

Abstract
Fat cadherins form a distinct subfamily of the cadherin gene superfamily, and are featured by their unusually large extracellular domain. In this work, we investigated the function of a mammalian Fat cadherin. Fat1 was localized at filopodial tips, lamellipodial edges, and cell–cell boundaries, overlapping with dynamic actin structures. RNA interference–mediated knockdown of Fat1 resulted in disorganization of cell junction–associated F-actin and other actin fibers/cables, disturbance of cell–cell contacts, and also inhibition of cell polarity formation at wound margins. Furthermore, we identified Ena/vasodilator-stimulated phosphoproteins as a potential downstream effector of Fat1. These results suggest that Fat1 regulates actin cytoskeletal organization at cell peripheries, thereby modulating cell contacts and polarity.
Keywords: Fat; cadherin; actin cytoskeleton; Ena/VASP; cell–cell interaction
Introduction
Cadherins are one of the major classes of cell–cell adhesion molecules, and they regulate many aspects of animal morphogenesis, including the formation of epithelial and synaptic junctions, morphogenetic cell movement, and cell growth (Gumbiner, 2000; Tepass et al., 2000; Hirano et al., 2003). The cadherins form a superfamily that can be categorized into several subfamilies, including classic cadherins, desmosomal cadherins, protocadherins, Flamingo/Celsr, and Fat. Classic and desmosomal cadherins are well characterized in their function as adhesion molecules, but the biological functions of other cadherins seem to have diverged. Classic cadherins have five repeated domains in the extracellular region (called extracellular cadherin [EC] domains), and a highly conserved cytoplasmic region, to which the catenin system binds. It links the classic cadherins to the actin cytoskeleton. Desmosomal cadherins (desmocollin and desmoglein) also have five EC domains, but they are linked with the intermediate filament cytoskeleton via plakoglobin/desmoplakin (Garrod et al., 2002). On the other hand, protocadherins, Flamingo/Celsr, and Fat cadherins show a large variety in their primary structures. They have diverse numbers of the EC domain, from 5 to 34. Furthermore, their cytoplasmic sequences are quite divergent. Thus, these cadherins might play roles distinct from those of the classic and desmosomal cadherins (Suzuki, 2000; Frank and Kemler, 2002; Hirano et al., 2003). In fact, Flamingo/Celsr was found to be important for both planar polarity regulation and neurite patterning (Chae et al., 1999; Usui et al., 1999; Gao et al., 2000; Curtin et al., 2003; Lee et al., 2003; Senti et al., 2003). Members of the protocadherin subfamily have likewise been implicated as playing roles in the establishment of synaptic connections (Kohmura et al., 1998), neuronal survival (Wang et al., 2002), cochlear function (Bolz et al., 2001; Bork et al., 2001; Di Palma et al., 2001), and other morphogenetic processes (Kim et al., 1998, 2000; Yamamoto et al., 1998; Rhee et al., 2003). However, the biological functions of many other members of the cadherin superfamily remain to be elucidated.
Vertebrate Fat cadherin is one such molecule. Fat is the largest among the members of the cadherin superfamily, commonly having 34 EC domains. The primary sequences of the cytoplasmic regions of Fat subtypes have almost no similarity to those of the classic cadherins. In Drosophila, fat was shown to be a gene responsible for hyperplastic overgrowth of imaginal discs (Bryant et al., 1988; Mahoney et al., 1991), and recent analyses revealed that Drosophila Fat regulates planar polarity patterning (Rawls et al., 2002; Strutt and Strutt, 2002; Yang et al., 2002; Fanto et al., 2003; Ma et al., 2003). In mammals, three subtypes of Fat, namely Fat1, 2, and 3, have been reported (Dunne et al., 1995; Ponassi et al., 1999; Cox et al., 2000; Inoue et al., 2001; Mitsui et al., 2002; Nakayama et al., 2002). We can predict that mammalian Fat might also play roles in cell proliferation or planar cell polarity. However, the cytoplasmic region is not highly conserved between the Drosophila and mammalian Fats, although their extracellular regions are similar. Thus, we cannot rule out the possibility that mammalian Fat might have acquired distinct roles from its Drosophila counterpart. Fat is expressed in various tissues at embryonic stages, especially in proliferating epithelial tissues such as the neural tube, lung epithelium, and proliferating layers in the skin (Dunne et al., 1995; Ponassi et al., 1999; Cox et al., 2000; Inoue et al., 2001; Mitsui et al., 2002; Nakayama et al., 2002).
In this report, we describe for the first time analyses at both molecular and cellular levels on the properties of mammalian Fat1. Our detailed examinations revealed Fat1 to be localized at filopodia, lamellipodia, and cell–cell contact sites. By performing RNA interference (RNAi) in PAM212 cells, we found that Fat1 was required for tight cell–cell association and proper actin organization. Furthermore, we found that in a wound-healing assay, Fat1 was required to regulate cell polarity at the wound margins. As for its molecular action, we identified Ena/vasodilator-stimulated phosphoproteins (VASPs) as a possible downstream effector of Fat1. Fat1 bound to these proteins via an Ena/VASP homology 1 (EVH1) domain–mediated interaction. We suggest that Fat1 regulates cell–cell adhesion and other cell behavior by controlling actin polymerization through the Ena/VASP system, at least in part.
Results
Localization of Fat1 at cell–cell boundaries
To understand the properties of Fat cadherin at the cellular level, we first examined the subcellular localization of endogenous Fat1 in various cell lines by using antibodies specific for Fat1 (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200403006/DC1). In DLD1 cells, a colon carcinoma line, Fat1 was detected at cell–cell junctions (Fig. 1 A). However, its distribution pattern was not identical to that of molecules of the classic cadherin system. β-Catenin, a classic cadherin-associated protein, was sharply concentrated at the apical portion of lateral cell contacts, where the adherens junction is located (Fig. 1 A). In contrast, Fat1 was not particularly strongly concentrated in the adherens junction region; rather, its staining was more intense in the lower portion of the cell contacts (Fig. 1 A). MDCK cells, a kidney epithelial line, showed a similar junctional staining for Fat1 (Fig. 1 B, low). However, in these cells Fat1 became barely detectable in the cell junctions of highly packed colonies (Fig. 1 B, high), indicating that junctional Fat1 does not persist in mature cell contacts in this cell line. Western blotting analysis showed that expression levels of Fat1 protein were reduced as the cell density increased in prolonged cultures (Fig. S1 F). In PAM212 cells, a transformed keratinocyte line, Fat1 was localized not only at cell junctions, but also at the free edges of cells (Fig. 1 C). As was the case for other cell lines, the staining of Fat1 at the cell junctions was not always identical to that of β-catenin (Fig. 1 C). Among these three cell lines, PAM212 cells expressed the highest level of Fat1 protein, and MDCK cells expressed the lowest level, as revealed by Western blotting analysis (Fig. S1 B).
Figure 1.
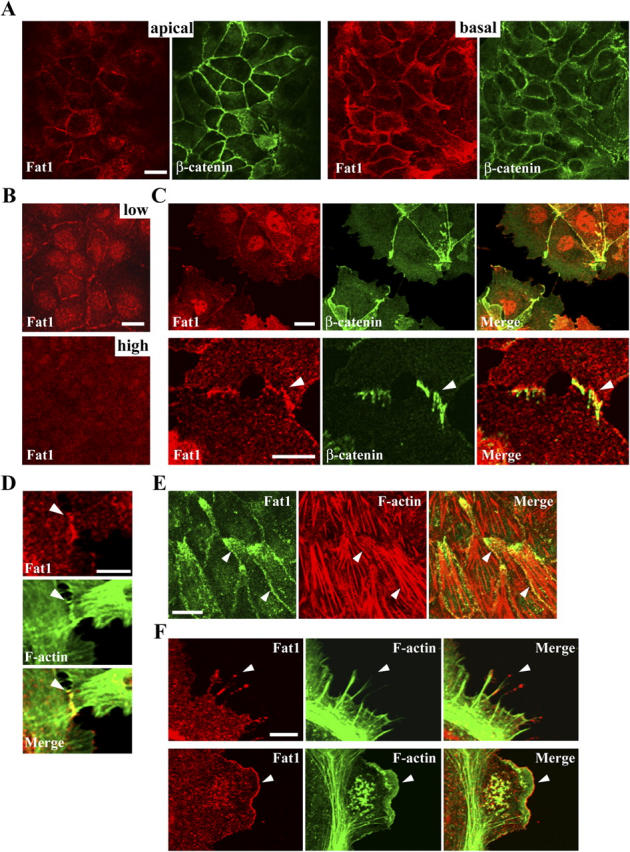
Subcellular localization of endogenous Fat1. (A) DLD1 cells doubly immunostained for Fat1 (red) and β-catenin (green). An apical and a basal confocal section are shown. Fat1 is localized at cell junctions, but is more abundant in the basal portion of the junctions. (B) MDCK cells immunostained for Fat1 at a low and high density. The junctional Fat1 tends to disappear at high cell densities. (C) PAM212 cells doubly immunostained for Fat1 (red) and β-catenin (green), showing localization at cell junctions (top panels). Bottom panels, close-up view of early cell–cell contacts. Fat1 is detected at cell contact sites, but its localization pattern is not identical to that of β-catenin (arrowhead). (D) Early contacts between PAM212 cells doubly immunostained for Fat1 (red) and F-actin (green). Fat1 colocalizes with junction-associated F-actin (arrowhead). (E) A confocal section of PAM212 cells at the basal level of the cell layer, doubly immunostained for Fat1 (green) and F-actin (red). Fat1 tends to accumulate at a protruded portion of cells as well as at cell–cell contacts (arrowhead), overlapping with F-actin. (F) Close-up views of the free surface of PAM212 cells doubly immunostained for Fat1 (red) and F-actin (green). Fat1 is localized to filopodia (top, arrowhead) and lamellipodia (bottom, arrowhead), overlapping with F-actin. Bars: 15 μm (A, B, and E), 10 μm (C, top), 5 μm (C, bottom; D and F).
In PAM212 cells, the Fat1 signals overlapped with those of cortical actin fibers. For example, Fat1 was colocalized with actin fibers underlying early cell–cell contacts (Fig. 1 D). At the basal level of confluent cultures, Fat1 often accumulated at a protruded portion of the cells, often overlapping with actin stress fibers (Fig. 1 E). Fat1 staining was also observed at the free edges of cells, including lamellipodia and filopodia (Fig. 1 F). Double staining for Fat1 and F-actin showed that their distributions overlapped in these structures, particularly at lamellipodia (see Fig. 4 E). Such colocalization of Fat1 and actin was observed in any other cell lines examined.
Figure 4.

Fat1 colocalized with VASP. (A) PAM212 cells were doubly immunostained for Fat1 (red) and VASP (green). Fat1 colocalized with VASP at lamellipodia. (B) A filopodium of PAM212 doubly immunostained for Fat1 (red) and VASP (green). (C) Fat1 and VASP colocalized at early cell–cell contact sites. (D) Effects of cytochalasin D treatment. PAM212 cells were treated with 150 nM cytochalasin D for 30 min, and were then doubly immunostained for Fat1 (red) and VASP or WAVE (green). The localization of Fat1 or WAVE was not affected, although that of VASP was perturbed. Bars: 15 μm (A), 2.5 μm (B and C), 10 μm (D).
Cell–cell associations are controlled by classic cadherins. To test their effects on Fat1 regulation, we treated PAM212 cells with blocking antibodies specific for E- and P-cadherins (Yoshida-Noro et al., 1984; Nose and Takeichi, 1986), the major classic cadherins expressed in this cell line. After a short period of incubation (30 min), β-catenin localization became diffuse, most extensively at the peripheral portions of cell colonies. Coincidently, the junctional concentration of Fat1 also became diffuse or was lost entirely (Fig. 2). Thus, we suggest that Fat1 requires the classic cadherin system to become localized at cell–cell junctions.
Figure 2.
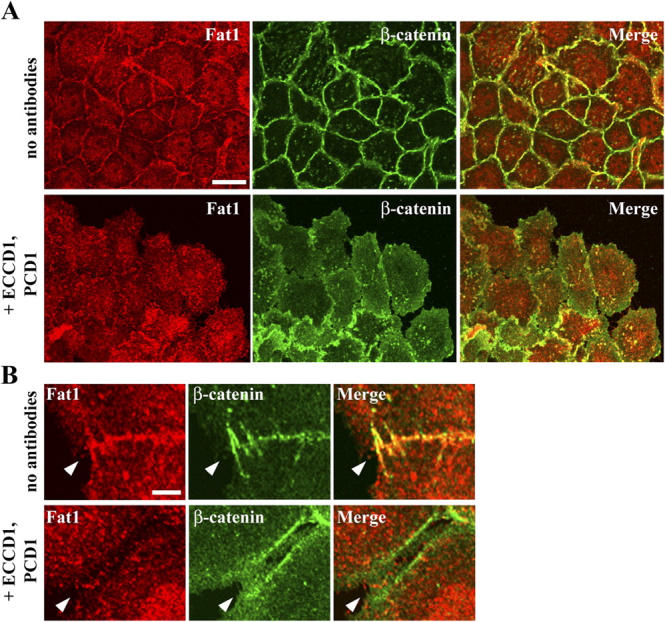
Junctional localization of Fat1 requires classic cadherins. (A) PAM212 cells were treated with blocking antibodies for E- and P-cadherin for 30 min. The cells were then doubly stained for Fat1 (red) and β-catenin (green). Ascites fluids of ECCD1 and PCD1 were used at a 250-fold dilution. Fat1 disappears from cell junctions, coinciding with the loss of junctional β-catenin. (B) Images at higher magnification. Arrowheads point to cell contact sites. Bars: 15 μm (A), 5 μm (B).
Fat1 binds to Ena/VASP proteins through an EVH1 domain–mediated interaction
We noticed that the Fat1 cytoplasmic tail has three consensus sequences to the binding site for the EVH1 domain of Ena/VASP proteins (residues 4310–4314, 4437–4441, and 4450–4454 of human Fat1; see also Fig. 3 A). Ena/VASP proteins are known to regulate the motility of the actin cytoskeleton by antagonizing capping proteins and/or by increasing the rate of dissociation of the branched junction (Bear et al., 2002; Krause et al., 2002; Renfranz and Beckerle, 2002; Samarin et al., 2003). In mammals, there are three known members of the Ena/VASP family, namely mammalian Enabled (Mena), VASP, and Ena/VASP-like (Reinhard et al., 1992; Gertler et al., 1996). They are thought to play redundant roles for regulating actin dynamics. The core amino acid sequence for binding to the EVH1 domain of Ena/VASP is F/L/Y/WPPPP (Niebuhr et al., 1997; Ball et al., 2000). Fat1 has three core sequences for the EVH1 domain, which are conserved across species from avian to mammalian, but not in Drosophila (Fig. 3 A; unpublished data). Among the three consensus sequences for the EVH1 binding, amino acid residues 4432–4443 and 4448–4456 of Fat1 are preferred candidate sequences for the high affinity binding to the EVH1 domain (Niebuhr et al., 1997; Carl et al., 1999; Ball et al., 2000). Furthermore, Ena/VASP proteins have been reported to localize at filopodia and lamellipodia (Reinhard et al., 1992; Gertler et al., 1996; Rottner et al., 1999). The above findings prompted us to test the possibility that Fat1 binds to Ena/VASP proteins.
Figure 3.
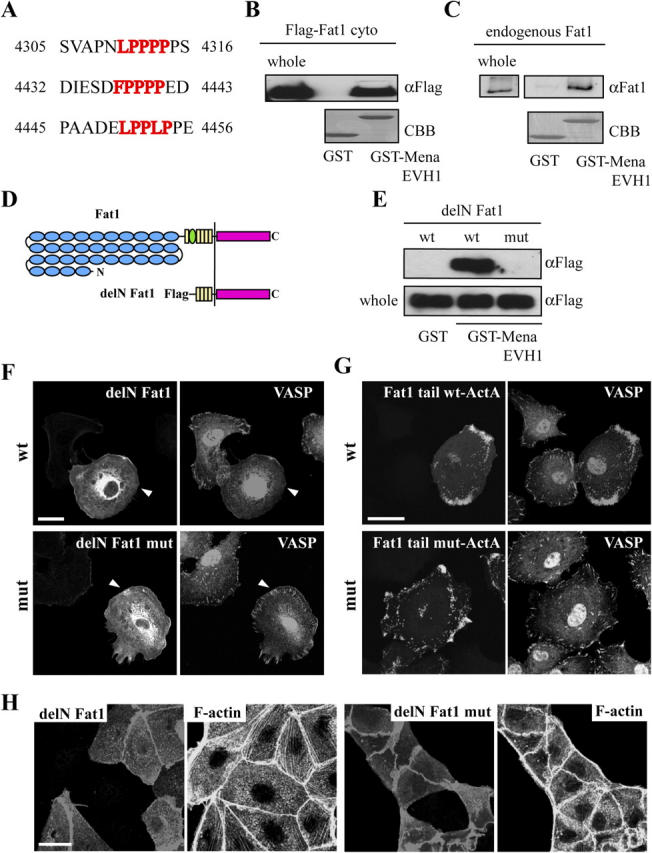
Fat1 binds to Ena/VASP protein and controls stress fiber formation. (A) Human Fat1 has three putative EVH1-binding sites in its cytoplasmic region. The minimum consensus sequence is F/L/Y/WPPPP. (B) Coprecipitation of the cytoplasmic region of Fat1 with a GST-Mena EVH1 domain. A GST fusion of the EVH1 domain of Mena was expressed in bacteria and purified by the standard method. An equal amount (10 μg) of GST or GST-Mena EVH1 domain was incubated with a lysate of MDCK cells expressing the Flag-tagged Fat1 cytoplasmic region. Coprecipitated Flag–Fat1 cytoplasmic region was detected with anti-Flag antibody (αFlag). Coomassie brilliant blue (CBB)–stained GST proteins are shown. Whole, whole-cell lysate. (C) Coprecipitation of endogenous Fat1 protein with a GST-Mena EVH1 domain. An equal amount (20 μg) of GST or GST-Mena EVH1 domain was incubated with a lysate of PAM212 cells collected from a confluent culture in a 100-mm dish. Coprecipitated Fat1 protein was detected with the rabbit anti-Fat1 antibody. CBB-stained GST proteins are shown. An entire image of the same blots is also shown in Fig. S2 A (available at http://www.jcb.org/cgi/content/full/jcb.200403006/DC1). (D) Schematic representation of Fat1. Blue circles, yellow boxes, and the green circle indicate the EC domain, EGF motif, and laminin A-G domain, respectively. A mutant form of Fat1, delN Fat1, is also shown. delN Fat1 contains four EGF repeats and the transmembrane and cytoplasmic regions, corresponding to residues 3987–4590 of human Fat1. (E) delN Fat1 wt or delN Fat1 mut, in which the binding sites for Ena/VASP proteins had been mutated, was examined for binding to the EVH1 domain of Mena. An equal amount (5 μg) of GST or GST-Mena EVH1 domain was incubated with a lysate of MDCK cells transfected with pFlag-CMV1-delN Fat1 wt or mut. The cells were collected from a 60-mm dish. Coprecipitated Fat1 proteins were detected using an anti-Flag antibody (top). A comparable amount of delN Fat1 proteins was present in the original whole-cell lysates (bottom). (F) NRK-52E cells were transfected with pFlag-CMV1-delN Fat1 wt or mut. The cells were doubly immunostained with anti-Flag antibody for detecting delN Fat1 and anti-VASP antibody. Note that VASP disappeared from focal adhesions in the cells overexpressing delN Fat1 (arrowheads in top panels), but not in those overexpressing delN Fat1 mut (arrowheads in bottom panels). (G) NRK-52E cells were transfected with pEGFP-Fat1 tail wt or mut-ActA. The cells were doubly immunostained with anti-EGFP antibody for detecting the Fat1 tail and anti-VASP antibody. Note that VASP was recruited to the mitochondria where EGFP-Fat1 tail wt-ActA concentrated, but not to the mitochondria where the mutant form of the Fat1 tail concentrated. (H) MDCK cells stably expressing Flag-tagged delN Fat1 or delN Fat1 mut were mixed with parent MDCK cells in a 1:1 ratio, cultured for 2 d, and doubly stained for delN Fat1 proteins and F-actin. Note the enhancement of stress fiber formation in cells with delN Fat1, but not in those with delN Fat1 mut. Bars, 15 μm.
To examine the binding between Fat1 and Ena/VASP proteins, we created a GST fusion protein containing the EVH1 domain of Mena (GST-Mena EVH1) and performed a GST pull-down assay. The EVH1 domain is necessary and sufficient for the interaction with ActA, zyxin, or vinculin (Niebuhr et al., 1997). As shown in Fig. 3 B, a Flag-tagged cytoplasmic portion of Fat1 (Fat1 cyto, see Materials and methods) exogenously expressed in MDCK cells was efficiently coprecipitated with the GST-Mena EVH1 protein, but not with a control GST protein. Furthermore, endogenous Fat1 protein was also coprecipitated with the same fusion protein (Fig. 3 C; see also Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200403006/DC1). To further confirm the direct binding between Fat1 and Ena/VASP, we used an NH2-terminal truncated form of Fat1, delN Fat1. This molecule contains four EGF motifs and transmembrane and cytoplasmic regions, to the NH2 terminus of which the signal sequence of preprotrypsin and a Flag tag are attached (Fig. 3 D). As it was reported that the first amino acid residue of F/L/Y/WPPPP was essential for the binding between Ena/VASP proteins and their binding partner (Niebuhr et al., 1997; Ball et al., 2000), we created a mutant form of delN Fat1 (delN Fat1 mut) in which phenylalanine and leucines of the putative Ena/VASP-binding sites (Leu 4310, Phe 4437, and Leu 4450 of human Fat1) were replaced by alanines. As shown in Fig. 3 E, the efficiency of coprecipitation with GST-Mena EVH1 was greatly reduced for delN Fat1 mut, as compared with that for wild-type delN Fat1.
To test the interaction between Fat1 and Ena/VASP at the cellular level, we overexpressed delN Fat1 in NRK-52E cells showing a clear focal adhesion distribution. In these cells, endogenous VASP disappeared from focal adhesions, where this protein normally localizes (Fig. 3 F, top panels), and was occasionally recruited to the sites where delN Fat1 was concentrated (unpublished data). Under these conditions, focal adhesions, as visualized by vinculin staining, still existed, suggesting that the effect of delN Fat1 was specific to Ena/VASP proteins (Fig. S2 B). However, the delN Fat1 mut, in which the Ena/VASP-binding sites had been mutated, did not exhibit such activity (Fig. 3 F, bottom panels). The expression levels of both delN Fat1 proteins and endogenous VASP in delN Fat1–transfected and delN Fat1 mut–transfected cells were comparable (unpublished data). Of the total number of transfectants, the percent showing the disappearance of VASP from focal adhesions was 88.8 ± 8.0% (mean ± SEM, from 10 independent experiments) for delN Fat1–expressing cells, and 8.0 ± 4.0% (from 12 independent experiments) for delN Fat1 mut–expressing cells. Furthermore, when the Fat1 cytoplasmic tail attached to the mitochondrial targeting sequence of Listeria protein ActA was expressed in NRK-52E cells, endogenous VASP was recruited to the mitochondria (Fig. 3 G, top panels). However, the mutant form of Fat1, in which the Ena/VASP-binding sites had been mutated, did not have such activity (Fig. 3 G, bottom panels). These results strongly support the idea that Fat1 binds to Ena/VASP proteins specifically through an EVH1 domain–mediated interaction. In addition, the Fat1 tail could not recruit N-WASP, WAVEs, vinculin, β-catenin, or ZO-1, indicating the specific interaction between Fat1 and Ena/VASP proteins (unpublished data). Notably, the Fat1 tail was not able to induce ectopic actin fiber formation at the mitochondria, although the NH2 terminus of zyxin, which also binds to Ena/VASP proteins, had such activity in the above assays (unpublished data).
Fat1 was colocalized with VASP at cell–cell contact and cell peripheries
Next, we used PAM212 cells to examine whether endogenous Fat1 was colocalized with Ena/VASP proteins. As shown in Fig. 4 (A–C), these two molecules did colocalize at lamellipodia, filopodial tips, or cell–cell contacts (see also Fig. 6 A). To investigate the relationship between the localization of Fat1 and Ena/VASP, we examined the behavior of these molecules in the presence of a low dose of cytochalasin D; for it had been reported that Ena/VASP disappears from lamellipodial edges by treatment with a low dose of cytochalasin D, whereas N-WASP remains (Bear et al., 2002). In the presence of 150 nM cytochalasin D, Mena and VASP all disappeared from the lamellipodial edges (Fig. 4 D, top panels; not depicted for Mena). However, the Fat1 distribution in these structures was not altered, and WAVE and N-WASP were also retained (Fig. 4 D, bottom panels; not depicted for N-WASP), suggesting that the localization of Fat1 might not be determined by Ena/VASP proteins.
Figure 6.
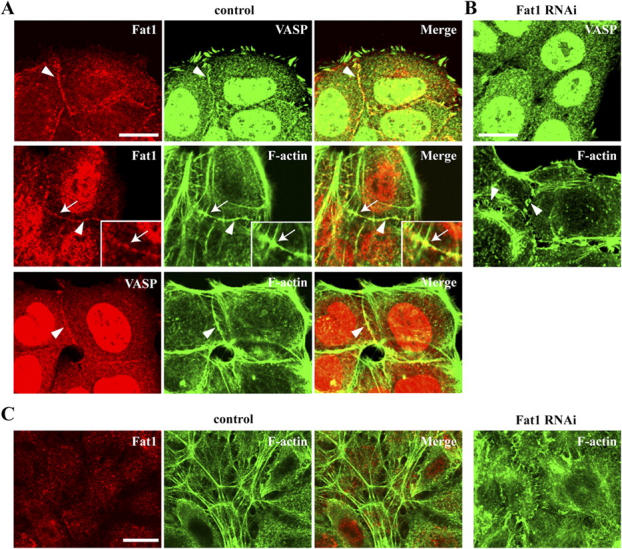
Fat1 regulates VASP localization and actin fiber formation in early cell–cell contacts. (A and B) PAM212 cells transfected with control or Fat1 RNAi plasmids were cultured for 72 h. The cells were then subjected to a Ca2+ switch assay, as described elsewhere (Fukuhara et al., 2002). 3 h after the medium had been changed from a low to high Ca2+, the cells were doubly immunostained for Fat1/VASP, Fat1/F-actin, or VASP/F-actin in control cells (A), or singly stained for VASP or F-actin in transfected cells (B). Insets, close-up views of part of the middle panels. Arrowheads point to representative cell–cell contact sites. The arrows indicate a site where a putative future radial actin cable meets with junction-associated F-actin. (C) 12 h after the medium change, cells were stained for F-actin. In control cells, radial actin cables are well formed at the apical plane of the cells. Fat1 is not colocalized with the radial actin cables. In Fat1 RNAi cells, such cable formation was suppressed. Bars: 10 μm (A and B), 15 μm (C).
Fat1 regulates actin dynamics through interaction with Ena/VASP proteins
To explore the intracellular molecular events regulated by Fat1, we examined the effects of Fat1 tail expression in MDCK cells, which had the lowest level of endogenous Fat1 among the cell types examined (Fig. S1 B). When either delN Fat1 or delN Fat1 mut was exogenously expressed in MDCK cells, these molecules became localized to cell junctions, showing a distribution similar to that of endogenous Fat1, and colocalizing with cortical F-actin bundles and VASP (Fig. 3 H, not depicted for VASP). We prepared multiple transfectant lines of MDCK cells constitutively expressing delN Fat1 or delN Fat1 mut (MDCK-delN Fat1 or MDCK-delN Fat1 mut cells, respectively). To compare the phenotypes of these transfected cells with those of parental cells with the highest degree of accuracy, we mixed each of the transfected cell lines with their parental cells. Notably, the formation of actin stress fibers was dramatically enhanced in the MDCK-delN Fat1 cells, but this enhancement did not occur in the MDCK-delN Fat1 mut cells (Fig. 3 H). The numbers of actin stress fibers per cell were 0.87 ± 0.22 (mean ± SEM; n = 138) for parental MDCK, 0.88 ± 0.32 (n = 144; P = 0.96) for the MDCK-delN Fat1 mut cells, and 11.7 ± 0.62 (n = 143; P < 0.001) for the MDCK-delN Fat1 cells, as determined in subconfluent 2-d cultures. These results suggest that the Fat1 cytoplasmic tail can regulate actin dynamics through interaction with Ena/VASP proteins if this protein is localized at junctional sites, although such activity was undetectable when Fat1 was accumulated at ectopic sites, such as mitochondria, in the cytoplasm, as mentioned in the previous section.
Fat1 is required for tight cell association and actin fiber organization
We used PAM212 cells, which express a high level of endogenous Fat1 (Fig. S1 B), to examine the effect of RNAi-mediated knockdown of Fat1 protein expression on cell behavior. After transfection with Fat1-RNAi plasmids, ∼95% of the cells in culture lost Fat1 protein, as judged by anti-Fat1 antibody staining. The remaining cells maintained high levels of Fat1 expression, allowing us to compare the properties of Fat1-positive and -negative cells side by side. Fat1-negative cells, in general, looked flatter than positive ones. Immunostaining for β-catenin in low density cultures showed that, in the absence of Fat1, cell–cell associations appeared to be significantly looser; i.e., the β-catenin signals along cell junctions became discontinuous and were interrupted by nonjunctional intercellular spaces (Fig. 5 A). Actin organization was also severely disrupted; junctional actin bundles were disorganized (compare arrow and arrowheads in Fig. 5 B), and the number of actin stress fibers was diminished in the Fat1 knockdown cells. These abnormal actin organizations were also observed in fully confluent cultures (Fig. 5 C).
Figure 5.
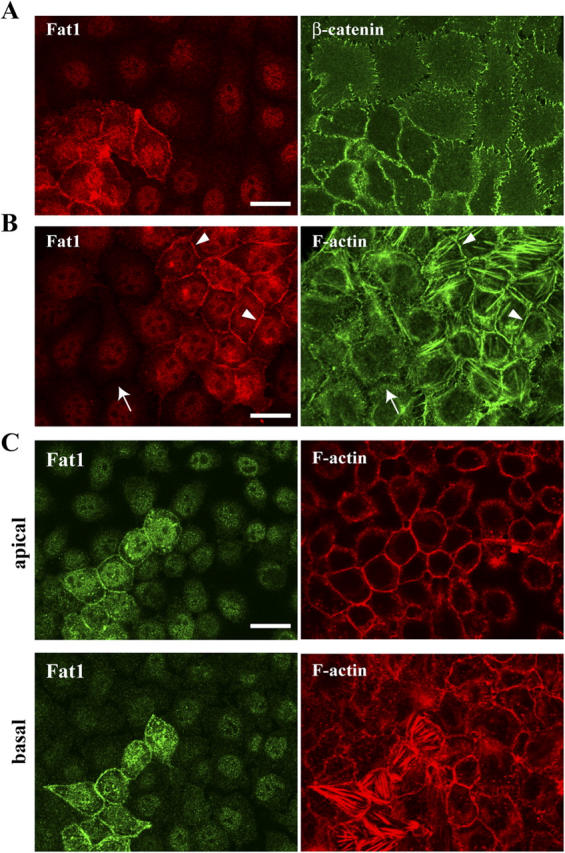
Fat1 is required for tight cell–cell association and actin organization. (A) Uncloned cells transfected with Fat1 RNAi plasmid were doubly immunostained for Fat1 (red) and β-catenin (green) 2 d after the transfection. Note the looser cell–cell associations for Fat1-negative cells compared with those for the positive cells. (B) A culture similar to that in A doubly immunostained for Fat1 (red) and F-actin (green). Arrowheads point to junctional accumulation of actin fibers. Arrow indicates the loss of F-actin–delineating cell junctions. (C) Uncloned cells transfected with Fat1 RNAi plasmid, plated in a high density, and cultured for 3 d. Apical and basal confocal sections, doubly stained for Fat1 (green) and F-actin (red), are shown. Bars, 15 μm.
To interrelate the above effect of Fat1 loss on actin fibers with its activity to interact with Ena/VASP proteins, we compared more closely the distributions of these proteins in control and Fat1 knockdown PAM212 cells, manipulating their adhesion by the Ca2+ switch method. As observed in primary keratinocytes (Vasioukhin et al., 2000), VASP proteins were localized to cell–cell contact sites at 1–8 h after the Ca2+ switch, and these junctional VASPs colocalized with Fat1 (Fig. 6 A). The VASP and Fat1 also colocalized with F-actin that had accumulated along these early cell–cell boundaries (Fig. 6 A). However, in Fat1 knockdown cells, VASP disappeared from cell boundaries, and the actin localization at early cell contact sites became punctate, instead of showing its linear organization in control cells (Fig. 6 B). These results are in accord with the idea that Fat1–VASP interactions may control actin organization. It is known that actins concentrated at initial cell contacts are gradually reorganized into radial actin cables (Vaezi et al., 2002), whose ends terminate at cell junctions, in keratinocytes. Formation of such radial actin cables was strongly suppressed in the Fat1 knockdown cells (Fig. 6 C). Previous work (Vaezi et al., 2002) showed that radial actin cable formation is initiated at the basolateral portion of the cells and then shifts to the apical portion. Fat1 was occasionally localized at the points where early radial actin cables appeared to “cross” the junctional actin fibers at the basolateral portion (Fig. 6 A, middle panels and insets). However, Fat1 did not particularly colocalize with radial actin cables fully organized at the apical portion (Fig. 6 C). These results suggest that Fat1 may participate in early phases of actin cable organization, but not directly in the later phases of this process.
Fat1 regulates early cell contacts and also cell polarity formation
As Fat1 was localized at free cell edges, we examined whether Fat1 had any role in initial cell–cell contacts by using the Ca2+ switch method. In control PAM212 cells, when their culture medium was switched from a Ca2+-chelated to a Ca2+-containing one, β-catenin as a marker of the classic cadherin–catenin system was promptly recruited to cell contact sites, with a concomitant induction of close cell–cell association (Fig. 7 A, top panels). However, in Fat1 knockdown cells, β-catenin accumulation was retarded (Fig. 7 A, bottom panels). These results suggest that Fat1 functions not only for maintenance of cell associations, but also for initiation of cell contacts.
Figure 7.
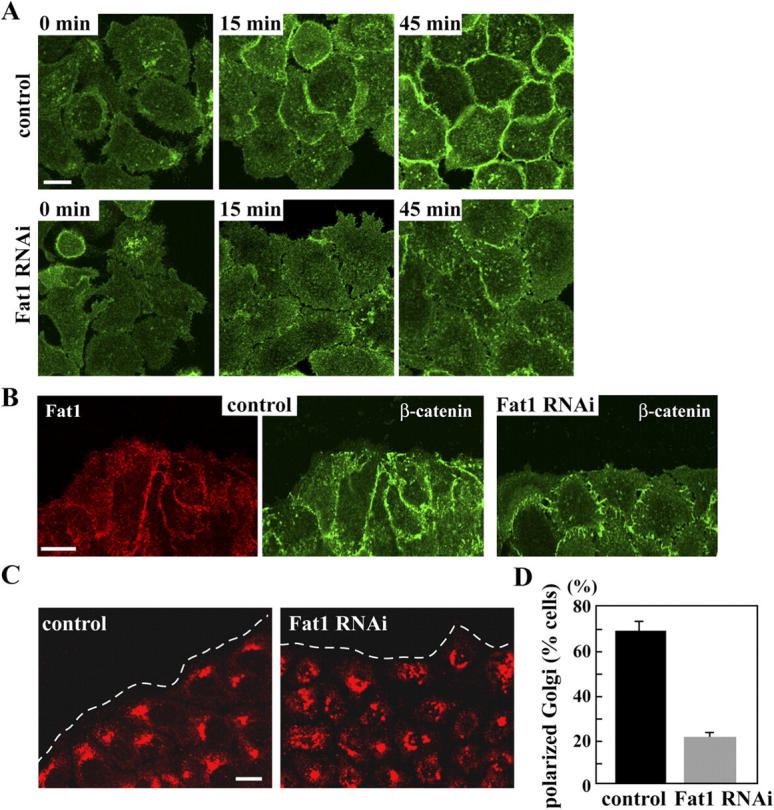
Fat1 regulates early cell contacts and also cell polarity formation. (A) PAM212 cells transfected with control or Fat1 RNAi plasmid were cultured for 72 h. Then, the cells were subjected to the Ca2+ switch, fixed at the indicated times, and stained for β-catenin. (B) PAM212 cells transfected with control RNAi plasmid or Fat1 RNAi plasmid were plated in a high density and were cultured for 72 h. The wound was made using a pipette tip. The cells were cultured for another 8 h, and then were stained for Fat1 and β-catenin. (C) 8 h after the wound had been made, the cells were stained for GM130 to visualize the Golgi apparatus. The wound margin is indicated by a dotted line. (D) Percentages of wound-edge cells with the Golgi apparatus oriented toward the wound front. Error bars denote SDs of six independent experiments. Bars, 10 μm.
Finally, we asked whether Fat1 had any activity to regulate dynamic cell rearrangement. As a test of this possible activity, we compared the behavior of PAM212 cells with that of their Fat1 knockdown counterparts in a wound-healing assay. As expected, Fat1 knockdown cells exhibited looser associations at wound margins compared with controls cells (Fig. 7 B). However, the overall wound closure rate was not significantly different between these cells (unpublished data). Then, we examined the distribution of the Golgi apparatus, which is known to be reoriented toward the edges of the wound in several cell lines (Nobes and Hall, 1999; Etienne-Manneville and Hall, 2001). In PAM212 cells, the Golgi apparatus surrounds the nucleus or accumulates at one side of the periphery of the nucleus; and it became reoriented to face the wound edge at ∼8 h after the wound had been made. When Fat1 was eliminated, the Golgi apparatus was no longer reoriented to the wound edges (Fig. 7, C and D), indicating that Fat1 was required for cellular polarization. To test whether the loss of actin stress fibers in Fat1 knockdown cells could be implicated in this phenomenon, we treated control PAM212 cells with Y27632, a Rock inhibitor, which can eliminate actin stress fibers. However, this treatment did not affect the reorientation of the Golgi apparatus (unpublished data), indicating that actin stress fibers were not involved in this process.
Discussion
Our present work has demonstrated novel cell biological properties of a vertebrate Fat cadherin, Fat1. This molecule was localized at both cell peripheries and cell–cell contact sites, overlapping with actin structures. Loss of Fat1 affected several classes of actin organization; not only junctional actin accumulation, but also the formation of radial actin cables and stress fibers. Furthermore, Fat1 deficiency resulted in looser cell association and abrogated cell polarity formation during wound healing. These findings suggest that Fat1 regulates cell contact and polarity, possibly by modulating actin polymerization at the cytoplasmic side.
How does Fat1 control the actin cytoskeleton? We identified Ena/VASP proteins as partners of Fat1. Ena/VASP proteins are localized to free cell edges, focal adhesions, and cell junctions, where they are thought to regulate the polymerization of actin (Laurent et al., 1999; Bear et al., 2000, 2002; Krause et al., 2002; Renfranz and Beckerle, 2002). Fat1 colocalized with Ena/VASP at these sites, with the exception of focal adhesions. Ena/VASP proteins have been reported to interact with several molecules, including zyxin, vinculin, semaphorin, Robo, and Fyb/Slap, via EVH1 domain–mediated interactions, and these interactions regulate the localization and activity of Ena/VASP proteins. We showed that Fat1 indeed bound to Ena/VASP proteins through an EVH1 domain–mediated interaction, and further demonstrated that the Fat1 tail promoted stress fiber formation in MDCK cells in a manner dependent on its EVH1-binding regions. When Fat1 protein was eliminated from PAM212 cells by RNAi, VASP was no longer localized to cell–cell contact sites; concomitantly, F-actin was reorganized into a punctate pattern at these sites. These findings suggest that the Fat1–Ena/VASP interaction is one possible mechanism for the regulation of actin organization.
Loss of α-catenin, a mediator for cadherin–actin interaction, also abolishes the localization of Ena/VASP proteins at cell–cell contacts (Vasioukhin et al., 2000). As Fat1 affected classic cadherin-mediated junctions, the effects of Fat1 deficiency on VASP localization and/or actin organization could have been indirectly produced through the perturbation of the classic cadherin system. However, in contrast to the case of α-catenin null mutants, in which junctional actin is eliminated (Vasioukhin et al., 2000), F-actin still accumulated at cell junctions in Fat1 knockdown cells, although its distribution pattern was altered, suggesting that the actin disorganization observed in Fat1 knockdown cells was not a simple consequence of the perturbation of the classic cadherin system. It is important to determine in future analyses how these two cadherin systems coordinate for actin organization. Our results also showed that the Fat1–VASP–actin colocalization occurred only at cell boundaries; nevertheless, Fat1 knockdown affected a broad rage of actin organization. There might be a cascade of actin reorganization that is triggered by cell–cell contacts, and Fat1-knockdown may block an early step of this cascade, indirectly affecting later steps. On the other hand, it is equally possible that the Fat1 tail may have other unidentified domains or functions to cover the regulation of multiple classes of actin organization, and the Fat1–VASP interaction is only part of the entire Fat1 system.
Our results indicated that the classic cadherin and Fat1 adhesion systems depended on one another for their full activities. They probably cooperate in cell–cell adhesion at different sites of the lateral cell membranes, as the apical adherens junction is enriched in the classic cadherin–catenin complex, whereas Fat1 is abundant at more basal sides of the lateral membranes. To elicit adhesive activity, Fat1 may undergo homophilic interactions via its cadherin motifs, as other members of the cadherin superfamily do. Such homophilic interactions may generate some intracellular signals to modulate cytoskeletal organization via the cytoplasmic tail. It remains to be elucidated what signals are elicited by Fat1 to control cell polarity, and how these are related to similar activities of the Drosophila homologue.
Fat1 is not ubiquitously expressed in the body, suggesting it has cell type–specific functions. For example, the level of Fat1 proteins greatly differed among epithelial lines, such as between PAM212 and MDCK cells. These observations suggest that this protein is not indispensable for the general control of cell–cell adhesion, but rather plays some cell type–specific roles. Recently, the phenotypes of transgenic mice lacking Fat1 have been reported (Ciani et al., 2003). These mice exhibit perinatal lethality, most probably caused by loss of renal glomerular slit junctions and fusion of glomerular epithelial cell processes (podocytes). Some Fat1−/− mice show holoprosencephaly and anophthalmia, which may be caused by defective cell–cell interactions. We are hopeful that the present cell biological findings may aid in the interpretation of such embryonic phenotypes in the future.
Materials and methods
Plasmids
A partial sequence of human Fat1 cDNA was obtained by the PCR method using total RNA from DLD1 cells as a template. The sequence corresponding to the EVH1 domain of mouse Mena (residues 1–175) was obtained by the PCR method using total RNA from the mouse brain as a template. pFlag-CMV1, a mammalian expression vector, was purchased from Sigma-Aldrich. This vector was used for extracellular secretion of NH2-terminal Flag tag fusion proteins. A DNA fragment encoding the signal sequence of preprotrypsin and the Flag tag of pFlag-CMV1 was amplified by the PCR method and was integrated into the pCA-IRES-Neo vector. This integrated vector was named after the pCA-Sig-IRES vector. pCA-IRES-Neo (constructed by T. Ichii in our laboratory) is a vector for stable expression in eukaryotic cells in which a neomycin resistance gene is driven by the IRES (internal ribosomal entry site) element. An NH2-terminal truncated form of Fat1 (Fat1 cyto, residues 3987–4590 of human Fat1), in which all the EC domains, one EGF motif, and the laminin A-G domain had been deleted, was subcloned into pFlag-CMV1 vector or pCA-Sig-IRES vector. The latter was used for establishing MDCK cells constitutively expressing the NH2-terminal truncated form of Fat1. Flag-tagged cytoplasmic region of Fat1 (residues 4201–4590 of human Fat1) was constructed using a pCMV-Tag1 vector (Stratagene). For the production of GST fusion proteins in Escherichia coli, pGEX-4T or pGEX-5X (Amersham Biosciences) was used. Mitochondrial targeting form of Fat1 cytoplasmic portion was constructed by attaching a mitochondrial-targeting sequence of Listeria ActA protein (LILAMLAIGVFSLGAFIKIIQLRKNN; a gift from Dr. R.M. Golsteyn, Institut de Recherches Servier, Suresnes, France) to the COOH terminus of Fat1 cytoplasmic tail. This sequence was integrated into pEGFP-C1 vector (CLONTECH Laboratories, Inc.).
RNAi
U6 promoter RNAi vector was provided by Dr. Y. Shi (Harvard Medical School, Boston, MA; Sui et al., 2002). The DNA sequences designed for construction of RNAi plasmid were as follows: 5′-GGACGACGGCCACTTCGAAGAG-3′, a mouse Fat1 sequence, for Fat1 RNAi; 5′-GGGTGTTTACATTACACATCCA-3′, a human Fat1 sequence, for control RNAi; and 5′-GGTCATGATGAGCGACTACGAG-3′, another mouse Fat1 sequence, for confirming the specific effect of Fat1 RNAi. Transfections were performed by using the Amaxa electroporation system (Amaxa). By using this system, >95% cells showed a reduction in the level of Fat1 protein ∼48 h after the transfection.
Mutagenesis
The mutants used were constructed by PCR-based mutagenesis. PCR was performed with PfuTurbo® polymerase (Stratagene). E. coli was transformed with the DpnI restriction enzyme (Stratagene)–treated PCR product. Positive clones were picked up, and mutagenesis was verified by sequencing.
Antibodies
Rabbit and rat anti-Fat1 antibodies were raised against the cytoplasmic portion of mouse Fat1 protein (residues 4201–4590). Affinity purification was performed using standard protocol. Other antibodies used were mouse or rabbit anti-Flag (Sigma-Aldrich), mouse anti-Mena (1:50; Transduction Laboratories), rabbit anti-VASP (1:250; Alexis), mouse anti-vinculin (1:250; Sigma-Aldrich), mouse anti-β-catenin (1:100; 5H10, a gift from Dr. M.J. Wheelock, University of Nebraska, Omaha, NE), mouse anti-GM130 (1:250; Transduction Laboratories), and rabbit anti-WAVE (1:250; a gift from Dr. T. Takenawa, University of Tokyo, Tokyo, Japan). For double-staining experiments, rat or rabbit anti-Fat1 antibodies were used in appropriate combinations with the above antibodies. For example, when rabbit anti-VASP protein was used for staining, rat (not rabbit) anti-Fat1 was used. To block E- and P-cadherin, antibodies ECCD1 (1:250; Yoshida-Noro et al., 1984) and PCD-1 (1:250; Nose and Takeichi, 1986), respectively, were used. F-actin was visualized by use of Alexa® 488– or 568–conjugated phalloidin (Molecular Probes, Inc.).
Cell cultures and transfection
MDCK, PAM212, and DLD1 cells were cultured in a 1:1 mixture of DME and Ham's F12 medium (Iwaki) supplemented with 10% FCS. NRK-52E cells (a rat renal epithelial cell line provided by S. Yonemura, RIKEN Center for Developmental Biology, Kobe, Japan) were cultured in DME (Nissui) containing 10% FCS. These cells were maintained in 5% CO2 at 37°C. Cells were transfected by using Effectine reagent (Qiagen) or FuGENE™ reagent (Roche) according to the manufacturer's protocol. Alternatively, cells were transfected by using the Amaxa electroporation system (Amaxa). PAM212 cells and NRK-52E cells were very efficiently transfected by using this apparatus. For the establishment of MDCK cells constitutively expressing NH2-terminal truncated forms of Fat1, we used the pCA-Sig-IRES vector (see above). 400 μg/ml G418 was used for selecting transfected cells. Selected cells were maintained as a noncloned population.
Immunostaining
Cells were fixed with 3.7% formaldehyde in PBS for 15 min at RT. The fixed cells were then permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked with 3% BSA in PBS for 30 min at 37°C. Thereafter, the cells were incubated with appropriate antibodies in 3% BSA in PBS for 1.5 h at 37°C. Next, the cells were washed three times with PBS and incubated with fluorochrome-conjugated secondary antibodies (1:500, Alexa Fluor® secondary antibodies; Molecular Probes, Inc.) in 3% BSA in PBS for 1 h at 37°C. For rat anti-Fat1 antibody, a Cy3-conjugated secondary antibody (1:500; CHEMICON International) was used. In double staining for Fat1 and other antigens, either the rat or rabbit anti-Fat1 antibody was chosen, depending on the antibody type used for the other antigen. After three washes with PBS and two washes with Milli-Q water (Millipore), coverslips were mounted with FluorSave™ reagent (Calbiochem). Preparations were analyzed using a laser-scanning confocal microscope (LSM 510 mounted on an Axiovert 100M microscope; Carl Zeiss MicroImaging, Inc.) on an inverted stand using C-Apochromat 40×/1.20 and Plan-Apochromat 63×/1.40 objectives (Carl Zeiss MicroImaging, Inc.) at RT. Images for presentation were prepared with Adobe Photoshop® software.
Western blotting
Fetal mouse brains or cells were lysed in 50 mM Hepes (pH 7.4) containing 2 mM EGTA, 2 mM MgCl2, 10% glycerol, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, and 20 μg/ml aprotinin. Proteins were fractionated by SDS-PAGE using a 2–15% gradient gel (Daiichi Pure Chemicals; in Fig. 3 C, Fig. S1 and Fig. S2) or a 10% gel (in Fig. 3). The fractionated proteins were electroblotted onto immobilion-P polyvinyldifluoride membranes (Millipore) by using either a wet or a semi-dry transfer apparatus (Bio-Rad Laboratories). The membrane was blocked with 5% skim milk for 30 min at RT. Then, proteins were probed for 16 h at 4°C with antibody against rat anti-Fat1 antibody (1:100), rabbit anti-Fat1 antibody (1:100), or anti-Flag antibody (1:500) in 20 mM Tris-HCl containing 3% BSA and 150 mM NaCl. The membrane was then washed three times at RT (15 min each time) in 20 mM Tris-HCl containing 150 mM NaCl and 0.5% Tween 20 (TBS-Tween), and was subsequently incubated for 2 h at RT with secondary antibody in 20 mM Tris-HCl containing 5% skim milk and 150 mM NaCl. After three washes with TBS-Tween, the proteins were detected using the ECL plus™ reagent (PerkinElmer) according to the manufacturer's protocol.
GST pull-down assay
Cells were lysed in 50 mM Hepes (pH 7.4) containing 2 mM EGTA, 2 mM MgCl2, 10% glycerol, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, and 20 μg/ml aprotinin. The lysates were then incubated with GST-recombinant proteins and GSH beads for 12 h. After the beads had been washed three times with lysis buffer, the bound proteins were separated by SDS-PAGE and analyzed by immunoblotting.
Wound-healing assay
PAM212 cells were plated in a high density and were cultured for 3 d. Monolayers of the cells were wounded by scraping with the tip of a plastic pipette.
Online supplemental material
Characterization of anti-Fat1 antibodies is described in detail. Fig. S1 shows characterization of antibodies raised against the cytoplasmic portion of Fat1. Fig. S2 shows that delN Fat1 displaces endogenous VASP, but not vinculin in Fig. 3 F. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200403006/DC1.
Acknowledgments
We are grateful to Dr. S. Yonemura for providing us with NRK-52E cells; to Dr. T. Takenawa for anti-WAVE and anti-N-WASP antibodies; to Dr. Y. Shi for the U6 RNAi vector; to Dr. R.M. Golsteyn for the Listeria ActA sequence; to Dr. Y. Saya (University of Kumamoto, Kumamoto, Japan) for zyxin cDNA; to Drs. A. Hall, T. Takenawa, S. Suetsugu, and M. Nakayama for valuable suggestions; and to Drs. S. Hirano, S. Nakagawa, S.C. Suzuki, T. Ichii, T. Ohtani, H. Togashi, and all other members of our laboratory for helpful discussions. We also thank M. Harata and H. Ishigami for technical support, and Douglas Sipp for critical reading of this manuscript.
This work was supported by a grant from the program Grants-in-Aid for Specifically Promoted Research of Ministry of Education, Science, Sports, and Culture of Japan to M. Takeichi, and by a grant-in-aid for scientific research from the Japan Society for Promotion of Science to T. Tanoue, who was also supported by the Special Postdoctoral Researcher program of RIKEN.
The online version of this article includes supplemental material.
Abbreviations used in this paper: EC, extracellular cadherin; EVH1, Ena/VASP homology 1; Mena, mammalian Enabled; RNAi, RNA interference; VASP, vasodilator-stimulated phosphoprotein.
References
- Ball, L.J., R. Kuhne, B. Hoffmann, A. Hafner, P. Schmieder, R. Volkmer-Engert, M. Hof, M. Wahl, J. Schneider-Mergener, U. Walter, et al. 2000. Dual epitope recognition by the VASP EVH1 domain modulates polyproline ligand specificity and binding affinity. EMBO J. 19:4903–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear, J.E., J.J. Loureiro, I. Libova, R. Fassler, J. Wehland, and F.B. Gertler. 2000. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 101:717–728. [DOI] [PubMed] [Google Scholar]
- Bear, J.E., T.M. Svitkina, M. Krause, D.A. Schafer, J.J. Loureiro, G.A. Strasser, I.V. Maly, O.Y. Chaga, J.A. Cooper, G.G. Borisy, and F.B. Gertler. 2002. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 109:509–521. [DOI] [PubMed] [Google Scholar]
- Bolz, H., B. von Brederlow, A. Ramirez, E.C. Bryda, K. Kutsche, H.G. Nothwang, M. Seeliger, M. del C-Salcedo Cabrera, M.C. Vila, et al. 2001. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 27:108–112. [DOI] [PubMed] [Google Scholar]
- Bork, J.M., L.M. Peters, S. Riazuddin, S.L. Bernstein, Z.M. Ahmed, S.L. Ness, R. Polomeno, A. Ramesh, M. Schloss, C.R. Srisailpathy, et al. 2001. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, P.J., B. Huettner, L.I. Jr. Held, J. Ryerse, and J. Szidonya. 1988. Mutations at the fat locus interfere with cell proliferation control and epithelial morphogenesis in Drosophila. Dev. Biol. 129:541–554. [DOI] [PubMed] [Google Scholar]
- Carl, U.D., M. Pollmann, E. Orr, F.B. Gertler, T. Chakraborty, and J. Wehland. 1999. Aromatic and basic residues within the EVH1 domain of VASP specify its interaction with proline-rich ligands. Curr. Biol. 9:715–718. [DOI] [PubMed] [Google Scholar]
- Chae, J., M.J. Kim, J.H. Goo, S. Collier, D. Gubb, J. Charlton, P.N. Adler, and W.J. Park. 1999. The Drosophila tissue polarity gene starry night encodes a member of the protocadherin family. Development. 126:5421–5429. [DOI] [PubMed] [Google Scholar]
- Ciani, L., A. Patel, N.D. Allen, and C. ffrench-Constant. 2003. Mice lacking the giant protocadherin mFAT1 exhibit renal slit junction abnormalities and a partially penetrant cyclopia and anophthalmia phenotype. Mol. Cell Biol. 23:3575–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, B., A.K. Hadjantonakis, J.E. Collins, and A.I. Magee. 2000. Cloning and expression throughout mouse development of mfat1, a homologue of the Drosophila tumour suppressor gene fat. Dev. Dyn. 217:233–240. [DOI] [PubMed] [Google Scholar]
- Curtin, J.A., E. Quint, V. Tsipouri, R.M. Arkell, B. Cattanach, A.J. Copp, D.J. Henderson, N. Spurr, P. Stanier, E.M. Fisher, et al. 2003. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 13:1129–1133. [DOI] [PubMed] [Google Scholar]
- Di Palma, F., R.H. Holme, E.C. Bryda, I.A. Belyantseva, R. Pellegrino, B. Kachar, K.P. Steel, and K. Noben-Trauth. 2001. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat. Genet. 27:103–107. [DOI] [PubMed] [Google Scholar]
- Dunne, J., A.M. Hanby, R. Poulsom, T.A. Jones, D. Sheer, W.G. Chin, S.M. Da, Q. Zhao, P.C. Beverley, and M.J. Owen. 1995. Molecular cloning and tissue expression of FAT, the human homologue of the Drosophila fat gene that is located on chromosome 4q34-q35 and encodes a putative adhesion molecule. Genomics. 30:207–223. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2001. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCζ. Cell. 106:489–498. [DOI] [PubMed] [Google Scholar]
- Fanto, M., L. Clayton, J. Meredith, K. Hardiman, B. Charroux, S. Kerridge, and H. McNeill. 2003. The tumor-suppressor and cell adhesion molecule Fat controls planar polarity via physical interactions with Atrophin, a transcriptional co-repressor. Development. 130:763–774. [DOI] [PubMed] [Google Scholar]
- Frank, M., and R. Kemler. 2002. Protocadherins. Curr. Opin. Cell Biol. 14:557–562. [DOI] [PubMed] [Google Scholar]
- Fukuhara, A., K. Irie, A. Yamada, T. Katata, T. Honda, K. Shimizu, H. Nakanishi, and Y. Takai. 2002. Role of nectin in organization of tight junctions in epithelial cells. Genes Cells. 7:1059–1072. [DOI] [PubMed] [Google Scholar]
- Gao, F.B., M. Kohwi, J.E. Brenman, L.Y. Jan, and Y.N. Jan. 2000. Control of dendritic field formation in Drosophila: the roles of flamingo and competition between homologous neurons. Neuron. 28:91–101. [DOI] [PubMed] [Google Scholar]
- Garrod, D.R., A.J. Merritt, and Z. Nie. 2002. Desmosomal cadherins. Curr. Opin. Cell Biol. 14:537–545. [DOI] [PubMed] [Google Scholar]
- Gertler, F.B., K. Niebuhr, M. Reinhard, J. Wehland, and P. Soriano. 1996. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell. 87:227–239. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B.M. 2000. Regulation of cadherin adhesive activity. J. Cell Biol. 148:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, S., S.T. Suzuki, and C.M. Redies. 2003. The cadherin superfamily in neural development: diversity, function and interaction with other molecules. Front. Biosci. 8:D306–D355. [DOI] [PubMed] [Google Scholar]
- Inoue, T., E. Yaoita, H. Kurihara, F. Shimizu, T. Sakai, T. Kobayashi, K. Ohshiro, H. Kawachi, H. Okada, H. Suzuki, et al. 2001. FAT is a component of glomerular slit diaphragms. Kidney Int. 59:1003–1012. [DOI] [PubMed] [Google Scholar]
- Kim, S.H., A. Yamamoto, T. Bouwmeester, E. Agius, and E.M. Robertis. 1998. The role of paraxial protocadherin in selective adhesion and cell movements of the mesoderm during Xenopus gastrulation. Development. 125:4681–4690. [DOI] [PubMed] [Google Scholar]
- Kim, S.H., W.C. Jen, E.M. De Robertis, and C. Kintner. 2000. The protocadherin PAPC establishes segmental boundaries during somitogenesis in Xenopus embryos. Curr. Biol. 10:821–830. [DOI] [PubMed] [Google Scholar]
- Kohmura, N., K. Senzaki, S. Hamada, N. Kai, R. Yasuda, M. Watanabe, H. Ishii, M. Yasuda, M. Mishina, and T. Yagi. 1998. Diversity revealed by a novel family of cadherins expressed in neurons at a synaptic complex. Neuron. 20:1137–1151. [DOI] [PubMed] [Google Scholar]
- Krause, M., J.E. Bear, J.J. Loureiro, and F.B. Gertler. 2002. The Ena/VASP enigma. J. Cell Sci. 115:4721–4726. [DOI] [PubMed] [Google Scholar]
- Laurent, V., T.P. Loisel, B. Harbeck, A. Wehman, L. Grobe, B.M. Jockusch, J. Wehland, F.B. Gertler, and M.F. Carlier. 1999. Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J. Cell Biol. 144:1245–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, R.C., T.R. Clandinin, C.H. Lee, P.L. Chen, I.A. Meinertzhagen, and S.L. Zipursky. 2003. The protocadherin Flamingo is required for axon target selection in the Drosophila visual system. Nat. Neurosci. 6:557–563. [DOI] [PubMed] [Google Scholar]
- Ma, D., C.H. Yang, H. McNeill, M.A. Simon, and J.D. Axelrod. 2003. Fidelity in planar cell polarity signalling. Nature. 421:543–547. [DOI] [PubMed] [Google Scholar]
- Mahoney, P.A., U. Weber, P. Onofrechuk, H. Biessmann, P.J. Bryant, and C.S. Goodman. 1991. The fat tumor suppressor gene in Drosophila encodes a novel member of the cadherin gene superfamily. Cell. 67:853–868. [DOI] [PubMed] [Google Scholar]
- Mitsui, K., D. Nakajima, O. Ohara, and M. Nakayama. 2002. Mammalian fat3: a large protein that contains multiple cadherin and EGF-like motifs. Biochem. Biophys. Res. Commun. 290:1260–1266. [DOI] [PubMed] [Google Scholar]
- Nakayama, M., D. Nakajima, R. Yoshimura, Y. Endo, and O. Ohara. 2002. MEGF1/fat2 proteins containing extraordinarily large extracellular domains are localized to thin parallel fibers of cerebellar granule cells. Mol. Cell. Neurosci. 20:563–578. [DOI] [PubMed] [Google Scholar]
- Niebuhr, K., F. Ebel, R. Frank, M. Reinhard, E. Domann, U.D. Carl, U. Walter, F.B. Gertler, J. Wehland, and T. Chakraborty. 1997. A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO J. 16:5433–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose, A., and M. Takeichi. 1986. A novel cadherin cell adhesion molecule: its expression patterns associated with implantation and organogenesis of mouse embryos. J. Cell Biol. 103:2649–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponassi, M., T.S. Jacques, L. Ciani, and C. ffrench Constant. 1999. Expression of the rat homologue of the Drosophila fat tumour suppressor gene. Mech. Dev. 80:207–212. [DOI] [PubMed] [Google Scholar]
- Rawls, A.S., J.B. Guinto, and T. Wolff. 2002. The cadherins fat and dachsous regulate dorsal/ventral signaling in the Drosophila eye. Curr. Biol. 12:1021–1026. [DOI] [PubMed] [Google Scholar]
- Reinhard, M., M. Halbrugge, U. Scheer, C. Wiegand, B.M. Jockusch, and U. Walter. 1992. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J. 11:2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfranz, P.J., and M.C. Beckerle. 2002. Doing (F/L)PPPPs: EVH1 domains and their proline-rich partners in cell polarity and migration. Curr. Opin. Cell Biol. 14:88–103. [DOI] [PubMed] [Google Scholar]
- Rhee, J., Y. Takahashi, Y. Saga, J. Wilson-Rawls, and A. Rawls. 2003. The protocadherin papc is involved in the organization of the epithelium along the segmental border during mouse somitogenesis. Dev. Biol. 254:248–261. [DOI] [PubMed] [Google Scholar]
- Rottner, K., B. Behrendt, J.V. Small, and J. Wehland. 1999. VASP dynamics during lamellipodia protrusion. Nat. Cell Biol. 1:321–322. [DOI] [PubMed] [Google Scholar]
- Samarin, S., S. Romero, C. Kocks, D. Didry, D. Pantaloni, and M.F. Carlier. 2003. How VASP enhances actin-based motility. J. Cell Biol. 163:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senti, K.A., T. Usui, K. Boucke, U. Greber, T. Uemura, and B.J. Dickson. 2003. Flamingo regulates r8 axon-axon and axon-target interactions in the Drosophila visual system. Curr. Biol. 13:828–832. [DOI] [PubMed] [Google Scholar]
- Strutt, H., and D. Strutt. 2002. Nonautonomous planar polarity patterning in Drosophila: dishevelled-independent functions of frizzled. Dev. Cell. 3:851–863. [DOI] [PubMed] [Google Scholar]
- Sui, G., C. Soohoo, E.B. Affar, F. Gay, Y. Shi, W.C. Forrester, and Y. Shi. 2002. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl. Acad. Sci. USA. 99:5515–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, S.T. 2000. Recent progress in protocadherin research. Exp. Cell Res. 261:13–18. [DOI] [PubMed] [Google Scholar]
- Tepass, U., K. Truong, D. Godt, M. Ikura, and M. Peifer. 2000. Cadherins in embryonic and neural morphogenesis. Nat. Rev. Mol. Cell Biol. 1:91–100. [DOI] [PubMed] [Google Scholar]
- Usui, T., Y. Shima, Y. Shimada, S. Hirano, R.W. Burgess, T.L. Schwarz, M. Takeichi, and T. Uemura. 1999. Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell. 98:585–595. [DOI] [PubMed] [Google Scholar]
- Vaezi, A., C. Bauer, V. Vasioukhin, and E. Fuchs. 2002. Actin cable dynamics and Rho/Rock orchestrate a polarized cytoskeletal architecture in the early steps of assembling a stratified epithelium. Dev. Cell. 3:367–381. [DOI] [PubMed] [Google Scholar]
- Vasioukhin, V., C. Bauer, M. Yin, and E. Fuchs. 2000. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 100:209–219. [DOI] [PubMed] [Google Scholar]
- Wang, X., J.A. Weiner, S. Levi, A.M. Craig, A. Bradley, and J.R. Sanes. 2002. Gamma protocadherins are required for survival of spinal interneurons. Neuron. 36:843–854. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., S.L. Amacher, S.H. Kim, D. Geissert, C.B. Kimmel, and E.M. De Robertis. 1998. Zebrafish paraxial protocadherin is a downstream target of spadetail involved in morphogenesis of gastrula mesoderm. Development. 125:3389–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C.H., J.D. Axelrod, and M.A. Simon. 2002. Regulation of Frizzled by fat-like cadherins during planar polarity signaling in the Drosophila compound eye. Cell. 108:675–688. [DOI] [PubMed] [Google Scholar]
- Yoshida-Noro, C., N. Suzuki, and M. Takeichi. 1984. Molecular nature of the calcium-dependent cell-cell adhesion system in mouse teratocarcinoma and embryonic cells studied with a monoclonal antibody. Dev. Biol. 101:19–27. [DOI] [PubMed] [Google Scholar]