

Abstract
Cofilin mediates lamellipodium extension and polarized cell migration by stimulating actin filament dynamics at the leading edge of migrating cells. Cofilin is inactivated by phosphorylation at Ser-3 and reactivated by cofilin-phosphatase Slingshot-1L (SSH1L). Little is known of signaling mechanisms of cofilin activation and how this activation is spatially regulated. Here, we show that cofilin-phosphatase activity of SSH1L increases ∼10-fold by association with actin filaments, which indicates that actin assembly at the leading edge per se triggers local activation of SSH1L and thereby stimulates cofilin-mediated actin turnover in lamellipodia. We also provide evidence that 14-3-3 proteins inhibit SSH1L activity, dependent on the phosphorylation of Ser-937 and Ser-978 of SSH1L. Stimulation of cells with neuregulin-1β induced Ser-978 dephosphorylation, translocation of SSH1L onto F-actin–rich lamellipodia, and cofilin dephosphorylation. These findings suggest that SSH1L is locally activated by translocation to and association with F-actin in lamellipodia in response to neuregulin-1β and 14-3-3 proteins negatively regulate SSH1L activity by sequestering it in the cytoplasm.
Keywords: LIM-kinase; cell polarity; 14-3-3; actin filaments; MCF-7
Introduction
For cells to migrate, an F-actin–rich lamellipodial membrane protrusion is first established in the direction of cell movement and is maintained at the leading edge throughout the migration (Ridley et al., 2003). Rapid turnover of actin filaments is essential to maintain and extend the lamellipodium for cell migration (Pollard and Borisy, 2003). Cofilin stimulates disassembly and severance of actin filaments at or near the pointed ends and thereby actin monomers are continuously supplied for polymerization and rapid turnover of actin filaments is given support (Chen et al., 2000). Thus, the activation of cofilin seems to play an essential role in maintaining and protruding lamellipodia at the leading edge of migrating cells.
Cofilin is inactivated by phosphorylation at Ser-3 by LIM-kinase (LIMK; Arber et al., 1998; Yang et al., 1998). Suppression of cofilin activity by LIMK overexpression abolished lamellipodium formation and polarized cell migration, which implicates cofilin in cell polarity formation and migration (Zebda et al., 2000; Dawe et al., 2003). Slingshot-1L (SSH1L) is a member of a Slingshot phosphatase family that dephosphorylates and reactivates an inactive Ser-3–phosphorylated cofilin (P-cofilin; Niwa et al., 2002). Previous studies showed that cofilin is dephosphorylated in response to various external stimuli that alter cell motility and morphology (Moon and Drubin, 1995). However, mechanisms that regulate SSH activity and cofilin dephosphorylation in response to external cues are poorly understood. Here, we provide evidence that SSH1L is locally activated in lamellipodia by associating with actin filaments and 14-3-3 proteins negatively regulate this activation by sequestering it in the cytoplasm.
Results and discussion
Neuregulin-1β (NRG) triggers lamellipodium formation and polarized cell migration of MCF-7 breast carcinoma cells (Spencer et al., 2000). To investigate mechanisms that regulate cofilin activity, we first determined if NRG stimulation would alter the level of P-cofilin. Using an anti–P-cofilin antibody, we found that NRG induced cofilin dephosphorylation (Fig. 1 A). During the time of cofilin dephosphorylation, actin remodeling continuously occurred and the cells altered their shapes to form a widespread lamellipodium (Fig. S1 and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200401136/DC1). Expression of dominant-negative Rac (RacN17) or phosphatase-dead SSH1L (SSH1L[CS]) blocked NRG-induced cofilin dephosphorylation (Fig. 1, B and C), suggesting that both Rac and SSH1L are involved in NRG-induced cofilin dephosphorylation.
Figure 1.
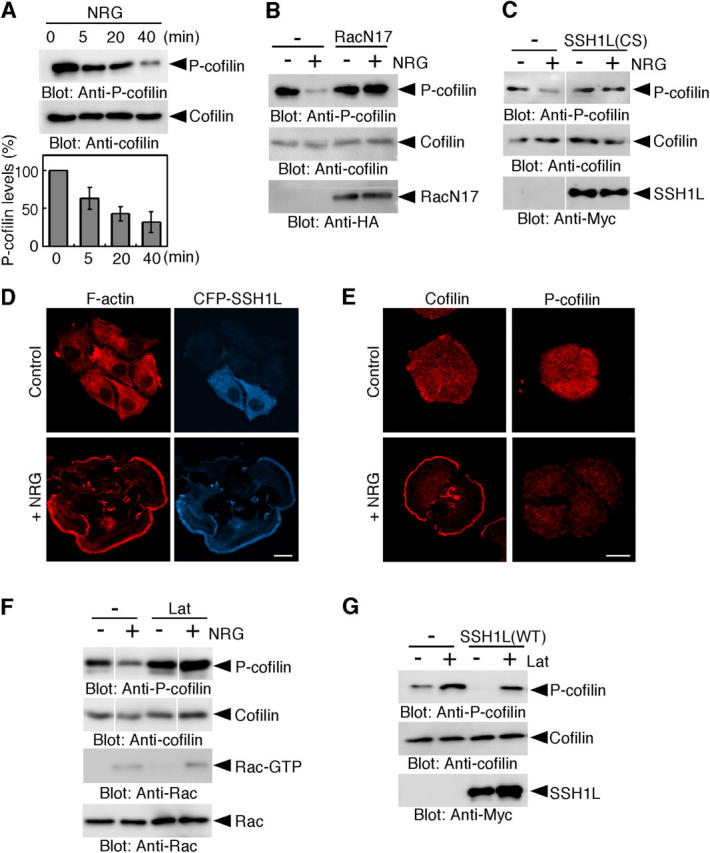
NRG stimulates cofilin dephosphorylation in a manner dependent on Rac, SSH1L, and actin assembly. (A) NRG induces cofilin dephosphorylation. MCF-7 cells were stimulated with NRG. Cell lysates were immunoblotted with antibodies against P-cofilin and cofilin. Relative P-cofilin levels are shown as means ± SEM of triplicate experiments. (B and C) Expression of RacN17 or SSH1L(CS) inhibits NRG-induced cofilin dephosphorylation. MCF-7 cells transfected with HA-RacN17 (B) or Myc-SSH1L(CS) (C) were stimulated with NRG for 20 min and the P-cofilin levels were analyzed as in A. (D) SSH1L accumulates in NRG-induced lamellipodia. MCF-7 cells expressing CFP-SSH1L were stimulated with NRG for 15 min and stained with rhodamine phalloidin for F-actin. Bar, 20 μm. (E) Cofilin, but not P-cofilin, accumulates in NRG-induced lamellipodia. MCF-7 cells were stimulated with NRG for 15 min and stained with antibodies against cofilin or P-cofilin. Bar, 20 μm. (F) Lat-A inhibits NRG-induced cofilin dephosphorylation. MCF-7 cells were treated with Lat-A for 10 min, then stimulated with NRG for 20 min, and the P-cofilin levels were analyzed. Active GTP-bound form of Rac was analyzed using pull-down assay with GST-PBD (p21-binding domain of PAK1; Nishita et al., 2002). (G) Lat-A inhibits SSH1L- induced cofilin dephosphorylation. MCF-7 cells transfected with Myc-SSH1L(WT) were treated with Lat-A for 30 min and the P-cofilin levels were analyzed as in A.
Next, we examined the distribution of SSH1L before and after NRG stimulation (Fig. 1 D). In the absence of NRG, CFP-tagged SSH1L was diffusely distributed in MCF-7 cells. In contrast, after exposure to NRG, CFP-SSH1L accumulated onto the F-actin–rich lamellipodium. Immunostaining with anticofilin antibody revealed that cofilin distributed uniformly in nonstimulated cells but accumulated in the lamellipodium after NRG stimulation (Fig. 1 E). In contrast, P-cofilin diffusely distributed before and after NRG stimulation and the level of P-cofilin decreased after NRG stimulation (Fig. 1 E). Thus, nonphosphorylated (active) cofilin is the major component of cofilin signals which accumulate in the lamellipodium. Distribution of cofilin, but not P-cofilin, in lamellipodia was also noted for migrating fibroblasts (Dawe et al., 2003). Together, these results suggest that SSH1L is recruited to the lamellipodium after NRG stimulation and is involved in the local activation of cofilin.
Pretreatment of cells with latrunculin-A (Lat-A), an inhibitor of actin assembly, blocked NRG-induced lamellipodium formation and SSH1L accumulation (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200401136/DC1). Lat-A increased the basal level of P-cofilin in MCF-7 cells and inhibited NRG-induced cofilin dephosphorylation (Fig. 1 F), which indicates that actin filament assembly is required for cofilin dephosphorylation. NRG induced Rac activation but this was not affected by Lat-A treatment (Fig. 1 F). Expression of SSH1L induced cofilin dephosphorylation in MCF-7 cells, but treatment of the cells with Lat-A suppressed SSH1L-induced cofilin dephosphorylation (Fig. 1 G), which suggests that cofilin-phosphatase activity of SSH1L depends on actin filament assembly.
As SSH1L has the potential to bind to F-actin (Niwa et al., 2002), we examined if actin filaments would directly control SSH1L activity. In vitro cofilin-phosphatase assays revealed that SSH1L activity was remarkably enhanced in the presence of F-actin, but not G-actin (Fig. 2 A). F-actin alone had no apparent effect. As reported previously (Ohta et al., 2003), full-length SSH1L and an NH2-terminal NP fragment have the potential to dephosphorylate P-cofilin, but a COOH-terminal PC fragment did not do so (Fig. 2 B). Addition of F-actin enhanced cofilin-phosphatase activity of full-length SSH1L, but not either the activity of NP or PC (Fig. 2 B). Kinetic analysis revealed that F-actin increased ∼10-fold the cofilin-phosphatase activity of full-length SSH1L, whereas it had no apparent effect on the activity of NP (Fig. 2 C). Because NP scarcely binds to F-actin (Ohta et al., 2003), SSH1L is likely activated by F-actin binding to the COOH-terminal region.
Figure 2.

F-actin activates the cofilin-phosphatase activity of SSH1L. (A) F-actin activates SSH1L. Myc-SSH1L expressed in COS cells was precipitated with anti-Myc antibody and subjected to in vitro phosphatase assay, using cofilin-(His)6 as a substrate, with or without F-actin (F) or G-actin (G). P-cofilin and total cofilin were measured by Pro-Q and Coomassie brilliant blue (CBB) staining, respectively. The bottom panel indicates the P-cofilin levels, as means ± SD of triplicate experiments. (B) F-actin activates SSH1L(WT), but not its NP or PC fragment. The conserved regions between a SSH family are indicated as A, B, P (phosphatase), and S domains. Cofilin-phosphatase activities of Myc-SSH1L(WT) and its NP and PC fragments with or without F- or G-actin were analyzed as in A. (C) Kinetic analyses of cofilin-phosphatase activity of WT and NP mutant of SSH1L with or without F-actin. Top panels show the P-cofilin levels at indicated times after incubation with WT or NP mutant of SSH1L, measured by Pro-Q staining. Bottom panels indicate the time courses of P-cofilin dephosphorylation as means ± SD of triplicate experiments.
As SSH1L accumulates onto the lamellipodia after NRG stimulation, we assumed that in nonstimulated cells SSH1L might be sequestered in the cytoplasm in a dormant form. To examine this possibility, we attempted to purify cytosolic proteins that interact with SSH1L. (Myc+His)-tagged SSH1L was expressed in COS-7 cells and the coprecipitating proteins were analyzed on SDS-PAGE (Fig. 3 A). The proteins (28–30 kD) that specifically bind to SSH1L (WT and CS) were identified as 14-3-3γ, τ, and ζ isoforms using mass spectrometry. Yeast two-hybrid screening also identified 14-3-3β as an SSH1L-binding protein (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200401136/DC1). In vitro pull-down assay revealed that full-length SSH1L efficiently bound to GST-14-3-3β but a truncated mutant Δ1 only weakly and Δ2 and Δ3 to an even lesser extent (Fig. 3 B), thus indicating that regions (961–1049) and (897–960) of SSH1L are responsible for the binding to 14-3-3β. Phosphatase treatment abrogated SSH1L binding to 14-3-3β (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200401136/DC1), indicating that the interaction depends on the phosphorylation. SSH1L contains the sequences RSSS937 and RSHS978, which accord with the 14-3-3–binding motif RSXpS (pS, phospho-Ser; Tzivion and Avruch, 2002). Therefore, we constructed point mutants of SSH1L (S937A, S978A and 2SA), in which either Ser-937 or Ser-978 or both were replaced by alanine. Pull-down assays revealed that 2SA mutant failed to bind to 14-3-3β and S978A and S937A mutants significantly reduced the binding ability (Fig. 3 C). In addition, (Myc+His)-SSH1L point mutants expressed in COS cells scarcely coprecipitated 14-3-3 proteins (Fig. 3 A). These findings suggest that SSH1L interacts with 14-3-3 proteins in a manner dependent on Ser-978 and Ser-937 phosphorylation. Decrease in the 14-3-3 binding potential of S978A and S937A mutants suggests that phosphorylation of both of these two sites is important for stable binding of the dimeric form of 14-3-3 proteins (Tzivion and Avruch, 2002).
Figure 3.

14-3-3 proteins bind to and inhibit SSH1L, dependent on Ser-937 and Ser-978 phosphorylation. (A) Purification of 14-3-3 proteins as SSH1L-interacting proteins. Lysates of COS cells expressing WT or CS mutant of (Myc+His)-SSH1L were precipitated with Ni-NTA agarose, run on SDS-PAGE, and stained by silver. Right panel shows similar experiments done for the indicated mutants of (Myc+His)-SSH1L. (B) The COOH-terminal region of SSH1L is required for binding to 14-3-3β. WT and deletion mutants of Myc-SSH1L were expressed in COS cells and subjected to in vitro pull-down assay with GST or GST-14-3-3β. (C) SSH1L interacts with 14-3-3β, dependent on Ser-937 and Ser-978 phosphorylation. WT and point mutants of Myc-SSH1L expressed in COS cells were subjected to in vitro pull-down assay. (D) 14-3-3γ inhibits SSH1L, but not its 2SA mutant, in cell-free assay. Myc-SSH1L or its 2SA mutant expressed in 293T cells were precipitated with an anti-Myc antibody and subjected to in vitro phosphatase assay, using cofilin-(His)6 as a substrate, with or without recombinant (Rec.) 14-3-3γ. Reaction mixtures were analyzed by blotting with anti–P-cofilin antibody. It is noted that SSH1L(WT), but not its 2SA mutant, coprecipitated endogenous (Endo.) 14-3-3 proteins (bottom). (E) 14-3-3γ protects SSH1L from F-actin– induced activation. Endogenous SSH1L in MCF-7 cells was precipitated with anti-SSH1L antibody or GST-14-3-3γ and subjected to in vitro phosphatase assay, as in Fig. 2 A. (F) Expression of 14-3-3γ increases the cellular P-cofilin level and suppresses SSH1L-induced cofilin dephosphorylation. Myc-14-3-3γ was expressed alone or with WT or 2SA mutant of Myc-SSH1L in COS cells, and lysates were analyzed by immunoblotting with indicated antibodies. The bottom panel indicates the relative levels of P-cofilin as means ± SD of triplicate experiments.
To examine the effect of 14-3-3 on SSH1L activity, SSH1L and its 2SA mutant expressed in 293T cells were immunoprecipitated and subjected to in vitro cofilin-phosphatase assays. As expected, endogenous 14-3-3 proteins were coprecipitated with SSH1L(WT), but not with its 2SA mutant (Fig. 3 D). Addition of recombinant 14-3-3γ to the reaction mixture significantly suppressed the activity of SSH1L(WT), but not that of SSH1L(2SA) (Fig. 3 D). Thus, 14-3-3γ inhibits the phosphatase activity of SSH1L by binding to the sequences surrounding pS937/978. Next, we asked if 14-3-3γ could protect SSH1L from F-actin–induced activation. SSH1L in MCF-7 cells was precipitated by GST-14-3-3γ and subjected to in vitro phosphatase assay with or without F-actin. SSH1L bound to GST-14-3-3γ was not activated by F-actin, whereas control SSH1L, which was precipitated with an anti-SSH1L antibody, was activated (Fig. 3 E). Thus, 14-3-3 proteins have the potential to prevent F-actin–induced activation of SSH1L.
Next, we examined the effects of 14-3-3γ expression on the P-cofilin level in cells. Overexpression of 14-3-3γ increased ∼1.2-fold the cellular P-cofilin level (Fig. 3 F), which could be explained by 14-3-3γ inhibition of the cofilin-phosphatase activity of SSH1L in the cells. Expression of SSH1L(WT) or SSH1L(2SA) in COS cells resulted in a decrease in the cellular P-cofilin level. Coexpression of 14-3-3γ suppressed the cofilin dephosphorylation caused by SSH1L(WT) but not that by SSH1L(2SA) (Fig. 3 F), which strongly suggests that 14-3-3γ increased the cellular P-cofilin level by binding to and inhibiting SSH1L. Recently, it was reported that 14-3-3ζ interacts with P-cofilin and thereby protects P-cofilin from dephosphorylation (Gohla and Bokoch, 2002). However, we were unable to detect the specific interaction between P-cofilin (or cofilin) and 14-3-3ζ (or β or γ isoform) in our assay system (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200401136/DC1). LIMK1 was also reported to bind to 14-3-3ζ (Birkenfeld et al., 2003), but we detected no interaction between LIMK1 and 14-3-3β under the conditions in which SSH1L and TESK1 (another cofilin kinase reported to bind to 14-3-3β; Toshima et al., 2001b) tightly bound to 14-3-3β (Fig. S5).
SSH1L accumulates in lamellipodia after NRG stimulation (Fig. 1 D). To examine the role of 14-3-3 for SSH1L localization, MCF-7 cells were coexpressed with 14-3-3γ and CFP-SSH1L. Expression of 14-3-3γ significantly suppressed NRG-induced lamellipodium formation and SSH1L accumulation to the lamellipodium (compare Fig. 4 A with Fig. 1 D). When a 2SA mutant was expressed, it colocalized with F-actin before and after NRG stimulation and accumulated in lamellipodia after NRG stimulation (Fig. 4 B). Coexpression of 14-3-3γ had no apparent effect on SSH1L(2SA) accumulation in lamellipodia (Fig. 4 C). These results suggest that 14-3-3γ suppresses SSH1L translocation to the lamellipodium, in a manner dependent on Ser-937/978 phosphorylation. We also examined the effects of 14-3-3γ on the level and localization of cofilin/P-cofilin in MCF-7 cells (Fig. 4 D). Expression of 14-3-3γ suppressed both cofilin accumulation in the lamellipodium and reduction in the P-cofilin level after NRG stimulation. Thus, 14-3-3 seems to inhibit SSH1L translocation to the lamellipodium and thereby suppress cofilin dephosphorylation in this region.
Figure 4.

14-3-3γ inhibits NRG-induced accumulation of SSH1L and cofilin in lamellipodia. (A–C) 14-3-3γ inhibits NRG-induced accumulation of SSH1L, but not its 2SA mutant, in lamellipodia. MCF-7 cells expressing CFP-SSH1L(WT) and HA-14-3-3γ (A), CFP-SSH1L(2SA) alone (B), or CFP-SSH1L(2SA) and HA-14-3-3γ (C), were stimulated with NRG for 15 min and stained with rhodamine phalloidin for F-actin. (D) 14-3-3γ inhibits NRG-induced cofilin dephosphorylation and accumulation in lamellipodia. MCF-7 cells transfected with HA-14-3-3γ were stimulated with NRG for 15 min and stained with anticofilin or anti–P-cofilin antibody. Arrowheads and arrows indicate 14-3-3γ–expressing and nonexpressing cells, respectively. Bar, 20 μm.
To quantitate the distribution of SSH1L before and after NRG stimulation, cell lysates were fractionated into detergent-soluble and -insoluble (cytoskeletal) fractions. The ratio of SSH1L and actin in the insoluble fraction increased after NRG stimulation (Fig. 5 A), which indicates that NRG induces actin assembly and translocation of SSH1L from cytosol to actin cytoskeletal fraction, as seen in Fig. 1 D. Lat-A treatment drastically reduced the amounts of SSH1L and actin in the insoluble fraction. When Myc-SSH1L was expressed in cells, the ratio of Myc-SSH1L in the insoluble fraction (58%) was much higher than that of endogenous SSH1L (16%), but was significantly decreased by coexpression of 14-3-3γ (18%; Fig. 5 B), which suggests that excess amounts of Myc-SSH1L (over the content of endogenous 14-3-3 proteins) localize onto the cytoskeletal fraction and 14-3-3γ can sequester overexpressed Myc-SSH1L in the soluble fraction. Myc-SSH1L in the insoluble fraction slightly increased after NRG stimulation, but coexpression of 14-3-3γ suppressed NRG-induced translocation of SSH1L into the insoluble fraction (Fig. 5 B). In addition, the ratio of SSH1L(2SA) in the insoluble fraction was extremely high (82%) and was not decreased by coexpression of 14-3-3γ (Fig. 5 B). Thus, 14-3-3 proteins appear to regulate subcellular distribution of SSH1L by associating with it through pS937/978 and thereby protecting it from translocation to the actin cytoskeleton. To determine if NRG induces dephosphorylation of SSH1L at Ser-978, we prepared an anti-pS978 antibody specific to Ser-978–phosphorylated SSH1L (Fig. S6, available at http://www.jcb.org/cgi/content/full/jcb.200401136/DC1). The level of Ser-978 phosphorylation significantly decreased after NRG stimulation (Fig. 5 C). Accordingly, NRG-induced translocation of SSH1L appears to be regulated by dephosphorylation of Ser-978.
Figure 5.

NRG induces translocation of SSH1L into the Triton X-100–insoluble cytoskeletal fraction and dephosphorylation at Ser-978. (A) NRG induces translocation of SSH1L to the Triton X-100–insoluble fraction. MCF-7 cells with or without pretreatment of Lat-A were exposed to NRG for 10 min and cell lysates were fractionated into Triton X-100–soluble (S) and –insoluble (I) fractions. Each fraction was run on SDS-PAGE and blotted using anti-SSH1L and anti–β-actin antibodies. Bottom panels indicate the ratio of the amounts of SSH1L and β-actin in the insoluble fraction, as means ± SD of triplicate experiments. (B) 14-3-3γ suppresses basal and NRG-induced translocation of SSH1L to the insoluble fraction. MCF-7 cells transfected with Myc-SSH1L(WT) or Myc-SSH1L(2SA) alone or cotransfected with 14-3-3γ were stimulated with NRG and cell lysates were fractionated and analyzed as in A. (C) NRG induces Ser-978 dephosphorylation of SSH1L. Lysates of MCF-7 cells before and after NRG stimulation were immunoprecipitated with anti-SSH1L antibody and blotted with anti-SSH1L and anti-pS978 antibodies. (D) A proposed model of the NRG-induced SSH1L activation and cofilin dephosphorylation.
Based on our observations, we propose a likely scenario for NRG-induced SSH1L activation and cofilin dephosphorylation as follows (Fig. 5 D): (a) NRG stimulates F-actin assembly to form a lamellipodial extension (through Rac activation); (b) NRG concomitantly induces SSH1L dephosphorylation at Ser-978 and/or Ser-937 and its dissociation from 14-3-3 proteins in the cytoplasm; (c) SSH1L released from 14-3-3 is translocated to the F-actin–rich lamellipodium and is activated by F-actin in this region; and (d) activated SSH1L induces cofilin dephosphorylation and thereby local activation of cofilin in the lamellipodium. Because the localization and activity of SSH1L are regulated by 14-3-3 binding, identification of protein kinase(s) and phosphatase(s) that control the SSH1L–14-3-3 interaction will be important. Furthermore, it must be answered how SSH1L is dephosphorylated when it is bound to 14-3-3.
During cell migration, the cell is polarized to form an F-actin–rich lamellipodial extension in the direction of cell movement. In our model, once the F-actin–rich structure is established in the front of migrating cells, SSH1L is recruited and activated in this region and supports local activation of cofilin in the lamellipodium. Given the essential role of cofilin in actin filament turnover, spatially restricted activation of SSH1L by F-actin will be an important mechanism for maintaining and extending lamellipodia in the front of the cell and sustaining polarized cell migration.
In addition, LIMK1 was also activated by NRG stimulation (Fig. S7, available at http://www.jcb.org/cgi/content/full/jcb.200401136/DC1), as reported previously (Vadlamudi et al., 2002). Previous studies showed that LIMK1 is required for lamellipodium formation and cell migration (Yang et al., 1998; Nishita et al., 2002). LIMK1 may play a role in the establishment of lamellipodia by transiently inactivating cofilin and inducing actin polymerization. LIMK1 may also contribute to stimulate actin turnover in lamellipodia by releasing free actin and cofilin from an actin–cofilin complex, which is produced from the pointed end of actin filaments by the action of cofilin (Rosenblatt and Mitchison, 1998). Because the released P-cofilin can be reused after dephosphorylation by SSH1L, coordinated activation of LIMK1 and SSH1L can accelerate the recycling of cofilin and thereby promote actin turnover. In contrast to the accumulation of SSH1L in lamellipodia after NRG stimulation, LIMK1 localizes diffusely in the cytoplasm. Therefore, the spatially distinct distribution of LIMK1 and SSH1L can generate the polarized pattern of cofilin activation and may contribute to polarized cell migration.
Materials and methods
Plasmids and antibodies
Plasmids for human SSH1L, 14-3-3 and their mutants were constructed as described previously (Toshima et al., 2001b; Niwa et al., 2002; Kaji et al., 2003; Ohta et al., 2003). Plasmids for 14-3-3γ and ζ were provided by T. Ichimura and T. Isobe (Tokyo Metropolitan University, Tokyo, Japan). Antibodies to P-cofilin, cofilin, and SSH1L were prepared as described previously (Toshima et al., 2001a; Kaji et al., 2003). An anti-pS978 antibody was raised against a synthetic phosphopeptide, LKRSH(pS)LAKLG. Antibodies against Myc (9E10; Roche), HA (3F10; Roche), Rac (23A8; Upstate Biotechnology), 14-3-3 (H-8; Santa Cruz Biotechnology, Inc.), and β-actin (AC-15; Sigma-Aldrich) were purchased commercially.
Cell culture and staining
MCF-7, COS-7, and HEK293T cells were maintained in DME supplemented with 10% FCS. Cells were transfected with expression plasmids using FuGENE6 (Roche). Serum-starved MCF-7 cells were treated with 50 ng/ml NRG (Genzyme) or 0.5 μM Lat-A (Molecular Probes). For staining, cells were fixed with 4% formaldehyde and incubated with 100% methanol. After blocking with 5 mg/ml BSA, cells were stained with anticofilin or anti–P-cofilin antibody. Rhodamine phalloidin was used to stain F-actin. Fluorescent images were obtained using a confocal microscope (model LSM510; Carl Zeiss MicroImaging, Inc.).
In vitro phosphatase assay
Cofilin-(His)6 expressed in Vero cells and purified with Ni-NTA agarose was used as a substrate. SSH1L was immunoprecipitated and incubated for 1 h at 30°C with 100 ng cofilin-(His)6 in 20 μl of lysis buffer containing 0.01% BSA (Niwa et al., 2002). Reaction mixtures were run on SDS-PAGE and P-cofilin was analyzed by immunoblotting with anti–P-cofilin antibody or staining with Pro-Q Diamond phosphoprotein gel stain kit (Molecular Probes), and cofilin was analyzed by Coomassie brilliant blue staining. To examine the effects of F-actin, purified rabbit muscle actin was polymerized in F-buffer (Niwa et al., 2002). G-actin was prepared in G-buffer (2 mM Tris-Cl, pH 8.0, 1 mM DTT, 0.2 mM ATP, and 0.2 mM CaCl2). SSH1L was incubated with 5 μg F- or G-actin and 100 ng cofilin-(His)6 in 20 μl of lysis buffer. To examine the effect of 14-3-3γ, GST-14-3-3γ expressed in Escherichia coli was used after thrombin cleavage.
In vitro kinase assay
MCF-7 cells were stimulated with NRG and LIMK1 was immunoprecipitated with anti-LIMK1 antibody and subjected to in vitro kinase reaction as described previously (Nishita et al., 2002).
Purification of SSH1L-binding proteins
Three 100-mm plates of COS-7 cells were transfected with (Myc+His)-SSH1L. Lysates were precleared with Sepharose-4B and then precipitated with Ni-NTA agarose. The pellets were washed, run on SDS-PAGE, and stained by silver. To identify SSH1L-binding proteins, the proteins resolved on SDS-PAGE were transferred onto a PVDF membrane and stained with Colloidal gold (Bio-Rad Laboratories). The protein bands were cut out, digested with lysyl endopeptidase, and analyzed by mass spectrometry.
In vitro pull-down assay
In vitro pull-down assay was done as described previously (Toshima et al., 2001b). Lysates of COS-7 cells expressing Myc-SSH1L or its mutants were incubated with GST or GST-14-3-3β and glutathione Sepharose. After centrifugation, the pellets were subjected to SDS-PAGE and analyzed by immunoblotting with an anti-Myc antibody.
Subcellular fractionation
MCF-7 cells treated with or without NRG were extracted for 3 min on ice with 1% Triton X-100 in PEM buffer (100 mM Pipes, pH 6.8, 1 mM EGTA, and 1 mM MgCl2) containing 2 μM phalloidin, 10 mM NaF, 1 mM Na3VO4, and 10 μg/ml leupeptin (Svitkina and Borisy, 1999). The soluble fraction was removed, then the residual insoluble fraction was washed once with PEM buffer and scraped with SDS-sample buffer. Equal amounts of soluble and insoluble fractions were subjected to SDS-PAGE and analyzed by immunoblotting.
Online supplemental material
Video 1 shows the time-lapse phase-contrast microscopy of MCF-7 cell shape changes for 38 min after NRG stimulation. Fig. S1 shows kinetics of actin filament reorganization after NRG stimulation. Fig. S2 shows that Lat-A inhibits NRG-induced lamellipod formation and SSH1L accumulation. Fig. S3 shows the interaction of SSH1L–14-3-3β in the yeast two-hybrid system. Fig. S4 shows the inhibition of the SSH1L–14-3-3β interaction by phosphatase treatment. Fig. S5 shows that 14-3-3 proteins interact with SSH1L and TESK1, but not with cofilin, P-cofilin, or LIMK1. Fig. S6 shows the specificity of an anti-pS978 antibody. Fig. S7 shows NRG-induced activation of LIMK1. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200401136/DC1.
Acknowledgments
We thank T. Ichimura and T. Isobe for plasmids for 14-3-3.
This work was supported by a grant for creative scientific research from the Japan Society of the Promotion of Science to K. Mizuno and a grant from Core Research for Evolution Science and Technology, Japan Science and Technology Corporation to T. Uemura.
The online version of this article contains supplemental material.
Abbreviations used in this paper: Lat-A, latrunculin-A; LIMK, LIM-kinase; NRG, neuregulin-1β; P-cofilin, Ser-3–phosphorylated cofilin; pS, phospho-Ser; SSH1L, Slingshot-1L.
References
- Arber, S., F.A. Barbayannis, H. Hanser, C. Schneider, C.A. Stanyon, O. Bernard, and P. Caroni. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 393:805–809. [DOI] [PubMed] [Google Scholar]
- Birkenfeld, J., H. Betz, and D. Roth. 2003. Identification of cofilin and LIM-domain-containing protein kinase 1 as novel interaction partners of 14-3-3ζ. Biochem. J. 369:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., B.W. Bernstein, and J.R. Bamburg. 2000. Regulating actin-filament dynamics in vivo. Trends Biochem. Sci. 25:19–23. [DOI] [PubMed] [Google Scholar]
- Dawe, H.R., L.S. Minamide, J.R. Bamburg, and L.P. Cramer. 2003. ADF/Cofilin controls cell polarity during fibroblast migration. Curr. Biol. 13:252–257. [DOI] [PubMed] [Google Scholar]
- Gohla, A., and G.M. Bokoch. 2002. 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Curr. Biol. 12:1704–1710. [DOI] [PubMed] [Google Scholar]
- Kaji, N., K. Ohashi, M. Shuin, R. Niwa, T. Uemura, and K. Mizuno. 2003. Cell cycle-associated changes in slingshot phosphatase activity and roles in cytokinesis in animal cells. J. Biol. Chem. 278:33450–33455. [DOI] [PubMed] [Google Scholar]
- Moon, A., and D.G. Drubin. 1995. The ADF/cofilin proteins: stimulus-responsive modulators of actin dynamics. Mol. Biol. Cell. 6:1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishita, M., H. Aizawa, and K. Mizuno. 2002. Stromal cell-derived factor 1α activates LIM kinase 1 and induces cofilin phosphorylation for T-cell chemotaxis. Mol. Cell. Biol. 22:774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa, R., K. Nagata-Ohashi, M. Takeichi, K. Mizuno, and T. Uemura. 2002. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 108:233–246. [DOI] [PubMed] [Google Scholar]
- Ohta, Y., K. Kousaka, K. Nagata-Ohashi, K. Ohashi, A. Muramoto, Y. Shima, R. Niwa, T. Uemura, and K. Mizuno. 2003. Differential activities, subcellular distribution and tissue expression patterns of three members of Slingshot family phosphatases that dephosphorylate cofilin. Genes Cells. 8:811–824. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., and G.G. Borisy. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 112:453–465. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., M.A. Schwartz, K. Burridge, R.A. Firtel, M.H. Ginsberg, G. Borisy, J.T. Parsons, and A.R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709. [DOI] [PubMed] [Google Scholar]
- Rosenblatt, J., and T.J. Mitchison. 1998. Actin, cofilin and cognition. Nature. 393:739–740. [DOI] [PubMed] [Google Scholar]
- Spencer, K.S.R., D. Grus-Porta, J. Leng, N.E. Hynes, and R.L. Klemke. 2000. ErbB2 is necessary for induction of carcinoma cell invasion by ErbB family receptor tyrosine kinases. J. Cell Biol. 148:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima, J., J.Y. Toshima, T. Amano, N. Yang, S. Narumiya, and K. Mizuno. 2001. a. Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol. Biol. Cell. 12:1131–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima, J.Y., J. Toshima, T. Watanabe, and K. Mizuno. 2001. b. Binding of 14-3-3β regulates the kinase activity and subcellular localization of testicular protein kinase 1. J. Biol. Chem. 276:43471–43481. [DOI] [PubMed] [Google Scholar]
- Tzivion, G., and J. Avruch. 2002. 14-3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. J. Biol. Chem. 277:3061–3064. [DOI] [PubMed] [Google Scholar]
- Vadlamudi, R.K., F. Li, L. Adam, D. Nguyen, Y. Ohta, T.P. Stossel, and R. Kumar. 2002. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat. Cell Biol. 4:681–690. [DOI] [PubMed] [Google Scholar]
- Yang, N., O. Higuchi, K. Ohashi, K. Nagata, A. Wada, K. Kangawa, E. Nishida, and K. Mizuno. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 393:809–812. [DOI] [PubMed] [Google Scholar]
- Zebda, N., O. Bernard, M. Bailly, S. Welti, D.S. Lawrence, and J. Condeelis. 2000. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J. Cell Biol. 151:1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]