

Abstract
The guanine nucleotide exchange factor Cdc24, the GTPase Cdc42, and the Cdc42 effectors Cla4 and Ste20, two p21-activated kinases, form a signal transduction cascade that promotes mitotic exit in yeast. We performed a genetic screen to identify components of this pathway. Two related bud cortex–associated Cdc42 effectors, Gic1 and Gic2, were obtained as factors that promoted mitotic exit independently of Ste20. The mitotic exit function of Gic1 was dependent on its activation by Cdc42 and on the release of Gic1 from the bud cortex. Gic proteins became essential for mitotic exit when activation of the mitotic exit network through Cdc5 polo kinase and the bud cortex protein Lte1 was impaired. The mitotic exit defect of cdc5-10 Δlte1 Δgic1 Δgic2 cells was rescued by inactivation of the inhibiting Bfa1-Bub2 GTPase-activating protein. Moreover, Gic1 bound directly to Bub2 and prevented binding of the GTPase Tem1 to Bub2. We propose that in anaphase the Cdc42-regulated Gic proteins trigger mitotic exit by interfering with Bfa1-Bub2 GTPase-activating protein function.
Keywords: cell polarity; Cdc14; Gic proteins; Lte1; MEN
Introduction
Yeast Cdc14 is a conserved phosphatase, which is entrapped in the nucleolus during most of the cell cycle (Shou et al., 1999; Visintin et al., 1999). This entrapment effectively inactivates the phosphatase by sequestering it away from its target substrates. At the beginning of anaphase, Cdc14 is activated through release from the nucleolus in a stepwise manner. First, in early anaphase, the cdc fourteen early anaphase release (FEAR) network, which includes separase Esp1, promotes a partial release of Cdc14 from the nucleolus (Stegmeier et al., 2002). In a second phase, the mitotic exit network (MEN), a GTPase-driven signaling cascade, triggers the release of the remaining Cdc14 (Shou et al., 1999; Visintin et al., 1999; Pereira et al., 2002). Full activation of Cdc14 is essential to dephosphorylate key cell cycle regulators such as the Cdk inhibitor Sic1 and Hct1/Cdh1, a specificity factor of the anaphase-promoting complex (APC/C; Schwob et al., 1994; Visintin et al., 1997, 1998). Together, these Cdc14 dephosphorylation events decrease Cdk activity, which is a prerequisite for cells to exit mitosis, not only in yeast but also in mammalian cells (Glotzer et al., 1991).
The small Ras-like GTPase Tem1 is one of the most upstream components of the MEN (Mah et al., 2001). The two-component GTPase-activating protein (GAP) Bfa1–Bub2 complex keeps Tem1 in the inactive GDP-bound form (Shirayama et al., 1994a; Geymonat et al., 2002). Tem1, Bfa1, and Bub2 preferentially localize to the yeast centrosome, the spindle pole body (SPB), that is destined to enter the bud in anaphase. The SPB that stays in the mother cell does not carry Bfa1, Bub2, or Tem1 (Fraschini et al., 1999; Pereira et al., 2001). The MEN activator Lte1 that functions upstream of Tem1 and shows homology to the Ras guanine nucleotide exchange factor protein Cdc25 associates in a polar fashion with the cell cortex of small- and medium-sized buds. This association of Lte1 is lost mid way through anaphase (Bardin et al., 2000; Pereira et al., 2000; Seshan et al., 2002; Yoshida et al., 2003). As a consequence of the polar cellular distribution of Bub2-Bfa1, Lte1, and Tem1, MEN activation only occurs after the anaphase spindle has extended into the bud (Bardin et al., 2000; Pereira et al., 2000). Mitotic exit and cytokinesis thus become dependent on the successful elongation of the anaphase spindle into the bud.
Although most MEN components are essential, the deletion of the MEN activator LTE1 has no obvious phenotype at temperatures between 30°C and 37°C (Shirayama et al., 1994a; Adames et al., 2001; Pereira et al., 2002) suggesting that alternative pathways control mitotic exit. For example, polo-like kinase Cdc5 activates the MEN through inhibitory phosphorylation of the Tem1 GAP component Bfa1 (Hu et al., 2001). In addition, the Rho-like GTPase Cdc42, a key regulator of polarized growth, activates mitotic exit by at least two mechanisms through the Cdc42 effectors Ste20 and Cla4, two p21-activated kinases (Höfken and Schiebel, 2002; Jensen et al., 2002; Seshan et al., 2002). Cla4 targets Lte1 to the bud cortex and regulates both activity and phosphorylation of Lte1 (Höfken and Schiebel, 2002; Seshan et al., 2002). In contrast, Ste20 facilitates mitotic exit in a pathway that is redundant with LTE1 (Höfken and Schiebel, 2002).
Here we show that the Cdc42 effectors Gic1 and Gic2 can promote mitotic exit. Gic proteins become essential for mitotic exit when MEN activation through Cdc5 polo kinase and Lte1 are impaired. Our data are consistent with a model in which Gic1 binds to Bub2 and prevents the assembly of the Bub2–Tem1 complex. Release of Gic1 from the bud cortex in anaphase is important for this regulation. This may provide an additional mechanism by which the elongation of the anaphase spindle into the bud is coordinated with the activation of mitotic exit.
Results
GIC1 and GIC2 promote mitotic exit
Cdc24, Cdc42, and Ste20 form a signaling cascade, which activates mitotic exit alongside the MEN component Lte1 (Shirayama et al., 1994a; Höfken and Schiebel, 2002). Although single deletion of either LTE1 or STE20 has no effect on cell cycle progression at 30°C, simultaneous deletion of both genes is lethal because of a defect in mitotic exit (Höfken and Schiebel, 2002). To gain further insight into the function of the Cdc42 pathway in mitotic exit, we screened for high dosage suppressors of the synthetic lethality of Δlte1 Δste20 cells. This screen should identify two classes of suppressing genes: (1) genes that act downstream of LTE1 or STE20; and (2) genes that share a function with LTE1 or STE20 in promoting mitotic exit.
TEM1 was identified as a class one suppressor (Table I) as it functions downstream of LTE1 in the MEN (Shirayama et al., 1994b). SIC1 was a class two suppressor because it promotes mitotic exit by inhibiting Cdc28-Clb2 (Schwob et al., 1994). SPO12 and PUP3 also allowed growth of Δlte1 Δste20 cells. SPO12 is a component of the FEAR network that is frequently obtained in genetic screens for mitotic exit components (Jaspersen et al., 1998; Stegmeier et al., 2002). PUP3 encodes a proteasome subunit (Finley et al., 1998). The mechanism by which PUP3 restored viability to Δlte1 Δste20 cells requires further studies. The most frequently isolated suppressor was BEM1 (Table I; Fig. 1 A). Bem1 acts as a scaffold protein that links Cdc42 with its guanine nucleotide exchange factor Cdc24 and downstream effectors such as Ste20 and Cla4. Bem1 stabilizes, as part of a positive feedback loop, active Cdc24 at sites of polarized growth and thus ensures continuous production of active Cdc42 (Bose et al., 2001; Butty et al., 2002). Therefore, it was not surprising that high gene dosage of CDC42 and of the Cdc42 activator CDC24 also suppressed Δlte1 Δste20 lethality (Table I, Fig. 1 A).
Table I. Suppressors of synthetic lethality of Δlte1 Δste20 cells.
| Gene | Frequency |
|---|---|
| BEM1 | 27 |
| TEM1 | 16 |
| STE20 | 13 |
| PUP3 | 7 |
| SPO12 | 5 |
| CDC42 | 4 |
| GIC1 | 3 |
| LTE1 | 2 |
| SIC1 | 2 |
Figure 1.


BEM1, CDC42, GIC1, and GIC2 are suppressors of the synthetically lethal phenotype of Δlte1 Δste20 cells. (A) Suppression of the synthetic lethality of Δlte1 Δste20 by BEM1, CDC24, CDC42, GIC1, and GIC2. Serial dilutions (1:10) of Δlte1 Δste20 pRS316-LTE1 cells carrying the indicated genes on the 2μm high copy plasmid pRS425 (Christianson et al., 1992) were spotted on YPDA and 5-FOA plates and incubated at 30°C. Note that only cells that spontaneously lost the URA3-based pRS316-LTE1 can grow on 5-FOA. (B) The CRIB domain of Gic1 is required for suppression of the growth defect of Δlte1 Δste20 cells. Serial dilutions (1:10) of Δlte1 Δste20 pRS316-LTE1 cells with the indicated pRS425 derivatives were spotted on YPDA and 5-FOA plates and incubated at 30°C. (C) Gic1 and Gic1crib− proteins are present at equal levels. Cells of B were grown in selective medium and analyzed by immunoblotting with antibodies specific for Gic1 and β-tubulin Tub2, which was used as loading control. Note the endogenous Gic1 present in the pRS425 cells was not detected by this assay. (D) GIC genes promote mitotic exit independently of SIC1. Dilutions of the indicated cell types were spotted on YPDA and 5-FOA plates and incubated at 30°C. (E) Overexpression of BEM1, CDC24, CDC42, GIC1, and GIC2 rescues the growth defect of Δlte1 cells at 10°C. Serial dilutions of Δlte1 cells transformed with the indicated pRS425 derivatives were grown on YPDA plates at 30°C and 10°C, respectively. (F) Overexpression of GIC1 and GIC2 suppresses the mitotic exit defect of Δlte1. Wild-type, Δlte1, and Δlte1 cells carrying GIC1, GIC1-pr, GIC1-pr-SS, or GIC2 on the high copy number plasmid pRS425 were arrested in G1 with α-factor. Cells progressed synchronously through the cell cycle at 14°C upon removal of α-factor by washing with precooled selective medium. Cell cycle progression was determined by following the number of cells with large buds and nucleolar Cdc14-GFP by fluorescence microscopy (n > 100).
The finding that high gene dosage of BEM1, CDC24, and CDC42 restored viability of the mitotic exit defective Δlte1 Δste20 cells suggested that additional Cdc42 effectors promote mitotic exit independently of Ste20 and Cla4/Lte1. Therefore, it was interesting that GIC1 and GIC2 were also identified in the suppressor screen (Fig. 1 A, Table I). Gic1 and Gic2 are two homologous and functionally redundant Cdc42-regulated proteins (Brown et al., 1997; Chen et al., 1997). We tested whether this suppression depended on the presence of Cdc42/Rac interactive binding (CRIB) motif within Gic1. This motif mediates interaction between Gic1 and Cdc42 (Brown et al., 1997; Chen et al., 1997). Mutating the residues I126A, S127A, and P129A (GIC1 crib−) or deleting codons 126–139 (GIC1 Δ crib) inactivates the CRIB domain and prevents the association of Gic1 with Cdc42 (Brown et al., 1997). Unlike the wild-type equivalent, neither GIC1 crib− (Fig. 1 B) nor GIC1 Δ crib (not depicted) could permit growth of Δlte1 Δste20 cells when present on 2μm-based plasmids. Immunoblot analysis revealed that 2μm-GIC1 and 2μm-GIC1 crib− were expressed at similar levels (Fig. 1 C). Thus, the Cdc42-regulated Gic1 promotes mitotic exit.
Taking the previously established interaction of Cdc24, Cdc42, and Gic1 (Brown et al., 1997; Chen et al., 1997; Butty et al., 2002) into consideration with our suppression data, it seemed likely that we had identified a pathway in which a signal is transmitted from Cdc24 to Cdc42 and hence to the Cdc42 effectors Gic1 and Gic2. The latter two would then promote mitotic exit.
Next, we investigated how GIC1 and GIC2 facilitate mitotic exit. Gic proteins could directly activate the APC/C specificity factor Hct1/Cdh1, the Wee1-like kinase Swe1 or the Cdk1 inhibitor Sic1 (Booher et al., 1993; Schwob et al., 1994; Schwab et al., 1997). However, 2μm-GIC1 allowed growth of Δlte1 Δste20 cells with similar efficiency whether HCT1/CDH1 or SWE1 function was present or not (unpublished data). In addition, the growth defect of Δsic1 and Δlte1 Δsic1 cells, which are delayed in mitotic exit (not depicted), was rescued by GIC1 and GIC2 to the same extent as by TEM1 (Fig. 1 D). This suggests that GIC1 and GIC2 promote mitotic exit independently of HCT1/CDH1, SIC1, and SWE1.
GIC genes could trigger the release of Cdc14 from the nucleolus. This possibility was addressed by turning to the well-characterized Δlte1 cells, which arrest in late anaphase due to an inability to activate the MEN (Shirayama et al., 1994b; Höfken and Schiebel, 2002). The cold sensitive growth defect of Δlte1 cells was suppressed by high gene dosage of BEM1, CDC24, CDC42, GIC1, and GIC2 (Fig. 1 E, 10°C). Then, we asked whether the failure of Δlte1 cells at 14°C to release Cdc14 from the nucleolus was rescued by GIC1 and GIC2. Cells were synchronized in G1 and allowed to progress into a new cell cycle at 14°C. Bud growth and release of Cdc14-GFP from the nucleolus were monitored as the synchronized populations divided. Consistent with published data (Shou et al., 1999; Visintin et al., 1999), Cdc14 was released from the nucleolus of wild-type cells as they entered anaphase (Fig. 1 F, t = 6 h) and was taken up by the nucleolus as cell exited mitosis (Fig. 1 F, t = 8–10 h). In contrast, in virtually all of the Δlte1 cells Cdc14 remained bound in the nucleolus and ∼90% of cells arrested as large budded, binucleated cells in late anaphase (Fig. 1 F). Δlte1 cells overexpressing GIC1 or GIC2 released Cdc14 from the nucleolus with similar kinetics and efficiency as wild-type cells. The decrease of cells with a large bud and the reuptake of Cdc14 into the nucleolus indicated that these cells successfully exited mitosis (Fig. 1 F). Thus, GIC1 and GIC2 suppressed the mitotic exit defect of Δlte1 cells by facilitating the release of Cdc14 from the nucleolus. This in turn implies that the Gic proteins activate either the FEAR or MEN networks which control Cdc14.
The mitotic exit function of Gic proteins becomes essential when Cdc5 and Lte1 are impaired
GIC1 and GIC2 promote mitotic exit by facilitating the release of Cdc14 from the nucleolus. Therefore, cells lacking GIC genes should, at least under certain conditions, have a mitotic exit defect. Single or double deletion of GIC genes did not reveal any obvious mitotic exit delay at 30°C (Fig. 2 C). At 37°C, Δgic1 Δgic2 cells arrest as unbudded cells due to a defect in actin polarization (Brown et al., 1997; Chen et al., 1997). One possible interpretation of this observation is that several mechanisms promote mitotic exit and due to this redundancy the function of Gic proteins in mitotic exit only becomes apparent when the other pathways are also impaired.
Figure 2.


GIC1 and GIC2 promote mitotic exit by releasing Cdc14 from the nucleolus. (A) Deletion of GIC1 and GIC2 in cdc5-10 Δlte1 is lethal. Serial dilutions (1:10) of the indicated cells all with pRS316-CDC5 were spotted on YPDA and 5-FOA plates and incubated at 30°C. (B) Serial dilutions (1:10) of the indicated strains with Gal1-BUB2 were spotted on glucose and galactose plates and incubated at 30°C. (C) Mitotic exit defect of cdc5-10 Δgic1 Δgic2 Δlte1. Wild-type, cdc5-10 Δlte1, Δgic1 Δgic2, and cdc5-10 Δgic1 Δgic2 Δlte1 all with CDC14-GFP and Gal1-BUB2 were grown in YP raffinose medium and arrested in G1 with α-factor. Cells progressed synchronously through the cell cycle upon removal of α-factor by washing with YP raffinose/galactose medium, which induced expression of Gal1-BUB2. The number of cells with large buds and nucleolar Cdc14-GFP (n > 100) was determined over time. (D) Immunoblots of protein extracts of wild-type, cdc5-10 Δlte1, Δgic1 Δgic2, and cdc5-10 Δgic1 Δgic2 Δlte1 cells of C with the indicated antibodies. (E) cdc5-10 Δgic1 Δgic2 Δlte1 CDC14-GFP cells (top) and cdc5-10 Δgic1 Δgic2 Δlte1 GFP-TUB1 cells (bottom) were treated as described in C and then fixed and stained with DAPI after 150 min incubation at 30°C. Bar, 5 μm.
We addressed this possibility by asking whether GIC1 and GIC2 became essential in mutants in which the regulation of mitotic exit was defective. Cdc5 polo kinase inactivates the Bub2–Bfa1 complex by phosphorylating Bfa1 (Hu et al., 2001). Lte1 activates the MEN upstream of the GTPase Tem1 (Shirayama et al., 1994b). cdc5-10 cells grew as wild-type cells at 30°C and 34°C but at 37°C they arrested in late anaphase in a manner that depended on the presence of a functional Bfa1–Bub2 GAP complex (unpublished data). Deletion of LTE1 in conditional lethal cdc5-10 cells was not lethal at 30°C (Fig. 2 A, row 2) but decreased the restrictive temperature of cdc5-10 cells from 37°C to 34°C (not depicted). Deletion of GIC1 and GIC2 in cdc5-10 or Δlte1 cells did not significantly affect growth of cells at 30°C (Fig. 2 A, rows 4 and 5). However, cdc5-10 Δgic1 Δgic2 Δlte1 mutant cells were unable to grow at 30°C (Fig. 2 A, row 6), a temperature that allowed growth of all single, double, and triple mutants. Deletion of the MEN inhibitor BUB2, or overexpression of TEM1 rescued the growth defect of cdc5-10 Δgic1 Δgic2 Δlte1 cells (Fig. 2 A, rows 7 and 8). Considering the established role of Bub2 in MEN inhibition (Pereira et al., 2000) and of Tem1 in MEN activation (Shou et al., 1999), this rescue shows that the cdc5-10 Δgic1 Δgic2 Δlte1 cells failed to grow because of a defect in mitotic exit and not because of another overlapping function of these four genes. Therefore, GIC1 and GIC2 become essential when the mitotic exit functions of Cdc5 and Lte1 are impaired.
To analyze the cell cycle phenotype of cdc5-10 Δgic1 Δgic2 Δlte1 cells, we generated conditional lethal cells by placing the BUB2 gene under control of the Gal1 promoter. cdc5-10 Δgic1 Δgic2 Δlte1 Gal1-BUB2 cells grown at 30°C in the presence of glucose (repression of Gal1-BUB2) were viable (Fig. 2 B) and progressed through the cell cycle similarly to wild-type cells (not depicted). Addition of galactose to induce expression of Gal1-BUB2 restored BUB2 function and the lethal phenotype of cdc5-10 Δgic1 Δgic2 Δlte1 cells (Fig. 2 B). We were now able to analyze the phenotype of cdc5-10 Δgic1 Δgic2 Δlte1 cells. α-Factor synchronized cells carrying CDC14-GFP and Gal1-BUB2 (Fig. 2 C) were released into a new cell cycle after the addition of galactose (t = 0) to induce BUB2 expression. Expression of BUB2 from the Gal1 promoter did not affect cell cycle progression of wild-type, cdc5-10, or cdc5-10 Δgic1 Δgic2 cells (not depicted) nor did it delay mitotic exit in these cells, which would become apparent through a plateau of cells with a large bud (Fig. 2 C). Remarkably, at 30°C, wild-type, cdc5-10 Δlte1, and Δgic1 Δgic2 cells released Cdc14-GFP from the nucleolus with similar kinetics and efficiency (maximum at ∼120 min) and then exited mitosis as indicated by the decrease of large-budded cells, the reuptake of Cdc14 into the nucleolus (Fig. 2 C), and the degradation of the mitotic cyclin Clb2 (Fig. 2 D). Moreover, mitotic exit was accompanied by the accumulation of the Cdk1-Clb inhibitor Sic1 in wild-type and, to a somewhat lesser extent, in cdc5-10 Δlte1 and Δgic1 Δgic2 cells (Fig. 2 D). In contrast, ∼90% cdc5-10 Δgic1 Δgic2 Δlte1 cells arrested in anaphase with a large bud, separated DAPI staining regions, a long anaphase spindle, Cdc14-GFP trapped in the nucleolus (Fig. 2, C and E), high Clb2 levels and no accumulation of Sic1 (Fig. 2 D). This combination of phenotypes is the hallmark of cells with a defect in mitotic exit (Pereira and Schiebel, 2001). Thus, cdc5-10 Δgic1 Δgic2 Δlte1 cells execute anaphase at 30°C similarly to wild-type cells but then fail to exit mitosis.
Gic1 disrupts the formation of the Bfa1–Bub2–Tem1 complex
We used the yeast two-hybrid system to test whether Gic1 interacted with proteins involved in mitotic exit. Strong two-hybrid interactions were detected between Gic1 and Cdc42 (Brown et al., 1997; Chen et al., 1997), Bfa1 and Cdc14 (Fig. 3 A). Consistent with published results (Uetz et al., 2000; Drees et al., 2001), we also observed a two-hybrid interaction between Gic1 and Bub2 (Fig. 3 A). In contrast, Gic2 constructs failed to show two-hybrid interactions even with Cdc42 (unpublished data). Thus, fusion to the Gal4 and LexA elements of the two-hybrid system probably impaired the activity of GIC2.
Figure 3.
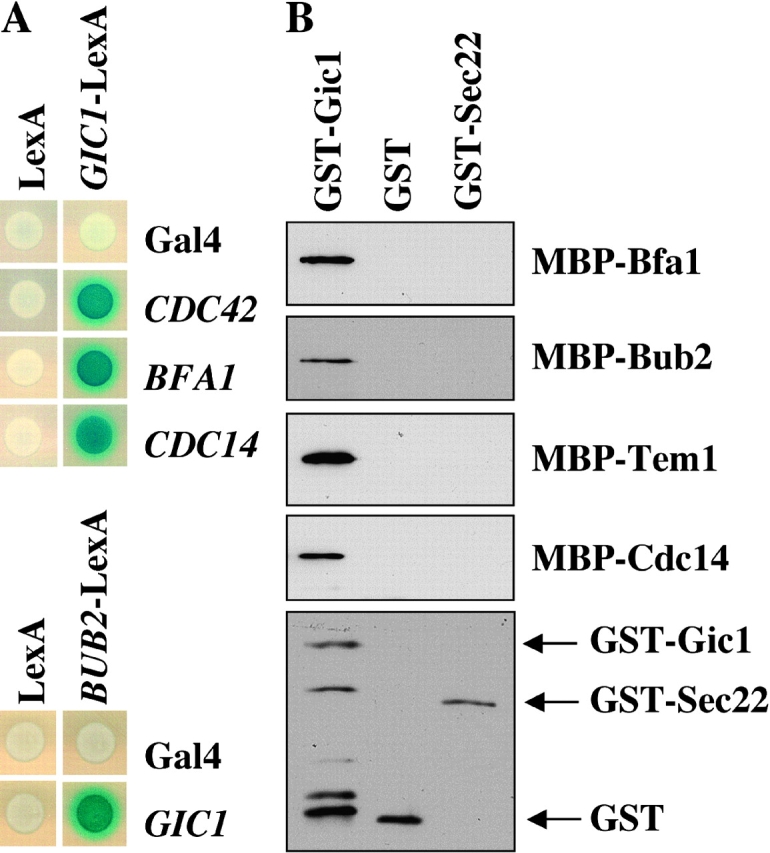
Gic1 interacts with MEN components. (A) Gic1 interacts with Cdc42, Bfa1, Bub2, and Cdc14 in the yeast two-hybrid system. Yeast strains containing the indicated two-hybrid plasmids were overlaid with top agar containing X-Gal and were incubated for 3 h at 30°. (B) Gic1 binds directly to Bfa1, Bub2, Tem1, and Cdc14. Purified GST, GST-Gic1, and GST-Sec22 (20 nM) were bound to Sepharose beads and incubated with the indicated MBP fusion proteins (20 nM). Eluted proteins were analyzed by immunoblotting using antibodies against MBP and GST. The bottom panel shows GST-Gic1, GST-Sec22, and GST eluted from the Sepharose beads of the MBP-Bfa1 experiment (visualized with the anti-GST antibodies). Similar blots were obtained for the other binding experiments using MBP-Bub2, MBP-Tem1, and MBP-Cdc14.
We confirmed the Gic1 two-hybrid interactions by an in vitro binding approach. Recombinant GST and GST fusion proteins with Gic1 and the unrelated protein Sec22 were purified from Escherichia coli and bound to glutathione-Sepharose beads. The immobilized GST proteins were incubated with yeast extracts containing Bfa1-3HA, Bub2-3HA, Tem1-3HA and Cdc14-3HA or with recombinant, E. coli–expressed proteins fused to maltose binding protein (MBP). Because both approaches gave similar results, we only show the outcome of the experiment with the purified MBP fusion proteins. Cdc14, Bfa1, Bub2, and Tem1 bound directly to GST-Gic1 but not to GST or GST-Sec22 (Fig. 3 B). In contrast, the NH2-terminal domain of Net11-600 that interacts with Cdc14 (Shou et al., 1999) failed to bind to GST-Gic1 (unpublished data). The interaction of Gic1 and Tem1 was probably not observed in the yeast two-hybrid assay due to the strong self-activation by Tem1 (unpublished data). Thus, Gic1 bound directly to Cdc14, Bfa1, Bub2, and Tem1.
Gic1 could activate mitotic exit by releasing Cdc14 from the inhibitory Net1–Cdc14 complex, however, two results suggest that this is unlikely. First, overexpression of GIC1 from the strong Gal1 promoter did not release Cdc14 from the nucleolus of cells arrested in metaphase by depletion of the APC/C subunit Cdc20 (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200309080/DC1). Second, when a preformed Cdc14–Net1 complex was incubated in vitro with increasing amounts of purified Gic1 protein, Cdc14 was not displaced from Net1 (Fig. S1 B). In contrast, increasing amounts of recombinant Net1 displaced Cdc14 from Gic1. Together, these results suggest that Gic1 does not promote mitotic exit by directly regulating Cdc14 localization. The functional relevance of the Cdc14–Gic1 interaction remains unclear.
A second possible mechanism by which Gic1 could regulate mitotic exit is by interfering with the formation of the inhibitory Bfa1–Bub2–Tem1 complex. We investigated whether Gic1 prevented the interaction of Tem1 with either Bub2 or Bfa1. Purified His6-Tem1 was preincubated with increasing amounts of GST-Gic1 or GST. The proteins were then incubated with purified, immobilized MBP, MBP-Bub2 or MBP-Bfa1. Tem1 and Gic1 failed to bind to MBP beads (unpublished data). Binding of Tem1 to Bub2 and Bfa1 was observed in the absence of Gic1 (Fig. 4, A and B, lane 1). The addition of GST-Gic1 (Fig. 4, A and B, lanes 2–6) but not GST (lanes 8–12) decreased His6-Tem1 binding to both Bub2 (Fig. 4 A, top) and Bfa1 (Fig. 4 B, top). It is important to note that Gic1 was ∼10 times more efficient in inhibiting Tem1 binding to Bub2 than inhibiting its association with Bfa1 (Fig. 4, A and B, compare lanes 1–4). Therefore, in vitro Gic1 preferentially inhibits Tem1 binding to Bub2.
Figure 4.


Gic1 disrupts the formation of the Bfa1–Bub2–Tem1 complex. (A) Gic1 disrupts the binding between Tem1 and Bub2. His6-Tem1 (20 nM final concentration) was incubated with increasing amounts of recombinant GST-Gic1 (0–200 nM) or GST (0–200 nM). The preincubated proteins were added to MBP-Bub2 beads (Bub2 was 20 nM) and incubated for 60 min. After washing, proteins bound to the beads were eluted and examined by immunoblotting using antibodies against Tem1. The fact that 2 nM Gic1 (lane 2) was able to completely neutralize 20 nm Tem1 indicates either that Tem1 is partially inactive, the protein concentration of Tem1 is underestimated or that Gic1 has multiple binding sites for Bub2 or Tem1. (B) Gic1 can disrupt binding of Tem1 to Bfa1. The experiment was carried out as in A but with MBP-Bfa1 beads. (C) Gic1 can disrupt binding between Bfa1 and Bub2. GST-Bub2 (20 nM) was incubated with increasing amounts of bacterial His6-Gic1 (0–200 nM). Proteins were added to MBP-Bfa1 beads (20 nM) and incubated for additional 60 min. After washing the beads bound proteins were eluted and examined by immunoblotting using antibodies against Bub2. (D) Gic1 can disrupt the Tem1–Bub2 complex in vivo. TEM1 BUB2-3HA (lanes 1 and 7) and TEM1-9Myc BUB2-3HA cells (lanes 2–6 and 8–12) carrying pRS425, pRS425-GIC1, pRS425-GIC1 crib−, pRS425-GIC1-pr, or pRS425-GIC1-pr-SS were lysed. Equal amounts of protein extract were incubated with anti-Myc antibodies. Immunoprecipitates were analyzed by immunoblotting with antibodies against the Myc and HA epitopes.
Next, we tested whether Gic1 was able to prevent formation of the Bfa1–Bub2 complex. GST-Bub2 was incubated with purified His6-Gic1 followed by the incubation with immobilized MBP-Bfa1. In the absence of Gic1, Bub2 interacted with Bfa1 (Geymonat et al., 2002). Addition of Gic1 decreased the efficiency with which Bfa1 and Bub2 formed a complex (Fig. 4 C).
Gic1-induced disruption of complex formation between Bub2 and Tem1 was also observed in vivo. Coimmunoprecipitation of Bub2 and Tem1 from yeast cell extracts has been reported previously (Pereira et al., 2000). We asked whether elevated Gic1 levels reduced the efficiency of this coimmunoprecipitation. In the presence of the pRS425 control plasmid the anti-Myc antibodies not only efficiently immunoprecipitated Tem1-9Myc but also Bub2-3HA (Fig. 4 D, lane 8). This coimmunoprecipitation was dependent on the presence of Myc-tagged Tem1 (Fig. 4 D, lane 7) indicating specificity of the precipitation. High gene dosage of GIC1 but not of the inactive 2μm-GIC1 CRIB− strongly reduced the efficiency of the coimmunoprecipitation of Tem1-9Myc and Bub2-3HA (Fig. 4 D, lanes 9 and 10). In contrast, 2μm-GIC1 did not affect the ability to coprecipitate Bfa1-Tem1 and Bfa1-Bub2 (unpublished data). This selectivity is consistent with our in vitro data indicating preferential inhibition of Tem1–Bub2 interaction by Gic1 (Fig. 4, A–C). Thus, Gic1 inhibits binding of Tem1 to Bub2 in vivo and in vitro.
Gic1 is a stable protein whose localization changes during the cell cycle
Gic2 has been reported to be subject to cell cycle–dependent degradation. In particular, Gic2 is absent from G2 cells (Brown et al., 1997). This raises the question as to how Gic1 and Gic2 can regulate mitotic exit when they are not present in mitosis. Reevaluation of Gic levels during the cell cycle revealed that Gic2 was partially degraded with bud emergence (Fig. 5 B, t = 70 min) but a considerable fraction persisted even after cells had entered mitosis. Gic1 protein levels did not fluctuate during the cell cycle (Fig. 5 A). The appearance of multiple bands in the SDS-PAGE indicated that Gic1 was subject to cell cycle–dependent modification in G1 when cells were predominantly unbudded (Fig. 5 A, t = 50 min). Thus, both Gic proteins are present in mitosis.
Figure 5.

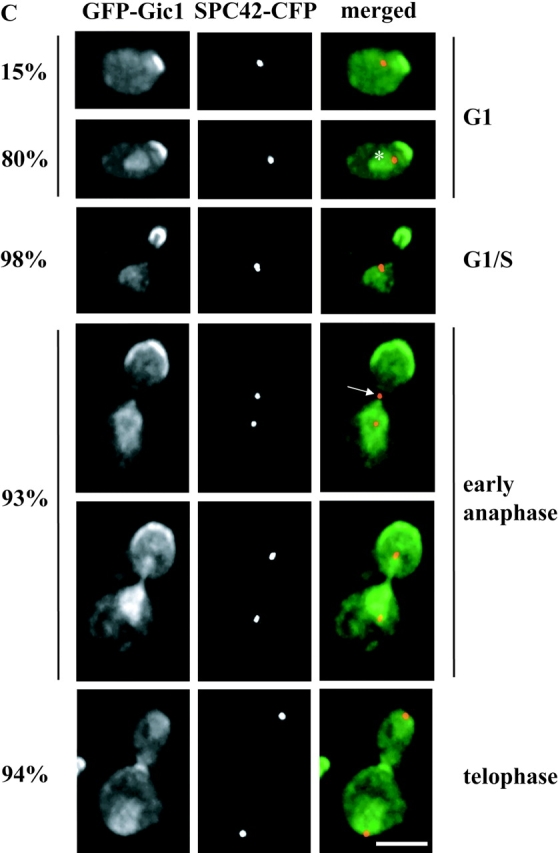

Gic1 was released from the bud cortex during anaphase. (A) Gic1 protein levels remain constant throughout the cell cycle. Gal1-CDC20 GIC1-9Myc cells were arrested in metaphase by incubating cells in YP raffinose medium (no induction of Gal1-CDC20). Galactose was added (t = 0) to the synchronized cells to induce CDC20 expression and to trigger anaphase onset. Samples were taken at the indicated time points after galactose addition. Cells were fixed, stained with DAPI and analyzed by fluorescence microscopy (n > 100). The circles in the cartoon cells indicate the DAPI staining regions. In addition, cell extracts were analyzed by immunoblotting with anti-Myc and anti-Tub2 (loading control) antibodies. (B) Gic2 is expressed in a cell cycle–dependent manner. Gal1-CDC20 GIC2-9Myc cells were treated and analyzed as in A. (C) Gic1 is localized to the bud cortex and the nucleus. Localization of Gic1 was determined using SPC42-CFP Gal1-GFP-GIC1 cells. Gal1-GFP-GIC1 of logarithmically growing cells was induced for 1 h by the addition of galactose. Fixed cells were analyzed by deconvolution fluorescence microscopy. The percentages indicate the frequencies of the various cell types at different stages of the cell cycle. In the remaining cells GFP-Gic1 did not show any specific cellular distribution. The asterisk indicates nuclear GFP-Gic1. The arrow points toward a cell in anaphase in which the SPB in the bud is exposed to Gic1. (D) Bud cortex association of Gic1 is independent of LTE1. Gal1-GFP-GIC1 of logarithmically growing LTE1 and Δlte1 cells was induced for 1 h by the addition of galactose. In most (98%) early anaphase cells GFP-Gic1 was associated with the bud cortex. Bars, 5 μm.
The Bfa1–Bub2–Tem1 complex is enriched at the SPB that is destined to enter the bud (Pereira et al., 2000). GFP-Gic1 has been reported to associate with distinct regions of the cell cortex (Chen et al., 1997). We reevaluated Gic1 localization in synchronized cells, to see whether the Gic1 signal overlaps with SPB-associated Bfa1–Bub2–Tem1 complex. The Gic1-GFP signal was increased by using either a functional, chromosomally integrated GIC1-4GFP gene fusion or a Gal1 promoter controlled GFP-GIC1. The outcome from both approaches was similar and we only present the Gal1-GFP-GIC1 experiment. In early G1 phase when cells had a single Spc42-CFP SPB signal, GFP-Gic1 associated as a patch with the cell cortex, probably at the presumptive bud site (Fig. 5 C, 95% of G1 cells). In 80% of G1 cells an additional Gic1 signal in the nucleus was apparent. With bud formation, when the two unseparated SPBs were still localized in the mother cell body, Gic1 became enriched at the bud cortex (Fig. 5 C, G1/S). This bud cortex association of Gic1 was independent of LTE1 (Fig. 5 D). At anaphase onset with the migration of one of the two SPBs into the bud, the Gic1 signal at the bud cortex (early anaphase) became less focused and the cytoplasmic Gic1, particularly in the bud, increased. This increase in cytoplasmic signal was probably caused by the release of Gic1 from the bud cortex. At this stage of the cell cycle the SPB in the bud carrying the Bfa1–Bub2–Tem1 complex (Pereira et al., 2000) became exposed to cytoplasmic Gic1 (Fig. 5 C, arrow). Because of the high GFP-Gic1 signal in the nucleus and cytoplasm it was difficult to evaluate whether Gic1 was enriched at SPBs. In telophase the signal of Gic1 on the bud cortex diminished further and a Gic1 signal appeared at the mother-bud neck. In summary, the SPB migrating into the bud in anaphase becomes exposed to Gic1.
The Gic1 which is associated with the bud cortex organizes the actin cytoskeleton but does not promote mitotic exit
The distribution of Gic1 suggests two possible mechanisms by which it may regulate mitotic exit. The bud cortex associated Gic1 could disrupt the inhibitory Bfa1–Bub2–Tem1 complex when the SPB carrying these proteins streaks along the bud cortex in anaphase (Adames and Cooper, 2000). Alternatively, Gic1 released from the bud cortex could prevent Tem1–Bub2 complex formation. To investigate whether the membrane associated Gic1 is sufficient to promote mitotic exit, we permanently anchor Gic1 to the plasma membrane. The COOH terminus of Ras2 (aa 301 to 322) can target a protein to the plasma membrane (Fig. 6 A, Gic1-pr; Pryciak and Huntress, 1998). This is because cysteine (Cys) 318 of Ras2 becomes palmitylated and Cys 319 becomes prenylated (Mitchell et al., 1994). To control for the possibility that the fusion of this sequence per se affects Gic1 function, Cys 318 and 319 of the Ras2 element were mutated to serine (Fig. 6 A, Gic1-pr-SS). These mutations prevent modification of Ras2301-322 and the permanent anchorage of Gic1-pr-SS to the plasma membrane.
Figure 6.


A permanently membrane-bound Gic1-pr promotes actin organization but not mitotic exit. (A) Schematic representation of the Gic1-Ras2301-322 fusion proteins. Full-length Gic1 (light gray) was fused to the most COOH-terminal part of wild-type and a mutated Ras2 (dark gray). The CCIIS region (CAAX box) of wild-type Ras2 or the mutated SSIIS (Cys residues 218 and 219 were replaced by Ser) are indicated. The C218S C219S mutations prevent palmitoylation and prenylation and thereby anchorage of Ras2 at the plasma membrane. (B) Localization of Gic1-pr and Gic1-pr-SS. Gal1-GFP-GIC1, Gal1-GFP-GIC1-pr, and Gal1-GFP-GIC1-pr-SS cells were grown in raffinose medium to mid-log phase. The Gal1 promoter was then induced for 1 h by the addition of galactose. Localization of fusion proteins was determined by deconvolution fluorescence microscopy. (C) Membrane-bound Gic1 rescued the growth defect of Δgic1 Δgic2 cells. A serial dilution of wild-type and Δgic1 Δgic2 cells harboring the indicated centromeric pRS315 derivatives (Sikorski and Hieter, 1989) were spotted onto YPDA plates. Plates were incubated at 30°C and 37°C. (D) GIC1-pr complements the actin polarization defect of Δgic1 Δgic2 cells. Δgic1 Δgic2 cells with either pRS315 or pRS315-GIC1-pr were grown in selective medium for 2 h at 37°C. Cells were fixed and the F-actin cytoskeleton was stained with rhodamine-phalloidin. (E) Membrane-bound Gic1 did not suppress lethality of Δlte1 Δste20 cells. Δlte1 Δste20 pRS316-LTE1 cells were transformed with the indicated multicopy pRS425 plasmids. Serial dilutions (1:10) of the cells were spotted onto YPDA and 5-FOA plates. Cells were incubated at 30°C. (F) Cells of E were grown in selective medium and analyzed by immunoblotting with antibodies against Gic1 and Tub2. (G) Gic1-pr and Gic1-pr-SS disrupt the binding between Tem1 and Bub2 in vitro. Experiment was performed as in Fig. 4 A using purified, recombinant proteins. Concentration of all proteins was 20 nM. Bars, 5 μm.
First, we analyzed the localization of Gic1-pr. GFP-Gic1-pr was highly enriched at the bud cortex and was not concentrated within the nucleus, as was the case for GFP-Gic1-pr-SS and GFP-Gic1 (Fig. 6 B). This suggests that palmitoylation and prenylation of Gic1-pr anchors the protein permanently to the plasma membrane. However, Gic1-pr did not simply assume the uniform cortical association around the entire cell that is characteristic of Ras2 (Yoshida et al., 2003), rather the presence of the Gic1 moiety led to the enrichment of Gic1-pr at the bud cortex.
The functionality of GIC1-pr and GIC1-pr-SS was tested. Δgic1 Δgic2 cells are unable to grow at 37°C because of a defect in actin polarization (Brown et al., 1997; Chen et al., 1997). We introduced wild-type and mutant GIC1 genes on the CEN-based low copy number vector pRS315 into Δgic1 Δgic2 cells. GIC1, GIC1-pr, and GIC1-pr-SS were equally efficient in complementing the growth defect of Δgic1 Δgic2 cells at 37°C (Fig. 6 C). Staining F-actin with rhodamine-phalloidin showed that most Δgic1 Δgic2 pRS315 cells accumulated as enlarged unbudded cells, which lacked a polarized actin cytoskeleton (Fig. 6 D). pRS315-GIC1-pr restored the actin cytoskeleton (Fig. 6 D) to the same degree as seen for GIC1 or GIC1-pr-SS (not depicted). Thus, the membrane bound Gic1-pr was able to fulfil the actin polarization function of Gic proteins.
Next, we investigated whether the membrane bound Gic1-pr suppressed the mitotic exit defect of Δlte1 Δste20 cells. Although GIC1-pr-SS on a 2μm high copy plasmid was as potent as GIC1 in suppressing the growth defect of Δlte1 Δste20 cells, GIC1-pr failed to support growth (Fig. 6 E). Consistently, only GIC1-pr-SS but not the membrane anchored GIC1-pr was able to suppress the mitotic exit defect of synchronized Δlte1 cells at 14°C (Fig. 1 F). Moreover, the lack of Gic1-pr to promote mitotic exit correlated with the incapability of this protein to disrupt the Bub2–Tem1 interaction in vivo (Fig. 4 D, lane 11). In contrast, the mitotic exit promoting GIC1-pr-SS interfered with Bub2–Tem1 interaction (Fig. 4 D, lane 12). The failure of Gic1-pr to stimulate mitotic exit and to interfere with Bub2–Tem1 interaction in vivo was not because the level of the Gic1-pr was preferentially decreased (Fig. 6 F) or because the pr-fusion enabled Gic1 to disrupt the Bub2–Tem1 interaction in vitro (Fig. 6 G). The latter result rather suggests that the reason why Gic1-pr did not reduce the Bub2–Tem1 interaction was the permanent association with the bud cortex. Thus, the membrane bound Gic1 is unable to promote mitotic exit and to disrupt the Bub2–Tem1 interaction.
Discussion
Previously, we have shown that the two Cdc42-regulated p21-activated kinases Cla4 and Ste20 play an important role in MEN activation (Höfken and Schiebel, 2002). Cla4 is required for the association of Lte1 with the bud cortex and activation of Lte1 that functions upstream of the small Ras-like GTPase Tem1 (Fig. 7, step 1; Shirayama et al., 1994a; Bardin et al., 2000; Pereira et al., 2000). Ste20 functions in a pathway parallel to Lte1 (Fig. 7, step 2; Höfken and Schiebel, 2002; Seshan et al., 2002). An alternative way to activate the MEN is the inactivation of the inhibitory Bub2–Bfa1 complex. Polo kinase Cdc5 inhibits Bub2-Bfa1 GAP activity through phosphorylation of Bfa1 (Fig. 7, step 3; Hu et al., 2001; Geymonat et al., 2003). Importantly, any single mechanism is not essential for mitotic exit. Mitotic exit is hardly affected by the absence of Lte1 or by a mutated Bfa1 that fails to become inactivated by Cdc5 (Adames et al., 2001; Hu et al., 2001). Mitotic exit is even close to normal in cdc5-10 Δlte1 cells grown at 30°C (Fig. 2 C). Therefore, it is likely that multiple mechanisms concertedly facilitate mitotic exit.
Figure 7.

Model for the function of Gic1. See Discussion for details.
Here, we show that Gic1 and Gic2 are two additional Cdc42 effectors that can promote mitotic exit independently of the functions of Lte1 and Ste20. This conclusion is supported by the following data: (a) GIC1 and GIC2 suppressed the synthetically lethal phenotype of Δlte1 Δste20 cells; (b) high dosage of either GIC gene was able to suppress the MEN defect of Δlte1 cells at 14°C; and (c) GIC1 and GIC2 became essential for mitotic exit in cdc5-10 Δlte1 cells (i.e., in cells in which these two MEN activation pathways are already impaired). cdc5-10 Δlte1 Δgic1 Δgic2 cells showed a cell cycle arrest phenotype reminiscent to MEN mutants (Pereira and Schiebel, 2001). This mitotic exit defect of Δgic1 Δgic2 cdc5-10 Δlte1 cells was rescued by MEN activation either through Bub2-Bfa1 GAP inactivation or TEM1 overexpression (Fig. 2 A).
Gic proteins triggered the nucleolar release of Cdc14 in Δlte1 cells at 14°C. Thus, Gic proteins activate either the MEN or the FEAR network, the two known pathways that regulate Cdc14 (Stegmeier et al., 2002), or directly regulate Cdc14. Gic1 interacted with Bfa1, Bub2, Tem1, and Cdc14 by two-hybrid and in vitro binding studies. Attempts to show these interactions by coimmunoprecipitation failed, however, this is likely because of the very transient nature of the interactions. Other established interactions of MEN components such as binding of Cdc14 to Bfa1 and of Cdc15 to Dbf2 cannot be seen by coimmunoprecipitation assays (Pereira et al., 2002; Visintin et al., 2003). Gic1 could disrupt the nucleolar Net1–Cdc14 complex and by this means release Cdc14 from the nucleus. However, the failure of Gic1 to release Cdc14 from Net1 in vivo and in vitro argues against this possibility. Instead, our in vitro and in vivo data suggest that Gic1 activates mitotic exit by inhibiting the interaction between Tem1 and Bub2. Disruption of the Bub2–Tem1 complex correlated with the capability of Gic1 to promote mitotic exit (Fig. 4 D). In contrast, a Gic1 protein lacking the activating CRIB domain failed both to promote mitotic exit and to disrupt the Bub2–Tem1 complex. A similar behavior was observed for the membrane anchored Gic1-pr. A function upstream or in parallel of TEM1 is also supported by the observation that GIC1 does not suppress the mitotic exit and Cdc14-release defect of tem1-3 cells (unpublished data).
The membrane bound, cytoplasmic, and nuclear forms of Gic1 could promote mitotic exit. The finding that a Gic1 protein permanently anchored to the plasma membrane was unable to promote mitotic exit (Fig. 1 F and Fig. 6 E) suggests that Gic1 has to be released from the bud cortex after its activation by Cdc42 in order to promote mitotic exit. In light of this dependency, we propose that upon anaphase onset Gic1 becomes activated by Cdc42 at the bud cortex and is then released into the cytoplasm. The soluble Gic1 then contributes to the inactivation of the Bub2-Bfa1 GAP. The half-life of Cdc42-activated Gic1 may be short after its release from the bud cortex. This would ensure that Gic1 only inactivates the Bub2–Bfa1–Tem1 complex in the bud. Such a mechanism could contribute to the coupling of anaphase spindle elongation into the bud and mitotic exit.
The observation that Gic1-pr polarizes the actin cytoskeleton but fails to promote mitotic exit demonstrates that Gic1 has two separate functions. The established role of Gic1 in bud formation and actin polarization (Brown et al., 1997; Chen et al., 1997) at the bud cortex is distinct from the cell cycle promoting function that requires soluble Gic1 (this paper). However, Cdc42 regulates both functions because they depend on the Cdc42-binding CRIB domain within Gic1 (Brown et al., 1997; Chen et al., 1997).
The question remains as to where the regulation of the Bub2-Bfa1 GAP and the GTPase Tem1 takes place? The Bub2–Bfa1–Tem1 complex is enriched at the SPB (Fraschini et al., 1999; Pereira et al., 2000) but the importance of this localization is unclear (Pereira and Schiebel, 2001). For example, Tem1 also was found in complexes with the bud cortex–associated kelch domain proteins Kel1 and Kel2 (Höfken and Schiebel, 2002). This Tem1 pool and not the SPB associated Tem1 may be important for mitotic exit. In this respect it is interesting that the Tem1 regulators Lte1 and Amn1 are also not enriched at SPBs (Pereira et al., 2000; Wang et al., 2003). Thus, it is possible that Gic1 activates Tem1 by disrupting the function of a soluble Bub2–Tem1 complex. Alternatively, the SPB associated Bub2-Bfa1-Tem1 could be the target of Gic1. Because of the high levels of cytoplasmic and nuclear Gic1, it is difficult to evaluate whether Gic1 is enriched at SPBs. In any case, it is likely that the cytoplasmic Gic1 at least transiently interacts with the Bub2–Bfa1–Tem1 complex at SPBs. This interaction could result in the displacement of Tem1 from SPBs, or simply inhibit Bub2-Bfa1 GAP activity without affecting Tem1 SPB binding. The finding that 2μm-GIC1 does not affect the Tem1-GFP fluorescence signal at SPBs (unpublished data) would favor the second possibility.
We propose that up until metaphase the MEN remains inactive because of the lack of Bfa1 phosphorylation and the binding of both Lte1 and Gic1 to the bud cortex. Upon anaphase onset, Cdc5, Gic1, and Lte1 concertedly activate the MEN (Fig. 7). The role of Lte1 in MEN activation is not fully understood. In fact, a recent report questioned a direct activation of Tem1 by Lte1 (Yoshida et al., 2003). Suppression of the Δlte1 mitotic exit defect by 2μm-GIC1 either means that GIC1 functions downstream or in parallel with LTE1. Thus, Gic1 could mediate MEN activation by Lte1. However, attempts to show interaction of Gic proteins and Lte1 by yeast two-hybrid, coimmunoprecipitation and in vitro binding failed (not depicted) and Gic1 associated with the bud cortex even in the absence of Lte1 (Fig. 5 D). Moreover, the observation that the Δlte1 cdc5-10 phenotype becomes more severe upon deletion of GIC1 and GIC2 excludes such a simple linear pathway. Therefore, we favor the branched pathway outlined in Fig. 7 (steps 1 and 4).
The mitotic spindle and the centrosome also regulate cell cycle progression in other organisms although the molecular mechanisms are much less understood. In fission yeast, a MAPK pathway seems to coordinate spindle positioning and cell cycle progression (Gachet et al., 2001). In mammalian cells, misoriented spindles caused a delay in anaphase onset (O'Connell and Wang, 2000). The centrosome regulates cell cycle–dependent mitotic exit in animal cells (Piel et al., 2001). It will be interesting to see whether conserved proteins found at the cell cortex, such as Cdc42 and its effectors, are common regulators that coordinate spindle alignment and anaphase onset with cell cycle progression. As a general principal, promoters of mitotic exit may be released from the cell cortex and thereby activated upon spindle elongation in anaphase.
Materials and methods
Growth conditions, yeast strains, and plasmid constructions
Basic yeast methods and growth media were prepared as described previously (Sherman, 1991). Yeast strains were grown in yeast extract, peptone, dextrose medium containing 100 mg/l adenine (YPDA medium). Synthetic complete medium was used to select for yeast plasmids. Plates were incubated for 2–10 d at the indicated temperatures. Yeast strains were derivatives of YPH499 (Sikorski and Hieter, 1989) and are listed in Table II. Plasmids and yeast strains were obtained from P. Pryciak (University of Massachusetts, Worcester, MA), S. Sedwick (National Institute for Medical Research, London, UK), W. Seufert (University of Stuttgart, Stuttgart, Germany), and A. Spang (Friedrich Miescher Laboratorium, MPI, Tübingen, Germany). Yeast strains were constructed using PCR amplified cassettes (Longtine et al., 1998; Knop et al., 1999; Maekawa et al., 2003). GIC1 crib− was constructed by introducing the I126A, S127A, and P129A mutations by site directed mutagenesis. GIC1 crib− fails to interact with Cdc42 (Brown et al., 1997).
Table II. Yeast strains and plasmids.
| Name | Source or reference | |
|---|---|---|
| Yeast strains | Genotype | |
| CLY269 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 cdc5-10 Δlte1::klTRP1a | this paper |
| ESM1192 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 | Höfken and Schiebel, 2002 |
| ESM1362 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 CDC14-GFP-klTRP1 | Höfken and Schiebel, 2002 |
| GPY130 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 BUB2-3HA-KanMX6 | G. Pereira |
| GPY146 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 BUB2-3HA-KanMX6 TEM1-9MYC-klTRP1 | G. Pereira |
| SGY37 | MATa ura3-52::URA3-lexA-op-LacZ trp1 his3 leu2 | Geissler et al., 1996 |
| THY87 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 Δste20::klTRP1 pSM903 | Höfken and Schiebel, 2002 |
| THY209 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-Gal1-CDC20 GIC1-9MYC-klTRP1 | this paper |
| THY210 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-Gal1-CDC20 GIC2-9MYC-klTRP1 | this paper |
| THY211 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δgic1::His3MX6 Δgic2::klTRP1 | this paper |
| THY320 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δgic1::His3MX6 Δgic2::klTRP Δlte1::KanMX6 | this paper |
| THY321 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 CDC14-GFP-klTRP1 | this paper |
| THY173 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-Gal1-GFP-GIC1 | this paper |
| THY430 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 cdc5-10 Δgic1::KanMX6 Δgic2::His3MX6 Δlte1::klTRP1 pCL33 | this paper |
| THY436 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 cdc5-10 Δgic1::KanMX6 Δgic2::His3MX6 Δlte1::klTRP1 Δbub2::hphNT1b | this paper |
| THY444 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 cdc5-10 Δgic1::His3MX6 Δgic2::KanMX6 | this paper |
| THY450 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δsic1::His3MX6 | this paper |
| THY455 | MATa lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 ura3-52-Gal1-BUB2-URA3 CDC14-GFP-LEU2 | this paper |
| THY456 | MATa lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 ura3-52-Gal1-BUB2-URA3 cdc5-10 Δlte1::klTRP1 CDC14-GFP-LEU2 | this paper |
| THY457 | MATa lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 ura3-52-Gal1-BUB2-URA3 Δgic1::His3MX6 Δgic2:: klTRP1 CDC14-GFP-LEU2 | this paper |
| THY458 | MATa lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 ura3-52-Gal1-BUB2-URA3 cdc5-10 Δgic1::KanMX6 Δgic2::His3MX6 Δlte1::klTRP1 Δbub2::hphNT1 CDC14-GFP-LEU2 | this paper |
| THY463 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-2TET-CDC20 CDC14-GFP-hphNT1 | this paper |
| THY471 | MATa lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 ura3-52-Gal1-BUB2-URA3 cdc5-10 Δgic1::KanMX6 Δgic2::His3MX6 Δlte1::klTRP1 Δbub2::hphNT1 ADE2-GFP-TUB1 | this paper |
| THY473 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-2TET-CDC20 CDC14-GFP-hphNT1 His3MX6-Gal1-3HA-GIC1 | this paper |
| THY474 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-2TET-CDC20 CDC14-GFP-hphNT1 His3MX6-Gal1-3HA-GIC2 | this paper |
| THY475 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-2TET-CDC20 CDC14-GFP-hphNT1 klTRP1-Gal1-FLAG-ESP1 | this paper |
| THY478 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-Gal1-GFP-GIC1 SPC42-CFP-klTRP1 | this paper |
| THY480 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-Gal1 GIC1-pr-SS | this paper |
| THY482 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-Gal1 GIC1-pr | this paper |
| THY485 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-Gal1-GFP-GIC1 Δlte1::klTRP1 | this paper |
| THY491 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 Δsic1::hphNT1 pSM903 | this paper |
| YPH499 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 | Sikorski and Hieter, 1989 |
| Plasmids | Construction | |
| pCL1 | pMM5 carrying BUB2 | this paper |
| pCL4 | pMM6 carrying BFA1 | this paper |
| pCL33 | pRS316 carrying CDC5 | this paper |
| pGP103 | pET28c carrying TEM1 | this paper |
| pMal-TEM1 | pMal-c2x carrying TEM1 | S. Sedgwick |
| pMal-BFA1 | pMal-c2x carrying BFA1 | S. Sedgwick |
| pMM5 | p423-pGal1-lexA-Myc | M. Knop |
| pMM6 | p425-pGal1-Gal4-HA | M. Knop |
| pSM770 | pRS425 carrying TEM1 | this paper |
| pSM890 | pMM6 carrying CDC14 | this paper |
| pSM903 | pRS316 carrying LTE1 | Höfken and Schiebel, 2002 |
| pSM922 | pRS425 carrying LTE1 | this paper |
| pSM731 | pGEX-5X-1 carrying BUB2 | this paper |
| pTH66 | pMM6 carrying CDC42 | this paper |
| pTH113 | pRS425 carrying CDC24 | this paper |
| pTH114 | pRS425 carrying CDC42 | this paper |
| pTH123 | pRS425 carrying GIC1 | this paper |
| pTH124 | pRS425 carrying GIC2 | this paper |
| pTH132 | pRS425 carrying BEM1 | this paper |
| pTH143 | pMM5 carrying GIC1 | this paper |
| pTH144 | pMM6 carrying GIC1 | this paper |
| pTH147 | pGEX-5X-1 carrying GIC1 | this paper |
| pTH158 | pMal-c2x carrying BUB2 | this paper |
| pTH175 | pET28a carrying NET1 1-600 | this paper |
| pTH178 | pET28c carrying GIC1 | this paper |
| pTH179 | pRS425 carrying GIC1 crib− | this paper |
| pTH185 | pRS425 carrying GIC1-pr | this paper |
| pTH186 | pRS425 carrying GIC1-pr-SS | this paper |
| pTH190 | pRS315 carrying GIC1-pr | this paper |
| pTH191 | pRS315 carrying GIC1-pr-SS | this paper |
| pTH194 | pRS315 carrying GIC1 | this paper |
| pTH205 | pGEX-5X-1 carrying GIC1-pr | this paper |
| pTH215 | pGEX-5X-1 carrying GIC1-pr-SS | this paper |
| pWS927 | pMal carrying CDC14 | W. Seufert |
| SEC22-GST | pGEX carrying SEC22 | A. Spang |
klTRP1 encodes the Kluyveromyces lactis TRP1 gene.
hphNT1 encodes the E. coli hph gene.
Suppression analysis and test for synthetic lethality
For the high copy suppression screen, Δlte1 Δste20 cells were transformed with a 2μm LEU2-based YEp13 library (a gift from K. Nasmyth, Institute of Molecular Pathology, Vienna, Austria) and grown on selective plates at 30°C for 3 d. Transformants were replica plated onto 5′-fluoroorotic acid (5-FOA) plates and incubated for 2 d at 30°C. Genes were subcloned after PCR amplification into 2μm pRS425 plasmids (Christianson et al., 1992). Shuffle strains were constructed by transforming strain YPH499 with a plasmid carrying the gene of interest on the URA3-based pRS316 (Sikorski and Hieter, 1989). The chromosomal gene was then disrupted. Cell density was determined with a counter (model Z1; Beckman Coulter). A final concentration of 106 cells/ml was serially diluted (1:10) and spotted on 5-FOA or selective plates.
To analyze cells for a mitotic exit defect, yeast cells with CDC14-GFP were incubated at 30°C for 3 h with 10 μg/ml of α-factor to arrest cells in G1 phase of the cell cycle. α-Factor was removed by washing the cells twice with medium (t = 0). Cells were incubated at the indicated temperature. The budding index and nucleolar Cdc14-GFP of fixed cells were determined by phase-contrast and fluorescence microscopy over time. DNA was stained with DAPI.
In vitro binding experiments and two-hybrid analysis
Expression of plasmids encoding GST, His6, and MBP gene fusions in E. coli BL21 DE3 was induced by the addition of 0.5 mM IPTG to L-broth. The cells were incubated for 6 h at 23°C. GST fusion proteins were incubated with glutathione-Sepharose beads (Amersham Biosciences). MBP fusion proteins were presented to amylose resin beads (New England BioLabs, Inc.) and His6 fusions were bound to Ni2+–NTA-agarose (QIAGEN). Proteins were affinity purified as recommended by the manufacturers. Protein concentration was determined using protein assay solution (Bio-Rad Laboratories) and was confirmed by PAGE followed by Coomassie blue staining. For in vitro binding experiments, 20 nM of bead-bound protein were incubated with 20 nM of soluble recombinant protein (total volume 1 ml) for 1 h at 4°C in binding buffer (PBS, 1 mM DTT, 5 mM MgCl2, 0.05% NP-40, 100 μM GTP). After three washes with binding buffer, the associated proteins were eluted with sample buffer and analyzed by immunoblotting. Two-hybrid interactions were determined in strain SGY37 with GIC1, BUB2, BFA1, CDC14, and CDC42 subcloned into pMM5 and pMM6 (Geissler et al., 1996).
Immunological techniques and microscopy
Mouse monoclonal anti-GST antibodies and polyclonal rabbit anti-Clb2, anti-Sic1, anti-Tem1, and anti-Tub2 antibodies have been described previously (Pereira et al., 2002). Monoclonal mouse anti-HA (12CA5) and anti-Myc (9E10) antibodies were obtained from Boehringer and mouse monoclonal anti-MBP antibodies were obtained from New England BioLabs, Inc. Secondary antibodies were obtained from Jackson ImmunoResearch Laboratories. Anti-Bub2 and anti-Gic1 antibodies were raised in sheep against the recombinant GST fusion proteins purified from E. coli. Antibodies were affinity purified with recombinant protein coupled to CNBr-Sepharose (Amersham Biosciences). Tem1-9Myc was immunoprecipitated from yeast cell extracts using anti-Myc antibodies coupled to ProA-Sepharose beads.
F-actin was stained with rhodamine-phalloidin (Höfken and Schiebel, 2002). For fluorescence microscopy, Z sequences were collected on an Axiophot microscope (Carl Zeiss MicroImaging, Inc.) controlled by Metamorph software (Universal Imaging Corp.) using a Coolsnap HQ camera (Photometrics). Images were deconvoluted with Huygens software (Scientific Volume Imaging), and colored and merged using Adobe Photoshop.
Online supplemental material
Fig. S1 shows that Gic1 does not disrupt the Net1–Cdc14 complex. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200309080/DC1.
Acknowledgments
We thank K. Nasmyth for the YEp13 library and P. Pryciak, S. Sedgwick, W. Seufert, and A. Spang for plasmids. We are grateful to S. Bagley for help with deconvolution microscopy, and I. Hagan and M. Donaldson for helpful discussion.
The work of E. Schiebel is supported by Cancer Research UK.
The online version of this article includes supplemental material.
Abbreviations used in this paper: APC/C, anaphase-promoting complex; CRIB, Cdc42/Rac interactive binding; Cys, cysteine; FEAR, cdc fourteen early anaphase release; GAP, GTPase-activating protein; MBP, maltose binding protein; MEN, mitotic exit network; SPB, spindle pole body.
References
- Adames, N.R., and J.A. Cooper. 2000. Microtubule interactions with the cell cortex causing nuclear movements in Saccharomyces cerevisiae. J. Cell Biol. 149:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adames, N.R., J.R. Oberle, and J.A. Cooper. 2001. The surveillance mechanism of the spindle position checkpoint in yeast. J. Cell Biol. 153:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin, A.J., R. Visintin, and A. Amon. 2000. A mechanism for coupling exit from mitosis to partitioning of the nucleus. Cell. 102:21–31. [DOI] [PubMed] [Google Scholar]
- Booher, R.N., R.J. Deshaies, and M.W. Kirschner. 1993. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 12:3417–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose, I., J.E. Irazoqui, J.J. Moskow, E.G. Bardes, T.R. Zyla, and D.J. Lew. 2001. Assembly of scaffold-mediated complexes containing Cdc42p, the exchange factor Cdc24p, and the effector Cla4p required for cell cycle-regulated phosphorylation of Cdc24p. J. Biol. Chem. 276:7176–7186. [DOI] [PubMed] [Google Scholar]
- Brown, J.L., M. Jaquenoud, M.-P. Gulli, J. Chant, and M. Peter. 1997. Novel Cdc42-binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes Dev. 11:2972–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butty, A.C., N. Perrinjaquet, A. Petit, M. Jaquenoud, J.E. Segall, K. Hofmann, C. Zwahlen, and M. Peter. 2002. A positive feedback loop stabilizes the guanine-nucleotide exchange factor Cdc24 at sites of polarization. EMBO J. 21:1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, G.C., Y.J. Kim, and C.S. Chan. 1997. The Cdc42 GTPase-associated proteins Gic1 and Gic2 are required for polarized cell growth in Saccharomyces cerevisiae. Genes Dev. 11:2958–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson, T.W., R.S. Sikorski, M. Dante, J.H. Shero, and P. Hieter. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene. 110:119–122. [DOI] [PubMed] [Google Scholar]
- Drees, B.L., B. Sundin, E. Brazeau, J.P. Caviston, G.C. Chen, W. Guo, K.G. Kozminski, M.W. Lau, J.J. Moskow, A. Tong, et al. 2001. A protein interaction map for cell polarity development. J. Cell Biol. 154:549–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley, D., K. Tanaka, C. Mann, H. Feldmann, M. Hochstrasser, R. Vierstra, S. Johnston, R. Hampton, J. Haber, J. McCusker, et al. 1998. Unified nomenclature for subunits of the Saccharomyces cerevisiae proteasome regulatory particle. Trends Biochem. Sci. 23:244–245. [DOI] [PubMed] [Google Scholar]
- Fraschini, R., E. Formenti, G. Lucchini, and S. Piatti. 1999. Budding yeast Bub2 is localized at the spindle pole bodies and activates the mitotic checkpoint via a different pathway from Mad2. J. Cell Biol. 145:979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachet, Y., S. Tournier, J.B.A. Millar, and J. Hyams. 2001. A MAP kinase-dependent actin checkpoint ensures proper spindle orientation in fission yeast. Nature. 412:352–355. [DOI] [PubMed] [Google Scholar]
- Geissler, S., G. Pereira, A. Spang, M. Knop, S. Souès, J. Kilmartin, and E. Schiebel. 1996. The spindle pole body component Spc98p interacts with the γ-tubulin-like Tub4p of Saccharomyces cerevisiae at the sites of microtubule attachment. EMBO J. 15:3899–3911. [PMC free article] [PubMed] [Google Scholar]
- Geymonat, M., A. Spanos, S.J. Smith, E. Wheatley, K. Rittinger, L.H. Johnston, and S.G. Sedgwick. 2002. Control of mitotic exit in budding yeast. In vitro regulation of Tem1 GTPase by Bub2 and Bfa1. J. Biol. Chem. 277:28439–28445. [DOI] [PubMed] [Google Scholar]
- Geymonat, M., A. Spanos, P.A. Walker, L.H. Johnston, and S.G. Sedgwick. 2003. In vitro regulation of budding yeast Bfa1/Bub2 GAP activity by Cdc5. J. Biol. Chem. 278:14591–14594. [DOI] [PubMed] [Google Scholar]
- Glotzer, M., A.W. Murray, and M.W. Kirschner. 1991. Cyclin is degraded by the ubiquitin pathway. Nature. 349:132–138. [DOI] [PubMed] [Google Scholar]
- Höfken, T., and E. Schiebel. 2002. A role for cell polarity proteins in mitotic exit. EMBO J. 21:4851–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, F., Y. Wang, D. Liu, Y. Li, J. Qin, and S.J. Elledge. 2001. Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell. 107:655–665. [DOI] [PubMed] [Google Scholar]
- Jaspersen, S.L., J.F. Charles, R.L. Tinker-Kulberg, and D.O. Morgan. 1998. A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol. Biol. Cell. 9:2803–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, S., M. Geymonat, A.L. Johnson, M. Segal, and L.H. Johnston. 2002. Spatial regulation of the guanine nucleotide exchange factor Lte1 in Saccharomyces cerevisiae. J. Cell Sci. 115:4977–4991. [DOI] [PubMed] [Google Scholar]
- Knop, M., K. Siegers, G. Pereira, W. Zachariae, B. Winsor, K. Nasmyth, and E. Schiebel. 1999. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 15:963–972. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., A. McKenzie, D.J. Demarini, N.G. Shah, A. Wach, A. Brachat, P. Philippsen, and J.P. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 14:953–961. [DOI] [PubMed] [Google Scholar]
- Maekawa, H., T. Usui, M. Knop, and E. Schiebel. 2003. Yeast Cdk1 translocates to the plus end of cytoplasmic microtubules to regulate bud cortex interactions. EMBO J. 22:438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah, A.S., J.J. Jang, and R.J. Deshaies. 2001. Protein kinase Cdc15 activates the Dbf2-Mob1 kinase complex. Proc. Natl. Acad. Sci. USA. 98:7325–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, D.A., L. Farh, T.K. Marshall, and R.J. Deschenes. 1994. A polybasic domain allows nonprenylated Ras proteins to function in Saccharomyces cerevisiae. J. Biol. Chem. 269:21540–21546. [PubMed] [Google Scholar]
- O'Connell, C.B., and Y. Wang. 2000. Mammalian spindle orientation and position respond to changes in cell shape in a dynein-dependent fashion. Mol. Biol. Cell. 11:1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, G., and E. Schiebel. 2001. The role of the yeast spindle pole body and the mammalian centrosome in regulating late mitotic events. Curr. Opin. Cell Biol. 13:762–769. [DOI] [PubMed] [Google Scholar]
- Pereira, G., T. Höfken, J. Grindlay, C. Manson, and E. Schiebel. 2000. The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol. Cell. 6:1–10. [PubMed] [Google Scholar]
- Pereira, G., T.U. Tanaka, K. Nasmyth, and E. Schiebel. 2001. Modes of spindle pole body inheritance and segregation of the Bfa1p/Bub2p checkpoint protein complex. EMBO J. 20:6359–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira, G., C. Manson, J. Grindlay, and E. Schiebel. 2002. Regulation of the Bfa1p–Bub2p complex at spindle pole bodies by the cell cycle phosphatase Cdc14p. J. Cell Biol. 157:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel, M., J. Nordberg, U. Euteneuer, and M. Bornens. 2001. Centrosome-dependent exit of cytokinesis in animal cells. Science. 291:1550–1553. [DOI] [PubMed] [Google Scholar]
- Pryciak, P.M., and F.A. Huntress. 1998. Membrane recruitment of the kinase cascade scaffold protein Ste5 by the G β γ complex underlies activation of the yeast pheromone response pathway. Genes Dev. 12:2684–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, M., A.S. Lutum, and W. Seufert. 1997. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 90:683–693. [DOI] [PubMed] [Google Scholar]
- Schwob, E., T. Bohm, M.D. Mendenhall, and K. Nasmyth. 1994. The B-type cyclin kinase inhibitor p40 (Sic1) controls the G1 to S transition in Saccharomyces cerevisiae. Cell. 79:233–244. [DOI] [PubMed] [Google Scholar]
- Seshan, A., A.J. Bardin, and A. Amon. 2002. Control of Lte1 localization by cell polarity determinants and Cdc14. Curr. Biol. 12:2098–2110. [DOI] [PubMed] [Google Scholar]
- Sherman, F. 1991. Getting started with yeast. Methods Enzymol. 194:3–21. [DOI] [PubMed] [Google Scholar]
- Shirayama, M., Y. Matsui, K. Tanaka, and A. Toh-e. 1994. a. Isolation of a CDC25 family gene, MSI2/LTE1, as a multicopy suppressor of ira1. Yeast. 10:451–461. [DOI] [PubMed] [Google Scholar]
- Shirayama, M., Y. Matsui, and A. Toh-e. 1994. b. The yeast TEM1 gene, which encodes a GTP-binding protein, is involved in termination of M-phase. Mol. Cell. Biol. 14:7476–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou, W., J.H. Seol, A. Shevchenko, C. Baskerville, D. Moazed, W.S. Chen, J. Jang, A. Shevchenko, H. Charbonneau, and R. Deshaies. 1999. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 97:233–244. [DOI] [PubMed] [Google Scholar]
- Sikorski, R.S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier, F., R. Visintin, and A. Amon. 2002. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 108:207–220. [DOI] [PubMed] [Google Scholar]
- Uetz, P., L. Giot, G. Cagney, T.A. Mansfield, R.S. Judson, J.R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart, et al. 2000. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 403:623–627. [DOI] [PubMed] [Google Scholar]
- Visintin, R., S. Prinz, and A. Amon. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 278:460–463. [DOI] [PubMed] [Google Scholar]
- Visintin, R., K. Craig, E.S. Hwang, S. Prinz, M. Tyers, and A. Amon. 1998. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell. 2:709–718. [DOI] [PubMed] [Google Scholar]
- Visintin, R., E.S. Hwang, and A. Amon. 1999. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature. 398:818–823. [DOI] [PubMed] [Google Scholar]
- Visintin, R., F. Stegmeier, and A. Amon. 2003. The role of polo kinase Cdc5 in the FEAR and mitotic exit networks. Mol. Biol. Cell. 14:4486–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., T. Shirogane, D. Liu, W. Harper, and S.J. Elledge. 2003. Exit from exit: resetting the cell cycle through Amn1 inhibition of G protein signalling. Cell. 112:697–709. [DOI] [PubMed] [Google Scholar]
- Yoshida, S., R. Ichihashi, and A. Toh-e. 2003. Ras recruits mitotic exit regulator Lte1 to the bud cortex in budding yeast. J. Cell Biol. 161:889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]