

Abstract
Transforming growth factor-βs (TGF-β) are secreted as inactive complexes containing the TGF-β, the TGF-β propeptide, also called the latency-associated protein (LAP), and the latent TGF-β binding protein (LTBP). Extracellular activation of this complex is a critical but incompletely understood step in TGF-β regulation. We have investigated the role of LTBP in modulating TGF-β generation by the integrin αVβ6. We show that even though αvβ6 recognizes an RGD on LAP, LTBP-1 is required for αVβ6-mediated latent TGF-β activation. The domains of LTBP-1 necessary for activation include the TGF-β propeptide-binding domain and a basic amino acid sequence (hinge domain) with ECM targeting properties. Our results demonstrate an LTBP-1 isoform-specific function in αVβ6-mediated latent TGF-β activation; LTBP-3 is unable to substitute for LTBP-1 in this assay. The results reveal a functional role for LTBP-1 in latent TGF-β activation and suggest that activation of specific latent complexes is regulated by distinct mechanisms that may be determined by the LTBP isoform and its potential interaction with the matrix.
Keywords: LTBP; TGF-β; integrin; αvβ6; latent TGF-β
Introduction
A growth factor that is released into the extracellular space must reach the appropriate target cell in biologically relevant concentrations at the correct time despite the random nature of diffusion. To augment the basic process of diffusion, multicellular organisms use the ECM to bind, concentrate, store and present growth factors. A common strategy used by numerous growth factors, including FGF and VEGF, to oppose entropy is to rely on charge-based association with ECM glycosaminoglycans and/or proteins (Flaumenhaft and Rifkin, 1992b; Taipale and Keski-Oja, 1997). The cytokine TGF-β exploits an interesting variation on this theme of ECM binding. TGF-β is secreted constitutively in covalent association with an ECM component, the latent TGF-β binding protein (LTBP). In addition, TGF-β is secreted in a latent state and must undergo a highly regulated activation process to be functional. An important and unresolved issue in TGF-β biology regards the connection between matrix incorporation and activation of the latent TGF-β complex.
The TGF-βs are multipotent cytokines that modulate cell growth, inflammation, matrix synthesis, and apoptosis (Taipale et al., 1998). Defects in TGF-β function are associated with a number of pathological states including tumor cell growth, fibrosis, emphysema, and autoimmune disease (Blobe et al., 2000; Morris et al., 2003). All three mammalian TGF-β isoforms: TGF-β1, 2, and 3 are synthesized as homodimeric proproteins (proTGF-β). The dimeric propeptides, also known as the latency-associated protein (LAP), are cleaved from the mature TGF-β dimer by furin-type enzymes but remain noncovalently associated with the mature cytokine (Dubois et al., 1995). The association between the TGF-β1, 2, and 3 prodomains (LAPs) and the corresponding mature growth factors prevents signaling through the TGF-β high affinity receptors (Lawrence et al., 1984). Thus, TGF-β bioactivity requires dissociation from LAP, a process termed latent TGF-β activation.
Early in the assembly of the TGF-β latent complex, disulfide linkages are formed between cysteine(s) of LAP and specific cysteines in the LTBP (Miyazono et al., 1991; Gleizes et al., 1996; Saharinen et al., 1996). The ternary complex of TGF-β, LAP, and LTBP is called the large latent complex (LLC; Fig. 1 A). Under most conditions, TGF-β is secreted as part of the LLC (Taipale et al., 1998), which is consistent with the observation that LTBP-1 and TGF-β1 expression are coregulated (Miyazono et al., 1991; Koski et al., 1999). The release of LAP–TGF-β, the small latent complex (SLC), without LTBP has been reported previously (Dallas et al., 1994), but this complex is inefficiently secreted (Miyazono et al., 1991). The biologic significance of these two different forms, LLC and SLC, of latent TGF-β is unclear.
Figure 1.
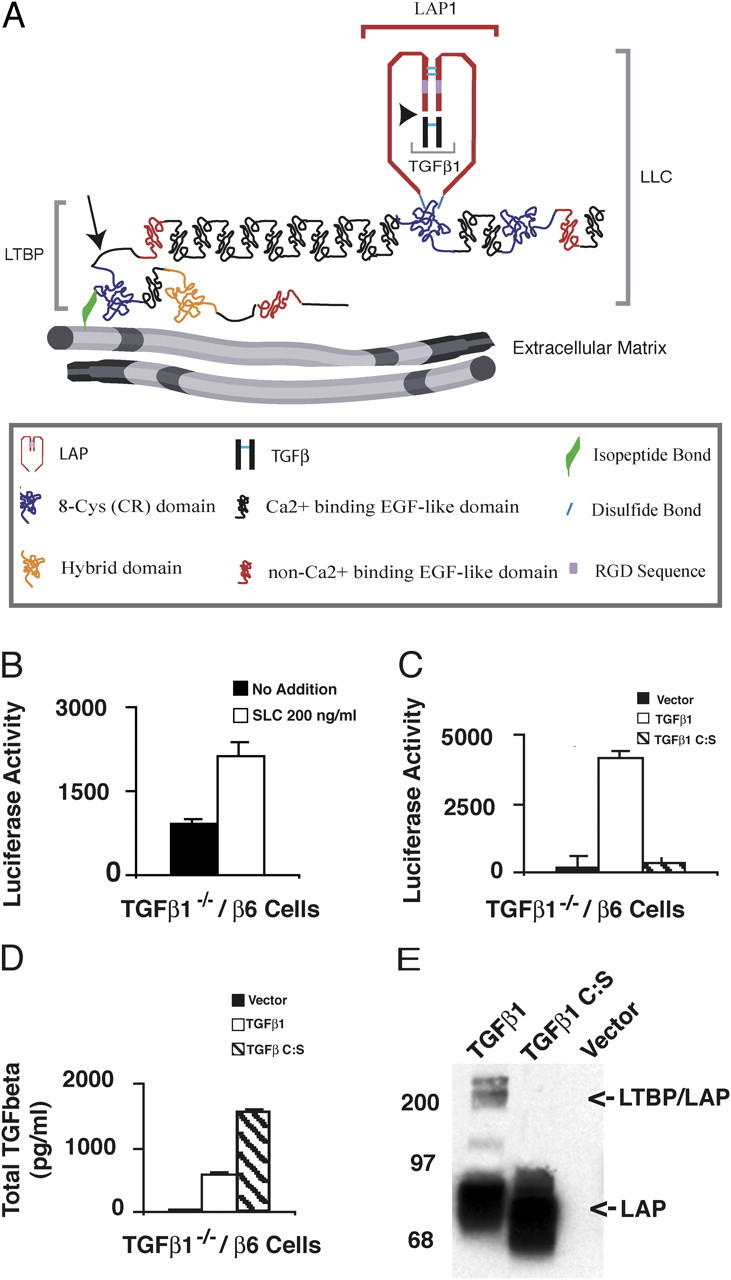
αVβ6-mediated latent TGF-β activation of SLC and TGF-β1 C:S. (A) The TGF-β large latent complex (LLC). The LLC is comprised of TGF-β (black), LAP (red) and LTBP. TGF-β and LAP are proteolytically processed at the site indicated by the arrowhead. LAP and LTBP are joined by disulfide bonds (light blue lines). The LLC is covalently linked by tTGase to the ECM via a glutamine-lysine isopeptide bond (green) near the NH2 terminus of LTBP. The hinge domain (arrow) of LTBP is a protease-sensitive region. (B) TGF-β1−/− cells that express the β-integrin subunit (TGF-β−/−/β6 cells) were co-cultured with TMLC that produce luciferase in response to TGF-β. Co-cultures contained either no addition or recombinant SLC (200 ng/ml). After 16–24 h, cell lysates were collected and luciferase activity measured. The errors bars represent the SEM from a single experiment performed in duplicate. This experiment was repeated multiple times with similar results. (C) TGF-β−/−/β6 cells were transfected with an empty vector, TGF-β1 cDNA, or TGF-β1 C:S cDNA and co-cultured with TMLCs for 16–24 h. The cell lysates were assayed for luciferase activity. (D) The transfected cells (C) were also used to generate conditioned media (CM) for 16–24 h. CM was collected, heated to 80°C for 10 min, diluted 10-fold, and added to TMLC. In separate wells, various concentrations of recombinant TGF-β were added to TMLCs to generate a TGF-β standard curve. The amount of TGF-β present in the CM was determined based upon the standard curve. Luciferase assays were performed in triplicate and the SD of a single experiment is shown. (C and D) The errors bars represent the SD of a single experiment that was performed in triplicate. These experiments were repeated multiple times with similar results. (E) The CM generated by the transfected cells was also used for Western blotting. Protein bands were revealed with Vb3A9 (anti-LAP).
LTBP-1 is a member of the LTBP/fibrillin protein family comprised of fibrillin-1, -2, and -3, and LTBP-1, -2, -3, and -4 (Ramirez and Pereira, 1999). These proteins contain multiple EGF-like repeats as well as unique domains containing eight cysteines or cysteine rich (CR) domain (Kanzaki et al., 1990; Tsuji et al., 1990; Sinha et al., 1998). LTBP-1, -3, and -4 form a subset within the family based on their ability to bind LAP. Only the third of the four CR domains within each of the LAP-binding LTBPs can disulfide bond to LAP (Saharinen and Keski-Oja, 2000; Lack et al., 2003). LTBP and its bound latent TGF-β are found primarily as components of the ECM (Taipale et al., 1994b). Previous work has implicated the NH2-terminal region of LTBP-1 in ECM binding (Nunes et al., 1997; Dallas et al., 2000; Unsold et al., 2001), and the NH2-terminal region of LTBP-1 may be cross-linked to ECM proteins by tissue transglutaminase (tTGase; Nunes et al., 1997). At present, the function of the various LTBP isoforms and their splice variants remains indeterminate.
The integrin αvβ6 is an in vivo activator of latent TGF-β1 and 3 (Munger et al., 1999; Annes et al., 2002). The expression of αvβ6 is restricted to epithelia, and in most epithelia the integrin is normally expressed at low levels (Breuss et al., 1993). Mice lacking the β6 subunit have persistent lung and skin inflammation but do not develop pulmonary fibrosis even when challenged with the profibrotic agent bleomycin (Huang et al., 1996; Munger et al., 1999). Analysis of gene expression in the lungs of bleomycin-treated mice suggests that lack of fibrosis in the β6-null mice is a consequence of decreased TGF-β activity because the overwhelming majority of TGF-β responsive genes up-regulated in control mice are not increased in the β6-null animals (Kaminski et al., 2000). The mechanism of integrin-mediated latent TGF-β activation is not well understood. However, a direct interaction between αvβ6 and the RGD amino acid sequence present in LAP is required, as mutation of this sequence to RGE eliminates activation. Importantly, integrin binding by itself to latent TGF-β is not sufficient to activate TGF-β as neither soluble αvβ6, LAP-β1–binding to integrins αVβ1, αvβ3 (Munger et al., 1998), nor α8β1 (Lu et al., 2002) activates latent TGF-β1.
Characterization of the mechanisms controlling the liberation of TGF-β from its latent complex is central to understanding TGF-β action. Although several activators (proteases, TSP-1, integrins) of latent TGF-β have been identified, the molecular basis for TGF-β activation remains only partially understood (Munger et al., 1997; Annes et al., 2003). For instance, there is strong evidence that LTBP plays an important role in protease-mediated latent TGF-β activation as both inhibitors of tTGase (Kojima and Rifkin, 1993) and antibodies raised against LTBP-1 block activation of latent TGF-β in several settings (Flaumenhaft et al., 1993; Nakajima et al., 1997; Gualandris et al., 2000). However, the function of LTBP in these systems is unclear. Unlike protease-mediated latent TGF-β activation, αvβ6-mediated latent TGF-β activation was reported not to require LTBP (Munger et al., 1999) based upon failure of anti-LTBP antibodies to block αvβ6-mediated activation. However, it is possible that the roles of LTBP in protease- and integrin-mediated activation are distinct and cannot be inhibited in the same way. An important next step in understanding latent TGF-β activation is to elucidate the function(s) of LTBP. Here, we present experimental results that define domains of LTBP-1 that are required for integrin-mediated latent TGF-β activation, demonstrate an isoform (LTBP-1) specific function and gain insight into the mechanism of LTBP-1 regulation of TGF-β generation.
Results
αVβ6-mediated activation of latent TGF-β requires LTBP
Our previous results suggesting that αVβ6-mediated activation of latent TGF-β involved the RGD of LAP led us to test if recombinant SLC would be activated when added to TGF-β1 null cells engineered to express the integrin αVβ6 (TGF-β−/−/β6 cells). To test this, TGF-β−/−/β6 cells were co-cultured with transformed mink lung epithelial cells (TMLC), which produce luciferase in the presence of TGF-β, and recombinant SLC added to the culture. At the end of the incubation, the monolayer was scraped and the amount of luciferase determined. Surprisingly, even when TGF-β−/−/β6 cells were incubated with rSLC at a final concentration of 200 ng/ml, which is 100-fold the typical endogenously secreted concentration of latent TGF-β, only a modest increase in active TGF-β, as determined by luciferase levels, was observed (only twofold above the background; Fig. 1 B). By contrast, a significant increase in TGF-β generation occurred when the same cells were transfected with an expression vector encoding the entire TGF-β precursor (Fig. 1 C, compare vector with TGF-β1) and subsequently co-cultured with TMLC, even though the total latent TGF-β concentration in the medium was only ∼700 pg/ml TGF-β (Fig. 1 D, TGF-β1; Annes et al., 2002). These results indicate that rSLC is a poor substrate for αVβ6-mediated activation. Two interpretations as to why rSLC is inefficiently activated are: (1) the mechanism of αVβ6-mediated activation limits cells to modifying only the TGF-β that they secrete; or (2) LTBP is a required component of this activation mechanism and therefore SLC is not a substrate. To distinguish between these possibilities, we took advantage of a form of TGF-β1 (Saharinen et al., 1996) that contains a point mutation in the LAP coding sequence (C33S) that prevents disulfide bonding to the third CR (CR3) domain of LTBP-1S. When TGF-β−/−/β6 cells were transfected with an expression vector encoding either wild-type TGF-β or the mutant form of TGF-β (TGF-β C:S), only wild-type TGF-β formed LLC, although TGF-β C:S LAP was well secreted (Fig. 1 E). These constructs contain coding sequence for TGF-β and its propeptide and should therefore code for latent TGF-β. The high secretion level of this protein probably reflects the absence of the otherwise unpaired C33. Assays for TGF-β generation by these transfected cells with a TGF-β reporter cell line revealed that cells transfected with wild-type TGF-β1 cDNA produced significant amounts of active TGF-β, but TGF-β-C:S transfected cells only formed active TGF-β at levels comparable to background (Fig. 1 D). Addition of an αVβ6-blocking antibody (10D5; 15 μg/ml) prevented TGF-β generation (unpublished data). The failure to activate TGF-β C:S did not reflect poor secretion of this mutant protein as these cells consistently secreted more total TGF-β than the wild-type TGF-β transfected cells (Fig. 1 D). These results led us to favor the hypothesis that αVβ6-mediated activation of latent TGF-β requires a covalent interaction between LAP and LTBP.
An LTBP-1–derived minimal TGF-β–binding construct does not support αVβ6-mediated activation of latent TGF-β
A covalent interaction between LTBP and LAP might be required for αVβ6-mediated activation because binding to LTBP may alter the structure of the SLC in a way that permits integrin-mediated activation, whereas in the absence of LTBP the SLC complex adopts a conformation unsuitable for integrin-mediated activation. If this hypothesis is correct, the small region of LTBP that covalently binds LAP might be sufficient to support αVβ6-mediated latent TGF-β activation. To test this supposition, we used an LTBP-1S–derived construct that encodes only the LAP binding domain of LTBP (CR3) plus the flanking EGF-like repeats (EGF13 and 14; ECR3E, Fig. 2 A; Gleizes et al., 1996). The EGF-like domains are not required for LAP binding but are included to enhance expression of the protein product. As a control, we generated a similar expression construct that encodes CR4, which does not bind LAP (Gleizes et al., 1996), plus its flanking EGF-like domains (EGF15 and 16; Fig. 2 A, ECR4E). The effect of these expression constructs on latent TGF-β activation was tested by using CHOβ6 cells transduced with ECR3E, ECR4E or empty virus (Fig. 3 A). In this experiment, the CHOβ6 cells (as opposed to the TGF-β1−/−/β6 cells in Fig. 1) activate their endogenously produced latent TGF-β (Fig. 3 A). Thus, the background levels of active TGF-β are higher than those observed in Fig. 1. Contrary to our hypothesis, CHOβ6 cells that expressed ECR3E demonstrated decreased latent TGF-β activation (∼50%), rather than unchanged or increased latent TGF-β activation compared with mock-transduced and ECR4E-transduced cells (Fig. 3 A). The empty vector and the ECR4E expression construct had similar effects on latent TGF-β activation.
Figure 2.
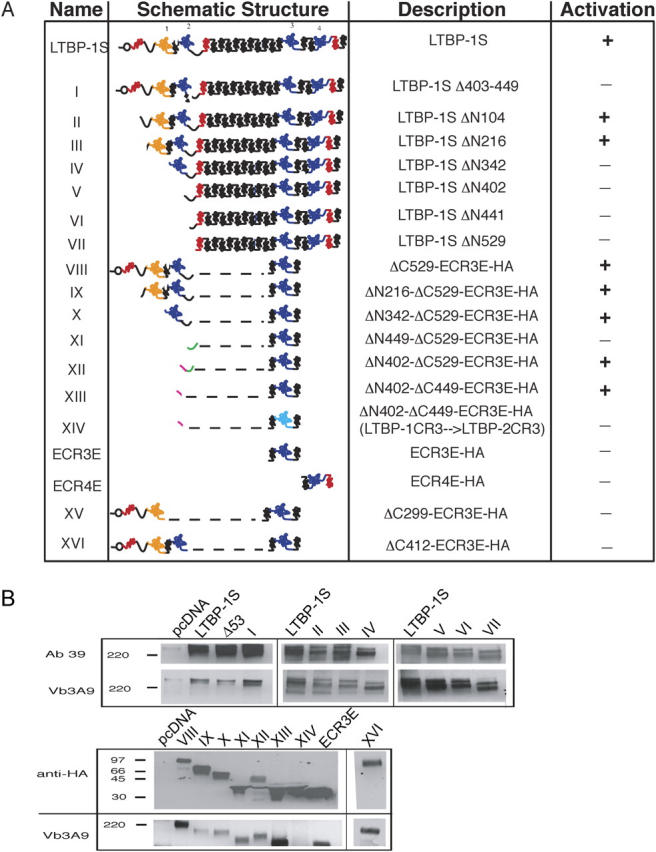
Structure and expression of LTBP constructs. A number of LTBP-1S–derived expression constructs are used throughout this paper. (A) These constructs are schematically represented with their name, description, and ability to support latent TGF-β activation given. The highly modular structure of LTBP-1 is represented with EGF-like domains in red (noncalcium binding) and black (calcium binding), and CR domains shown in yellow (hybrid) and blue. The mutated CR3 domain of construct XIV (light blue) has been altered to resemble the non TGF-β–binding CR3 domain of LTBP-2. (B) The expression constructs shown schematically in A were cotransfected with simian proTGF-β1 into CHO cells and the conditioned media were collected and probed with Ab39 (anti–LTBP-1S), Vb3A9 (anti-LAP) and anti-HA, as appropriate, to demonstrate secretion and complex formation with LAP.
Figure 3.

Affect of ECR3E on αVβ6-mediated latent TGF-β activation and LLC formation. CHO/β6 cells were transduced with empty, ECR3E- or ECR4E-expressing viruses. (A) The transduced cells were co-cultured with TGF-β-reporter TMLCs for 16–24 h before harvesting cell lysates and measuring luciferase activity. Experiments were performed in triplicate and the SDs of a single experiment are given. The errors bars represent the SD of a single experiment that was performed in triplicate. This experiment was repeated multiple times with similar results. (B) The transduced cells were transiently transfected with a TGF-β1 cDNA expression vector and allowed to generate CM for 16–24 h. The media were used for Western blotting. The reactive bands were revealed with an anti-LAP antibody (Vb3A9).
The above results suggested that ECR3E blocked activation by preventing the formation of the LLC. To test this possibility, we transiently transfected CHOβ6, CHOβ6/ECR3E, and CHOβ6/ECR4E cells with a full-length TGF-β expression construct. The conditioned media generated with these cells was collected and used to assess LLC formation by blotting with an anti-LAP antibody (Fig. 3 B). Whereas empty virus-transduced cells and ECR4E-transduced cells secreted both free LAP and LAP complexed with LTBP (Fig. 3 B, vector and ECR4E-2HA), ECR3E-transduced cells produced mostly LAP–ECR3E complex and barely detectable levels of LAP–LTBP complex (Fig. 3 B). Therefore, the inhibitory effect of ECR3E on latent TGF-β activation correlated with a decrease in the formation of LLC. These results suggested that the function of LTBP in αVβ6-mediated activation cannot be substituted by a minimal TGF-β binding cassette.
A fragment of the hinge domain rescues ECR3E function
Based upon the preceding results that (1) αVβ6-mediated activation requires a covalent interaction between LTBP and latent TGF-β and that (2) ECR3E is not sufficient to support αVβ6-mediated activation, we suspected that an additional domain of LTBP-1S is required for latent TGF-β activation by this mechanism. To define the domain of LTBP-1 involved in the activation process by a gain of function rather than a loss of function method, we required an experimental system in which αVβ6-mediated activation of latent TGF-β was ineffective unless rescued by the introduction of LTBP-1S. The development of such a system was hindered by endogenous LTBP expression in every cell line tested (unpublished data). However, we circumvented this problem by taking advantage of CHO-β6 cells that stably express ECR3E. These cells have an impaired ability to activate latent TGF-β due to a block in LLC formation (similar to Fig. 3 B). Therefore, by transiently transfecting LTBP-1S into CHO-β6/ECR3E cells, we could restore LLC formation and αVβ6-mediated latent TGF-β activation.
We mapped the LTBP-1S domains required for αVβ6-mediated latent TGF-β activation by testing expression constructs with increasing NH2-terminal deletions (Fig. 2 A, constructs II–VII). The initial results of these experiments indicated a requirement for a region between amino acids 216 and 342 (CR1 and EGF2; Fig. 4 A) based upon the fact that construct III supports integrin-mediated activation, but construct IV, which lacks CR1 and EGF2, does not. Further NH2-terminal deletion of LTBP-1 (constructs VI and VII) did not restore αVβ6-mediated activation of latent TGF-β (Fig. 4 A and not depicted). We attempted to identify the minimal LTBP-1S–derived construct that sustained activation (Fig. 4 B) by fusing the NH2-terminal portion of LTBP-1S (construct VIII, amino acids 1–529) to ECR3E. This construct retained the ability to bind latent TGF-β (Fig. 2 B), although the LTBP lacked the long EGF-like stretch and the COOH-terminal portion of LTBP-1S. Surprisingly, this construct rescued latent TGF-β activation (Fig. 4 B, construct VIII). Further NH2-terminal deletions of construct VIII (constructs IX, X, and XII) did not prevent latent TGF-β activation (Fig. 4 B). Indeed, a short amino acid sequence (amino acids 402–529) derived from the hinge domain, when fused to ECR3E, was sufficient to restore activation by CHO-β6/ECR3E cells (Fig. 4 B, construct XII). The apparent discrepancy between the results of the NH2-terminal deletion study that implicates amino acids 216–342 (CR1 and EGF2) and the minimal domain experiment that identifies amino acids 402–529 (hinge) as necessary for latent TGF-β activation raises the possibility that amino acids 216–342 modulate the function of the hinge domain by interacting with a distal portion of LTBP-1S, although other explanations are possible.
Figure 4.
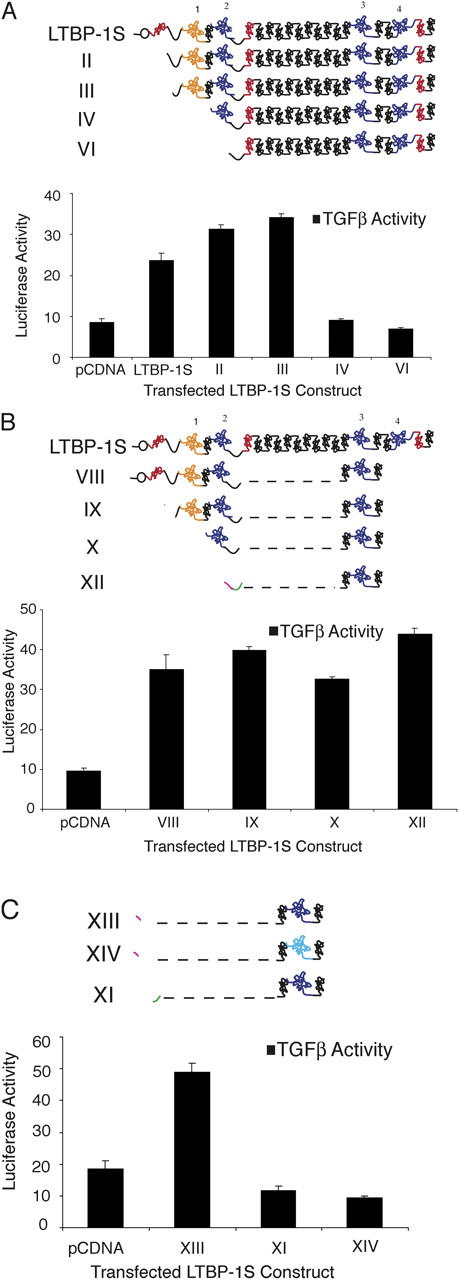
LTBP-1S–derived expression constructs rescue TGF-β activation. (A) CHO- β6/ECR3E cells were cotransfected with NH2-terminal deletion constructs and wild-type TGF-β1 before co-culture with TGF-β reporter TMLCs. After 16–24 h, the cell lysates were collected and luciferase activity measured. Similar experiments were conducted with additional LTBP-1S–derived expression constructs in B and C. In C, the size of the active hinge region (green) has been enlarged relative to other sequences for illustration purposes. In all cases, secretion of the transfected constructs and TGF-β complex formation was demonstrated (not depicted). Experiments were performed in triplicate. The error bars represent the SD from a single experiment.
To identify the required hinge domain, two additional LTBP-1–derived constructs were synthesized (Fig. 2 A, constructs XI and XIII) and tested. The results revealed that amino acids 402–449 (construct XIII), but not amino acids 449–529 (construct XI), from the hinge domain are sufficient to rescue ECR3E function (Fig. 4 C). Thus, the NH2-terminal portion of the hinge domain functions in a capacity that is not mimicked by the COOH-terminal portion of the hinge domain. In addition, we wanted to demonstrate that the ability of construct XIII (ΔN402-449-ECR3E) to support αVβ6-mediated latent TGF-β activation required a covalent interaction between latent TGF-β and the CR3 domain. To eliminate the disulfide interaction between LAP and the CR3 domain, we took advantage of the fact that the CR3 of LTBP-2 does not covalently bind LAP because of a two amino acid deletion compared with LTBP-1 (Saharinen and Keski-Oja, 2000). By deleting the phenylalanine and proline at positions 1060 and 1061 in the CR3 domain of construct XIII, such that it resembled the CR3 domain of LTBP-2, we generated construct XIV that fails to complex with latent TGF-β (Fig. 2 A; see Materials and methods; for review see Saharinen and Keski-Oja, 2000). As anticipated, construct XIV failed to support αVβ6-mediated latent TGF-β activation (Fig. 4 C). Thus, amino acids 402–449-ECR3E are sufficient to fulfill the function of LTBP-1S in αVβ6-mediated latent TGF-β activation, and this function requires covalent association between LAP and ECR3E.
Next, we sought to determine if regions of LTBP-1S other than the hinge domain could support integrin-mediated activation. To accomplish this, we tested a mutant form of LTBP-1 that lacked amino acids 402–449 for the ability to support activation (Fig. 2 A, construct I). Indeed, construct I failed to support αVβ6-mediated activation (Fig. 5 B). Interestingly, the hinge domain of LTBP-1 is not conserved among the other LTBP isoforms (Fig. 5 A). This fact raised the possibility that LTBP-1 performs a function that is not fulfilled by the other LTBP isoforms. To test this hypothesis, we transfected CHO-ECR3E cells with an LTBP-3 expression construct and measured latent TGF-β activation (Fig. 5 B). Despite the fact that LTBP-3 was secreted in complex with latent TGF-β at levels similar to LTBP-1 (Fig. 5 C, compare LTBP-1S with LTBP-3 [LTBP1 403-449]), LTBP-3 did not support latent TGF-β activation (Fig. 5 B). As a further test of possible isoform specificity, an LTBP-3 expression construct in which the native hinge domain was replaced with that of LTBP-1 (LTBP-3 with LTBP-1 hinge) was tested for the ability to support αVβ6-mediated activation. Strikingly, LTBP-3 with LTBP-1 hinge supported TGF-β generation (Fig. 5 B). This result indicates that the hinge domain of LTBP-1 confers an isoform-specific function within the LTBP family.
Figure 5.
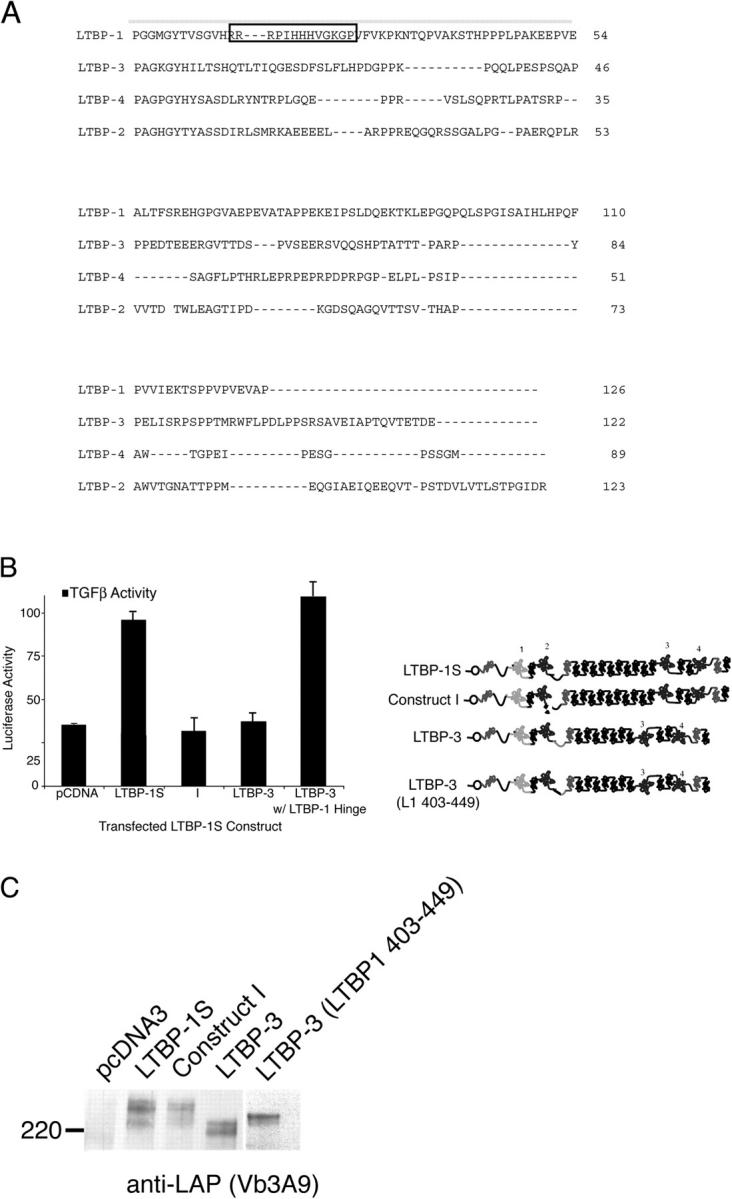
Activation and LLC formation of LTBP-1S and LTBP-3. (A) Alignment of the hinge domains of LTBP-1, -2, -3, and -4 using MAP (http://searchlauncher.bcm.tmc.edu/multi-align/multi-align.html). Amino acids 403–449 of LTBP-1S are overlined. The boxed amino acid sequence identifies a putative GAG-binding sequence. (B) CHO-β6/ECR3E cells were cotransfected with either full-length TGF-β1 and LTBP-1S–derived expression constructs or LTBP-3–based expression constructs before co-culture with TGF-β reporter TMLCs. After 16–24 h, cell lysates were harvested and luciferase activity measured. The error bars represent the SD of a single experiment that was performed in triplicate. This experiment was repeated multiple times with similar results. (C) Conditioned media from the same transfected cells were collected and subjected to Western blotting using Vb3A9 to reveal reactive protein bands.
αVβ6-mediated activation requires fixed latent TGF-β
Previous work in our laboratory has implicated the NH2-terminal portion of LTBP-1 in matrix incorporation of LTBP-1 (Nunes et al., 1997). This finding led us to test the possibility that amino acids 402–449 are capable of targeting the latent TGF-β complex to the ECM. CHO K7 cells were stably transduced with either PMX empty virus, construct XI, or construct XIII, subsequently transfected with latent TGF-β cDNA, and allowed to synthesize a matrix for 24 h. The cells were removed with EDTA, and the amount of TGF-β incorporated into the ECM was measured (see Materials and methods). Whereas construct XI reduced ECM targeting of TGF-β two- to threefold, construct XIII supported ECM association of latent TGF-β similar to empty virus-transduced cells (unpublished data). (Similar results were obtained with 2T3 and SW480 cells; unpublished data.) This result is consistent with our hypothesis that amino acids 402–449 are sufficient for ECM association.
The cosegregation of two activities for amino acids 402–449 of LTBP-1, namely support of latent TGF-β activation and ECM association, led us to suspect that fixation of latent TGF-β is required for αVβ6-mediated latent TGF-β activation. According to this hypothesis, expression of the minimal TGF-β binding construct, ECR3E, blocks integrin-mediated activation by preventing LLC formation thereby disrupting localization of the latent TGF-β complex. Therefore, we reasoned that artificial targeting of ECR3E-bound latent TGF-β to the vicinity of the ECM should restore αVβ6-mediated latent TGF-β activation. To test this prediction, we took advantage of the fact that the ECR3E constructs encode tandem COOH-terminal HA epitopes. Wells of a 96-well microtiter plate were either mock coated or anti-HA Ab coated (25 μg/ml). These wells were subsequently used to test latent TGF-β activation by CHO K7 cells engineered to express (1) β6 cDNA (Fig. 6 A, bars 1 and 5), (2) ECR3E and β6 cDNAs (Fig. 6 A, bars 2 and 6), (3) ECR3E cDNA (Fig. 6 A, bars 3 and 7), or (4) empty vectors (Fig. 6 A, bars 4 and 8). The β6-expressing cells activated their endogenous latent TGF-β when plated on either surface (Fig. 6 A, bars 1 and 5). However, by comparing bars 2 and 6 in Fig. 6 A, it is clear that the CHO-ECR3E/β6 cells demonstrate an enhanced (approximately four times) activation of latent TGF-β on wells coated with anti-HA Ab compared with mock-coated wells. In fact, on mock coated wells CHO-ECR3E/β6 activated approximately half as much latent TGF-β as CHO-β6 cells (Fig. 6 A, compare bar 1 with bar 2); on anti-HA Ab-coated wells CHO-ECR3E/β6 cells activated approximately two times more latent TGF-β than CHO-β6 (Fig. 6 A, compare bar 5 with bar 6). Additional controls demonstrate that expression of both ECR3E and αVβ6 are required for enhanced activation on anti-HA Ab-coated wells (Fig. 6 A, bars 3, 4, 7, and 8). We also observed a direct relationship between the HA Ab-coating concentration and TGF-β generation by CHO-ECR3E/β6 cells (Fig. 6 B). In all cases, the generation of active TGF-β was blocked by an αVβ6-specific blocking Ab (Fig. 6 B). Similar results were obtained with SW-480 cells (not depicted). These and the previous results suggest that LTBP-1S–derived expression constructs that support αVβ6-mediated activation do so because they effectively localize latent TGF-β, whereas constructs that fail to support activation do so because they cannot associate with the ECM.
Figure 6.
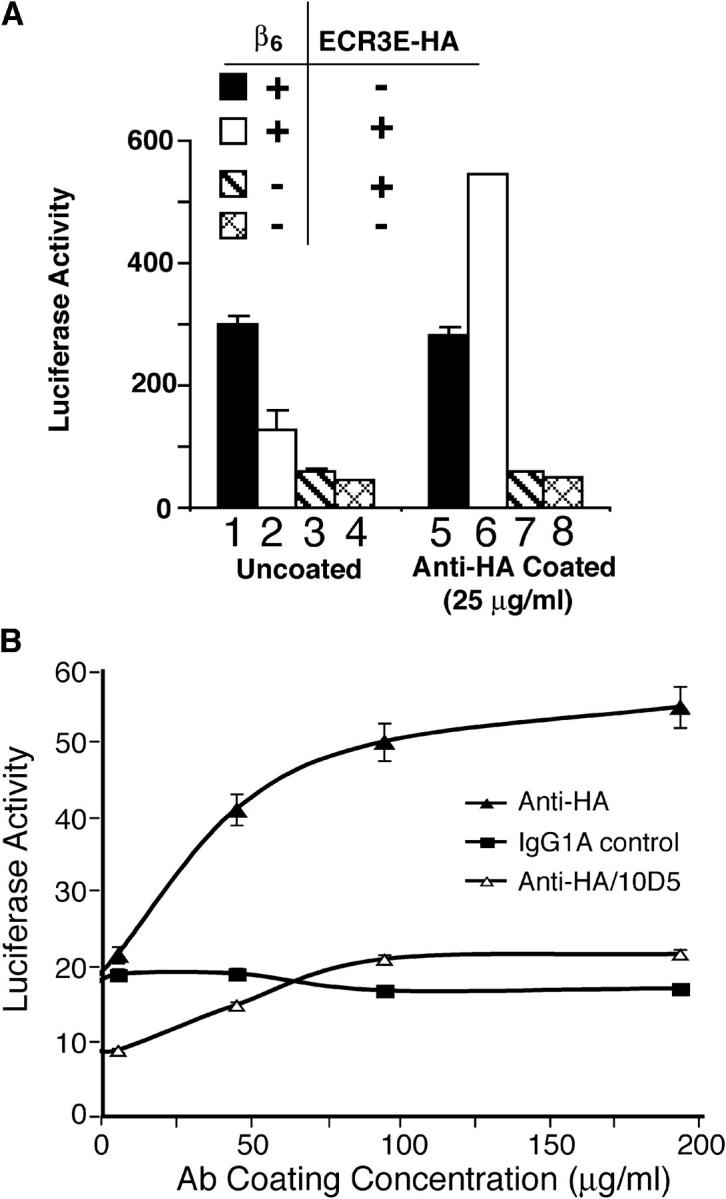
Activation of artificially localized latent TGF-β. (A) CHO cells that stably express β6-integrin (bars 1 and 5), β6-integrin and ECR3E (bars 2 and 6), ECR3E (bars 3 and 7), or untransfected (bars 4 and 8) were co-cultured with TMLCs on mock-coated or HA-antibody–coated wells. After 16–24 h, cell lysates were collected and assayed for luciferase activity. (B) The wells of a 96-well microtiter plate were coated with an anti-HA or control antibody, or anti-αvβ6 at various concentrations. CHO-ECR3E/β6 cells and TMLCs were co-cultured on these wells. After 16–24 h, the cell lysates were collected and the luciferase activity measured. Each experimental condition was performed in triplicate. The SD from a single experiment is shown.
An extension of the hypothesis that localization is required for αVβ6-mediated latent TGF-β activation is that latent TGF-β secreted and deposited in the ECM by one cell can be activated by a second cell. To test this prediction, we allowed CHO cells engineered to express either LTBP-1S or ECR3E to generate a matrix for 48 h. The matrix-producing cells (CHO-LTBP-1S and CHO-ECR3E cells) were removed and latent TGF-β1 activation measured by co-culture (Fig. 7). These cell types were selected because they do not efficiently activate endogenously produced latent TGF-β (Fig. 3 A and not depicted). Both SW480-ECR3E/β6 and CHO-ECR3E/β6 cells activate TGF-β when placed on a matrix produced by CHO-LTBP-1 cells. If these cells are plated on a matrix produced by CHO-ECR3E cells, considerably less TGF-β is formed (Fig. 7). This decrease in active TGF-β presumably represents the diminished amount of latent TGF-β incorporated into the matrix in the presence of ECR3E. This result indicates that latent TGF-β in the form of the LLC can be usefully stored and subsequently activated in the ECM.
Figure 7.

Activation of ECM deposited latent TGF-β. CHO cells stably transfected with ECR3E or LTBP-1S (5 × 104) were plated in a 96-well plate for 48 h before removal with PBS/20 mM EDTA. SW480-ECR3E/β6 or CHO-ECR3E/β6 cells and TMLCs were added to these wells. After 16–24 h, cell lysates were collected and luciferase activity measured. All experimental conditions were performed in triplicate. The SD from a single experiment is shown.
Discussion
LTBPs regulate TGF-β action through participation in secretion (Miyazono et al., 1991), ECM localization (Taipale et al., 1992, 1994b), and activation (Flaumenhaft et al., 1993; Nakajima et al., 1997; Gualandris et al., 2000) of latent TGF-β. How these distinct functions of LTBP relate to one another is not understood. Using αVβ6-mediated latent TGF-β activation as the model system, we have investigated the function of LTBP by exploring the relationship between localization and activation of latent TGF-β. Several lines of evidence support the hypothesis that αVβ6-mediated latent TGF-β activation requires LTBP to localize and fix latent TGF-β. First, recombinant SLC, which lacks LTBP, is not an effective substrate for activation, whereas latent TGF-β incorporated into the ECM by one cell may be used by another cell at a later time. Second, TGF-β that cannot covalently interact with LTBP fails to be activated by αVβ6. Third, a minimal TGF-β–binding cassette (ECR3E), derived from LTBP-1S, that cannot bind the ECM obstructs αVβ6-mediated activation. Fourth, activation of ECR3E-bound latent TGF-β can be accomplished through artificial localization and fixation of this complex to the pericellular environment.
The requirement of LTBP-dependent localization of latent TGF-β for activation suggests that TGF-β activity is regulated not only by the expression of latent TGF-β and an activator, but also by the different forms of LLC, the availability of a binding partner and the supply of ECM-stored latent TGF-β. For instance, removal of the NH2-terminal region of LTBP with plasmin generates a form of LTBP that resembles platelet LTBP and fails to be incorporated into the ECM (Taipale et al., 1994a). Consistent with our hypothesis, expression constructs that encode forms of LLC similar to platelet LLC (Fig. 2 A, constructs V, VI, VII) result in protein products that fail to support activation in our system (Kanzaki et al., 1990; Miyazono et al., 1991). In addition, the various isoforms of LTBP may differentially regulate TGF-β activity, as latent TGF-β bound to LTBP-3 is not a substrate for αVβ6-mediated activation. It is interesting that the greatest degree of sequence divergence between LTBP-1 and LTBP-3 is within their hinge domains and that it is this region of the molecule that confers an LTBP-1–specific function. This specificity for LTBP-1 hinge domain in αVβ6-mediated activation of latent TGF-β suggests that the hinge domains of LTBP-3, -2, and -4 may have distinct functions and cannot substitute for each other under all conditions. Furthermore, the activities of different LTBP isoforms and splice variants may relate to their distinct patterns of intra- and extracellular localization.
Our work indicates that processes that influence matrix deposition of latent TGF-β are predicted to affect αVβ6-dependent TGF-β generation both positively and negatively. Interestingly, matrix association of latent TGF-β may differentially influence protease-mediated activation versus αVβ6-mediated activation. Protease-mediated latent TGF-β activation may require release of latent TGF-β from the ECM for subsequent cell surface localization and activation (Flaumenhaft and Rifkin, 1992a; Nunes et al., 1997), whereas αVβ6-mediated latent TGF-β activation may occur while latent TGF-β is fixed. To better understand latent TGF-β activation and TGF-β activity in general, it will be important to answer questions such as: (1) Is LTBP required for other activation mechanisms? (2) What are the roles of the various LTBP isoforms in these processes? (3) How is activation of SLC accomplished/regulated?
Mapping identified a portion of the hinge domain, amino acids 402–449, and the TGF-β–binding domain as the minimal LTBP-1–derived domains that support αVβ6-mediated latent TGF-β activation. (The relationship of the hinge domain and the tTGase cross-linking site is unclear.) This hinge region may function by targeting latent TGF-β to an extracellular location. The hinge sequence of LTBP-1 contains a putative heparin-binding site that might facilitate ECM or cell surface targeting of the latent TGF-β complex (Oklu et al., 1998; Dallas et al., 2002; Isogai et al., 2003). Currently, we are trying to identify the hinge domain binding partner.
To understand the mechanism of αVβ6-mediated activation, it is necessary to appreciate the function of LTBP in this process. The results presented here suggest that LTBP promotes αVβ6-mediated activation by both concentrating and fixing the latent complex. The requirement for integrin–cytoskeleton interaction for latent TGF-β activation suggests that force generation by the integrin may be necessary. Indeed, the mechanism by which integrins generate force may explain why fixation of latent TGF-β in the ECM is important: integrin-dependent force generation across the cell membrane increases with increasing resistance (Choquet et al., 1997). We hypothesize that fixation of latent TGF-β provides resistance to integrin pulling, allowing focal contact reinforcement and force generation sufficient to release TGF-β from latency. Our current conception of αVβ6-mediated latent TGF-β activation is represented schematically in Fig. 8. In this model, LTBP-1 but not LTBP-3 is able to fix the latent complex to the ECM. Consequently, the LTBP-1–TGF-β complex is incorporated into the ECM and, therefore, when bound by αVβ6, promotes focal adhesion formation, force generation, and release of TGF-β from the latent complex. By contrast, integrin binding to latent the LTBP-3–TGF-β complex does not result in adhesion complex formation, force generation, and latent TGF-β activation. Consistent with this model, we predict that protease-mediated release of a soluble latent TGF-β restricts TGF-β generation by the integrin αVβ6. Thus, the function of LTBP-1 in αVβ6-mediated latent TGF-β activation is to concentrate and fix latent TGF-β so that integrin pulling is opposed. This resistance alters the conformation of LAP and releases TGF-β. The necessity of localization and force generation provide mechanisms for spatially restricting TGF-β activity and raises the threshold of latent TGF-β activation to limit inappropriate cytokine activity.
Figure 8.

Schematic representation of αVβ6-mediated latent TGF-β activation. TGF-β is secreted in a complex with a variety of LTBP isoforms and splice variants. The highly variable primary sequence of the hinge domain localizes latent TGF-β in the extracellular environment. Importantly, the hinge domain of LTBP-1 functions in a capacity that is not replicated by the hinge domain of LTBP-3. Once latent TGF-β is fixed in the ECM, the integrin αVβ6 binds LAP and generates a retractile force. The magnitude of this force is related to the resistance garnered through association of the latent complex with the ECM. Once the force generated by the integrin exceeds a threshold, biologically active TGF-β is made available. Release of the latent complex from its association with the ECM, for example by proteases, is predicted to prevent αVβ6-mediated latent TGF-β activation as integrin retraction will no longer be resisted.
Materials and methods
Cell lines and reagents
CHO K7 cells were obtained from the ATCC. CHO-ECR3E-2HA, CHO-ECR4E-2HA, and CHO-LTBP cells were generated by stable transfection of CHO K7 cells with a pcDNA3 (Invitrogen) expression vector containing ECR3E-2HA, ECR4E-2HA, and LTBP-1S coding sequences, respectively. The generation of these expression constructs is described below. Stable cell lines were selected with G418 Sulfate (Mediatech) and cloned by limiting dilution. CHO-ECR3E-2HA/β6, CHO-ECR4E-2HA/β6 and CHO-LTBP/β6 cells were generated by retroviral transduction of the stable lines with PMX/β6 retrovirus. Retroviral production was accomplished by using the Pantropic Retroviral Expression System following the manufacturer's instructions (CLONTECH Laboratories, Inc.). TMLC, which produce luciferase in response to TGF-β, were used as described previously (Abe et al., 1998). TGF-β1−/− liver fibroblasts were a gift from A. Roberts (National Institutes of Health, Bethesda, MD) were stably transfected with either pCDNA3.1/Hygro (−) (TGF-β1−/− cells) or pCDNA3.1/Hygro (−) containing the β6-subunit cDNA (TGF-β1−/−/β6 cells) as described previously (Annes et al., 2002). β6-Integrin transfected SW480 (SW480/β6 cells) and CHO cells (CHO/β6 cells) were a gift from D. Sheppard (University of California, San Francisco [UCSF], San Francisco, CA; Weinacker et al., 1994). SW480-ECR3E-2HA/β6 were generated by retroviral transduction of SW480/β6 cells with PMX-ECR3E-2HA.
Cells were cultured in Dulbecco's minimum essential medium (DMEM) containing 10% heat-inactivated FCS. Mouse mAb HA.11 against the HA epitope was purchased from Covance Research Products (Berkeley Antibody Company). VB3A9 is an mAb directed against the LAP portion of human TGF-β1 (Munger et al., 1998). Ab39, rabbit polyclonal antisera raised against platelet LTBP-1, was provided by C.-H. Heldin (Ludwig Institute for Cancer Research, Uppsala, Sweden). Mouse anti-αVβ6 mAb 10D5 (Huang et al., 1998) was a gift of D. Sheppard. Recombinant simian TGF-β1 LAP was produced as described previously (Munger et al., 1998). All molecular biology enzymes were purchased from Roche Diagnostics. Recombinant human TGF-β1 SLC was purchased from R&D Systems.
Constructs and vectors
pcDNA3 and pSecTag 2C vectors were obtained from Invitrogen. PMX retroviral vector was provided by T. Kitamura (Tokyo University, Tokyo, Japan). Simian TGF-β1 cDNA was a gift from R. Derynck (UCSF). Human TGF-β1 C33S cDNA was a gift from J. Keski-Oja (University of Helsinki, Helsinki, Finland). Human LTBP-1S cDNA (pSV7d-BP13) was a gift of K. Miyazono and C.-H. Heldin (Ludwig Institute for Cancer Research). β6-Integrin cDNA was a gift of D. Sheppard.
Generation of pcDNA3-LTBP-1S and pcDNA3-ΔN441 expression vectors was described previously (Nunes et al., 1997). In brief, pcDNA3-LTBP-1S was constructed by fusing the DraI–DrI fragment of pSV7d-BP13 (nucleotides 68–4543) in frame with the baculovirus glycoprotein GP67 signal sequence in pcDNA3. Similarly, pcDNA3-ΔN441 was generated by fusing the HpaI–DraI fragment (nucleotides 1414–4543) of pSV7d-BP13 in frame with the GP67 signal sequence in the pcDNA3 vector.
The PCR primers and templates used for generation of the expression constructs described below are given in Table I. The expression construct pcDNA3-ECR3E-2HA (nucleotides 2872–3408) was generated by PCR amplification (primers 1, 2) using pcDNA3-LTBP-1S as the template. This PCR product was fused to the BM40 signal sequence the pRcCMV/Ac7 vector (gift of R. Timpl, Max-Plank Institute, Martinsried, Germany). The pRcCMV/Ac7–ECR3E vector was digested with HpaI–XbaI and an adapter cassette coding for two copies of the HA-epitope was subcloned in frame and downstream of the ECR3E coding sequence. The ECR3E-2HA coding sequence was subsequently transferred from pRcCMV/Ac7 to an intermediate vector (pKN185; gift of Y. Yamada, National Institutes of Health) using an EagI digest of both vectors. The ECR3E-2HA coding sequence was subsequently released from PKN185-ECR3E-2HA by a HindIII–BamHI digest and subcloned into pCDNA3 (HindIII–BamHI) and pBluescript II KS+ (Strategene). ECR3E-2HA was transferred from pBluescript II KS+ -ECR3E-2HA (pBlue-ECR3E-2HA) to PMX (HindIII–NotI digest). To make PMX-ECR4E-2HA, the ECR4E coding sequence was amplified (primers 3, 4) using pcDNA3-LTBP-1S as the template and subcloned into the pBlue-ECR3E-2HA vector backbone, which retained the BM40 signal sequence and the 2HA epitopes. This was accomplished by SpeI–HpaI digest of both the ECR4E PCR product and pBlue-ECR3E-2HA vector and ligation of the digested ECR4E PCR product into the pBlue-ECR3E-2HA vector backbone. ECR4E-2HA was subsequently transferred to the PMX retroviral vector by HindIII–NotI digest of pBlue-ECR4E-2HA, purification of the ECR4E-2HA insert, and ligation into similarly digested PMX retrovirus vector DNA.
Table I. Construction of expression vectors.
| Description | Primers |
|---|---|
| pCDNA3 LTBP-1S | a gift from K. Miyazono and C-H. Heldin (Ludwig Institute for Cancer Research, Uppsala, Sweden) |
| pCDNA3 LTBP-1S ΔN441 | described previously in Nunes et al., 1997 |
| pCDNA3 ECR3E-HA | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S) |
| pBS ECR3E-HA | primer 1: cggggatccactagtggatgtgaatgaatgtgaact |
| PMX ECR3E-HA | primer 2: aacaagcactgcagtttcacagg; *A 3′ HA epitope tag was added via ligation of an adapter cassette; 5′-aacctacccctacgacgtgcccgactacgcctacccctacgacgtgcccgactacgcct; gaagatcttgattggaattccggccgt-3′ |
| PMX ECR4E | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S) primer 3: cactagtggatatggatgaatgtcaagaccccagt; primer 4: gggtaggttaacagatcttcttcagaaataagtttttgttccacgtggaaacaggtcatcttggcagtatc |
| pcDNA3 ΔC529-ECR3E-HA | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S); primer 9: atttacgaattcatggatactaagctgatgtgtttg; primer 10: cacattcattcacatcaggagctacttcaacaggcacaggagg; Reaction 2 (template = pCDNA3 ECR3E); primer 11: gaagtagctcctgatgtgaatgaatgtgaactgctcagtgg; primer 12: tcaaatgcggccgctcaggcgtagtcgggcacgtcg |
| pcDN3 ΔC412-ECR3E-HA | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S); primer 13: atttacgaattcatggatactaagctgatgtgtttg; primer 14: attcattcacatcgccagaaaccgtataaccc; Reaction 2 (template = pCDNA3 ECR3E); primer 15: acggtttctggcgatgtgaatgaatgtgaactgc; primer 16: tcaaatgcggccgctcaggcgtagtcgggcacgtcg |
| pSecTag2C LTBP-1S ΔN104 | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S); primer 17: ctatcagcggccgcctcagatcccagtccatggtgcc; primer 18: tgtttactcgagactccaggtcactgtctttctctaaattcaagg |
| pSecTag2C LTBP-1S ΔN216 | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S); primer 19: ctatcagcggccgcctgtcattcctcacgtctaccc; primer 18: as above |
| pSecTag2C LTBP-1S ΔN342 | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S); primer 20: ctatcagcggccgcctccccctgtgatctcggaagagaaaggg; primer 18: as above |
| pSecTag2C LTBP-1S ΔN402 | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S); primer 21: ctatcagcggccgcctccccctgtgatctcggaagagaaaggg; primer 18: as above |
| pSecTag2C LTBP-1S ΔN441 | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S); primer 22: ctatcagcggccgcctactcatcctccacctctcccagcc; primer 18: as above |
| pSecTag2C LTBP-1S ΔN529 | PCR amplification: Reaction 1 (template = pCDNA3 LTBP-1S); primer 23: ctatcagcggccgcctgcttctacgtctagtgccagcc; primer 18: as above |
| pSecTag 2C ΔN216-ΔC529-ECR3E-HA |
PCR amplification: Reaction 1 (template = pcDNA3 ΔC529-ECR3E-HA); primer 24: ctatcagcggccgcctgtcattcctcacgtctaccc; primer 25: cacattcattcacatcaggagctacttcaacaggcacaggagg; Reaction 2 (pcDNA3 ΔC529-ECR3E-HA); primer 11/primer 12 |
| pSecTag 2C ΔN342-ΔC529-ECR3E-HA |
PCR amplification: Reaction 1 (template = pcDNA3 ΔC529-ECR3E-HA); primer 26: ctatcagcggccgcctccccctgtgatctcggaagagaaaggg; primer 25: as above; Reaction 2 (pcDNA3 ΔC529-ECR3E-HA); primer 11/primer 12 |
| pSecTag 2C ΔN402-ΔC529-ECR3E-HA |
PCR amplification: Reaction 1 (template = pcDNA3 ΔC529-ECR3E-HA); primer 27: ctatcagcggccgcctcctggtggaatgggttatacggtttctggcg; primer 25: as above; Reaction 2 (pcDNA3 ΔC529-ECR3E-HA); primer 11/primer 12 |
| pSecTag 2C ΔN449-ΔC529-ECR3E-HA |
PCR amplification: Reaction 1 (template = pcDNA3 ΔC529-ECR3E-HA); primer 28: ctatcagcggccgcctaaggaagagccagtggaggccctgacc; primer 25: as above; Reaction 2 (pcDNA3 ΔC529-ECR3E-HA); primer 11/primer 12 |
| pSecTag 2C ΔN402-ΔC449-ECR3E-HA |
PCR amplification: Reaction 1 (template = pcDNA3 LTBP-1S); primer 29: cccaagcttggcctggtggaatgggttatacg; primer 30: cgggatcccggctgggagaggtggag |
| pSecTag 2C ΔN19-ΔC449-ECR3E-HA |
PCR amplification: Reaction 1 (template = pcDNA3 LTBP-1S); primer 31: cccaagcttggagtaaccacactggccgcatc; primer 32: cgggatcccttggcaaaaggtgttgttaaccc |
| pcDNA3 LTBP-1S Δ403-449 |
PCR amplification: Reaction 1 (template = pcDNA3 LTBP-1S); primer 33: gcagctatcgatgtacctgc; primer 34: gggatatcacagatttccttaaaagcagctgt; Reaction 2 (template = pcDNA3 LTBP-1S); primer 35: gggttaacaaggaagagccagtggaggc; primer 36: cccctgtccacaggtgcac |
| pcDNA3 LTBP-1S-HA |
PCR amplification: Reaction 1 (template = respective pcDNA3 untagged); primer 37: cccaagcttgggttcatagacgcaggccaatc |
| pcDNA3 LTBP-1S Δ403-449-HA |
primer 38: ctagtctagactaagcgtagtctgggacgtcgtatgggtactccaggtcactgtctttctc |
| pcDNA3; LTBP-1S ΔN402-HA | |
| PMX ΔN402-ΔC449-ECR3E-HA |
PCR amplification: Reaction 1 (template = respective pSecTag2C vectors); primer 39: ttagtagaattcatggagacagacacactcctgc |
| PMX ΔN402-ΔC449-ECR3E-HA; L1→L2 |
primer 40: ccgctcgagcggttatcaggcgtagtcgggcacgtc |
| PMX; ΔN449-ΔC529-ECR3E-HA | |
| pcDNA3 LTBP-3 with LTBP-1 hinge |
PCR amplification: Reaction 1 (template = pcDNA3 LTBP-3); primer 41: gccaggatatcaacgaatgtgcgatgccc; primer 42: cccattccaccagggcagatctccttgaaggc; Reaction 2 (template = pcDNA3 LTBP-1); primer 43: caaggagatctgccctggtggaatgggttatacgg; primer 44: ttcaatcggcattcattgatttctgtcacttgagtagg; Reaction 4 (template = pcDNA3 LTBP-3); primer 45: aaatcaatgaatgccgattgaaccagaatatctgtgg; primer 46: gcaggtgctagggtcgcgacactc |
A variety of constructs were synthesized using the technique of strand overlap extension in which two separate PCR reactions (reactions 1 and 2) were set up to generate overlapping DNA products that were subsequently purified, mixed, and sewn together in a third PCR reaction (reaction 3). The extreme 5′ and 3′ primers from the first PCR reactions were used in reaction 3. The expression vectors pcDNA3-ΔC529/ECR3E-2HA and pcDNA3-ΔC412/ECR3E-2HA were constructed using strand overlap extension PCR (primers 9/10, 11/12 and 13/14, 15/16, respectively) with pcDNA3-LTBP-1S as the template. The products were digested with EcoRI–NotI and ligated into similarly digested pcDNA3. Several NH2-terminal deletion constructs were made by fusing PCR amplified portions of LTBP-1S downstream and in frame with the murine Ig κ-chain leader sequence present in the pSecTag2C vector (Invitrogen; pSecTag2C-ΔN104 primers 17, 18; pSecTag2C-ΔN216 primers 19, 18; pSecTag2C-ΔN342: primers 20, 18; pSecTag2C-ΔN402 primers 21, 18 pSecTag2C-ΔN441 primers 22, 18; pSecTag2C-ΔN529 primers 23, 18). These PCR products were digested with NotI–XhoI and subcloned into similarly digested pSecTag2C.
A number of LTBP-1S deletion constructs were made in pSecTag2C using strand overlap extension PCR reactions pSecTag2C-ΔN216/ΔC529-ECR3E-2HA (primers 24/25,11/12); pSecTag2C-ΔN342/ΔC529-ECR3E-2HA (primers 26/25, 11/12); pSecTag2C-ΔN402/ΔC529-ECR3E-2HA (primers 27/25, 11/12); pSecTag2C-ΔN449/ΔC529-ECR3E-2HA (primers 28/25, 11/12). In all cases, the amplified products of reaction 3 were NotII–XhoI digested before being subcloned into similarly digested pSecTag2C vector. pSecTag2C-ΔN402/ΔC449-ECR3E-HA L1→L2 was generated by two successive QuickChange Site-Directed Mutagenesis reactions (Stratagene). The first reaction deleted the phenylalanine and proline (amino acids 1060 and 1061 of LTBP-1S) that are found in CR3 of human LTBP-1 but not in CR3 of human LTBP-2. This was accomplished using a sense primer (5′-GGGGAGATAACTGCGAAATCTGCCCGGTCTTGGGAACTGC-3′) and an equivalent antiparallel primer. The second reaction mutated the glutamate and isoleucine of human LTBP-1 CR3 (amino acids 1058 and 1059 of LTBP-1S) to aspartate and leucine, which are found in the homologous region of human CR3 of LTBP-2. This was accomplished using a sense primer (5′-GGGGAGATAACTGCGACCTCTGCCCGGTCTTGGGAACTGC-3′) and an equivalent antiparallel primer. pSecTag2C–ΔN402/ΔC449-ECR3E-HA was generated by PCR amplification (primers 29, 30). The PCR fragment was digested with HindIII and BamHI and cloned into a similarly digested pSecTag2C-ECR3E-HA vector. pSecTag2C-ΔN19/ΔC449-ECR3E-HA was constructed by PCR amplification (primers 31, 32). The PCR product was digested with HindIII and BamHI and cloned into similarly digested pSecTag2C-ECR3E-HA vector.
pcDNA-LTBP1 Δ402-449 was constructed by PCR amplification (primers 33/34, 35/36) of 5′ and 3′ fragments that were digested (reaction 1, ClaI–EcoRV; reaction 2, HpaI–EcoRI), purified and cloned into pcDN3-LTBP-1S digested with ClaI–EcoRI. HA tagged versions of pcDNA3-LTBP-1, -LTBP-1S-ΔN402-449, and -LTBP-ΔN402 were generated by digesting these constructs with EcoRI–XbaI and cloning in a COOH-terminal HA tagged amplified product from pcDNA3-LTBP-1S (primers 37, 38). PMX-ΔN402/ΔC449-ECR3E-HA, PMX-ΔN402/ΔC449-ECR3E-HA L1→2, and PMX-ΔN449/ΔC529-ECR3E-HA were generated by PCR amplification using the respective pSecTag2C vectors as templates (primers 39, 40). The PCR products were digested with EcoRI–XhoI and cloned into PMX vector digested with EcoRI–SalI. PMX-β6 retroviral vector was prepared by isolating the Pme digestion fragment of pHygrobeta6 and cloning into filled-in EcoRI digested PMX virus (Annes et al., 2002). The LTBP-3 with the LTBP-1 hinge expression construct was synthesized by a series of strand-overlap extension PCR reactions that sewed the LTBP-1 hinge domain into the LTBP-3 coding sequence. First, overlapping PCR products derived from LTBP-3 (upstream of the hinge domain) and LTBP-1 were synthesized separately (primers 41/42 and 43/44). These products were then combined together for reaction 3. A fourth PCR reaction was set up to generate an LTBP-3–derived product (downstream of the hinge domain) that also overlapped with the LTBP-1 hinge domain (primers 45/46). This product was combined with the product of reaction 3 and sewn together in a fifth PCR reaction (primers 41/46). The product of this PCR reaction was digested with EcoRV and Nru and cloned into similarly digested pcDNA3-LTBP-3. All constructs were checked by automated sequencing.
TGF-β bioassays
TGF-β activation was measured using CHO cells stably transfected with an ECR3E-2HA expression construct and the β6-integrin subunit (CHO-ECR3E-2HA/β6) subsequently transiently transfected with proTGF-β1 and various LTBP-1S–derived cDNAs. Before transient transfection, test cells were plated at 4 × 105 cells per 35-mm well in DMEM/10% FCS. After 16 h, cells were transfected with the LTBP-1S expression constructs (1 μg per well) and the TGF-β1 expression construct (400 ng per well) using LipofectAMINE Plus. After 16 h, the cells were collected in 3 ml of DMEM/10% FCS and replated in 96-well and 24-well plates (50 and 500 μl per well, respectively). TMLC (1.5 × 104) were added to the 96-well plates (final volume, 100 μl per well). When appropriate, 10D5 (20 μg/ml) or LAP (100 μg/ml) was added to the co-culture. Conditioned media were generated in the 24-well plate monocultures. After 16–24 h, TGF-β activation was assessed by measuring luciferase activity in the cell lysates from co-cultures (Abe et al., 1994). In addition, the conditioned media from monoculture wells was collected after 16–24 h and analyzed by immunoblotting (Ab39 or HA.11) for secretion of the various LTBP forms as well as by TMLC assay for total TGF-β secretion by heat activating latent TGF-β (80°C for 10 min). The samples were incubated with TMLC overnight and luciferase activity measured.
To test TGF-β1 SLC as a substrate for αVβ6-mediated latent TGF-β activation, TGF-β1−/− cells and TGF-β1−/−/β6 cells (1.5 × 104 cells per well) were co-cultured with TMLC (1.0 × 104 cells per well) in 96-well plates in the presence of TGF-β1 SLC (0–200 ng/ml of TGF-β1 SLC). After 16–24 h, luciferase activity in the cell lysates was measured. To test if αVβ6-mediated latent TGF-β activation requires TGF-β binding to LTBP, TGF-β1−/−/β6 cells (8 × 104 cells per 35-mm well) were transiently transfected with cDNAs encoding either wild-type human TGF-β1 (400 ng per well) or human TGF-β1C:S (400 ng per well). The establishment of co-cultures to measure TGF-β activation and monocultures to measure total TGF-β secretion was as described previously (Abe et al., 1994).
ECM deposition of latent TGF-β was testing by using, CHO-K7, SW-480, or 2T3 osteoblast precursor cells transduced with PMX virus, 402-449ECR3E virus, or 450529ECR3E virus and subsequently transfected with human TGF-β1 cDNA. 2 d after transfection, cells were replated in 96-well plates, 20,000 cells per well. 24 h later, cells in 96-well plates were washed once with PBS, and detached from the cell culture plate with 20 mM EDTA/PBS at 37°C for 30–40 min. Wells were washed two more times with PBS, and 100 μl DMEM was added into each well. The 96-well plate was incubated at 80°C for 20 min to activate and release matrix-bound latent TGF-β. The media were collected and put onto TMLC to measure TGF-β activity.
To test the ability of CHO-ECR3E-2HA/β6 cells to activate latent TGF-β deposited in the ECM, CHO cells stably transfected with ECR3E (CHO-ECR3E) or LTBP-1S (CHO-LTBP) were allowed to synthesize a matrix in 96-wells (5.0 × 104 cells per well) for 48 h before they were removed with PBS/15 mM EDTA. CHO-ECR3E-2HA/β6 cells (2.0 × 104 cells per well) or SW480-ECR3E-2HA/β6 cells (2.5 × 104 cells per well) were plated on the preformed matrices with reporter cells (1.5 × 104 cells per well; 100 μl final volume) to measure TGF-β activity. A TGF-β neutralizing antibody (1D11; 15 μg/ml) or an αVβ6-blocking antibody (10D5; 20 μg/ml) was added to co-culture wells as appropriate.
To test if artificially targeting ECR3E-2HA–bound latent TGF-β to the vicinity of the ECM restored latent TGF-β activation, the wells of a 96-well plate were either coated with the mouse mAb HA.11 (25 μg/ml) in PBS or mock coated for 1.5 h at 37°C. The wells were washed with DMEM/10% FCS, and various cell types (SW480/β6, SW480-ECR3E-2HA/β6, SW480-ECR3E-2HA, SW480-PMX) were added (2.0 × 104 cells per well) to both anti-HA–coated and mock-coated wells. TMLC (1.5 × 104 cells per well) were added to each well to a final volume of 100 μl per well. TGF-β neutralizing antibody (1D11; 15 μg/ml) was added as appropriate. After 16–24 h, luciferase activity was assayed.
Acknowledgments
The authors thank Ms. Melinda Vassallo and Eric D. Annes for their expert technical assistance.
This work was supported by grants CA34282, CA78422, DE-13742 (to D.B. Rifkin), and T32 GM07308 (to J.P. Annes).
Abbreviations used in this paper: CR, cysteine rich; CR3, third CR; DMEM, Dulbecco's minimum essential medium; LAP, latency-associated protein; LLC, large latent complex; LTBP, latent TGF-β binding protein; SLC, small latent complex; TMLC, transformed mink lung cells; tTGase; tissue transglutaminase.
References
- Abe, M., J.G. Harpel, C.N. Metz, I. Nunes, D.J. Loskutoff, and D.B. Rifkin. 1994. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216:276–284. [DOI] [PubMed] [Google Scholar]
- Abe, M., N. Oda, and Y. Sato. 1998. Cell-associated activation of latent transforming growth factor-beta by calpain. J. Cell. Physiol. 174:186–193. [DOI] [PubMed] [Google Scholar]
- Annes, J.P., J.S. Munger, and D.B. Rifkin. 2003. Making sense of latent TGFbeta activation. J. Cell Sci. 116:217–224. [DOI] [PubMed] [Google Scholar]
- Annes, J.P., D.B. Rifkin, and J.S. Munger. 2002. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett. 511:65–68. [DOI] [PubMed] [Google Scholar]
- Blobe, G.C., W.P. Schiemann, and H.F. Lodish. 2000. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 342:1350–1358. [DOI] [PubMed] [Google Scholar]
- Breuss, J.M., N. Gillett, L. Lu, D. Sheppard, and R. Pytela. 1993. Restricted distribution of integrin beta 6 mRNA in primate epithelial tissues. J. Histochem. Cytochem. 41:1521–1527. [DOI] [PubMed] [Google Scholar]
- Choquet, D., D.P. Felsenfeld, and M.P. Sheetz. 1997. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 88:39–48. [DOI] [PubMed] [Google Scholar]
- Dallas, S.L., D.R. Keene, S.P. Bruder, J. Saharinen, L.Y. Sakai, G.R. Mundy, and L.F. Bonewald. 2000. Role of the latent transforming growth factor beta binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J. Bone Miner. Res. 15:68–81. [DOI] [PubMed] [Google Scholar]
- Dallas, S.L., S. Park-Snyder, K. Miyazono, D. Twardzik, G.R. Mundy, and L.F. Bonewald. 1994. Characterization and autoregulation of latent transforming growth factor beta (TGF beta) complexes in osteoblast-like cell lines. Production of a latent complex lacking the latent TGF beta-binding protein. J. Biol. Chem. 269:6815–6821. [PubMed] [Google Scholar]
- Dallas, S.L., J.L. Rosser, G.R. Mundy, and L.F. Bonewald. 2002. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 277:21352–21360. [DOI] [PubMed] [Google Scholar]
- Dubois, C.M., M.H. Laprise, F. Blanchette, L.E. Gentry, and R. Leduc. 1995. Processing of transforming growth factor beta 1 precursor by human furin convertase. J. Biol. Chem. 270:10618–10624. [DOI] [PubMed] [Google Scholar]
- Flaumenhaft, R., M. Abe, Y. Sato, K. Miyazono, J. Harpel, C.H. Heldin, and D.B. Rifkin. 1993. Role of the latent TGF-beta binding protein in the activation of latent TGF-beta by co-cultures of endothelial and smooth muscle cells. J. Cell Biol. 120:995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaumenhaft, R., and D.B. Rifkin. 1992. a. Cell density dependent effects of TGF-beta demonstrated by a plasminogen activator-based assay for TGF-beta. J. Cell. Physiol. 152:48–55. [DOI] [PubMed] [Google Scholar]
- Flaumenhaft, R., and D.B. Rifkin. 1992. b. The extracellular regulation of growth factor action. Mol. Biol. Cell. 3:1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleizes, P.E., R.C. Beavis, R. Mazzieri, B. Shen, and D.B. Rifkin. 1996. Identification and characterization of an eight-cysteine repeat of the latent transforming growth factor-beta binding protein-1 that mediates bonding to the latent transforming growth factor-beta1. J. Biol. Chem. 271:29891–29896. [DOI] [PubMed] [Google Scholar]
- Gualandris, A., J.P. Annes, M. Arese, I. Noguera, V. Jurukovski, and D.B. Rifkin. 2000. The latent transforming growth factor-beta-binding protein-1 promotes In vitro differentiation of embryonic stem cells into endothelium. Mol. Biol. Cell. 11:4295–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X., J. Wu, S. Spong, and D. Sheppard. 1998. The integrin alphavbeta6 is critical for keratinocyte migration on both its known ligand, fibronectin, and on vitronectin. J. Cell Sci. 111:2189–2195. [DOI] [PubMed] [Google Scholar]
- Huang, X.Z., J.F. Wu, D. Cass, D.J. Erle, D. Corry, S.G. Young, R.V. Farese, and D. Sheppard. 1996. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J. Cell Biol. 133:921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai, Z., R.N. Ono, S. Ushiro, D.R. Keene, Y. Chen, R. Mazzieri, N.L. Charbonneau, D.P. Reinhardt, D.B. Rifkin, and L.Y. Sakai. 2003. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 278:2750–2757. [DOI] [PubMed] [Google Scholar]
- Kaminski, N., J.D. Allard, J.F. Pittet, F. Zuo, M.J. Griffiths, D. Morris, X. Huang, D. Sheppard, and R.A. Heller. 2000. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc. Natl. Acad. Sci. USA. 97:1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki, T., A. Olofsson, A. Moren, C. Wernstedt, U. Hellman, K. Miyazono, L. Claesson-Welsh, and C.H. Heldin. 1990. TGF-beta 1 binding protein: a component of the large latent complex of TGF-beta 1 with multiple repeat sequences. Cell. 61:1051–1061. [DOI] [PubMed] [Google Scholar]
- Kojima, S., and D.B. Rifkin. 1993. Mechanism of retinoid-induced activation of latent transforming growth factor-beta in bovine endothelial cells. J. Cell. Physiol. 155:323–332. [DOI] [PubMed] [Google Scholar]
- Koski, C., J. Saharinen, and J. Keski-Oja. 1999. Independent promoters regulate the expression of two amino terminally distinct forms of latent transforming growth factor-beta binding protein-1 (LTBP-1) in a cell type-specific manner. J. Biol. Chem. 274:32619–32630. [DOI] [PubMed] [Google Scholar]
- Lack, J., J.M. O'Leary, V. Knott, X. Yuan, D.B. Rifkin, P.A. Handford, and A.K. Downing. 2003. Solution structure of the third TB domain from LTBP1 provides insight into assembly of the large latent complex that sequesters latent TGF-beta. J. Mol. Biol. 334:281–291. [DOI] [PubMed] [Google Scholar]
- Lawrence, D.A., R. Pircher, C. Kryceve-Martinerie, and P. Jullien. 1984. Normal embryo fibroblasts release transforming growth factors in a latent form. J. Cell. Physiol. 121:184–188. [DOI] [PubMed] [Google Scholar]
- Lu, M., J.S. Munger, M. Steadele, C. Busald, M. Tellier, and L.M. Schnapp. 2002. Integrin alpha8beta1 mediates adhesion to LAP-TGFbeta1. J. Cell Sci. 115:4641–4648. [DOI] [PubMed] [Google Scholar]
- Miyazono, K., A. Olofsson, P. Colosetti, and C.H. Heldin. 1991. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO J. 10:1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, D.G., X. Huang, N. Kaminski, Y. Wang, S.D. Shapiro, G. Dolganov, A. Glick, and D. Sheppard. 2003. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature. 422:169–173. [DOI] [PubMed] [Google Scholar]
- Munger, J.S., J.G. Harpel, P.E. Gleizes, R. Mazzieri, I. Nunes, and D.B. Rifkin. 1997. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int. 51:1376–1382. [DOI] [PubMed] [Google Scholar]
- Munger, J.S., J.G. Harpel, F.G. Giancotti, and D.B. Rifkin. 1998. Interactions between growth factors and integrins: latent forms of transforming growth factor-beta are ligands for the integrin alphavbeta1. Mol. Biol. Cell. 9:2627–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger, J.S., X. Huang, H. Kawakatsu, M.J. Griffiths, S.L. Dalton, J. Wu, J.F. Pittet, N. Kaminski, C. Garat, M.A. Matthay, et al. 1999. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 96:319–328. [DOI] [PubMed] [Google Scholar]
- Nakajima, Y., K. Miyazono, M. Kato, M. Takase, T. Yamagishi, and H. Nakamura. 1997. Extracellular fibrillar structure of latent TGF beta binding protein-1: role in TGF beta-dependent endothelial-mesenchymal transformation during endocardial cushion tissue formation in mouse embryonic heart. J. Cell Biol. 136:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes, I., P.E. Gleizes, C.N. Metz, and D.B. Rifkin. 1997. Latent transforming growth factor-beta binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-beta. J. Cell Biol. 136:1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oklu, R., J.C. Metcalfe, T.R. Hesketh, and P.R. Kemp. 1998. Loss of a consensus heparin binding site by alternative splicing of latent transforming growth factor-beta binding protein-1. FEBS Lett. 425:281–285. [DOI] [PubMed] [Google Scholar]
- Ramirez, F., and L. Pereira. 1999. The fibrillins. Int. J. Biochem. Cell Biol. 31:255–259. [DOI] [PubMed] [Google Scholar]
- Saharinen, J., and J. Keski-Oja. 2000. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell. 11:2691–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen, J., J. Taipale, and J. Keski-Oja. 1996. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 15:245–253. [PMC free article] [PubMed] [Google Scholar]
- Sinha, S., C. Nevett, C.A. Shuttleworth, and C.M. Kielty. 1998. Cellular and extracellular biology of the latent transforming growth factor-beta binding proteins. Matrix Biol. 17:529–545. [DOI] [PubMed] [Google Scholar]
- Taipale, J., and J. Keski-Oja. 1997. Growth factors in the extracellular matrix. FASEB J. 11:51–59. [DOI] [PubMed] [Google Scholar]
- Taipale, J., K. Koli, and J. Keski-Oja. 1992. Release of transforming growth factor-beta 1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. J. Biol. Chem. 267:25378–25384. [PubMed] [Google Scholar]
- Taipale, J., S. Matikainen, M. Hurme, and J. Keski-Oja. 1994. a. Induction of transforming growth factor beta 1 and its receptor expression during myeloid leukemia cell differentiation. Cell Growth Differ. 5:1309–1319. [PubMed] [Google Scholar]
- Taipale, J., K. Miyazono, C.H. Heldin, and J. Keski-Oja. 1994. b. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J. Cell Biol. 124:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale, J., J. Saharinen, and J. Keski-Oja. 1998. Extracellular matrix-associated transforming growth factor-beta: role in cancer cell growth and invasion. Adv. Cancer Res. 75:87–134. [DOI] [PubMed] [Google Scholar]
- Tsuji, T., F. Okada, K. Yamaguchi, and T. Nakamura. 1990. Molecular cloning of the large subunit of transforming growth factor type beta masking protein and expression of the mRNA in various rat tissues. Proc. Natl. Acad. Sci. USA. 87:8835–8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsold, C., M. Hyytiainen, L. Bruckner-Tuderman, and J. Keski-Oja. 2001. Latent TGF-beta binding protein LTBP-1 contains three potential extracellular matrix interacting domains. J. Cell Sci. 114:187–197. [DOI] [PubMed] [Google Scholar]
- Weinacker, A., A. Chen, M. Agrez, R.I. Cone, S. Nishimura, E. Wayner, R. Pytela, and D. Sheppard. 1994. Role of the integrin alpha v beta 6 in cell attachment to fibronectin. Heterologous expression of intact and secreted forms of the receptor. J. Biol. Chem. 269:6940–6948. [PubMed] [Google Scholar]