

Abstract
We have found that key mitotic regulators show distinct patterns of degradation during exit from mitosis in human cells. Using a live-cell assay for proteolysis, we show that two of these regulators, polo-like kinase 1 (Plk1) and Aurora A, are degraded at different times after the anaphase-promoting complex/cyclosome (APC/C) switches from binding Cdc20 to Cdh1. Therefore, events in addition to the switch from Cdc20 to Cdh1 control the proteolysis of APC/CCdh1 substrates in vivo. We have identified a putative destruction box in Plk1 that is required for degradation of Plk1 in anaphase, and have examined the effect of nondegradable Plk1 on mitotic exit. Our results show that Plk1 proteolysis contributes to the inactivation of Plk1 in anaphase, and that this is required for the proper control of mitotic exit and cytokinesis. Our experiments reveal a role for APC/C-mediated proteolysis in exit from mitosis in human cells.
Keywords: Aurora; cytokinesis; mitosis; APC/C; Cdh1
Introduction
In animal cells, the regulated proteolysis of cyclin A, cyclin B1, and securin during mitosis are all essential for the proper timing of events leading up to separation of sister chromatids at the onset of anaphase (den Elzen and Pines, 2001; Geley et al., 2001; Stemmann et al., 2001; Hagting et al., 2002; Leismann and Lehner, 2003). Proteolysis of these key mitotic regulators is mediated by the anaphase-promoting complex/cyclosome (APC/C) ubiquitin ligase and requires the activating subunit Cdc20/fizzy (Morgan, 1999; Peters, 2002). Cdc20 activity is replaced by that of Cdh1/fizzy-related during mitotic exit, and the role of Cdh1 in suppressing mitotic cyclins is essential to establish the G1 phase of the cell cycle (for review see Peters, 2002). However, this switch from Cdc20 to Cdh1 is thought to allow degradation of many additional substrates because APC/CCdh1 has been shown to have broader substrate specificity than APC/CCdc20 (Fang et al., 1998; Pfleger and Kirschner, 2000; Hagting et al., 2002; Zur and Brandeis, 2002). Amongst the regulators degraded during mitotic exit in mammalian cells are Cdc20, the polo-like kinase 1 (Plk1), the Aurora kinases, and the CENP-E motor protein (Brown et al., 1994; Weinstein, 1997; Ferris et al., 1998; Honda et al., 2000). However, it has not been shown whether these substrates are all degraded as soon as the APC/C switches from its Cdc20- to Cdh1-activated form, or whether they are degraded at distinct times, perhaps to coordinate exit from mitosis. In budding yeast, mitotic exit is under the tight control of the mitotic exit network (Morgan, 1999), a signaling cascade required for the activation of Cdh1 by the Cdc14 phosphatase that can be restrained by a Bub2-dependent checkpoint that monitors the position of the spindle (Li, 1999; Pereira et al., 2000). An equivalent network in mammalian cells has yet to be identified, although homologues of some of the components, such as Cdc14, have been identified. In human cells, a homologue of the spindle checkpoint protein Mad2 (Mad2B) has been shown to inhibit Cdh1 in vitro (Chen and Fang, 2001), but the role of Mad2B in mitotic exit, if any, is not known.
Here, we have begun to examine the role and regulation of proteolysis during mitotic exit in mammalian cells, through studying fluorescent protein (FP)–tagged substrates in living cells. We find that different mitotic regulators are degraded at different times, indicating that APC/CCdh1 activity may be modulated to coordinate mitotic exit and cytokinesis.
Results
Initially, we investigated whether it was possible to identify differences in the degradation patterns of substrates by analyzing immunoblots of endogenous protein levels in HeLa cell extracts. Analysis of extracts from cells synchronized through mitotic exit by release from nocodazole-induced arrest showed that the degradation of APC/CCdh1 substrates was not concerted (Fig. 1). Plk1 disappeared from cell extracts earlier than Aurora A and CENP-E, whereas Aurora B appeared to be degraded later and was still detectable in G1 cell extracts (Fig. 1 A). In further experiments, p55Cdc20 appeared to be degraded earlier than Plk1 (Fig. 1 B). PRC1 and BubR1, proposed to be late mitotic substrates (Jiang et al., 1998; Chan et al., 1999), were not significantly degraded after release from nocodazole (Fig. 1 A). Although this analysis confirmed that specific mitotic regulators were degraded during exit from mitosis and indicated that they were degraded with different kinetics, it did not show exactly when degradation of the substrates began. Moreover, we found that the rate of recovery from the nocodazole block varied between cells. Therefore, we tagged substrates with FPs to use as markers for the endogenous substrates in single-cell analyses of degradation.
Figure 1.

The disappearance of mitotic regulators at exit from mitosis is not concerted. (A and B) HeLa cells were synchronized in prometaphase by a nocodazole block after a thymidine/aphidicolin double block/release (see Materials and methods). Mitotic cells were harvested by shake-off, washed, and replated in the absence of nocodazole. At the time points indicated, samples were processed for immunoblot analysis of the indicated proteins. (A) Parallel samples were processed for analysis of DNA content by flow cytometry, and the M-phase/G1 distribution of each cell population is indicated. Protein loading was assayed by Ponceau S staining and is equivalent for each time point. (B) Extracts were probed with antibodies against p55Cdc20 and Plk1, and with β-tubulin as a loading control. We found that the time required for recovery from nocodazole varied between each of the three experiments we performed (compare Plk1 degradation in A and B), but that Cdc20 levels always fell before those of Plk1. ns, extracts from nonsynchronized cells. Note that comparing levels in unsynchronized cells to mitotic cells in A indicates that PRC1 (but not BubR1) levels are cell cycle regulated.
We began by analyzing Plk1. A GFP-Plk1 chimera had previously been validated as a marker for the localization of endogenous human Plk1 (Arnaud et al., 1998), so we examined whether it was also a suitable marker for the mitotic degradation of Plk1. We injected G2 phase HeLa cells with a cDNA encoding Plk1 tagged at its amino terminus with EYFP (YFP-Plk1), and recorded the fluorescence of mitotic cells by time-lapse imaging. YFP-Plk1 fluorescence started to decline at the beginning of anaphase (Fig. 2), timing that was clearly distinguishable from the decrease in cyclin B1-CFP in metaphase (Clute and Pines, 1999; Hagting et al., 2002; Fig. 2 A; Video 1 and supplemental data, available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1). The onset of Plk1 degradation correlated with its relocalization from the kinetochores to the midzone of the mitotic spindle (Arnaud et al., 1998; Fig. 2 A, time point 3). The decline in YFP-Plk1 fluorescence was indeed a result of proteolysis because it was blocked by the proteasome inhibitor MG132 (Fig. 2 B). The onset and rate of YFP-Plk1 degradation were highly reproducible in this assay (Fig. 2 C), and in almost all cells, 60–75% of YFP-Plk1 was degraded during mitotic exit. Cells expressing very high levels of YFP-Plk1 were not able to degrade the protein properly and were unable to exit normally from mitosis (see Fig. 5). Using a U20S cell line stably expressing YFP-Plk1 (Jackman et al., 2003), we have estimated that the usual levels of expression of YFP-Plk1 achieved during injection experiments varied from less than that of endogenous Plk1 to approximately threefold the endogenous level. The timing of YFP-Plk1 degradation was identical in U20S cells (unpublished data).
Figure 2.

Plk1 degradation begins at the start of anaphase and is proteasome dependent. (A) HeLa cells synchronized in late G2 phase were coinjected with cDNAs encoding YFP-Plk1 and cyclin B1-CFP, and fluorescence in mitotic cells was recorded by time-lapse imaging. Fluorescence in whole cells was measured and plotted as pixel values. The degradation curves shown are representative of at least seven cells in at least two separate experiments. YFP-Plk1–associated fluorescence at the various stages of mitosis is shown (images taken from Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1). 1, prophase; 2, prometaphase; 3, early anaphase; 4, late anaphase; 5, telophase. (B) MG132 at a final concentration of 50 μM was added to a cell in anaphase during time-lapse imaging of cells expressing YFP-Plk1 as in A. Its degradation curve has been superimposed on that of a control cell in the dish, which had completed mitosis before addition of MG132. (C) YFP fluorescence in cells expressing different levels of YFP-Plk1. Degradation of YFP-Plk1 at anaphase was measured in >20 cells, with or without coexpression of cyclin B1-CFP, in >10 separate experiments. Here, natural log values of fluorescence from representative cells have been plotted to show that the rate of degradation does not vary between cells, and is not affected by coexpression of cyclin B1. The vertical line in C indicates the average time of anaphase onset and is accurate within 90 s for each cell.
Figure 5.
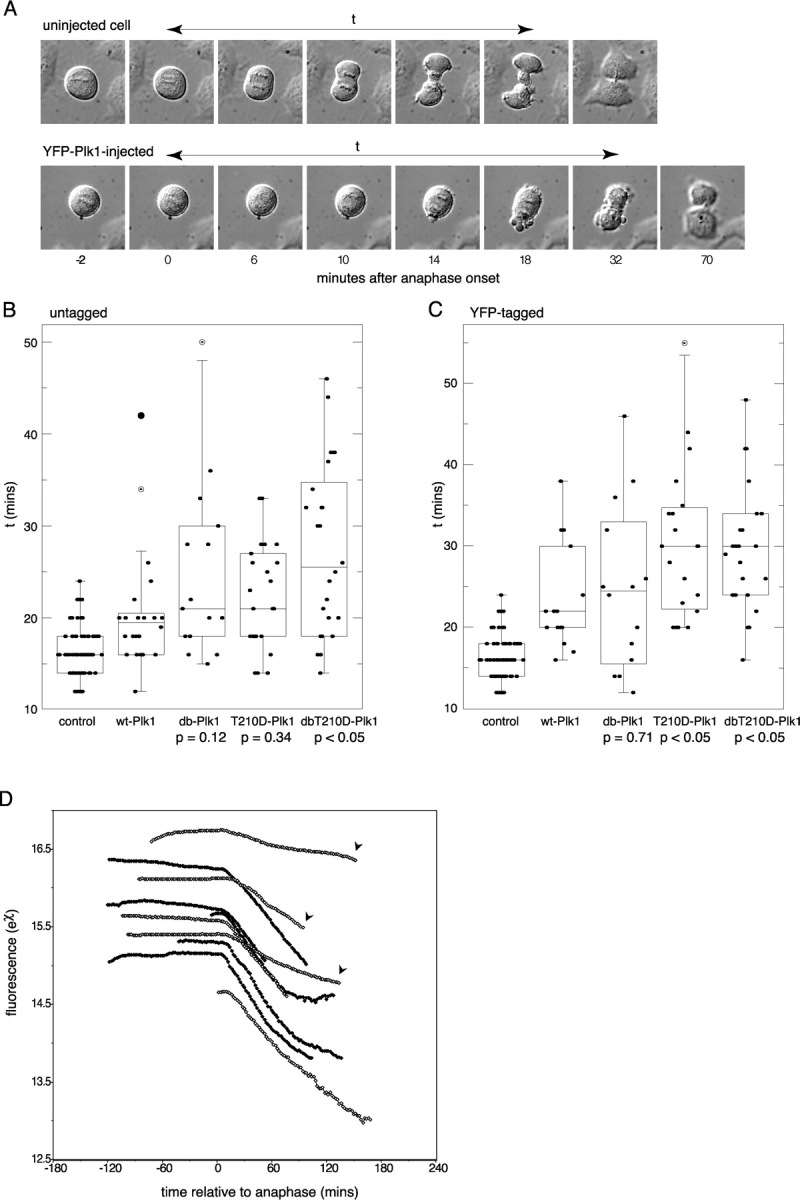
Nondegradable Plk1 causes a delay in mitotic exit. (A) Time-lapse DIC images of mitotic exit for a normal (uninjected) HeLa cell and for one injected in G2 phase with a YFP-Plk1 expression plasmid. These cells were filmed concurrently in the same dish (see Materials and methods). t, mitotic exit time (see below). (B and C) Box plot distributions of mitotic exit times for uninjected cells and cells injected with cDNAs encoding untagged wt-, db-, T210D-, dbT210D-Plk1 (B), or YFP-tagged versions of the same constructs (C). A YFP expression plasmid was used as a marker for cells injected with untagged constructs (B). In these distributions, the maximum and minimum values, the interquartile range (marked by boxes), and the median value (marked by horizontal line) for mitotic exit times in each population are shown. Suspected outliers are shown as open circles, an outlier as a filled circle. P values for each population of cells expressing a Plk1 mutant, compared with that expressing the wild-type version, are indicated. The software used is available online upon request. (D) Degradation curves (plotted as natural log values) for YFP-Plk1 and YFP-T210D-Plk1 in cells showing impaired mitotic exit. Cells expressing either YFP-Plk1 (filled circles) or YFP-T210D-Plk1 (open circles) were compared from the same experiment. Cells in which mitotic exit took >24 min are indicated by arrowheads. This figure is representative of three separate experiments.
The time at which YFP-Plk1 degradation began appeared to reflect the switch from APC/CCdc20 to APC/CCdh1 at anaphase (Hagting et al., 2002). Therefore, we compared the proteolysis of YFP-Plk1 with an in vivo marker for APC/CCdh1. We used a CFP-tagged destruction box (D-box) mutant of securin (db-securin-CFP) that was a substrate for APC/CCdh1 but not APC/CCdc20 in vitro, and whose degradation in vivo depended on the KEN box motif present in the amino terminus, and on a decline in cyclin B-Cdk1 activity (Hagting et al., 2002). db-securin-CFP degradation preceded that of YFP-Plk1 by several minutes (Fig. 3 A), indicating that the activation of APC/CCdh1 is not sufficient to initiate YFP-Plk1 destruction. Our finding that APC/CCdh1 was active before Plk1 degradation began was consistent with our observation that another APC/CCdh1 substrate, p55Cdc20, appeared to be degraded earlier than Plk1 after release from a nocodazole arrest (Fig. 1 B). However, we were unable to verify this in vivo because FP-Cdc20 was a very poor substrate for APC/CCdh1 (unpublished data).
Figure 3.

Different APCCdh1 substrates are degraded at different times. (A and B) HeLa cells synchronized in late G2 phase were coinjected with cDNAs encoding YFP-Plk1 and db-securin-CFP (A; securin Δ61-68) or Aurora A-YFP and CFP-Plk1 (B), and fluorescence in mitotic cells was recorded by time-lapse imaging. Fluorescence in whole cells was measured and plotted as pixel values. In A, the timing of anaphase onset has been estimated as 6 min before spindle elongation in anaphase B (because these cells do not separate sister chromatids). The degradation curves shown are representative of at least four cells in two separate experiments (A) and ten cells in three separate experiments (B). Aurora A-YFP–associated fluorescence at the various stages of mitosis is shown. 1, metaphase; 2, early anaphase; 3, mid anaphase; 4, late anaphase. See also Videos 2 and 3 (available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1) for further examples.
We were interested in comparing the timing of destruction of other APC/CCdh1 substrates with Plk1, and selected Aurora A, which was recently shown to be an APC/CCdh1 substrate both in vitro and in vivo (Castro et al., 2002; Littlepage and Ruderman, 2002; Taguchi et al., 2002). Degradation of Aurora A-YFP began 6–12 min after the start of anaphase, always after that of CFP-Plk1 (Fig. 3 B). Aurora A consistently began to be degraded at the moment of maximum spindle elongation in anaphase B (Fig. 3 B, see also Fig. 4 C), when microtubule-associated Aurora A-YFP appeared on the central spindle, and fluorescence at the spindle pole diminished (Fig. 3 B, time point 2; Videos 2 and 3, and supplemental data, available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1). In contrast to Plk1 degradation, which stopped once cells entered G1 phase, Aurora A-YFP degradation continued to completion (Fig. 3 B, see also Fig. 2 C). The distinct patterns of disappearance of Plk1 and Aurora A indicate that the degradation of different APC/CCdh1 substrates is tied to specific events during mitotic exit.
Figure 4.

Plk1 degradation in anaphase depends on a D-box-like motif. (A) The sequences of Plk1 homologues were aligned (supplemental data and Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1). R337xxL is the only candidate D-box not conserved in Plk2 and Plk3. R337 and L340 were mutated to alanine (R337A, L340A) to create YFP-db-Plk1. (B and C) HeLa cells synchronized in late G2 phase were coinjected with cDNAs encoding YFP-db-Plk1 and CFP-Plk1 wild-type (B) or CFP-db-Plk1 and Aurora A-YFP (C), and fluorescence in mitotic cells was recorded by time-lapse imaging. Fluorescence in whole cells was measured and plotted as pixel values. YFP-db-Plk1 was found to be stable in at least 20 anaphase cells in eight separate experiments, one of which is shown in Video 4 (available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1). The experiment shown in C is representative of five cells in two separate experiments.
Having found that there was an ordered proteolysis of APC/CCdh1 substrates during anaphase, we wished to determine whether APC/CCdh1-mediated proteolysis played a role in mitotic exit by blocking degradation of one of its substrates. We examined the Plk1 sequence for motifs that might direct its mitotic degradation and identified an RxxL D-box-like motif (Glotzer et al., 1991) between the kinase and polo-box domains of Plk1 (Fig. 4 A). This motif is conserved in all mammalian orthologues of Plk1, in Xenopus Plx1, and in Drosophila polo, but is not present in fission yeast plo1, whose levels do not fluctuate during the cell cycle (Mulvihill et al., 1999). Moreover, this motif is not conserved in mammalian Plk3/Prk that is not degraded at mitosis (Ouyang et al., 1997; Ferris et al., 1998; Fig. S1 and supplemental data, available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1). Therefore, this motif appeared to be a good candidate to act as a Plk1 D-box. We made alanine substitutions at the conserved RxxL positions of the putative D-box sequence (R337A, L340A), and found that this mutant of Plk1 (YFP-db-Plk1) was stable in anaphase (Fig. 4 B), and the dynamic localization of this protein during mitosis was indistinguishable from that of the wild-type protein (Video 4 and supplemental data, available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1). Mutating an adjacent sequence (E348A, N349A) had no effect (unpublished data). Immunofluorescence analysis confirmed that the untagged version of the R337A, L340A mutant (db-Plk1) was stable in mitosis because it was present at high levels in newly divided cells (Fig. S2 and supplemental data). We conclude that R337 defines a D-box that directs Plk1 destruction in anaphase. YFP-db-Plk1 did not prevent Aurora A-YFP being degraded, showing that the degradation of later substrates did not depend on prior degradation of Plk1, and confirming that the stabilization of Plk1 was not due to a general inhibition of APC/CCdh1 (Fig. 4 C).
Nondegradable Plk1 frequently interfered with exit from mitosis. Furthermore, cells expressing high levels of YFP-Plk1 frequently were unable to degrade the protein properly, and in these cells mitotic exit was perturbed (Fig. 5). In these cells, anaphase and cytokinesis were slowed down; spindle elongation was impaired and cleavage furrow ingression was delayed, with many cells exhibiting extensive blebbing of the plasma membrane during this delay (Fig. 5 A). Although spindle elongation was frequently slow, maximum elongation (measured as the extent of sister chromatid separation observed by differential interference contrast [DIC] microscopy) was usually unaffected. There was frequently a delay after maximum spindle elongation before the cleavage furrow began to ingress, whereas these events occurred concurrently in uninjected cells. We did not observe chromosome decondensation during this delay; therefore, both mitotic exit and cytokinesis appeared to be delayed. The delay in mitotic exit in cells unable to degrade Plk1 was frequently accompanied by abnormal movement of the anaphase spindle during prolonged cleavage furrow ingression (Videos 5 and 6 and supplemental data, available at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1), indicating that the coordination between microtubule and actin cytoskeletons might be compromised in these cells. Overexpression of Plk1 or YFP-Plk1 also delayed mitosis before anaphase, and this delay was aggravated by a constitutively active form of the kinase, but not by nondegradable forms. However, the delay in mitotic exit was independent of the total time spent in mitosis (unpublished data).
We sought to quantify the delay in mitotic exit by measuring the total time (t) taken from sister chromatid separation to the completion of cleavage in cells injected with either YFP-tagged or untagged versions of Plk1 (Fig. 5 A). Injecting cells with YFP alone had no effect on mitotic exit (unpublished data). We found that there was a large variation in exit times in cells injected with nondegradable Plk1, best illustrated by box plots (Fig. 5, B and C), with some cells taking three times longer than the control mean value. Untagged, nondegradable Plk1 (Fig. 5 B; db-Plk1, untagged) consistently had a more pronounced effect on mitotic exit than wild-type Plk1, indicating that Plk1 proteolysis might be required for normal mitotic exit.
In budding yeast, redundant pathways have been demonstrated to inactivate mitotic cyclin-Cdks after mitosis: APCCdh1-mediated degradation is not required for cell viability as long as the Cdk inhibitor Sic1 is present (Schwab et al., 1997; Visintin et al., 1997). Thus, we considered the possibility that there might be parallel pathways to inactivate APCCdh1 targets during exit from mammalian mitosis. Because Plks are activated in mitosis by phosphorylation at a threonine residue in the activation loop (Kelm et al., 2002), we inserted the activating T210D mutation (Qian et al., 1999) at this site to generate a version of Plk1 that could not be inactivated by dephosphorylation. We found that, like nondegradable db-Plk1, the constitutively active T210D-Plk1 delayed mitotic exit compared with wild-type Plk1. Furthermore, a double mutant of Plk1 that was constitutively active and nondegradable, dbT210D-Plk1, significantly increased the frequency with which we observed this phenotype (Fig. 5 B, see P values).
We thought that the large variation in mitotic exit times (Fig. 5 B) might arise from variability between cells in the level of Plk1, which we were unable to measure because the proteins were not fluorescently tagged. Therefore, we examined mitotic exit times in cells expressing YFP-tagged Plk1, where we could select cells expressing comparable levels of the different constructs. In these cells, we observed similar delays in mitotic exit, but also saw a similar variation in mitotic exit times (Fig. 5 C). Additionally, we found an increased delay in cells expressing wild-type YFP-Plk1 compared with untagged Plk1. This indicated that the YFP tag could reduce the rate of degradation of the protein. Perhaps because of this, mutating the D-box alone in YFP-Plk1 had a less significant effect on mitotic exit times (Fig. 5 C). To confirm the link between Plk1 degradation and mitotic exit times in cells expressing YFP-tagged Plk1, we examined the rates of YFP-Plk1 and YFP-T210D-Plk1 degradation and correlated this with the time taken to exit mitosis. We found that degradation was systematically impaired in cells displaying very prolonged mitotic exit times (Fig. 5 D). This occurred more frequently in YFP-T210D-Plk1 cells than in YFP-Plk1 cells, indicating that inactivation of Plk1 in anaphase may be required for its proper degradation.
Discussion
We have presented evidence that normal exit from mitosis requires the inactivation of Plk1, and that this can be either by degradation or by dephosphorylation. Thus, APC/CCdh1-dependent proteolysis appears to play an important role in the proper exit from mitosis. However, because there are likely to be parallel pathways to inactivate mitotic kinases, there may not be any one mitotic regulator whose degradation is essential for mitotic exit. Indeed, in early embryos, mitotic cycles occur in the absence of Cdh1 expression (Sigrist and Lehner, 1997; Lorca et al., 1998). We suggest that degradation directed by APC/CCdh1 increases the efficiency with which mitotic regulators are inactivated at the end of mitosis. Consistent with this suggestion, Cdh1−/− DT40 cells, although viable, show prolonged mitotic exit (Sudo et al., 2001).
Why should there be a requirement for Plk1 inactivation during anaphase? This appears to contradict the well-documented requirement for Plk activity in cytokinesis and mitotic exit (for review see Bahler et al., 1998; Carmena et al., 1998; Heitz et al., 2001; Lee et al., 2001; Song and Lee, 2001; Mulvihill and Hyams, 2002; Seong et al., 2002). We propose that in addition to being required for assembly of the contractile ring, Plk1 may inhibit cleavage furrow ingression in mammalian cells, perhaps to ensure that ingression cannot occur before anaphase (Shuster and Burgess, 2002). According to the model most recently proposed (Dechant and Glotzer, 2003), this inhibition could be achieved by regulating microtubule bundling in the central spindle. Work published by Neef et al. (2003) during the preparation of this manuscript allows us to identify the kinesin-like protein MKLP-2 as a candidate to mediate the inhibitory effect of Plk1 on cell cleavage. MKLP-2 localizes to the central spindle during anaphase in a complex with Plk1, and has microtubule bundling activity in vitro that can be negatively regulated by Plk1.
Although previous work by others has described the appearance of multinucleated cells after transfection of Plk1 (Mundt et al., 1997), a failure of cytokinesis is a rare event under our experimental conditions, perhaps reflecting the lower levels of expression achieved with microinjection. We find that almost all cells attempt cytokinesis within 50 min of anaphase. This 50-min interval corresponds to the previously described period of “cortical contractility,” or C-phase (Martineau et al., 1995; Canman et al., 2000). C-phase can be extended by inhibiting proteolysis with MG132 in the presence of blebbistatin, an inhibitor of myosin II (Straight et al., 2003), indicating that the length of C-phase is regulated by proteolysis. Our results show that the relevant substrate does not appear to be Plk1 because nondegradable Plk1 delays cytokinesis without altering the length of C-phase (Fig. 5, B and C). Moreover, cells starting with excess Plk1 almost always eventually perform cytokinesis. Thus, we suggest that C-phase is not just the window of opportunity during which cytokinesis can occur, but also represents the maximum period for which cytokinesis can be delayed in response to inappropriate conditions.
Our preliminary analyses indicate that the delay in cytokinesis correlates with delayed recruitment of cleavage furrow components (unpublished data). An understanding of the pathways that coordinate cytokinesis with mitotic exit remains an important challenge, and our finding that there is ordered degradation of mitotic regulators as cells exit from mitosis may provide important clues to these pathways.
Materials and methods
Cell culture and synchronization
HeLa cells were cultured and synchronized for microinjection in G2 phase as described previously (Clute and Pines, 1999). Mitotic cells for the experiment shown in Fig. 1 were prepared by a modified version of the synchronization regime, where cells were released from aphidicolin (Sigma-Aldrich) into medium containing 400 ng/μl nocodazole (Sigma-Aldrich) and incubated for 12 h before harvesting by shake-off. Mitotic cells were washed three times in ice-cold PBS and replated in medium prewarmed to 37°C. Cell cycle distributions of cell populations at different time points were calculated by analysis of DNA content on a FACsort™ Flow Cytometer (Becton Dickinson) after propidium iodide staining as described previously (Lindon et al., 2000). MG132 was obtained from Calbiochem.
Immunoblotting
Extracts were made from cell populations at the indicated time points after nocodazole release. Cells were washed with PBS. Extracts prepared by addition of boiling SDS sample buffer directly to culture dishes (for reattached cells in G1), and to cells pelleted from the culture medium (for unattached mitotic cells), were pooled. Samples were heated at >95°C for 3 min and then sheared through 21G needles. Approximately 5 μg of each sample was blotted by the standard semi-dry transfer technique onto Immobilon™-P (Millipore). Filters were processed for immunoblotting using standard techniques. Rabbit polyclonal antisera used were raised against (1) human Plk1 amino-terminal peptide (Upstate Biotechnology); (2) cyclin B1 (Hagting et al., 1998); (3) human AIK1 (Aurora A) and mouse AIK2 amino terminus (Aurora B; both gifts of Peter Donovan, Thomas Jefferson University, Philadelphia, PA); (4) BubR1 (a gift of Gordon Chan, Fox Chase Cancer Center, Philadelphia, PA); (5) CENP-E (a gift of Tim Yen, Fox Chase Cancer Center, Philadelphia, PA); and (6) PRC1 (a gift of Tony Hunter, Salk Institute, La Jolla, CA). Goat polyclonal anti–human p55Cdc20 was obtained from Santa Cruz Biotechnology, Inc.
Construction of cDNA plasmids
Plk1 cDNA was cloned from pCMX-GFP10c-Plk1 (Arnaud et al., 1998) into pEYFP-C3 (CLONTECH Laboratories, Inc.) for expression as a fusion protein with YFP at the amino terminus, and into pcDNA3 for expression as an untagged protein.
New point mutations were constructed by whole-plasmid PCR using Pfu DNA polymerase (Stratagene) and confirmed by automated sequencing. Constitutively active versions of Plk1 were constructed by swapping in sequences from pRcCMV-Plk1-T210D (a gift of Erich Nigg, Max Planck Institute, Martinsried, Germany).
Human Aurora A sequence was generated by PCR from pCRUZ-myc-Aurora A (a gift of Claude Prigent, University of Rennes, Rennes, France) and was cloned into pEYFP-N1, and is expressed as a fusion protein linked at its carboxy terminus to YFP. pECFP-N1-cyclin B1 and pECFP-N1-securinΔ61-68 have been described previously (Hagting et al., 2002). Histone 2B-YFP was the gift of Claire Acquaviva (Wellcome Trust/Cancer Research UK Institute, Cambridge, UK). Further details of all constructs used are available upon request.
Microinjection and time-lapse imaging and analysis
Cells were injected and analyzed using time-lapse DIC fluorescence microscopy with different filter cubes to distinguish YFP- and CFP-associated fluorescence as described previously (Clute and Pines, 1999; Hagting et al., 1999, 2002), but with the addition of a programmable XY stage (Prior Scientific Instruments Ltd.) to allow concurrent filming of several fields of cells. Images were collected every 2 or 3 min and saved in IP Lab Spectrum (Scanalytics) format as 16-bit data using a reference look-up table with a preset linear pixel intensity scale. ImageJ software (National Institutes of Health; modified by Jean-Yves Thuret) was used for quantifying CFP and YFP fluorescence. Fluorescence levels in whole cells were measured as pixel values within a region of interest (ROI) drawn around each cell and applied to all images in a series. The ROI drawn in each case was large enough to allow for changing cell shape during mitotic exit. Because we subtracted background pixel values from our measured values, this method gave accurate measurements of total cell fluorescence. DIC images were used to determine the onset of anaphase. Images were then converted to PICT format and exported to Adobe Photoshop®, or processed via ImageJ to make QuickTime® movies.
Online supplemental material
A sequence alignment of human Plk family members, showing the position of the nonconserved D-box motif, is shown in Fig. S1. Fig. S2 shows anti-Plk1 staining of G1 cells injected with untagged versions of Plk1, and confirms that untagged db-Plk1 is not degraded in mitosis and/or G1 phase. Videos available online show examples of cells degrading Plk1 (Video 1), Aurora A (Video 2), or both (Video 3), cells expressing nondegradable Plk1 (Video 4), and cells exhibiting delayed mitotic exit in response to nondegraded Plk1 (Videos 5 and 6). All supplemental videos, supplemental figures, and an associated Materials and methods section are available online at http://www.jcb.org/cgi/content/full/jcb.200309035/DC1.
Acknowledgments
We thank Claude Prigent for the Aurora A cDNA and Tim Yen, Peter Donovan, and Tony Hunter for antibodies. We are grateful to Rob Wolthuis for valuable discussions, to Anja Hagting for advice on immunofluorescence, to Jean-Yves Thuret for his adaptation of ImageJ software, and to Claire Acquaviva and Anja Hagting for useful comments on the manuscript. All members of the J. Pines Lab contributed invaluable advice.
This work was made possible through a Wellcome Trust Advanced Training Fellowship to C. Lindon, by an FP5 RTN from the European Union (contract number QLG1-CT-2001-02026), and by Programme Grant C29/A1782 to J. Pines from Cancer Research UK.
The online version of this article includes supplemental material.
Abbreviations used in this paper: APC/C, anaphase-promoting complex/cyclosome; D-box, destruction box; DIC, differential interference contrast; FP, fluorescent protein; Plk1, polo-like kinase 1.
References
- Arnaud, L., J. Pines, and E.A. Nigg. 1998. GFP tagging reveals human polo-like kinase 1 at the kinetochore/centromere region of mitotic chromosomes. Chromosoma. 107:424–429. [DOI] [PubMed] [Google Scholar]
- Bahler, J., A.B. Steever, S. Wheatley, Y. Wang, J.R. Pringle, K.L. Gould, and D. McCollum. 1998. Role of polo kinase and Mid1p in determining the site of cell division in fission yeast. J. Cell Biol. 143:1603–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, K.D., R.M.R. Coulson, T.J. Yen, and D.W. Cleveland. 1994. Cyclin-like accumulation and loss of the putative kinetochore motor CENP-E results from coupling of continuous synthesis with specific degradation at the end of mitosis. J. Cell Biol. 125:1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman, J.C., D.B. Hoffman, and E.D. Salmon. 2000. The role of pre- and post-anaphase microtubules in the cytokinesis phase of the cell cycle. Curr. Biol. 10:611–614. [DOI] [PubMed] [Google Scholar]
- Carmena, M., M.G. Riparbelli, G. Minestrini, A.M. Tavares, R. Adams, G. Callaini, and D.M. Glover. 1998. Drosophila polo kinase is required for cytokinesis. J. Cell Biol. 143:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro, A., Y. Arlot-Bonnemains, S. Vigneron, J.C. Labbe, C. Prigent, and T. Lorca. 2002. APC/Fizzy-related targets Aurora-A kinase for proteolysis. EMBO Rep. 3:457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, G.K., S.A. Jablonski, V. Sudakin, J.C. Hittle, and T.J. Yen. 1999. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 146:941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., and G. Fang. 2001. MAD2B is an inhibitor of the anaphase-promoting complex. Genes Dev. 15:1765–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute, P., and J. Pines. 1999. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol. 1:82–87. [DOI] [PubMed] [Google Scholar]
- Dechant, R., and M. Glotzer. 2003. Centrosome separation and central spindle assembly act in redundant pathways that regulate microtubule density and trigger cleavage furrow formation. Dev. Cell. 4:333–344. [DOI] [PubMed] [Google Scholar]
- den Elzen, N., and J. Pines. 2001. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153:121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell. 2:163–171. [DOI] [PubMed] [Google Scholar]
- Ferris, D.K., S.C. Maloid, and C.C. Li. 1998. Ubiquitination and proteasome mediated degradation of polo-like kinase. Biochem. Biophys. Res. Commun. 252:340–344. [DOI] [PubMed] [Google Scholar]
- Geley, S., E. Kramer, C. Gieffers, J. Gannon, J.-M. Peters, and T. Hunt. 2001. APC/C-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 153:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer, M., A.M. Murray, and M.W. Kirschner. 1991. Cyclin is degraded by the ubiquitin pathway. Nature. 349:132–138. [DOI] [PubMed] [Google Scholar]
- Hagting, A., C. Karlsson, P. Clute, M. Jackman, and J. Pines. 1998. MPF localisation is controlled by nuclear export. EMBO J. 17:4127–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagting, A., M. Jackman, K. Simpson, and J. Pines. 1999. Translocation of cyclin B1 to the nucleus at prophase requires a phosphorylation-dependent nuclear import signal. Curr. Biol. 9:680–689. [DOI] [PubMed] [Google Scholar]
- Hagting, A., N. den Elzen, H.C. Vodermaier, I.C. Waizenegger, J.-M. Peters, and J. Pines. 2002. Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J. Cell Biol. 157:1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz, M.J., J. Petersen, S. Valovin, and I.M. Hagan. 2001. MTOC formation during mitotic exit in fission yeast. J. Cell Sci. 114:4521–4532. [DOI] [PubMed] [Google Scholar]
- Honda, K., H. Mihara, Y. Kato, A. Yamaguchi, H. Tanaka, H. Yasuda, K. Furukawa, and T. Urano. 2000. Degradation of human Aurora2 protein kinase by the anaphase-promoting complex-ubiquitin-proteasome pathway. Oncogene. 19:2812–2819. [DOI] [PubMed] [Google Scholar]
- Jackman, M., C. Lindon, E.A. Nigg, and J. Pines. 2003. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 5:143–148. [DOI] [PubMed] [Google Scholar]
- Jiang, W., G. Jimenez, N.J. Wells, T.J. Hope, G.M. Wahl, T. Hunter, and R. Fukunaga. 1998. PRC1: A human mitotic spindle-associated CDK substrate protein required for cytokinesis. Mol. Cell. 2:877–885. [DOI] [PubMed] [Google Scholar]
- Kelm, O., M. Wind, W.D. Lehmann, and E.A. Nigg. 2002. Cell cycle-regulated phosphorylation of the Xenopus polo-like kinase Plx1. J. Biol. Chem. 277:25247–25256. [DOI] [PubMed] [Google Scholar]
- Lee, S.E., S. Jensen, L.M. Frenz, A.L. Johnson, D. Fesquet, and L.H. Johnston. 2001. The Bub2-dependent mitotic pathway in yeast acts every cell cycle and regulates cytokinesis. J. Cell Sci. 114:2345–2354. [DOI] [PubMed] [Google Scholar]
- Leismann, O., and C.F. Lehner. 2003. Drosophila securin destruction involves a D-box and a KEN-box and promotes anaphase in parallel with Cyclin A degradation. J. Cell Sci. 116:2453–2460. [DOI] [PubMed] [Google Scholar]
- Li, R. 1999. Bifurcation of the mitotic checkpoint pathway in budding yeast. Proc. Natl. Acad. Sci. USA. 96:4989–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindon, C., O. Albagli, P. Domeyne, D. Montarras, and C. Pinset. 2000. Constitutive instability of muscle regulatory factor Myf5 is distinct from its mitosis-specific disappearance, which requires a D-box-like motif overlapping the basic domain. Mol. Cell. Biol. 20:8923–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlepage, L.E., and J.V. Ruderman. 2002. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev. 16:2274–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorca, T., A. Castro, A.M. Martinez, S. Vigneron, N. Morin, S. Sigrist, C. Lehner, M. Doree, and J.C. Labbe. 1998. Fizzy is required for activation of the APC/cyclosome in Xenopus egg extracts. EMBO J. 17:3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau, S.N., P.R. Andreassen, and R.L. Margolis. 1995. Delay of HeLa cell cleavage into interphase using dihydrocytochalasin B: retention of a postmitotic spindle and telophase disc correlates with synchronous cleavage recovery. J. Cell Biol. 131:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, D. 1999. Regulation of the APC and the exit from mitosis. Nat. Cell Biol. 1:E47–E53. [DOI] [PubMed] [Google Scholar]
- Mulvihill, D.P., and J.S. Hyams. 2002. Cytokinetic actomyosin ring formation and septation in fission yeast are dependent on the full recruitment of the polo-like kinase Plo1 to the spindle pole body and a functional spindle assembly checkpoint. J. Cell Sci. 115:3575–3586. [DOI] [PubMed] [Google Scholar]
- Mulvihill, D.P., J. Petersen, H. Ohkura, D.M. Glover, and I.M. Hagan. 1999. Plo1 kinase recruitment to the spindle pole body and its role in cell division in Schizosaccharomyces pombe. Mol. Biol. Cell. 10:2771–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundt, K.E., R.M. Golsteyn, H.A. Lane, and E.A. Nigg. 1997. On the regulation and function of human polo-like kinase 1 (PLK1): effects of overexpression on cell cycle progression. Biochem. Biophys. Res. Commun. 239:377–385. [DOI] [PubMed] [Google Scholar]
- Neef, R., C. Preisinger, J. Sutcliffe, R. Kopajtich, E.A. Nigg, T.U. Mayer, and F.A. Barr. 2003. Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J. Cell Biol. 162:863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang, B., H. Pan, L. Lu, J. Li, P. Stambrook, B. Li, and W. Dai. 1997. Human Prk is a conserved protein serine/threonine kinase involved in regulating M phase functions. J. Biol. Chem. 272:28646–28651. [DOI] [PubMed] [Google Scholar]
- Pereira, G., T. Hofken, J. Grindlay, C. Manson, and E. Schiebel. 2000. The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol. Cell. 6:1–10. [PubMed] [Google Scholar]
- Peters, J.M. 2002. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell. 9:931–943. [DOI] [PubMed] [Google Scholar]
- Pfleger, C.M., and M.W. Kirschner. 2000. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14:655–665. [PMC free article] [PubMed] [Google Scholar]
- Qian, Y.W., E. Erikson, and J.L. Maller. 1999. Mitotic effects of a constitutively active mutant of the Xenopus polo-like kinase Plx1. Mol. Cell. Biol. 19:8625–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, M., A.S. Lutum, and W. Seufert. 1997. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 90:683–693. [DOI] [PubMed] [Google Scholar]
- Seong, Y.S., K. Kamijo, J.S. Lee, E. Fernandez, R. Kuriyama, T. Miki, and K.S. Lee. 2002. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J. Biol. Chem. 277:32282–32293. [DOI] [PubMed] [Google Scholar]
- Shuster, C.B., and D.R. Burgess. 2002. Transitions regulating the timing of cytokinesis in embryonic cells. Curr. Biol. 12:854–858. [DOI] [PubMed] [Google Scholar]
- Sigrist, S.J., and C.F. Lehner. 1997. Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell. 90:671–681. [DOI] [PubMed] [Google Scholar]
- Song, S., and K.S. Lee. 2001. A novel function of Saccharomyces cerevisiae CDC5 in cytokinesis. J. Cell Biol. 152:451–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmann, O., H. Zou, S.A. Gerber, S.P. Gygi, and M.W. Kirschner. 2001. Dual inhibition of sister chromatid separation at metaphase. Cell. 107:715–726. [DOI] [PubMed] [Google Scholar]
- Straight, A.F., A. Cheung, J. Limouze, I. Chen, N.J. Westwood, J.R. Sellers, and T.J. Mitchison. 2003. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 299:1743–1747. [DOI] [PubMed] [Google Scholar]
- Sudo, T., Y. Ota, S. Kotani, M. Nakao, Y. Takami, S. Takeda, and H. Saya. 2001. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J. 20:6499–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi, S., K. Honda, K. Sugiura, A. Yamaguchi, K. Furukawa, and T. Urano. 2002. Degradation of human Aurora-A protein kinase is mediated by hCdh1. FEBS Lett. 519:59–65. [DOI] [PubMed] [Google Scholar]
- Visintin, R., S. Prinz, and A. Amon. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 278:460–463. [DOI] [PubMed] [Google Scholar]
- Weinstein, J. 1997. Cell cycle-regulated expression, phosphorylation, and degradation of p55Cdc. A mammalian homolog of CDC20/Fizzy/slp1. J. Biol. Chem. 272:28501–28511. [DOI] [PubMed] [Google Scholar]
- Zur, A., and M. Brandeis. 2002. Timing of APC/C substrate degradation is determined by fzy/fzr specificity of destruction boxes. EMBO J. 21:4500–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]