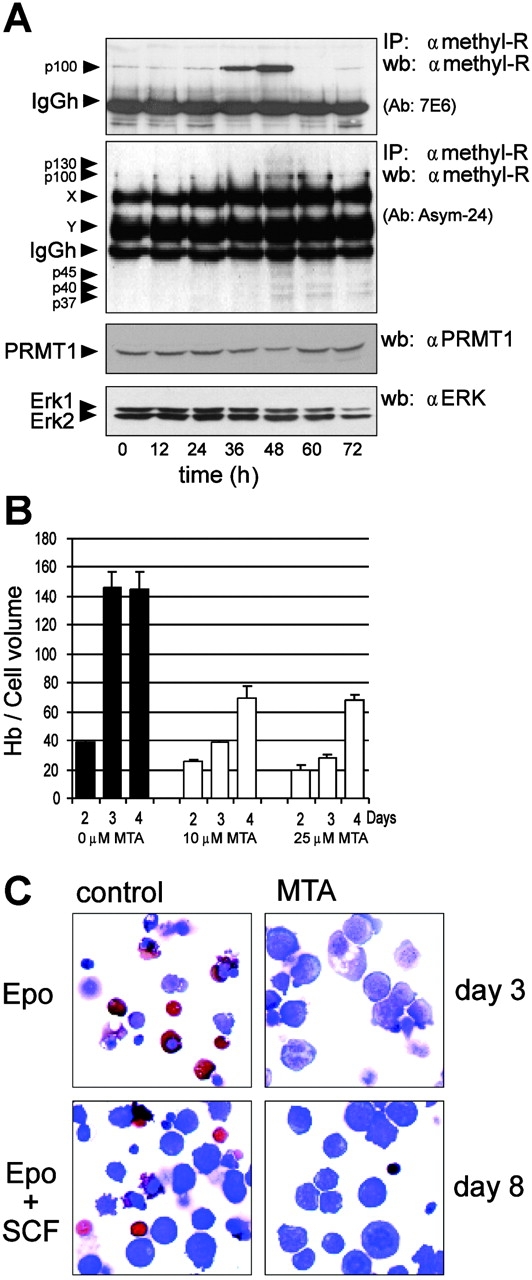
Protein methylation is associated with differentiation but not with renewal of erythroid progenitors. (A) Arginine-methylated proteins were immunoprecipitated and stained with two anti–methyl-arginine antibodies (E76 and ASYM24) from cell lysates taken at 12-h intervals during differentiation of I/11 cells. The size of specific proteins is indicated in kilodaltons. X and Y represent uncharacterized proteins detected by ASYM24 at all stages of differentiation. The bottom panels represent whole cell extract probed for total levels of PRMT1 and ERK (loading control). (B) During differentiation of I/11 cells in the presence of Epo and in the absence or presence of MTA (10 and 25 mM), hemoglobin accumulation was quantified at days 2–4. Hemoglobin per cell volume is measured in arbitrary units. Values represent mean ± SD of three measurements. (C) I/11 cells were seeded in differentiation medium in the presence of Epo (top) or Epo plus SCF (bottom), in the absence (left) or presence (right) of the methyl-transferase inhibitor MTA (25 mM). At day 3 (Epo) or day 8 (Epo/SCF), cell morphology was analyzed in cytospins stained for hemoglobin (hemoglobinized cells stain orange/brown) and histological dyes. In the presence of MTA, cells retained a blast morphology and failed to accumulate hemoglobin.
To examine whether inhibition of methylation affects the balance between expansion and differentiation of erythroid progenitors, I/11 erythroblasts were seeded in differentiation medium containing Epo or Epo plus SCF to analyze differentiation. Aliquots were exposed to the methyl-transferase inhibitor 5′-deoxy-5′-methylthioadenosine (MTA; 10 and 25 μM) or its solvent. Cell numbers, cell volume, hemoglobinization, and cell morphology were monitored daily. In the presence of Epo, I/11 cells achieve full hemoglobinization in 3 d, but addition of MTA (both 10 and 25 μM) impairs hemoglobin accumulation (Fig. 7 B). In cytospins generated at day 3, the control cells are hemoglobinized and enucleated, whereas MTA-treated cells maintained blast morphology (Fig. 7 C). In the presence of Epo and SCF, erythroblasts reduced cell size and accumulated hemoglobin at day 8, whereas they retained blast morphology and continued to proliferate in the presence of MTA. This showed that inhibition of methyl transferases impairs differentiation but fails to affect proliferation of erythroid progenitors. Although the inhibitor is not specific for PRMT1, these results support a potential role of PRMT1 in terminal erythroid differentiation.
Discussion
FoxO3a expression increases sharply during erythroid differentiation, followed by loss of phosphorylation and increased transcriptional activity. Inappropriate activation of FoxO3a accelerates differentiation of erythroid progenitors to erythrocytes. BTG1 was identified as a novel target gene of FoxO3a, repressed by SCF and exerting a negative effect on erythroid progenitor renewal. Deletion of BoxC in BTG1, a domain known to bind PRMT1, abrogated proliferation inhibition by BTG1, suggesting that protein arginine methylation might interfere with renewal. This finding is supported by the observation that inhibition of methyl transferase activity does not interfere with progenitor expansion, but blocks differentiation into mature erythrocytes.
PI3K and FoxO3a control the balance between renewal divisions and differentiation of erythroid progenitors
PI3K-dependent signaling is associated with expansion of erythroid progenitors (Klingmuller et al., 1997; Haseyama et al., 1999; von Lindern et al., 2001). Both Epo and SCF promote erythroid proliferation by inhibition of FoxOs, whereas active FoxO3a was shown to cause cell cycle arrest and apoptosis in erythroblasts unless inhibited by phosphorylation through PKB (Kashii et al., 2000; Mahmud et al., 2002). We find FoxO1a, FoxO3a, and FoxO4 to be expressed in renewing erythroid progenitors. The disappearance of FoxO1a and FoxO4 in differentiation suggests that these FoxO members may specifically have a role in the PI3K-dependent control of progenitor expansion, possibly via previously reported effector pathways (Dijkers et al., 2000a,b; Medema et al., 2000; Kops et al., 2002; Ramaswamy et al., 2002). Actually, the three FoxO members may have overlapping functions in the control of progenitor expansion. However, because not only activation of FoxO3a but also inhibition of PI3K activity induced differentiation rather than apoptosis, all three FoxO members may be able to abrogate renewal and initiate the differentiation program. Once the differentiation program is started, only FoxO3a is up-regulated and only FoxO3a may control gene expression in late differentiation, including BTG1. Although activation of FoxO3a enhanced differentiation, RNA interference (RNAi) suppressing FoxO3a impaired differentiation as monitored by reduced hemoglobinization. However, inhibition of FoxO3a did not abrogate differentiation. Possibly, the remaining expression of FoxO3a is sufficient to allow the differentiation process to proceed. Alternatively, the tight control of erythroid cell numbers, evidenced by the very rare occurrence of erythroid leukemia, has recruited several complementary mechanisms that guarantee proper differentiation of erythroid progenitors. The observation that FoxO3a-deficient mice develop a compensated anemia with reticulocytosis (Castrillon et al., 2003) illustrates the importance of FoxO3a in erythroid differentiation. The reticulocytosis suggests that the anemia is caused by instability of erythrocytes rather than by increased apoptosis of progenitors.
The role of FoxO3a in erythroid differentiation
After induction of differentiation, erythroid progenitors undergo three to four cell divisions while maturing to hemoglobinized, enucleated erythrocytes (Dolznig et al., 2001; von Lindern et al., 2001). Activated FoxO3a induced expression of p27Kip and p130-Rb2 in erythroblasts as has been shown for other cell types (confirmed by quantitative PCR [unpublished data]; Dijkers et al., 2000b; Medema et al., 2000; Kops et al., 2002). Forced activation of FoxO3a accelerated differentiation and reduced the number of cell divisions. FoxO3a-induced expression of p27KIP and p130-Rb2 most likely contributed to G1 arrest. However, cell cycle arrest caused by exogenous p27Kip fails to induce terminal differentiation of erythroid progenitors, causing apoptosis instead (Hofmann et al., 2001). Furthermore, erythroid progenitors lacking p27Kip did not show any alterations in erythroid differentiation (unpublished data). Thus, erythroid differentiation requires more than a (p27Kip-mediated) cell cycle arrest. In contrast to other cells, erythroid differentiation requires three to four cell divisions without size control to mature into erythrocytes. We cannot conclude at present whether the newly identified FoxO3a target BTG1 contributes to differentiation related phenotypic changes like an altered cytoskeleton organization, cell cycle arrest, chromatin condensation, and enucleation, or whether it directly contributes to the early control of the balance between renewal and differentiation. However, the fact that BTG1 controls protein methylation in erythroid differentiation, and that this may contribute to control of renewal versus differentiation, identifies a novel FoxO3a-dependent mechanism that may regulate many aspects of the differentiation process. The importance of BTG function is underscored by the fact that the homologous family member BTG2 is similarly suppressed by SCF and induced during differentiation (Kolbus et al., 2003), although via a distinct mechanism. This complementary regulation underscores the biological importance of BTG activation but precludes conclusive experiments on the requirement for BTG1 by underexpression.
The role of BTG1 in protein methylation and differentiation
Screening of a cDNA array enriched for hematopoietic transcripts identified BTG1 as a major FoxO3a target. Others also reported BTG1 to be a potential Forkhead target (Ramaswamy et al., 2002), but so far, its role and regulation were not studied in detail.
Activation of FoxO3a in expanding erythroblasts induced a greater than twofold up-regulation of BTG1 within 2 h, indicating a rapid induction. A more pronounced up-regulation is detected during differentiation. However, FoxO3a(A3):ER protein can only be expressed at low levels in expanding cells, resulting in FoxO3a(A3):ER concentrations at best similar to endogenous levels. In contrast, FoxO3a levels rise considerably during differentiation. This difference on FoxO3a expression may explain the difference in BTG1 expression in these experimental conditions. Expression of BTG1 is not solely regulated by FoxO3a, we also reported BTG1 to be regulated by glucocorticoids (Kolbus et al., 2003), and it was shown to be a putative vitaminD3 target (Savli et al., 2002). Moreover, preliminary data suggest that CREB and FoxO3a have to cooperate in BTG1 induction (unpublished data). Thus, regulation of BTG1 may be complex, suggesting an important role in cell fate determination.
The BTG1 protein lacks enzymatic activity but contains several protein interaction domains (Fig. 6 A), suggesting a function as an adaptor molecule for enzymes and their targets or as a regulatory cofactor. BTG1 has been shown to interact with PRMT1 via its BoxC domain (Lin et al., 1996; Berthet et al., 2002), resulting in positive regulation of PRMT1 activity. Thus, BTG1 may direct the associated methyl transferase activity toward substrates binding to its NH2-terminal domains (e.g., HOXB9 and the carbon catabolite repressor [CCR4]–associated factor 1, CAF1; Bogdan et al., 1998; Rouault et al., 1998; Prevot et al., 2000, 2001). Although expression of BTG1 in mouse bone marrow cells fully blocked expansion of erythroid progenitors, deletion of the PRMT1-associated BoxC domain largely abolished this negative effect, indicating that recruitment of PRMT1 is essential to the function of BTG1 in erythroid differentiation. Erythroid progenitors may be more sensitive to this function of BTG1 because BTG1 expression only marginally affected myeloid colony formation induced by GM-CSF. Erythroid differentiation seemed to require arginine methylation because its inhibition by MTA completely blocked terminal erythroid differentiation without affecting the proliferation of immature erythroblasts. Despite the fact that MTA inhibits other S-adenosyl-l-methionine–dependent methyl transferases, we can still conclude that methylation is required for terminal differentiation and not for renewal of erythroblasts. Similarly, global inhibitors of methylation also inhibited differentiation in the PC12 cell line (Cimato et al., 2002) and PC12 cells loaded with a penetratin–BTG1/BoxC fusion peptide failed to differentiate, suggesting that neuronal differentiation involves PRMT1 regulation by BTG1 (Berthet et al., 2002). Overexpression of BTG1 inhibited myoblast proliferation and induced differentiation (Rodier et al., 2001). BTG2, the closest homologue of BTG1 was shown to be up-regulated in neuronal differentiation (Bradbury et al., 1991; Corrente et al., 2002) and to play a role in germ cell and muscle cell differentiation (for review see Tirone, 2001).
It has become increasingly clear that arginine methylation functions as a molecular switch, promoting or preventing specific protein–protein interactions (for review see McBride and Silver, 2001). PRMT1 contributes to 90% of the total cellular arginine methyl transferase activity (Tang et al., 2000), and mice lacking PRMT1 die at day 6.5 of development, just before gastrulation occurs (Pawlak et al., 2000). We showed that increased BTG1 expression after induction of differentiation is accompanied by arginine methylation of multiple proteins, ranging from 37 to 100 kD. The p100 detected by 7E6 is detectable at 36 h, maximal at 48 h, and not detected at later time points, although expression of BTG1 and PRMT1 persist. Possibly, its expression is only transient, or the BTG1–PRMT1 complex is directed to other substrates in the course of the differentiation program. PRMT1 was reported to induce methylation of STAT1, but STAT1, STAT3, or STAT5 antibodies did not recognize the 100-kD proteins. So far, we do not know the targets of PMRT1 in erythroid differentiation. Interestingly, BTG1 interacts with CAF1, and the CCR4–CAF1 complex is involved in heterochromatin formation, gene silencing, and negative regulation of mRNA stability (Bogdan et al., 1998; Tucker et al., 2001). In erythroid cells, both PRMT1 and CAF1 are expressed throughout differentiation at constant levels (this paper and unpublished data), leaving the possibility that BTG1-mediated activation of PRMT1 and CAF1 contributes to epigenetic gene regulation including condensation of the nucleus and enucleation late in erythroid differentiation.
Materials and methods
Cells and reagents
COS, 3T3, and ecotropic Phoenix cells were cultured in DME (Life Technologies) supplemented with 10% FCS (Life Technologies); BA/F3 were cultured in RPMI 1640 supplemented with 10% FCS and 10 ng/ml murine IL-3. LY294002 was obtained from Qbiogene, 4OHT and MTA were obtained from Sigma-Aldrich.
Expansion and differentiation of erythroid progenitors
The erythroid cell line I/11 was cultured as described previously (von Lindern et al., 2001). Cell numbers and cell size were determined using an electronic cell counter (model CASY1; Schärfe-System). Cell morphology was analyzed in cytospins stained with histological dyes and neutral benzidine (Beug et al., 1982) using a microscope (model Bx4040; Olympus; 40× objective, NA 0.65), CCD camera (model Dp50; Olympus), and Viewfinder Lite (1.0) acquisition software. Images were cropped using Adobe photoshop 6.0. Hemoglobin was measured as described by Kowenz et al. (1987). In short, 2–4 × 104 cells were washed in PBS, lysed in 20 μl H2O, and frozen until all samples were collected. 100 μl of reagent mix (0.5 mg/ml o-phenylenediamine [Sigma-Aldrich] and 0.03% H2O2 in 0.1 M citrate/phosphate buffer, pH 5.0) was added to thawed samples, the reaction was stopped after 3 min with 20 μl 8 N H2SO4, and the extinction of the reaction product was read on an ELISA photometer at 492 nM, using the extinction at 690 nM as a control. Extinction/cell number or extinction/cell volume was taken as a measure for hemoglobinization. Apoptosis was determined using the TUNEL assay according to the manufacturer's protocol (Roche).
Generation of stable FoxO3a(A3):ER or FoxO3a-RNAi–expressing I/11 clones
The FoxO3a(A3):ER construct (Dijkers et al., 2002) was cloned in the retroviral expression vector pBABE-puro. Four RNAi constructs were cloned in pSuper-retro (provided by Anton Berns, Netherlands Cancer Institute, Amsterdam, Netherlands): FOXi1 AATGAAGGCACGGGCAAGAGCTCTT; FOXi2 AACCAGACACTCCAAGACCTGCTT; FOXi3 AGTGACTTGGACCTGGACATGTT; and FOXi4 AGCCAGCTCGGCCATGGTGAT. These constructs were transiently expressed in Phoenix cells, together with the FoxO3a(A3):ER construct. Only the FOXi2 sequence suppressed FoxO3a expression and was used for stable expression in I/11 cells. To obtain stable expression, 0.5 × 106 ecotropic Phoenix cells, seeded in 60-mm dishes, were transfected with 16 μg of plasmid DNA using calcium phosphate coprecipitation. 40 h after transfection, cells were treated with 10 μg/ml mitomycinC (Kyowa Hakko Kogyo) for 1 h and washed three times with PBS, twice with an interval of >4 h. I/11 cells (0.5 × 106/ml) were added and cocultured for 24 h in StemPro-34™ plus factors. I/11 cells were removed and grown in 2 μg/ml puromycin containing (Sigma-Aldrich) semisolid medium (Methocel-containing StemPro-34™ [Invitrogen], supplemented with factors). After 7 d, well-separated colonies were picked, expanded, and analyzed for FoxO3a(A3):ER expression.
Western blotting and antibodies
I/11 cells were growth factor deprived for 4 h in plain IMDM (Life Technologies) and stimulated at 37°C with 100 ng/ml SCF or 5 U/ml Epo. Reactions were stopped by the addition of ice-cold PBS. Cell lysis, immunoprecipitation, SDS-PAGE, and Western blotting were performed as described previously (van Dijk et al., 2000). Antibodies used in this study were as follows: α-HA (F-7; Santa Cruz Biotechnology, Inc.), α-FoxO4a (N-19; Santa Cruz Biotechnology, Inc.), α-FoxO1a (Cell Signaling Technology), α-FoxO3a (Upstate Biotechnology), α-phospho–FoxO3a (S-253; Upstate Biotechnology), α-p27Kip1 (BD Biosciences), α-ERK1/2 (K-23; Santa Cruz Biotechnology, Inc.), α-STAT3 (C-20; Santa Cruz Biotechnology, Inc.), α-mono- and dimethylarginine (7E6; Abcam), α-dimethylarginine (ASYM24; Upstate Biotechnology), and α-PRMT1 (a gift from J.P. Rouault, Hôpital Edouard Herriot, Lyon, France).
RNA isolation and real-time quantitative PCR
After 2-h stimulation (50 nM 4OHT, 5 U/ml Epo, and 100 ng/ml SCF), cells were lysed, nuclei were removed, and RNA was isolated as described in Kolbus et al. (2003). 1 μg of total RNA was used to synthesize cDNA exactly as described by Kolbus et al. (2003). The cDNA was diluted 1:10 to 1:200 before PCR amplification. The primer sequences used for the amplification of mBTG1 were as follows: forward, 5′-TGC AGG AGC TGC TGG CAG-3′, and reverse, 5′-TGC TAC CTC CTG CTG GTG A-3′; murine ribonuclease inhibitor, forward, 5′-TCC AGT GTG AGC AGC TGA G-3′, and reverse, 5′-TGC AGG CAC TGA AGC ACC A-3′. The real-time PCR assay involves TaqMan technology (model 7700 or 7900 sequence detector; PE Corp.). The reactions were performed as described by Kolbus et al. (2003). The amplification program consisted of one cycle of 50°C with 2-min hold, 1 cycle of 95°C with 10-min hold, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 62°C for 30 s, and extension at 62°C for 30 s. The CT values of RNase inhibitor were used to normalize the BTG1 values.
cDNA array hybridizations and analysis
Total RNA was used to hybridize a custom-made hematopoietic microarray containing ∼9,000 cDNAs and enriched for erythroid and T cell–specific cDNAs by subtracting cDNA of expanding I/11 cells and quiescent CD4+ T cells from cDNAs prepared from 3T3 fibroblasts and EpH4 epithelial cells. A full description of the array and the array hybridization is available as supplemental data. The quality of RNA was determined with a bioanalyzer (model 2100; Agilent Technologies) according to the manufacturers instructions. For a single hybridization, 30 μg total RNA was reverse transcribed into cDNA using Cy5-UTP (CyDye; Amersham Biosciences), whereas control total RNA was labeled with Cy3-UTP. The microarrays were hybridized and analyzed as described previously (Kolbus et al., 2003). The scanning was performed using a Genepix 400A scanner (Axon Instruments, Inc.), and the analysis was performed using the GenePix program.
Cloning of the BTG1 promoter and luciferase reporter assays
The mBTG1 cDNA clone L16846 was aligned to the mouse BTG1 genomic sequence (CELERA) and the human BAC clone AC025164. The −1033/+82 BTG1 promoter fragment was cloned into the pGL3-Basic vector (Promega) after expansion by PCR using a 5′ oligo (5′-GTG GTG TGT ATT GCA TCT GAT GAC C-3′), a 3′ oligo (5′-CAC ATC GCT CGG ACC TCC CCA GCC-3′), and the Expand High Fidelity PCR system (Roche). The −314/+82 and the −67/+82 promoter fragments were obtained using the internal NheI and SmaI sites, respectively. The DBE1 was mutated using the Quickchange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol with primer 5′-CGG GGG GTT TAT TTA AAT ACA AGC AGA TTA CG-3′ and its complementary sequence.
For reporter assays, COS cells were seeded at 2.5 × 105 cells/35-mm well (Costar) and transfected with 4 μg DNA by calcium phosphate coprecipitation. After 24 h, cells were washed with PBS and subsequently lysed in 25 mM Tris-phosphate (pH 7.8, 15% glycerol, 1% Triton X-100, 1 mM DTT, and 8 mM MgCl2). Luciferase activity was measured using the Steady-Glo system (Promega). LacZ determination was used to correct for transfection efficiency.
Online supplemental material
Table S1 is an excel file that contains the data of the microarray analysis. A MIAME compliant description of the microarray, the samples, and the hybridization and scanning procedures is given as a separate pdf file. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200307056/DC1.
Acknowledgments
We want to thank Dr. I.P. Touw for many critical discussions regarding the work presented. We thank Herbert Auer for his patient help with the array analysis, Claudia Antonissen for excellent technical assistance preparing colony assays, and Dr. J.P. Rouault for his gift of anti-PRMT1 and for his advice on experiments concerning BTG1.
This work was supported by grants from the Dutch Cancer Society (EUR 2000-2230), the European Union (HPRN-CT-2000-00083), the Netherlands Organization for Scientific Research (NWO 901-08-338), and fellowships of the Erasmus University Rotterdam to T.B. van Dijk and the Dutch Academy of Arts and Sciences to M. von Lindern.
The online version of this article contains supplemental material.
A. Kolbus's present address is Dept. of Gynecologic Endocrinology and Reproductive Medicine, University of Vienna Medical School, A-1090 Vienna, Austria.
T.B. van Dijk's present address is Dept. of Cell Biology and Genetics, Erasmus MC, 3015 GE Rotterdam, Netherlands.
Abbreviations used in this paper: 4OHT, 4-hydroxytamoxifen; BTG1, B cell translocation gene 1; DBE, Daf-16 binding element; Dex, dexamethasone; Epo, erythropoietin; ER, estrogen receptor; FoxO, Forkhead box, class O; GM-CSF, granulocyte–macrophage colony-stimulating factor; MTA, 5′-deoxy-5′-methylthioadenosine; PI3K, phosphatidylinositol-3-kinase; PRMT1, protein arginine methyl transferase 1; RNAi, RNA interference; SCF, stem cell factor.
References
- Bao, H., S.M. Jacobs-Helber, A.E. Lawson, K. Penta, A. Wickrema, and S.T. Sawyer. 1999. Protein kinase B (c-Akt), phosphatidylinositol 3-kinase, and STAT5 are activated by erythropoietin (EPO) in HCD57 erythroid cells but are constitutively active in an EPO-independent, apoptosis-resistant subclone (HCD57-SREI cells). Blood. 93:3757–3773. [PubMed] [Google Scholar]
- Berthet, C., F. Guehenneux, V. Revol, C. Samarut, A. Lukaszewicz, C. Dehay, C. Dumontet, J.P. Magaud, and J.P. Rouault. 2002. Interaction of PRMT1 with BTG/TOB proteins in cell signalling: molecular analysis and functional aspects. Genes Cells. 7:29–39. [DOI] [PubMed] [Google Scholar]
- Beug, H., S. Palmieri, C. Freudenstein, H. Zentgraf, and T. Graf. 1982. Hormone-dependent terminal differentiation in vitro of chicken erythroleukemia cells transformed by ts mutants of avian erythroblastosis virus. Cell. 28:907–919. [DOI] [PubMed] [Google Scholar]
- Bogdan, J.A., C. Adams-Burton, D.L. Pedicord, D.A. Sukovich, P.A. Benfield, M.H. Corjay, J.K. Stoltenborg, and I.B. Dicker. 1998. Human carbon catabolite repressor protein (CCR4)-associative factor 1: cloning, expression and characterization of its interaction with the B-cell translocation protein BTG1. Biochem. J. 336:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury, A., R. Possenti, E.M. Shooter, and F. Tirone. 1991. Molecular cloning of PC3, a putatively secreted protein whose mRNA is induced by nerve growth factor and depolarization. Proc. Natl. Acad. Sci. USA. 88:3353–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil, D.P., and B.A. Hemmings. 2001. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem. Sci. 26:657–664. [DOI] [PubMed] [Google Scholar]
- Broudy, V.C. 1997. Stem cell factor and hematopoiesis. Blood. 90:1345–1364. [PubMed] [Google Scholar]
- Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- Castrillon, D.H., L. Miao, R. Kollipara, J.W. Horner, and R.A. DePinho. 2003. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 301:215–218. [DOI] [PubMed] [Google Scholar]
- Cimato, T.R., J. Tang, Y. Xu, C. Guarnaccia, H.R. Herschman, S. Pongor, and J.M. Aletta. 2002. Nerve growth factor-mediated increases in protein methylation occur predominantly at type I arginine methylation sites and involve protein arginine methyltransferase 1. J. Neurosci. Res. 67:435–442. [DOI] [PubMed] [Google Scholar]
- Corrente, G., D. Guardavaccaro, and F. Tirone. 2002. PC3 potentiates NGF-induced differentiation and protects neurons from apoptosis. Neuroreport. 13:417–422. [DOI] [PubMed] [Google Scholar]
- Datta, S.R., A. Brunet, and M.E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905–2927. [DOI] [PubMed] [Google Scholar]
- Dijkers, P.F., R.H. Medema, J.W. Lammers, L. Koenderman, and P.J. Coffer. 2000. a. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 10:1201–1204. [DOI] [PubMed] [Google Scholar]
- Dijkers, P.F., R.H. Medema, C. Pals, L. Banerji, N.S. Thomas, E.W. Lam, B.M. Burgering, J.A. Raaijmakers, J.W. Lammers, L. Koenderman, and P.J. Coffer. 2000. b. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol. 20:9138–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkers, P.F., K.U. Birkenkamp, E.W. Lam, N.S. Thomas, J.W. Lammers, L. Koenderman, and P.J. Coffer. 2002. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B–enhanced cell survival through maintenance of mitochondrial integrity. J. Cell Biol. 156:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolznig, H., F. Boulme, K. Stangl, E.M. Deiner, W. Mikulits, H. Beug, and E.W. Mullner. 2001. Establishment of normal, terminally differentiating mouse erythroid progenitors: molecular characterization by cDNA arrays. FASEB J. 15:1442–1444. [DOI] [PubMed] [Google Scholar]
- Dolznig, H., B. Habermann, K. Stangl, E.M. Deiner, R. Moriggl, H. Beug, and E.W. Mullner. 2002. Apoptosis protection by the epo target bcl-x(l) allows factor-independent differentiation of primary erythroblasts. Curr. Biol. 12:1076–1085. [DOI] [PubMed] [Google Scholar]
- Erkeland, S.J., M. Valkhof, C. Heijmans-Antonissen, R. Delwel, P.J. Valk, M.H. Hermans, and I.P. Touw. 2003. The gene encoding the transcriptional regulator Yin Yang 1 (YY1) is a myeloid transforming gene interfering with neutrophilic differentiation. Blood. 101:1111–1117. [DOI] [PubMed] [Google Scholar]
- Furuyama, T., T. Nakazawa, I. Nakano, and N. Mori. 2000. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 349:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseyama, Y., K. Sawada, A. Oda, K. Koizumi, H. Takano, T. Tarumi, M. Nishio, M. Handa, Y. Ikeda, and T. Koike. 1999. Phosphatidylinositol 3-kinase is involved in the protection of primary cultured human erythroid precursor cells from apoptosis. Blood. 94:1568–1577. [PubMed] [Google Scholar]
- Hofmann, J.F., M. Sykora, N. Redemann, and H. Beug. 2001. G1-Cdk activity is required for both proliferation and viability of cytokine-dependent myeloid and erythroid cells. Oncogene. 20:4198–4208. [DOI] [PubMed] [Google Scholar]
- Kashii, Y., M. Uchida, K. Kirito, M. Tanaka, K. Nishijima, M. Toshima, T. Ando, K. Koizumi, T. Endoh, K. Sawada, et al. 2000. A member of Forkhead family transcription factor, FKHRL1, is one of the downstream molecules of phosphatidylinositol 3-kinase-Akt activation pathway in erythropoietin signal transduction. Blood. 96:941–949. [PubMed] [Google Scholar]
- Klingmuller, U., H. Wu, J.G. Hsiao, A. Toker, B.C. Duckworth, L.C. Cantley, and H.F. Lodish. 1997. Identification of a novel pathway important for proliferation and differentiation of primary erythroid progenitors. Proc. Natl. Acad. Sci. USA. 94:3016–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbus, A., M. Blazques-Domingo, S. Carotta, W. Bakker, S. Luedemann, M. von Lindern, P. Steinlein, and H. Beug. 2003. Cooperative signaling between cytokine receptors and the glucocorticoid receptor in expansion of erythroid progenitors: molecular analysis by expression profiling. Blood. 102:3136–3146. [DOI] [PubMed] [Google Scholar]
- Kops, G.J., N.D. de Ruiter, A.M. De Vries-Smits, D.R. Powell, J.L. Bos, and B.M. Burgering. 1999. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 398:630–634. [DOI] [PubMed] [Google Scholar]
- Kops, G.J., R.H. Medema, J. Glassford, M.A. Essers, P.F. Dijkers, P.J. Coffer, E.W. Lam, and B.M. Burgering. 2002. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol. Cell. Biol. 22:2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowenz, E., A. Leutz, G. Doderlein, T. Graf, and H. Beug. 1987. ts-oncogene-transformed erythroleukemic cells: a novel test system for purifying and characterizing avian erythroid growth factors. Modern Trends in Human Leukemia VII. R. Neth, R.C. Gallo, M.F. Greaves, and H. Kabisch, editors. Springer Verlag, Heidelberg. 199–209. [DOI] [PubMed]
- Lin, W.J., J.D. Gary, M.C. Yang, S. Clarke, and H.R. Herschman. 1996. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J. Biol. Chem. 271:15034–15044. [DOI] [PubMed] [Google Scholar]
- Mahmud, D.L., M. G-Amlak, D.K. Deb, L.C. Platanias, S. Uddin, and A. Wickrema. 2002. Phosphorylation of forkhead transcription factors by erythropoietin and stem cell factor prevents acetylation and their interaction with coactivator p300 in erythroid progenitor cells. Oncogene. 21:1556–1562. [DOI] [PubMed] [Google Scholar]
- McBride, A.E., and P.A. Silver. 2001. State of the arg: protein methylation at arginine comes of age. Cell. 106:5–8. [DOI] [PubMed] [Google Scholar]
- Medema, R.H., G.J. Kops, J.L. Bos, and B.M. Burgering. 2000. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 404:782–787. [DOI] [PubMed] [Google Scholar]
- Pawlak, M.R., C.A. Scherer, J. Chen, M.J. Roshon, and H.E. Ruley. 2000. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol. Cell. Biol. 20:4859–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevot, D., T. Voeltzel, A.M. Birot, A.P. Morel, M.C. Rostan, J.P. Magaud, and L. Corbo. 2000. The leukemia-associated protein Btg1 and the p53-regulated protein Btg2 interact with the homeoprotein Hoxb9 and enhance its transcriptional activation. J. Biol. Chem. 275:147–153. [DOI] [PubMed] [Google Scholar]
- Prevot, D., A.P. Morel, T. Voeltzel, M.C. Rostan, R. Rimokh, J.P. Magaud, and L. Corbo. 2001. Relationships of the antiproliferative proteins BTG1 and BTG2 with CAF1, the human homolog of a component of the yeast CCR4 transcriptional complex: involvement in estrogen receptor alpha signaling pathway. J. Biol. Chem. 276:9640–9648. [DOI] [PubMed] [Google Scholar]
- Ramaswamy, S., N. Nakamura, I. Sansal, L. Bergeron, and W.R. Sellers. 2002. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2:81–91. [DOI] [PubMed] [Google Scholar]
- Raught, B., and A.C. Gingras. 1999. eIF4E activity is regulated at multiple levels. Int. J. Biochem. Cell Biol. 31:43–57. [DOI] [PubMed] [Google Scholar]
- Rodier, A., P. Rochard, C. Berthet, J.P. Rouault, F. Casas, L. Daury, M. Busson, J.P. Magaud, C. Wrutniak-Cabello, and G. Cabello. 2001. Identification of functional domains involved in BTG1 cell localization. Oncogene. 20:2691–2703. [DOI] [PubMed] [Google Scholar]
- Rouault, J.P., D. Prevot, C. Berthet, A.M. Birot, M. Billaud, J.P. Magaud, and L. Corbo. 1998. Interaction of BTG1 and p53-regulated BTG2 gene products with mCaf1, the murine homolog of a component of the yeast CCR4 transcriptional regulatory complex. J. Biol. Chem. 273:22563–22569. [DOI] [PubMed] [Google Scholar]
- Savli, H., Y. Aalto, B. Nagy, S. Knuutila, and S. Pakkala. 2002. Gene expression analysis of 1,25(OH)2D3-dependent differentiation of HL-60 cells: a cDNA array study. Br. J. Haematol. 118:1065–1070. [DOI] [PubMed] [Google Scholar]
- Tang, E.D., G. Nunez, F.G. Barr, and K.L. Guan. 1999. Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274:16741–16746. [DOI] [PubMed] [Google Scholar]
- Tang, J., P.N. Kao, and H.R. Herschman. 2000. Protein-arginine methyltransferase I, the predominant protein-arginine methyltransferase in cells, interacts with and is regulated by interleukin enhancer-binding factor 3. J. Biol. Chem. 275:19866–19876. [DOI] [PubMed] [Google Scholar]
- Tang, T.T., D. Dowbenko, A. Jackson, L. Toney, D.A. Lewin, A.L. Dent, and L.A. Lasky. 2002. The forkhead transcription factor AFX activates apoptosis by induction of the BCL-6 transcriptional repressor. J. Biol. Chem. 277:14255–14265. [DOI] [PubMed] [Google Scholar]
- Tirone, F. 2001. The gene PC3(TIS21/BTG2), prototype member of the PC3/BTG/TOB family: regulator in control of cell growth, differentiation, and DNA repair? J. Cell. Physiol. 187:155–165. [DOI] [PubMed] [Google Scholar]
- Tucker, M., M.A. Valencia-Sanchez, R.R. Staples, J. Chen, C.L. Denis, and R. Parker. 2001. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell. 104:377–386. [DOI] [PubMed] [Google Scholar]
- van Dijk, T.B., E. van Den Akker, M.P. Amelsvoort, H. Mano, B. Lowenberg, and M. von Lindern. 2000. Stem cell factor induces phosphatidylinositol 3′-kinase-dependent Lyn/Tec/Dok-1 complex formation in hematopoietic cells. Blood. 96:3406–3413. [PubMed] [Google Scholar]
- von Lindern, M., E.M. Deiner, H. Dolznig, M. Parren-Van Amelsvoort, M.J. Hayman, E.W. Mullner, and H. Beug. 2001. Leukemic transformation of normal murine erythroid progenitors: v- and c-ErbB act through signaling pathways activated by the EpoR and c-Kit in stress erythropoiesis. Oncogene. 20:3651–3664. [DOI] [PubMed] [Google Scholar]