

Abstract
The Saccharomyces cerevisiae mitogen-activated protein kinases (MAPKs) Fus3 and Kss1 bind to multiple regulators and substrates. We show that mutations in a conserved docking site in these MAPKs (the CD/7m region) disrupt binding to an important subset of their binding partners, including the Ste7 MAPK kinase, the Ste5 adaptor/scaffold protein, and the Dig1 and Dig2 transcriptional repressors. Supporting the possibility that Ste5 and Ste7 bind to the same region of the MAPKs, they partially competed for Fus3 binding. In vivo, some of the MAPK mutants displayed reduced Ste7-dependent phosphorylation, and all of them exhibited multiple defects in mating and pheromone response. The Kss1 mutants were also defective in Kss1-imposed repression of Ste12. We conclude that MAPKs contain a structurally and functionally conserved docking site that mediates an overall positively acting network of interactions with cognate docking sites on their regulators and substrates. Key features of this interaction network appear to have been conserved from yeast to humans.
Keywords: mitogen-activated protein kinases; signal transduction; phosphorylation; binding sites; protein binding
Introduction
The components of intracellular signaling pathways are dynamically interconnected into complex networks, where the proteins correspond to the nodes of the network and the protein–protein and enzyme–substrate interactions are the links between them. The links are a key to understanding not only the overall flow of information, but also systems level questions of why particular network structures have evolved, and how network structure contributes to function (Vespignani, 2003). In addition, the links are potential drug targets that might yield highly specific therapeutic effects. For example, it would be beneficial to be able to block the ability of a kinase to phosphorylate some of its substrates while not interfering with its ability to phosphorylate others.
The mating pheromone response pathway of the yeast Saccharomyces cerevisiae provides an advantageous model system for exploring these issues because it is one of the best characterized multitiered signaling pathways in eukaryotes (for review see Dohlman and Thorner, 2001). Mating is initiated when peptide pheromone secreted by a cell of one mating type binds to a receptor on the surface of a cell of the opposite mating type. Receptor occupancy activates a heterotrimeric G protein, and the Gβγ subunit then transmits the mating signal to downstream components, leading to the activation of a MAPK cascade.
The MAPK cascade module consists of three sequentially acting protein kinases. The last in the sequence is the MAPK (also termed extracellular signal–regulated kinase; ERK) that is phosphorylated and thereby activated by a MAPK/ERK kinase (MEK, or MKK). MEK activity is regulated, in turn, via phosphorylation by the first member of the module, a MEK kinase (MEKK). In the yeast mating pathway, the MEKK is Ste11, which activates the MEK Ste7, which then activates two MAPKs, Kss1, and Fus3. The Ste5 adaptor/scaffold protein binds to all three tiers of this module, first enabling Ste11MEKK activation, and then assisting signal transmission from MEKK to MEK to MAPK (Elion, 2001).
The downstream targets of the active MAPKs include the Ste12 transcription factor and its associated repressors Dig1 and Dig2 (Elion et al., 1993; Cook et al., 1996; Tedford et al., 1997). MAPK-mediated phosphorylation of Ste12, Dig1, and Dig2 is thought to underlie the transcriptional induction of a battery of over 200 genes (Roberts et al., 2000). In addition, unphosphorylated Kss1MAPK binds directly to Ste12 and, with the help of Dig1 and Dig2, represses the ability of Ste12 to activate the transcription of certain genes, particularly those involved in filamentous invasive growth (Cook et al., 1997; Madhani et al., 1997; Bardwell et al., 1998b). Filamentous invasive growth, although distinct from mating, is regulated by many of the same components that regulate mating, including the MAPK cascade (Dohlman and Thorner, 2001).
The component proteins of the MAPK cascade have been well conserved throughout eukaryotic evolution, for example, there is 50% sequence identity between human ERK2 and yeast Fus3MAPK; so too has been their arrangement into a three-tier module. In contrast, eukaryotes have devised a plethora of ways to channel signals into and out of the MAPK cascade, many of which do not appear to be highly conserved (Widmann et al., 1999). For example, Ste5, Ste12, Dig1, and Dig2 homologues are not found outside of fungi. Nevertheless, there may be conserved molecular mechanisms by which the members of the core MAPK module interact with upstream regulators and downstream substrates. One possibility is the relatively high affinity binding of MAPKs to their substrates and regulators via “docking” sites that are distinct from the enzyme–substrate interactions that occur during catalysis. For example, Ste7MEK binds with high affinity to Fus3MAPK and Kss1MAPK via a short docking site in its NH2-terminal, noncatalytic domain (Bardwell et al., 1996). Similar docking sites, or “D-sites” (consensus (R/K)2-3–(X)2-6–L/I–X–L/I) are found at the N termini of many other MEK family members (Bardwell and Thorner, 1996; Bardwell et al., 2001), and have been shown to mediate the interaction of most human MEKs with their cognate MAPKs (Enslen et al., 2000; Bardwell et al., 2001; Ho et al., 2003). D-sites and related docking motifs were also identified in transcription factors, phosphatases, scaffolds, other kinases, and other proteins (for reviews see Sharrocks et al., 2000; Enslen and Davis, 2001). Such findings suggest that animal MAPKs may participate in a widespread network of interactions mediated by D-sites and other modular docking sites such as FXFP (Jacobs et al., 1999). However, even though one of the first D-sites was discovered in S. cerevisiae, it has heretofore been unclear if yeast MAPKs, like their mammalian counterparts, are part of such an extensive network.
Another unresolved question concerns the function of this network of docking interactions. Studies showing that D-site–dependent interactions enhance substrate phosphorylation indicate that docking plays a positive role in signal transmission (Sharrocks et al., 2000; Enslen and Davis, 2001). Confoundingly, the opposite answer has come from genetic studies of MAPK mutants defective in docking. These mutants have amino acid substitutions in a patch of acidic residues designated the conserved docking (CD; Tanoue et al., 2000) region, originally identified as the site of the sevenmaker (7m) mutation in the Drosophila MAPK rolled. This allele was found in a screen for mutants that would trigger R7 photoreceptor cell differentiation in the absence of upstream signaling events (Brunner et al., 1994). Hence, the MAPK7m mutant had a hyperactive, gain of function phenotype, which was later shown to be, in part, a consequence of decreased binding to a D-site–containing MAPK phosphatase (Karim and Rubin, 1999). CD/7m-region mutations, when introduced into mammalian ERK2 or yeast Fus3, likewise compromise the interaction of these MAPKs with their deactivating phosphatases (Bott et al., 1994; Chu et al., 1996; Zhan and Guan, 1999). Moreover, the Fus3 mutants have a gain of function phenotype characterized by increased levels of signaling or signal crossover (Hall et al., 1996; Zhan and Guan, 1999). Thus, these studies do not appear to support the conception of an extensive network of MAPK-docking interactions that have a predominantly positive role in signal transmission. Here, we report our analysis of the CD/7m region in Fus3MAPK and Kss1MAPK.
Results
Sevenmaker-region mutants of Kss1 and Fus3
To determine the region(s) of the MAPKs required for interaction with the D-site in Ste7MEK, site-directed mutagenesis was used to introduce amino acid substitutions into candidate regions of Fus3MAPK and Kss1MAPK. The D-site of Ste7 is contained within its first 19 residues; within this region, two dibasic submotifs (R4 K5 and R9 R10), and a hydrophobic submotif (L15 L17), are involved in MAPK interaction (Bardwell et al., 2001). Because basic submotifs are a prominent feature of the D-site of Ste7, and of other D-sites, acidic patches on the surface of the MAPKs were prioritized for mutagenesis; one such patch was the CD/7m region, a cluster of negatively charged surface residues on the back side of the kinase, opposite the catalytic cleft. We found that the Ste7–MAPK interaction was essentially abolished by mutations in this region.
The CD/7m alleles constructed for this paper are shown in Fig. 1; most consist of single or multiple substitutions of charged residues with alanines; the exception is Fus3-7m2, which contains a D317N substitution, mimicking the original sevenmaker allele of Drosophila rolled. A description of our mutants in other regions of the MAPKs, and their effects on MAPK protein–protein interactions will appear separately (unpublished data).
Figure 1.

Yeast MAPK protein interactions and the CD/sevenmaker region. (A and B) Subset of the protein interactions in which the yeast MAPKs Kss1 and Fus3 participate. (C) Sequences of D. melanogaster rolled (GenBank/EMBL/DDBJ accession no. P40417), H. sapiens ERK2 (NP_620407), and S. cerevisiae Kss1 and Fus3 proteins (P14681 and S28548), in the CD/7m region. The COOH-terminal–most residue shown is indicated on the right. The site of the sevenmaker mutation in rolled (D334N) is underlined. Residues identical in all four proteins are denoted at the bottom by an asterisk. (D) Altered amino acids in the CD/7m-region mutants of Kss1 and Fus3 are underlined.
The mutations diminish binding to Ste7 and Ste5
The CD/7m mutants of Kss1MAPK and Fus3MAPK were tested for in vitro binding to Ste7MEK, Ste5, and Dig1 (Fig. 2). The MAPK-interacting domains of these proteins were fused to S. japonicum GST, and the resulting fusion proteins were expressed in bacteria and purified by adsorption to glutathione-agarose beads. These fusions (or GST alone as a control) were then incubated with Kss1 or Fus3 (or mutants thereof) that had been produced in radiolabeled form by in vitro translation, and partially purified by ammonium sulfate precipitation. Bead-bound complexes were collected by sedimentation and analyzed by SDS-PAGE and autoradiography, and quantified on a phosphorimager.
Figure 2.


Impaired binding of MAPK CD/7m-region mutants to Ste7 and Ste5. (A) MAPK-binding domains of Ste7, Ste5 and Dig1 were fused to GST and tested for binding to 35S-labeled Fus3, Kss1, or mutants thereof. (B) In vitro binding assay. MAPK proteins (∼1 pmole; ‘Input’ (top) shows 5% of this amount) were incubated with 100 ng of GST-Ste71-98, 2 μg of GST-Ste5241-336, or 10 μg of GST-Dig1213-452. Glutathione-Sepharose beads were then added, and the resulting bead-bound protein complexes were isolated, resolved by 10% SDS-PAGE, and analyzed by staining with Coomassie blue (CB) for visualization of the bound GST fusion protein, and by autoradiography for visualization of the bound radiolabeled (35S) protein. No Coomassie blue staining is shown for GST-Ste7 because the small amount used (100 ng) was difficult to photograph (but was verified by eye). The numbers below show the normalized relative binding, with the binding of the wild-type set at 100. The percent of the input of wild-type Fus3 bound to Ste7, Ste5, and Dig1 was 14.0, 12.6, and 0.6, respectively. For Kss1: 9.0, 3.7, and 1.3, respectively. (C) Dissociation constants (K ds) for MAPK interactions with GST-Ste7 and GST-Ste5. The mean K d (in micromolar) in shown, as well as the SEM (s.e.) and the number of experiments performed to determine the mean (N). (D) The Ste7 D-site competes with Ste5241-336 for Fus3 binding. 35S-labeled, wild-type Fus3 protein (∼1 pmole) was incubated with 6 μg of purified GST or GST-Ste5241-336 prebound to glutathione-Sepharose beads, and with either no peptide, 25 or 100 μM of Ste72-22 peptide (Ste7ds) or 25 or 100 μM of a scrambled control for this peptide (scram7; (Bardwell et al., 2001)). Other details as in B, above. The bar graph shows the quantification of relative binding, as determined using a phosphorimager. After subtracting the nonspecific background adsorption to GST alone, 19.7% of the input Fus3 bound to wild-type GST-Ste5 in the absence of peptide. Results were normalized by setting this value as 100%. Data show the mean ± SD of two experiments.
A fusion of GST to the first 98 residues of Ste7 exhibited high affinity binding to wild-type Fus3 and Kss1 (Fig. 1 B), as we have reported previously (Bardwell et al., 2001). In contrast, all three of the Fus3-7m mutants exhibited undetectable binding to GST-Ste71-98 (<1% of wild-type). Similar results were obtained in the case of Kss1, except that the single mutants in the CD/7m region had reduced, but still detectable, binding to Ste7 (9–18% of wild-type); the Kss1-7m3 mutant, which contains both substitutions, did not bind at all to Ste7 (<1% of wild-type).
The minimal MAPK-binding domain of Ste5 is contained within residues 241–336, as determined by a two-hybrid analysis (Choi et al., 1994). A GST fusion protein containing this region bound to wild-type Fus3 and Kss1 (Fig. 2 B). Unexpectedly, the CD/7m mutants of Fus3 and Kss1 exhibited decreased binding to GST-Ste5241-336. As was seen for binding to Ste7, the Kss1 single-substitution mutants still manifested significant binding to Ste5, whereas the binding of the double and triple Kss1 mutants, like all three Fus3 mutants, were more substantially impaired.
Dig1 and Dig2 were initially identified in a two-hybrid screen using Kss1 as bait (Cook et al., 1996). A GST fusion protein containing the shortest Dig1 region isolated in this screen (residues 213–452) bound to Kss1, as reported previously (Cook et al., 1996), and also bound to Fus3 (Fig. 2 B). In contrast to the results seen when MAPK binding to Ste7 and Ste5 was examined, MAPK binding to GST-Dig1213-452 was not significantly impaired by the CD/7m-region mutations (Fig. 2 B). Hence, the mutations compromise some, but not all, of the protein–protein interactions in which the MAPKs participate. In support of this conclusion, the Kss1-7m1, 7m2, and 7m3 mutants, like wild-type Kss1 protein (Printen and Sprague, 1994), exhibited a robust interaction with full-length Ste11MEKK in the two-hybrid assay (unpublished data). Subsequent experiments revealed that the MAPKs also bind to another region of Dig1 (see below).
We quantified formation of the Ste7 and Ste5 complexes using a method described previously (Fig. 2 C; Bardwell et al., 1996; Ho et al., 2003). Both Kss1 and Fus3 bound to GST-Ste7 with high affinity (K d ≈ 0.1 μM). Mutations in the CD/7m region decreased the Ste7–MAPK binding affinity by >500-fold. Compared with the Ste7–Fus3 interaction, the Ste5–Fus3 interaction was weaker (K d ≈ 1 μM), and was less severely impaired by the CD/7m-region mutations (∼12-fold decrease in binding affinity). Interestingly, the Ste5–Kss1 interaction (K d ≈ 6.5 μM) was weaker than the Ste5–Fus3 interaction. The affinity of the Ste5–Kss1 interaction was decreased approximately sixfold by the CD/7m-region mutations.
Ste5 and Ste7 partially compete for docking to Fus3
The observation that the CD/7m-region mutations reduced MAPK binding to both Ste7MEK and Ste5 suggested that these two proteins might compete for MAPK docking. Consistent with this possibility, a peptide version of the Ste7 docking site (residues 2–22) inhibited the binding of wild-type Fus3 protein to GST-Ste5241-336 by almost 80% (Fig. 2 D). A control peptide consisting of residues 2–22 of Ste7 in a scrambled order was not inhibitory.
The CD/7m mutations abolish MEK–MAPK complex formation in cell extracts
To assess the effects of the CD/7m-regions mutations on protein stability in vivo, the wild-type and mutant coding sequences were subcloned into a low copy (CEN) expression vector, driven by their endogenous promoter, and expressed in a strain lacking endogenous Kss1 and Fus3 (i.e., a kss1Δ fus3Δ strain). All four of the Kss1 mutant proteins were produced at levels comparable to wild type (not depicted and see Fig. 4 A). The Fus3-7m1 and 7m2 mutant proteins, however, were produced at only ∼40% of the wild-type level and the Fus3-7m3 protein could not be detected (not depicted and see Fig. 4 D). The pheromone response pathway positively regulates FUS3 expression; however, decreased levels of Fus3-7m mutants was also observed in a KSS1+ strain (unpublished data), suggesting that a signaling defect is not the underlying cause. Hence, the mutations appear to destabilize Fus3 protein to a variable extent depending on the precise mutation.
Figure 4.


Activation-loop phosphorylation and pheromone-induced gene expression of MAPK mutants. Strains YDM230 (MAT a fus3Δ kss1Δ) or YDM200 (MAT a fus3Δ KSS1 +) were transformed with plasmids carrying alleles of FUS3 or KSS1 to generate the indicated genotypes, and with a plasmid (YEpH-FUS1Z) carrying a FUS1-lacZ reporter gene. The fus3 alleles were carried on two different plasmids (YCpU and YCpT). (A) In vivo pheromone-stimulated phosphorylation of Kss1 and mutants. Cultures of strain YDM230, carrying an “empty” YCpU vector (−), or expressing from the same plasmid Kss1, or the 7m1 or 7m3 mutants thereof, were exposed to 0 or 500 nM α-factor mating pheromone for 5 min. Cell extracts were prepared, and 100 μg/lane of TCA-precipitated protein was separated on a 10% SDS-PAGE gel, and analyzed by immunoblotting with a phosphorylation-state specific antibody (top). The immunoblot was then stripped and reprobed with anti-Kss1 antisera (bottom). (B) FUS1-lacZ expression measured after a 2-h treatment with 500 nM α-factor mating pheromone (phm). (C and D) FUS1-lacZ expression (C) and activation-loop phosphorylation (D) of Fus3-7m mutants after treatment with 500 nM α-factor for 60 min. (B and C) Error bars are shown (three to six experiments).
Wild-type Kss1 and Fus3 proteins are found in complexes with full-length Ste7MEK protein in yeast cell extracts (Bardwell et al., 1996). When assessed in this assay, both of the Kss1 mutants tested (7m2 and 7m3), and both of the Fus3 mutants tested (7m1 and 7m2), were unable to form complexes with Ste7 (Fig. 3), consistent with the results obtained using in vitro–translated and bacterially expressed proteins (Fig. 2).
Figure 3.

Decreased complex formation of MAPK CD/7m mutants with Ste7 in yeast cell extracts. (A) Kss1 or Fus3 was coimmunoprecipitated from yeast cell extracts with full-length Ste7 tagged at its COOH terminus with the c-Myc epitope. (B) Cultures of strain YDM230 overexpressing Kss1, or mutants thereof, from the KSS1 promoter on multi-copy plasmid (YEpT), with (lanes 3–8) or without (lanes 1 and 2) cooverexpression of tagged Ste7, were grown and harvested. Cell-free extracts were prepared and portions equivalent to 5% of the amount added in the immunoprecipitation reactions (T) were subjected to SDS-PAGE in a 10% polyacrylamide gel (lanes 1, 3, 5, and 7). Samples of the extracts were immunoprecipitated with the anti-c-Myc mAb 9E10, solubilized, divided into two equal portions (‘IP’, lanes 2, 4, 6, and 8), resolved by SDS-PAGE and analyzed by immunoblotting with either anti-Kss1 antiserum (top) or anti-Ste7 antiserum (bottom). (C) Same experiment as in B, with Fus3 instead of Kss1.
Phosphorylation of the Kss1-7m3 mutant is reduced
Ste7MEK phosphorylates the Fus3 and Kss1 MAPKs on a threonine and tyrosine residue located in a surface loop (the activation loop) situated near the MAPK active site. The reduced ability of the CD/7m-region mutants of the MAPKs to bind to Ste7 and Ste5 suggested that they might also have a defect in their ability to be phosphorylated, and thereby activated, in response to pheromone stimulation. To examine this, the wild-type and mutant Kss1 alleles were expressed at endogenous levels in a strain lacking Fus3. As assessed using an antibody that specifically recognizes the dually phosphorylated MAPKs (Sabbagh et al., 2001), wild-type Kss1 protein was hyperphosphorylated after a 5-min treatment with 500 nM pheromone (Fig. 4 A). Phosphorylation of the Kss1-7m1 mutant, which exhibited a moderate defect in binding to Ste7 in vitro and only a modest defect in binding to Ste5 (Fig. 2), was undiminished relative to wild-type Kss1. In contrast, phosphorylation of the Kss1-7m3 mutant, which exhibited a substantial defect in binding to Ste7 and Ste5 in vitro (Fig. 2), was significantly reduced relative to wild type (Fig. 4 A).
In parallel, as a marker for pheromone-induced transcription, the pheromone-stimulated expression of a FUS1-lacZ reporter gene was monitored. FUS1 is representative of a battery of genes that is induced after exposure to mating pheromone (Roberts et al., 2000). As shown in Fig. 4 B, the Kss1-7m mutants had a substantially reduced ability to drive the expression of the reporter gene. Surprisingly, this was even somewhat true of the Kss1-7m1 mutant, which was phosphorylated normally (Fig. 4 A).
Similarly, the ability of the Fus3-7m mutants to support pheromone-induced transcription was assessed. We compensated for the somewhat reduced stability of the Fus3-7m mutants by expressing them from two different low copy plasmids (each with a different selectable marker) in the same strain. Using this procedure, it was possible to produce the mutants at a level that was 80% of that obtained when wild-type Fus3 was expressed from either selectable plasmid (not depicted and Fig. 4 D). Such strains were used for all bioassays. When expressed in this manner in a strain lacking Kss1, both Fus3-7m1 and Fus3-7m2 showed dramatic defects in their ability to drive FUS1-lacZ expression in pheromone-stimulated cells (Fig. 4 C). In contrast, these mutants were dually phosphorylated to a level comparable to wild-type Fus3 (Fig. 4 D), despite their inability to dock with Ste7MEK and reduced binding to Ste5 (Figs. 2 and 3).
Kss1 is phosphorylated and activated during mating in a wild-type cell, where it acts with Fus3 to promote pheromone-induced transcription (Sabbagh et al., 2001); moreover, Kss1 can partially support pheromone-induced transcription in the absence of functional Fus3 (Roberts et al., 2000). Indeed, pheromone-induced reporter gene expression was ∼50% of wild-type in the fus3-7m1 KSS1 + strain (Fig. 4 C). In addition, Kss1 was hyperphosphorylated in this strain (Fig. 4 D, compare lane 1 with lane 3). When three independent experiments like that shown in Fig. 4 D were averaged, Kss1 phosphorylation was 3.1 (± 0.6)-fold higher in the fus3-7m1 KSS1+ strain relative to wild-type. Active Fus3 initiates a feedback loop that limits the magnitude and duration of Kss1 phosphorylation (as well as its own phosphorylation; Sabbagh et al., 2001). Fus3-7m1 is apparently unable to initiate this feedback loop.
The mutants have pheromone response defects
To further determine the net effect of protein–protein interactions mediated by the CD/7m region of Kss1 and Fus3 on the transmission of the mating pheromone signal, selected mutants of Kss1 and Fus3 were tested in several different measures of the mating pheromone response. The Fus3-7m1 and Kss1-7m3 mutants were the focus of this analysis, because they exhibited a clear binding defect to Ste7 and Ste5 (Fig. 2), and because they contained equivalent amino acid substitutions (Fig. 1 D).
First, as a measure of the long-term effects of pheromone action, pheromone-imposed cell-cycle arrest and recovery from arrest were monitored using the standard halo bioassay. As shown in Fig. 5 A, compared with wild type, the fus3-7m1 kss1Δ strain exhibited smaller and much more turbid haloes, indicating a marked reduction in the initial strength of pheromone-imposed arrest and/or a substantially accelerated rate of recovery.
Figure 5.

The CD/7m MAPK mutants have multiple defects in the mating pheromone response. Strains were generated as in Fig. 4. (A) Pheromone-imposed cell-cycle arrest. A lawn of cells was grown for 48 h in the presence of disks containing 12 μg (left) or 3 μg (right) of α-factor mating pheromone. Pheromone-imposed cell-cycle arrest is indicated by the zone of growth inhibition (“halo”) surrounding a disk. (B) Pheromone-induced morphogenesis. Cells were treated for 4 h with 5 μM α-factor, fixed, and photographed at a magnification using a microscope and imaging system. (C) Mating. The strains analyzed in B were scored for mating to a tester strain (DC17) using a quantitative assay. Mating efficiencies (diploids formed per input MATa haploid) are normalized to the value (0.13) obtained for cells expressing wild-type Fus3 and Kss1. Each experiment was repeated three to six times; error bars are shown. The photos at the bottom show the results of a qualitative mating assay performed in parallel.
Second, to assess pheromone-regulated morphogenesis, the formation of a mating projection (shmoo formation) was monitored 4 h after initial exposure to pheromone. This treatment induced wild-type cells to form substantial mating projections (Fig. 5 B, compare a with b); in contrast, cells lacking both MAPKs did not form projections as expected (Fig. 5 B, c). Cells lacking Fus3 underwent some pheromone-induced shape changes but did not form obvious projections (Fig. 5 B, d), whereas cells lacking Kss1 formed projections that were smaller than wild type (Fig. 5 B, e). These controls confirm that both MAPKs are required for normal pheromone-induced morphogenesis, with Fus3 playing a primary role, in agreement with the work of the Elion lab (Farley et al., 1999). In cells lacking Kss1, the Fus3-7m1 mutant was unable to support any morphogenesis, so that the cells resembled those lacking both MAPKs (Fig. 5 B, f). In contrast, the cells containing Fus3-7m1 and wild-type Kss1 were able to undergo substantial morphogenesis, although not with wild-type efficiency (Fig. 5 B, g). Hence, Kss1 was able to almost completely suppress the morphogenesis defect of the fus3-7m1 strain, but not of the fus3Δ strain.
Finally, to assess the role of CD/7m-mediated interactions in overall mating proficiency, mating assays were performed (Fig. 5 C). In a quantitative mating assay, cells lacking Kss1 (but containing Fus3) mated with essentially wild-type efficiency (96%), whereas cells lacking Fus3 (but containing Kss1) mated with reduced efficiency (9%), and cells lacking both MAPKs were unable to mate (<0.02%), as has been extensively document elsewhere (Farley et al., 1999). In cells lacking Fus3, replacing wild-type Kss1 with Kss1-7m3 resulted in an 18-fold drop in mating efficiency. Kss1-7m2 was equally defective in mating, whereas Kss1-7m1 was only twofold down (unpublished data). Similarly, in cells lacking Kss1, replacing wild-type Fus3 with Fus3-7m1 resulted in a 12-fold decrease in mating; Fus3-7m2 was less severely compromised (fourfold decrease). Thus, the double-substitution mutants of the CD/7m-region of the MAPKs have significantly diminished functionality relative to wild type.
As was seen in the analyses of pheromone-regulated morphogenesis (see above), the mating defect of Fus3-7m1 was substantially enhanced when Kss1 was absent, such that the fus3-7m1 kss1Δ strain mated much worse than the fus3-7m1 KSS1+ strain (Fig. 5 C). Conversely, Fus3-7m1 substantially suppressed the mating defect of the fus3Δ KSS1+ strain. This striking mutual suppression was even more evident when mating was performed at 37°C (unpublished data). The presence of Kss1 also partially suppressed (or the absence of Kss1 enhanced) the cell-cycle arrest defect of Fus3-7m1 (unpublished data). These data suggest that Fus3-7m1 is defective in a function that Kss1 can provide (most likely, pheromone-induced transcription; Fig. 4 C), but is still able (to some extent) to execute cell-cycle arrest and morphogenesis functions that Kss1 cannot provide, perhaps because the CD/7m region of Fus3 is not involved in its ability to bind the relevant substrates.
The mutants are also defective in downstream interactions
The previous results demonstrate that several of the CD/7m mutants are phosphorylated (and presumably activated) to near wild-type levels, but are unable to support Ste12-driven expression of mating genes. This suggested that the CD/7m mutants must also be defective in interacting with some of their downstream substrates. We focused on the transcriptional regulators Ste12, Dig1, and Dig2. All three proteins are known substrates of the MAPKs, although the target residues critical for regulation are undetermined.
First, the Kss1–Ste12 interaction was scrutinized (the Fus3–Ste12 interaction is much weaker, and hence was not examined). In an in vitro binding assay (Fig. 6 A), unphosphorylated Kss1 bound to GST-Ste12 with reasonably high affinity (K d ≈ 2 μM). The Kss1-7m3 mutant bound with a somewhat reduced affinity (K d ≈ 8 μM), but still exhibited clearly detectable binding to Ste12. It is unclear whether this modest drop in binding affinity has a significant functional consequence in vivo.
Figure 6.
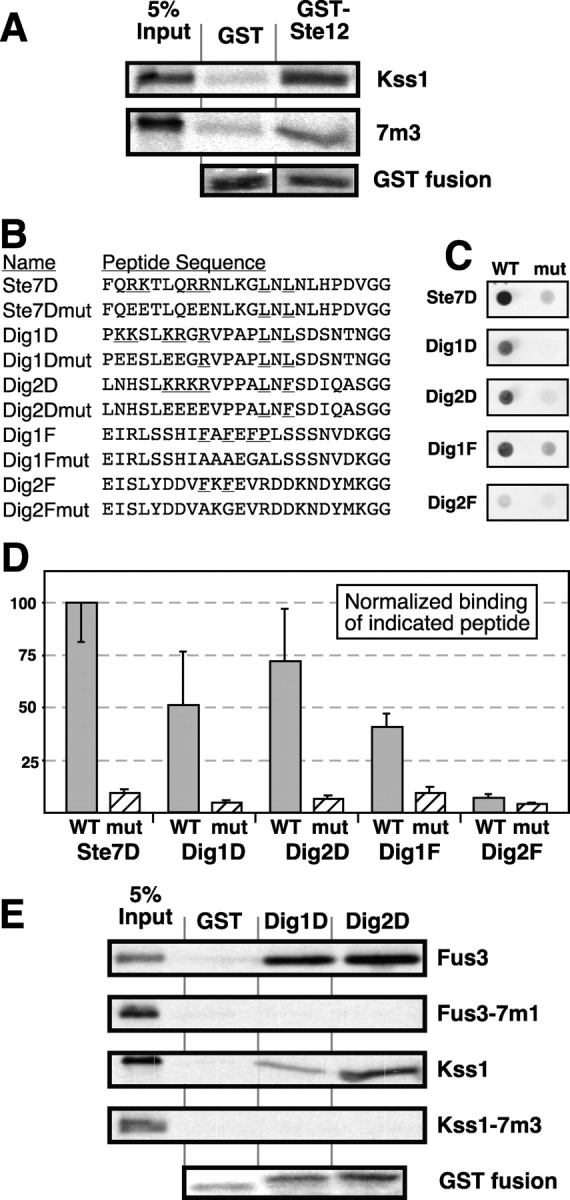
The mutants are defective in binding to D-sites in Dig1 and Dig2. (A) Binding of ∼1 pmole 35S-labeled Kss1 or Kss1-7m3 to 2 μg of GST or 1 μg of GST-Ste12298–473. Experimental design as in Fig. 2. The bottom panel shows a representative loading control for the GST fusion proteins, stained with Coomassie blue. (B) Sequences of the peptides covalently attached to cellulose membrane for peptide array experiment. Conserved residues of the D-site or FXFP-site are underlined. The peptides correspond to the following portions of the full-length proteins, where the NH2-terminal methionine is considered to be residue 1: Ste7D, residues 2–23; Dig1D, 92-113; Dig2D, 95–116; Dig1F, 402-423; Dig2F, 288–309. (C) Autoradiogram of representative peptide spots bound to 35S-labeled Fus3. (D) Graphical representation of relative peptide binding, as quantified on a phosphorimager. The average fold above background binding to the Ste7 peptide was set to 100, and the other peptides were normalized relative to it. Data obtained with Fus3 and Kss1 were not significantly different, and thus were pooled. Four experiments like that shown in D were averaged; error bars are shown. (E) Binding of ∼1 pmole 35S-labeled Fus3, Fus3-7m1, Kss1 or Kss1-7m3 to 1 μg of GST, GST-Dig193-111 (Dig1D), or GST-Dig298–114 (Dig2D). Experimental design as in Fig. 2.
Second, the interaction of Kss1 with Ste12, Dig1, and Dig2 was evaluated in the two-hybrid assay. A fusion of wild-type Kss1, or the Kss1-7m3 mutant, to the Gal4 DNA-binding domain (GBD) was tested against fusions of Ste12, Dig1, or Dig2 to the Gal4 transcriptional activation domain (GAD). In this assay, wild-type Kss1-GBD displayed a minimal background activation of the two-hybrid reporter gene, and the interaction with Ste12, Dig1, or Dig2 was 4-, 3-, and 23-fold above this background, respectively, consistent with our previous reports (Cook et al., 1996; Bardwell et al., 1998a). For reasons that are unclear, the background level of expression driven by Kss1-7m3-GBD was substantially higher than that driven by wild-type Kss1-GBD. Coexpression of Kss1-7m3-GBD with full-length Dig1-GAD or Dig2-GAD did not increase reporter gene expression above this already high background level, whereas coexpression of Kss1-7m3-GBD with Ste12-GAD resulted in a further about sixfold increase in signal (unpublished data). These results suggest that Kss1-7m3 has a defect in binding to full-length Dig1 or Dig2 but is still able to bind to Ste12.
The observations that Kss1-7m3 was compromised in its ability to bind to full-length Dig1 or Dig2 in the two-hybrid assay, but was able to bind to Dig1213-452 (Fig. 2), appeared to be contradictory. Although DIG1(213-452) was the shortest DIG1 coding sequence to be isolated from the two-hybrid screen that resulted in the initial cloning of the DIG1 and DIG2 genes (Cook et al., 1996), its two-hybrid interaction with Kss1 is significantly weaker than that of full-length Dig1 (unpublished data). One possibility that would explain all these data is the existence of an additional MAPK-binding domain NH2-terminal to residue 213 of Dig1. Indeed, inspection revealed that both Dig1 and Dig2 contain apparently homologous D-sites located ∼100 residues from their N termini (Fig. 6 B).
To determine whether these putative D-sites in Dig1 and Dig2 were capable of binding to Kss1 and Fus3, we used a peptide array binding assay (Reineke et al., 2001). Peptides chosen (Fig. 6 B) included the well-characterized D-site of Ste7, the putative D-sites of Dig1 and Dig2, and mutants thereof in which basic residues were altered to acidic residues. In addition, wild-type and mutant peptides containing putative FXFP-like docking sites (Jacobs et al., 1999) near the COOH termini of Dig1 and Dig2 were also included. A membrane containing the bound peptides was incubated with a solution containing radiolabeled Fus3 or Kss1, and the binding of the labeled MAPK to the individual peptide spots was quantified (Fig. 6, C and D). Both MAPKs bound strongly to the wild-type Ste7 D-site peptide, and to both the Dig1 and Dig2 D-site peptides, but not to the corresponding mutant peptides. In addition, both MAPKs bound to the Dig1 FXFP-containing peptide, but not to the corresponding mutant peptide. Finally, both MAPKs did not appreciably bind to the Dig2 FXF-containing peptide; moreover, what little binding there was to this peptide was not lessened by mutation of the two phenylalanine residues. Thus, the putative D-sites in Dig1 and Dig2 can bind to Kss1 and Fus3, as can the putative FXFP site in Dig1.
To determine if the CD/7m-region mutations impaired the binding of the MAPKs to the D-sites in Dig1 and Dig2, the binding of radiolabeled Kss1 and Fus3, and the Fus3-7m1 and Kss1-7m3 mutants, to GST-Dig193-111 and GST-Dig298-114 was measured. Wild type Kss1 and Fus3 bound with high affinity to these fusion proteins, whereas the CD/7m-region mutants did not (Fig. 6 E). The strongest interaction (Fus3–Dig2) had a K d of roughly 1 μM. Two other interesting observations emerged from the quantification of this experiment. First, Fus3 bound to both the Dig1 and Dig2 D-sites with a much higher affinity than did Kss1 (∼17-fold). The functional significance of this difference in the relative binding affinity of Kss1 and Fus3 is unclear, but one possibility is that the higher affinity of Fus3 for the D-sites in Dig1 and Dig2 may compensate for its lower affinity for Ste12 (Bardwell et al., 1998a) and for the COOH terminus of Dig1 (Fig. 2). Second, both Kss1 and Fus3 bound to the Dig2 D-site better than the Dig1 D-site (approximately threefold higher affinity). The higher affinity of the MAPKs for the Dig2 D-site may serve to counteract weakened/absent binding to the Dig2 FXFP-like site.
In summary, the results presented in Fig. 6 collectively suggest that Kss1 and Fus3 bind to Dig1 and Dig2 via an interaction between the CD/7m regions of the MAPKs and D-sites in the NH2-terminal portions of Dig1 and Dig2. Furthermore, both MAPKs may have a secondary, weaker interaction with an FXFP docking site near the COOH terminus of Dig1.
CD/7m mutants of Kss1 are defective in Kss1-imposed repression of Ste12
Unphosphorylated Kss1 binds to Ste12 and potently represses the transcription of filamentation genes (Madhani et al., 1997; Bardwell et al., 1998a). Unphosphorylated Kss1 also represses the basal expression of pheromone-responsive genes (Bardwell et al., 1998b). This Kss1-imposed repression requires Kss1-Ste12 binding, because both Kss1-imposed repression and Kss1-Ste12 binding are reduced by a point mutation (Y231C) in Kss1 (Bardwell et al., 1998a), as well as by mutations in a different region of Kss1 known as the MAPK-insertion region (Madhani et al., 1997; unpublished data). Kss1-imposed repression also requires the Dig1 and Dig2 repressors, and we have proposed that Kss1, by virtue of its ability to bind to both Ste12 and Dig1/2, stabilizes the Ste12–Dig complex, thereby potentiating Dig-dependent repression of Ste12 (Bardwell et al., 1998b). Kss1-imposed repression of Ste12 does not require that Kss1 be activated. In fact, activation-loop phosphorylation of Kss1 weakens Kss1-Ste12 binding and thereby relieves repression (Bardwell et al., 1998a). Hence it is possible to completely separate Kss1-imposed repression from upstream signaling. The model of Kss1-imposed repression is shown in Fig. 7 A.
Figure 7.

Kss1-7m3 is defective in Kss1-imposed repression of Ste12-dependent transcription. (A) Model of Kss1-imposed repression of gene expression (Bardwell et al., 1998a, 1998b). Unphosphorylated Kss1 binds to Ste12 and Dig1/Dig2, thereby stabilizing the repressive interaction of Dig1/Dig2 with Ste12. Phosphorylation of Kss1 by Ste7 weakens Kss1-Ste12 binding, consequently destabilizing the repressive complex. (B, left) Expression of a filamentation response element (FRE)-driven reporter (YEpL-FT1Z, containing the TEC1 FRE) in haploid strain JCY137 (ste7Δ kss1Δ fus3Δ) carrying either an empty vector (Δ), or expressing from plasmid YCpU either wild-type (W.T.) KSS1, or kss1-7m1, 7m2 or 7m3, or kss1-Y231C (Bardwell et al., 1998a). (Right) Expression of FRE-driven reporter (YEpL-FTyZ, containing the Ty1 FRE) in diploid strain JCY119 (ste7Δ/ste7Δ kss1Δ/kss1Δ) expressing the indicated KSS1 alleles, measured after 52 h growth on low nitrogen plates. (C) 20 μg of selected protein extracts used in B were immunoblotted with anti-Kss1 antibody.
Because Kss1-7m3 was deficient in Dig1 and Dig2 binding (Fig. 6), we reasoned that if Kss1-Dig binding is required for Kss1-imposed repression, then Kss1-7m3 should be defective in repression. To test this hypothesis, the Kss1-7m mutants were expressed in place of wild-type Kss1 in a filamentation-competent strain; Ste12-dependent transcription of filamentation genes was then monitored in a quantitative assay, using a reporter gene driven by a filamentation response element (FRE; Madhani and Fink, 1997). This experiment was performed in a strain lacking Ste7MEK, so as to separate any potential repression defect of Kss1-7m3 from the effects of its reduced phosphorylation by Ste7 (Fig. 4). As shown in Fig. 7 B, in both haploids and diploids, FRE-driven transcription was low in a ste7Δ KSS1+ strain, as Ste12 protein was constitutively repressed by Kss1 protein that was locked in its repressive (i.e., unphosphorylated) state. Removal of Kss1 (ste7Δ kss1Δ) resulted in a substantial (>10-fold) derepression of FRE-driven transcription as described previously (Cook et al., 1997; Madhani et al., 1997). Replacement of Kss1 with Kss1-7m2 or Kss1-7m3 also depressed FRE-driven transcription, to a level that was ∼60% of that obtained in the absence of Kss1; Kss1-7m1 had only a slight repression defect. A bioassay of haploid invasive growth supported the conclusions drawn from the reporter gene data (unpublished data). In summary, the CD/7m-region mutants of Kss1 are defective in Kss1-imposed repression, likely due to decreased Dig binding.
Discussion
Common D-sites in MAPK-interacting proteins
Here, we found that mutations in an evolutionarily conserved surface patch of the MAPKs, the CD/7m region, compromised the binding of the yeast MAPKs Fus3 and Kss1 to multiple partners, including the Ste7 MEK, the Ste5 scaffold protein, and the Dig1 and Dig2 transcriptional repressors. Significantly, however, only some of the protein–protein interactions that the MAPKs participate in were impaired. Of those that were, most (the exception being Ste5) were mediated a common docking site on the MAPK-interacting partner, the D-site, that has the rough consensus (K/R)1-3-X1-6-ϕ-X-ϕ where ϕ represents a hydrophobic residue, typically L or I. The basic and hydrophobic submotifs of the D-site apparently bind to the MAPK in a highly cooperative fashion, as mutation of either can dramatically diminish binding (Zúñiga et al., 1999; Bardwell et al., 2001). It is likely that the basic submotif of the D-site interacts directly with the acidic residues of the CD/7m region (Tanoue et al., 2000). The MAPK-binding domain of Ste5 (Ste5241-336) does not contain a clear consensus D-site. However, it does contain two dibasic submotifs. Furthermore, a Ste5318-339 peptide, which contains both of these submotifs, binds to Fus3 in an array assay (unpublished data).
That the same region of MAPKs is required for binding to many MAPK-interacting proteins suggests that these proteins may compete with each other for binding to their cognate MAPKs, binding in a competitive and perhaps mutually exclusive fashion (Bardwell et al., 2003). Indeed, we showed here that the Ste7 D-site competed with Ste5 for binding to Fus3MAPK (Fig. 2 D). This result suggests that, in a Ste5–Ste7–MAPK complex, the MAPK can bind with high affinity either to Ste5 or Ste7, but not to both simultaneously. Similarly, mammalian c-Jun N-terminal kinase (JNK) MAPKs bind to D-sites in scaffolds such as JIP1-3 and β-arrestin 2 (Enslen et al., 2000; Sharrocks et al., 2000), and also bind to a D-site in the MKK4 (Ho et al., 2003). The competitive binding of MEKs and scaffolds to MAPKs may influence the dynamics of scaffold-assisted MAPK activation in a manner that is not presently understood.
Binding to Ste7
The interaction between the Ste7 MEK and its cognate MAPKs Kss1 and Fus3 has the highest affinity of any MEK–MAPK interaction yet measured in any organism (K d ≈ 5–100 nM, depending on the assay; Bardwell et al., 1996, 2001). This affinity was decreased more than 500-fold by the CD/7m region mutations (Fig. 2). In cells, this is predicted to decrease the fraction of Ste7 bound to the MAPKs at any instant in time from greater than 90% to less than 5%. Consistent with this prediction, the Kss1-7m3 mutant was inefficiently phosphorylated by Ste7 in vivo (Fig. 4). On the other hand, there was no obvious decrease in the phosphorylation of the Fus3 mutants, perhaps because other protein interactions compensated for the loss of Ste7-dependent docking (Bardwell et al., 2001). In addition, any defects in MAPK activation caused by the CD/7m-region mutations may have been counterbalanced by defects in negative regulatory mechanisms, for example, decreased binding of the mutants to MAPK phosphatases, or the defect in the Fus3-dependent feedback circuit.
Binding to Dig1 and Dig2
We found that the CD/7m mutants of Kss1 and Fus3 exhibited reduced binding to full-length Dig1 and Dig2 in the two-hybrid assay. Accordingly, these MAPK mutants also lost the ability to bind to isolated versions of putative D-sites found in the NH2-terminal halves of Dig1 and Dig2 (Fig. 6). Binding of the mutants to the COOH-terminal half of Dig1, perhaps to an FXFP-type docking site located there, was undiminished. Collectively, these data indicate that the interaction of the MAPKs with Dig1 and Dig2 is bimodal, with the D-site playing a major role in stable complex formation. The importance of both the D-site and the FXFP-like site for Dig1 function in vivo has been delineated recently by Thorner's lab (J.X. Zhu-Shimoni and J. Thorner, personal communication).
What is the cellular function of the CD/7m-region–mediated interactions of the MAPKs with Dig1 and Dig2? Our data indicate that the CD/7m mutants are defective in both positive and negative components of MAPK-regulated transcription. The defect in positive regulation was exemplified by the Fus3-7m1 and 7m2 mutants, which were significantly compromised in their ability to support pheromone-induced transcription (Fig. 4). The MAPKs are thought to positively regulate transcription by phosphorylating Ste12 and/or Dig1 and Dig2, thereby destabilizing the Ste12–Dig complex. It is likely that the CD/7m-region–mediated docking of Fus3 and Kss1 to this complex potentiates these phosphorylation events. This model is consistent with a series of studies showing that the docking of animal MAPKs to transcription factors potentiates the phosphorylation of nearby target residues (Sharrocks et al., 2000).
The MAPKs, particularly Kss1, negatively regulate gene expression by a mechanism termed “MAPK-imposed repression” (Fig. 7 A). We proposed a model for this process in which Kss1 stabilizes the interaction of the Dig repressors with their target, Ste12, by virtue of its ability to bind to all three proteins (Bardwell et al., 1998b). This model was supported by the finding that two mutants of Kss1, Y231C and Δloop, that had reduced binding to Ste12 (but undiminished binding to Dig1 and Dig2) were partially deficient in repression (Bardwell et al., 1998a). This paper provides important support for another key tenet of this model. The Kss1-7m3 mutant, deficient in binding Dig1 and Dig2, but still able to bind Ste12 (Fig. 6), was defective in Kss1-imposed repression (Fig. 7). Hence, Kss1-Dig binding is indeed required for repression, as proposed.
A caveat to the above interpretation is that Kss1-7m3 was partially compromised in its ability to bind to Ste12298-473 in vitro (Fig. 6). We discount the significance of this modest effect on Ste12 binding, because Kss1-Y231C and Kss1-Δloop both bind to Ste12 much worse than Kss1-7m3, but support Kss1-imposed repression better than Kss1-7m3 (Bardwell et al., 1998a; unpublished data).
CD/7m-region–mediated interactions have an overall positive role
Most of the previous genetic and cell-based studies of 7m-region mutants of MAPKs emphasized a hyperactive, gain of function phenotype that was attributed to a decreased interaction with negative regulators such as deactivating phosphatases (Bott et al., 1994; Brunner et al., 1994; Chu et al., 1996; Hall et al., 1996; Karim and Rubin, 1999; Zhan and Guan, 1999). These findings are not entirely consistent with subsequent evidence, here extended to yeast, that the CD/7m region also mediates interactions with positively acting upstream regulators and downstream substrates (Tanoue et al., 2000). Hence, heretofore, genetic studies of the consequences of mutating the MAPK sevenmaker region contradicted the far-reaching implications of the biochemical studies.
Hall et al. (1996) isolated a 7m-region mutant, Fus3D317G,F329S, that protected overproduced Fus3 from repression by the HOG–MAPK pathway. We did not specifically examine this phenotype. In contrast to our results, however, Hall et al. (1996) did not find that their allele had a signaling defect, perhaps due to the particular allele they used. Zhan and Guan (1999) observed that Fus3D317N and Fus3D317G conferred a slightly hyperactive phenotype when expressed in wild-type cells; they did not examine the phenotype of these mutants in fus3Δ cells. The tissue culture studies of ERK2D319N also were based on expression of the mutant in cells containing endogenous ERK2 (Bott et al., 1994; Chu et al., 1996). There is also some evidence that the ERK2D319N mutant may compromise the interaction with phosphatases more so than it does with other targets (Yung et al., 2001; unpublished data); perhaps the same is true of Drosophila rolledD324N. Differences in expression levels might also account for some of the above discrepancies, because we found that multi-copy expression of the Fus3-7m mutants suppressed their signaling defect (unpublished data).
Here, we showed that, when expressed at near-endogenous levels in a fus3Δ kss1Δ strain, the Fus3 and Kss1 CD/7m-region mutants exhibited deficiencies in multiple mating end points (Figs. 4 and 5). These observations provide important genetic evidence that the network of interactions mediated by the MAPK CD/7m region has overall a positive role in signal transmission. Our results are in agreement with those of Farley et al. (1999), who isolated and studied a series of Fus3 reduced-function alleles, two of which had mutations in the CD/7m region.
Conclusions
It is apparent that an interaction network involving MAPKs and multiple binding partners has been structurally and functionally conserved from yeast to humans (Fig. 8). Indeed, it seems that the network architecture has been conserved to a greater degree than the component nodes, as no homologues of Ste5, Dig1, or Dig2 can be recognized in any animal genome. Thus, the links in the MAPK network have apparently been preserved, even though some of the component nodes have changed completely, replaced by scaffolds and transcription factors that may be partial analogs of Ste5 and Ste12/Dig1/Dig2, but are clearly not orthologues. Evidently, the network itself is more evolutionarily stable than the nodes that constitute it.
Figure 8.

A conserved MAPK protein interaction network. Shown are selected components of the yeast mating (A), mammalian ERK1/2 (B), and mammalian JNK (C) MAPK cascades. Components shown include the MAPKs themselves (circles), MEKs (ovals), phosphatases (unshaded rectangles), scaffolds (eight-sided polygons), and transcription factors (shown bound to DNA). D sites are indicated by a boxed or circled ‘D’. FXFP docking sites are indicated by a boxed or circled ‘F’. The CD/7m region of the MAPKs is indicated by a circled ‘7’. The potential involvement of this region in the binding of ERK to Elk-1 is supported by a recent paper (Yung et al., 2001); its involvement in binding to KSR or MP1 has not, to our knowledge, been assessed; KSR does have an FXFP site (Jacobs et al., 1999). Likewise, the CD/7m region of JNK has so far been shown only to play a role in the interaction with MKK4/SEK1 and MKP5 (Tanoue et al., 2000). Studies of its role in binding to scaffolds, c-Jun or ATF-2 have not yet been reported, although all of these JNK-interacting proteins contain functional D-sites.
Materials and methods
Yeast strains used in this paper are described in Table S1. Standard yeast media and culture conditions, and bioassays for pheromone response were prepared as described previously (Bardwell et al., 1996). Two-hybrid analysis was performed as described previously (Bardwell et al., 1998a). Plasmids used in this work are described in Tables S2–S8.
Oligonucleotide-directed, PCR-based mutagenesis was performed using the Quickchange kit (Stratagene), and confirmed by sequencing. In vitro transcription and translation were performed in a coupled system (SPT3) purchased from Novagen; [35S]methionine was used to label the translation products. Partial purification of translation products, and determination of dissociation constants from binding assay results were performed as described previously (Bardwell et al., 1996, 2001). GST fusion proteins were produced in bacteria as described previously (Cook et al., 1996; Bardwell et al., 2001), except GST-Ste12298-473, which was produced in yeast (Bardwell et al., 1998a). The peptide array binding assay was performed as described previously (Ho et al., 2003). GST cosedimentation assays (Cook et al., 1996; Bardwell et al., 2001), coimmunoprecipitation from yeast cell extracts and quantification of reporter gene expression (Bardwell et al., 1996), and analysis of Kss1 and Fus3 phosphorylation levels in vivo (Sabbagh et al., 2001), were also performed as described previously. Binding assay results were quantified using a PhosphorImagerTM (Molecular Dynamics, Inc.). Immunoblots were quantified using an Odyssey infrared imaging system (LI-COR Inc.).
Microscopy
For Fig. 5 B, formalin-fixed cells were mounted under a standard coverslip and visualized using a microscope (model Axioskop 2 MOT; Carl Zeiss MicroImaging, Inc.) equipped with a 100× oil-immersion objective, NA 7, at RT. The images were acquired using a camera (model Axiocam HRc; Carl Zeiss MicroImaging, Inc.) and Axiovision V3.04E software.
Online supplemental material
Yeast strains used in this paper are described in Table S1. Plasmids used here are described in Tables S2–S8. All supplemental tables are available at http://www.jcb.org/cgi/content/full/jcb.200310021/DC1.
Acknowledgments
For gifts of strains and reagents with thank Gerald Fink (Massachusetts Institute of Technology, Cambridge, MA) and Jeremy Thorner (University of California, Berkeley, Berkeley, CA). We also thank Judy Zhu-Shimoni and Jeremy Thorner for communicating results before publication.
This work was supported by a Burroughs Wellcome Foundation New Investigator award, by a Beckman Foundation Young Investigator award, and by National Institutes of Health Research (NIH) grant GM60366 (all to L. Bardwell). W. Sabbagh was partially support by NIH GM69013.
A.B. Kusari and D.M. Molina contributed equally to this work.
The online version of this article includes supplemental material.
A.B. Kusari's present address is Keck Graduate Institute of Applied Life Sciences, 535 Watson Dr., Claremont, CA 91711.
Abbreviations used in this paper: CD, conserved docking; ERK, extracellular signal–regulated kinase; FRE, filamentation response element; GBD, Gal4 DNA-binding domain; JNK, c-Jun N-terminal kinase; MEK, MAPK/ERK kinase; MEKK, MEK kinase; MKK, MAPK kinase.
References
- Bardwell, L., and J. Thorner. 1996. A conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem. Sci. 21:373–374. [PubMed] [Google Scholar]
- Bardwell, L., J.G. Cook, E.C. Chang, B.R. Cairns, and J. Thorner. 1996. Signaling in the yeast pheromone response pathway: specific and high-affinity interaction of the mitogen-activated protein (MAP) kinases Kss1 and Fus3 with the upstream MAP kinase kinase Ste7. Mol. Cell. Biol. 16:3637–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell, L., J.G. Cook, D. Voora, D.M. Baggott, A.R. Martinez, and J. Thorner. 1998. a. Repression of yeast Ste12 transcription factor by direct binding of unphosphorylated Kss1 MAPK and its regulation by the Ste7 MEK. Genes Dev. 12:2887–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell, L., J.G. Cook, J.X. Zhu-Shimoni, D. Voora, and J. Thorner. 1998. b. Differential regulation of transcription: repression by unactivated mitogen-activated protein kinase Kss1 requires the Dig1 and Dig2 proteins. Proc. Natl. Acad. Sci. USA. 95:15400–15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell, A.J., L.J. Flatauer, K. Matsukuma, J. Thorner, and L. Bardwell. 2001. A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J. Biol. Chem. 276:10374–10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell, A.J., M. Abdollahi, and L. Bardwell. 2003. Docking sites on mitogen-activated protein kinase (MAPK) kinases, MAPK phosphatases and the Elk-1 transcription factor compete for MAPK binding and are crucial for enzymic activity. Biochem. J. 370:1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott, C.M., S.G. Thorneycroft, and C.J. Marshall. 1994. The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett. 352:201–205. [DOI] [PubMed] [Google Scholar]
- Brunner, D., N. Oellers, J. Szabad, W.H. Biggs, III, S.L. Zipursky, and E. Hafen. 1994. A gain-of-function mutation in Drosophila MAP kinase activates multiple receptor tyrosine kinase signaling pathways. Cell. 76:875–888. [DOI] [PubMed] [Google Scholar]
- Choi, K.-Y., B. Satterberg, D.M. Lyons, and E.A. Elion. 1994. Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae. Cell. 78:499–512. [DOI] [PubMed] [Google Scholar]
- Chu, Y., P.A. Solski, R. Khosravi-Far, C.J. Der, and K. Kelly. 1996. The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J. Biol. Chem. 271:6497–6501. [DOI] [PubMed] [Google Scholar]
- Cook, J.G., L. Bardwell, S.J. Kron, and J. Thorner. 1996. Two novel targets of the MAP kinase Kss1 are negative regulators of invasive growth in the yeast Saccharomyces cerevisiae. Genes Dev. 10:2831–2848. [DOI] [PubMed] [Google Scholar]
- Cook, J.G., L. Bardwell, and J. Thorner. 1997. Inhibitory and activating functions for MAPK Kss1 in the S. cerevisiae filamentous growth signaling pathway. Nature. 390:85–88. [DOI] [PubMed] [Google Scholar]
- Dohlman, H.G., and J.W. Thorner. 2001. Regulation of G protein–initiated signal transduction in yeast: paradigms and principles. Annu. Rev. Biochem. 70:703–754. [DOI] [PubMed] [Google Scholar]
- Elion, E.A. 2001. The Ste5p scaffold. J. Cell Sci. 114:3967–3978. [DOI] [PubMed] [Google Scholar]
- Elion, E., B. Satterberg, and J. Kranz. 1993. Fus3 phosphorylates multiple components of the mating signal transduction cascade: evidence for Ste12 and Far1. Mol. Biol. Cell. 4:495–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enslen, H., and R.J. Davis. 2001. Regulation of MAP kinases by docking domains. Biol. Cell. 93:5–14. [DOI] [PubMed] [Google Scholar]
- Enslen, H., D.M. Brancho, and R.J. Davis. 2000. Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 19:1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley, F.W., B. Satterberg, E.J. Goldsmith, and E.A. Elion. 1999. Relative dependence of different outputs of the Sacchromyces cerevisiae pheromone response pathway on the MAP kinase Fus3p. Genetics. 151:1425–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, J.P., V. Cherkasova, E. Elion, M.C. Gustin, and E. Winter. 1996. The osmoregulatory pathway represses mating pathway activity in Saccharomyces cerevisiae: isolation of a FUS3 mutant that is insensitive to the repression mechanism. Mol. Cell. Biol. 16:6715–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, D.T., A.J. Bardwell, M. Abdollahi, and L. Bardwell. 2003. A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J. Biol. Chem. 278:32662–32672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, D., D. Glossip, H. Xing, A.J. Muslin, and K. Kornfeld. 1999. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13:163–175. [PMC free article] [PubMed] [Google Scholar]
- Karim, F.D., and G.M. Rubin. 1999. PTP-ER, a novel tyrosine phosphatase, functions downstream of Ras1 to downregulate MAP kinase during Drosophila eye development. Mol. Cell. 3:741–750. [DOI] [PubMed] [Google Scholar]
- Madhani, H.D., and G.R. Fink. 1997. Combinatorial control required for the specificity of yeast MAPK signaling. Science. 275:1314–1317. [DOI] [PubMed] [Google Scholar]
- Madhani, H.D., C.A. Styles, and G.R. Fink. 1997. MAP kinases with distinct inhibitory functions impart signaling specificity during yeast differentiation. Cell. 91:673–684. [DOI] [PubMed] [Google Scholar]
- Printen, J.A., and G.F. Sprague, Jr. 1994. Protein-protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics. 138:609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke, U., R. Volkmer-Engert, and J. Schneider-Mergener. 2001. Applications of peptide arrays prepared by the SPOT-technology. Curr. Opin. Biotechnol. 12:59–64. [DOI] [PubMed] [Google Scholar]
- Roberts, C.J., B. Nelson, M.J. Marton, R. Stoughton, M.R. Meyer, H.A. Bennett, Y.D. He, H. Dai, W.L. Walker, T.R. Hughes, et al. 2000. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 287:873–880. [DOI] [PubMed] [Google Scholar]
- Sabbagh, W., L.J. Flatauer, A.J. Bardwell, and L. Bardwell. 2001. Specificity of MAP kinase signaling in yeast differentiation involves transient vs. sustained MAPK activation. Mol. Cell. 8:683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrocks, A.D., S.H. Yang, and A. Galanis. 2000. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci. 25:448–453. [DOI] [PubMed] [Google Scholar]
- Tanoue, T., M. Adachi, T. Moriguchi, and E. Nishida. 2000. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2:110–116. [DOI] [PubMed] [Google Scholar]
- Tedford, K., S. Kim, D. Sa, K. Stevens, and M. Tyers. 1997. Regulation of the mating pheromone and invasive growth responses in yeast by two MAP kinase substrates. Curr. Biol. 7:228–238. [DOI] [PubMed] [Google Scholar]
- Vespignani, A. 2003. Evolution thinks modular. Nat. Genet. 35:118–119. [DOI] [PubMed] [Google Scholar]
- Widmann, C., S. Gibson, M.B. Jarpe, and G.L. Johnson. 1999. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79:143–180. [DOI] [PubMed] [Google Scholar]
- Yung, Y., Z. Yao, D.M. Aebersold, T. Hanoch, and R. Seger. 2001. Altered regulation of ERK1b by MEK1 and PTP-SL and modified Elk1 phosphorylation by ERK1b are caused by abrogation of the regulatory C-terminal sequence of ERKs. J. Biol. Chem. 276:35280–35289. [DOI] [PubMed] [Google Scholar]
- Zhan, X.-L., and K.-L. Guan. 1999. A specific protein-protein interaction accounts for the in vivo substrate selectivity of Ptp3 towards the Fus3 MAP kinase. Genes Dev. 13:2811–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zúñiga, A., J. Torres, J. Ubeda, and R. Pulido. 1999. Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J. Biol. Chem. 274:21900–21907. [DOI] [PubMed] [Google Scholar]