

Abstract
Humans with mutations in either DCX or LIS1 display nearly identical neuronal migration defects, known as lissencephaly. To define subcellular mechanisms, we have combined in vitro neuronal migration assays with retroviral transduction. Overexpression of wild-type Dcx or Lis1, but not patient-related mutant versions, increased migration rates. Dcx overexpression rescued the migration defect in Lis1 +/− neurons. Lis1 localized predominantly to the centrosome, and after disruption of microtubules, redistributed to the perinuclear region. Dcx outlined microtubules extending from the perinuclear “cage” to the centrosome. Lis1 +/− neurons displayed increased and more variable separation between the nucleus and the preceding centrosome during migration. Dynein inhibition resulted in similar defects in both nucleus–centrosome (N-C) coupling and neuronal migration. These N-C coupling defects were rescued by Dcx overexpression, and Dcx was found to complex with dynein. These data indicate Lis1 and Dcx function with dynein to mediate N-C coupling during migration, and suggest defects in this coupling may contribute to migration defects in lissencephaly.
Keywords: migration; Lis1; doublecortin; centrosome; nucleus
Introduction
One of the most distinctive features in mammalian brain development is that newly born postmitotic neurons migrate hundreds of cell body distances from their sites of origin to reach their final destination (Hatten, 2002). Hemizygous mutations in DCX or heterozygous mutations in LIS1 lead to nearly identical neuronal migration defects in humans known as lissencephaly (Dobyns and Truwit, 1995; Dobyns et al., 1996; Lo Nigro et al., 1997), which is characterized by a smooth surface of the brain, four abnormal cortical layers, and a variably hypoplastic cerebellum, associated with intractable epilepsy, hypotonia, and profound mental retardation from infancy.
LIS1, located on chromosome 17p13.3, encodes the LIS1 protein. LIS1 functions on an evolutionarily conserved pathway regulating microtubule (MT) function and dynein motor activity. In Aspergillus nidulans, the LIS1 homologue nudF is required for proper nuclear transport into and within hyphal processes (Xiang et al., 1995), and other nud (nuclear distribution defect) genes such as nudA, nudG, and nudK encode for dynein components (Xiang et al., 1994, 1999). The mammalian orthologues of NUD proteins bind to LIS1 (Faulkner et al., 2000; Feng et al., 2000; Niethammer et al., 2000; Sasaki et al., 2000; Smith et al., 2000), suggesting that the fungal nuclear migration pathway may be conserved in mammals to mediate nuclear translocation. The fact that the main defect observed in cultured Lis1 +/− mouse neurons is in nuclear movement within the limits of the cell membrane, but not in neurite length (Hirotsune et al., 1998; unpublished data), also supports the recapitulation of LIS1 in mammalian nuclear translocation.
DCX, located on Xq22.3-q23, encodes for an MT-associated protein, doublecortin (DCX). DCX directly polymerizes purified tubulin into MTs, and misexpression in heterologous cells leads to the formation of depolymerization-resistant MTs (Francis et al., 1999; Gleeson et al., 1999a; Horesh et al., 1999). Similarly, DCX has evolutionarily conserved orthologues, where in Caenorhabditis elegans mutations in zyg-8 result in defects in MT stability and karyokinesis during asymmetric division at the one-cell stage (Gönczy et al., 2001), suggesting that DCX also functions in migration on an evolutionarily conserved pathway. Although Dcx knockout mice do not display a major disruption in migration, acute inactivation in rodents produces significant migration defects (Corbo et al., 2002; Bai et al., 2003).
Recent data suggest that Lis1 and Dcx display overlapping localization and may interact. In fixed cells of various types, Lis1 localizes to the centrosome (Feng et al., 2000; Sasaki et al., 2000; Smith et al., 2000), the perinuclear region (Coquelle et al., 2002), kinetochores (Faulkner et al., 2000), the plus end of MTs (Coquelle et al., 2002; Lee et al., 2003; Xiang, 2003), and the leading cell cortex (Swan et al., 1999; Dujardin et al., 2003). Evidence suggests Dcx localizes to MT structures in both the leading process (Friocourt et al., 2003) and the perinuclear region (Gleeson et al., 1999a). Dcx and Lis1 coimmunoprecipitate from brain lysate, and purified Dcx and Lis1 bind cooperatively to MTs (Caspi et al., 2000). These data suggest that they may share functionality during migration. However, their roles have not thus far been tested in mammalian migrating neurons, the cells directly affected in lissencephaly. Here, we use mouse cerebellar granule neurons and demonstrate that Lis1 and Dcx function with dynein to mediate nucleus–centrosome (N-C) coupling in neuronal migration. We propose that proper N-C coupling may be critical in neuronal migration.
Results
A genetically modifiable neuronal migration system
To define the mechanistic roles of Dcx and Lis1 in mammalian neuronal migration, we used mouse cerebellar granule neurons in an in vitro migration assay combined with retroviral-mediated transgene expression (Hatten, 1985; Bix and Clark, 1998; Hirotsune et al., 1998; Gambello et al., 2003). The rationale to use this system is: (1) there is a clear cerebellar migration defect in humans with LIS1 or DCX mutations (Berg et al., 1998; Dobyns et al., 1999), being the most commonly mutated genes in individuals with lissencephaly with cerebellar hypoplasia (Ross et al., 2001); (2) migration is robust, quantifiable, and reproducible; (3) as glia are removed during the purification steps, granule neurons migrate along the neurites of other neurons in a nonglial guided fashion (Lois et al., 1996), thus ensuring the analysis of a single mode of migration (i.e., elimination of glial-based migration); and (4) this assay was used previously to demonstrate a cell-autonomous migration defect in Lis1-deficient neurons (Hirotsune et al., 1998; Gambello et al., 2003).
Cerebellar granule neurons were dissociated from postnatal d 5 mice and cultured with retrovirus, resulting in spherical cellular reaggregates that were transferred to poly-d-lysine– and laminin-coated slides (Liang and Crutcher, 1992). A fraction of neurons migrated radially from each. After 12 h of migration, the distance between transduced cell bodies and the edge of the reaggregate was measured, allowing for an integrated estimate of the migration rate (Fig. 1).
Figure 1.
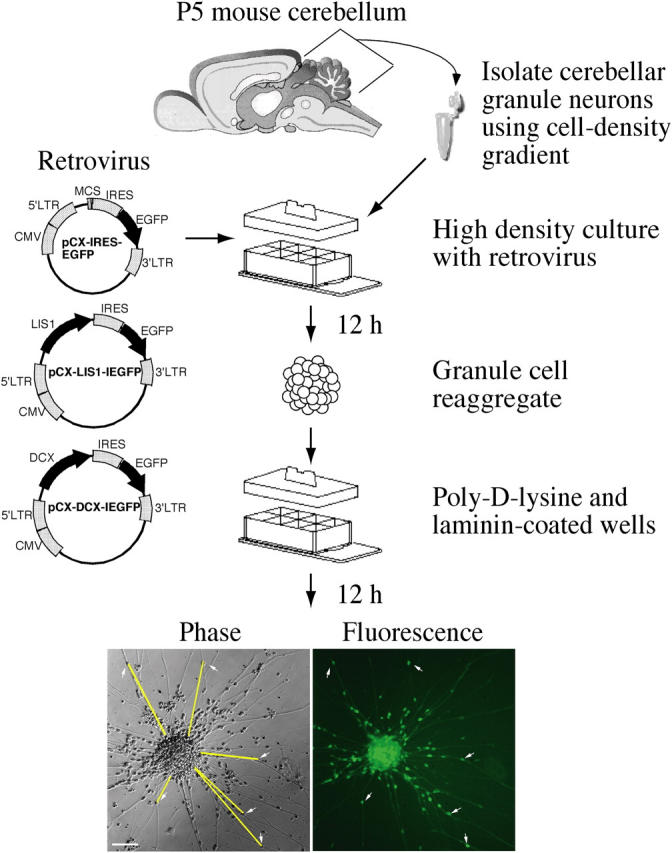
Genetically modifiable neuronal migration assay. White arrows in the images indicate some of the transduced neurons, and yellow lines indicate corresponding migration distances from the edge of the reaggregate. Retroviral constructs encoding GFP alone, Lis1;GFP, and Dcx;GFP are shown.
Overexpression of Dcx or Lis1 leads to an increase in neuronal migration
Neuronal migration is suggested to be sensitive to LIS1 dosage, as heterozygous mutation leads to lissencephaly in humans, and graded reduction of Lis1 results in graded migration defects in mice (Hirotsune et al., 1998; Gambello et al., 2003). Patients with hypomorphic DCX missense mutations display a less severe phenotype than those with truncation mutations (Gleeson et al., 1999b; Matsumoto et al., 2001). These dose-dependent negative effects of LIS1 or DCX on migration when mutated or deleted suggested that overexpression may have positive regulatory effects on migration. Therefore, we determined if neuronal migration is enhanced by overexpression of either protein.
To quantitate the amount of retrovirally transduced proteins, lysates were collected from infected cells and analyzed by SDS-PAGE/Western blotting for the presence of endogenous and epitope-tagged Dcx or Lis1 (Fig. 2 A). Quantitation of band intensity and correction for percentage of transduced cells indicated the epitope-tagged Dcx and Lis1 proteins were produced at ∼85% of the endogenous level (Fig. 2 A, see table), meaning transduction increased protein levels a little less than twofold.
Figure 2.
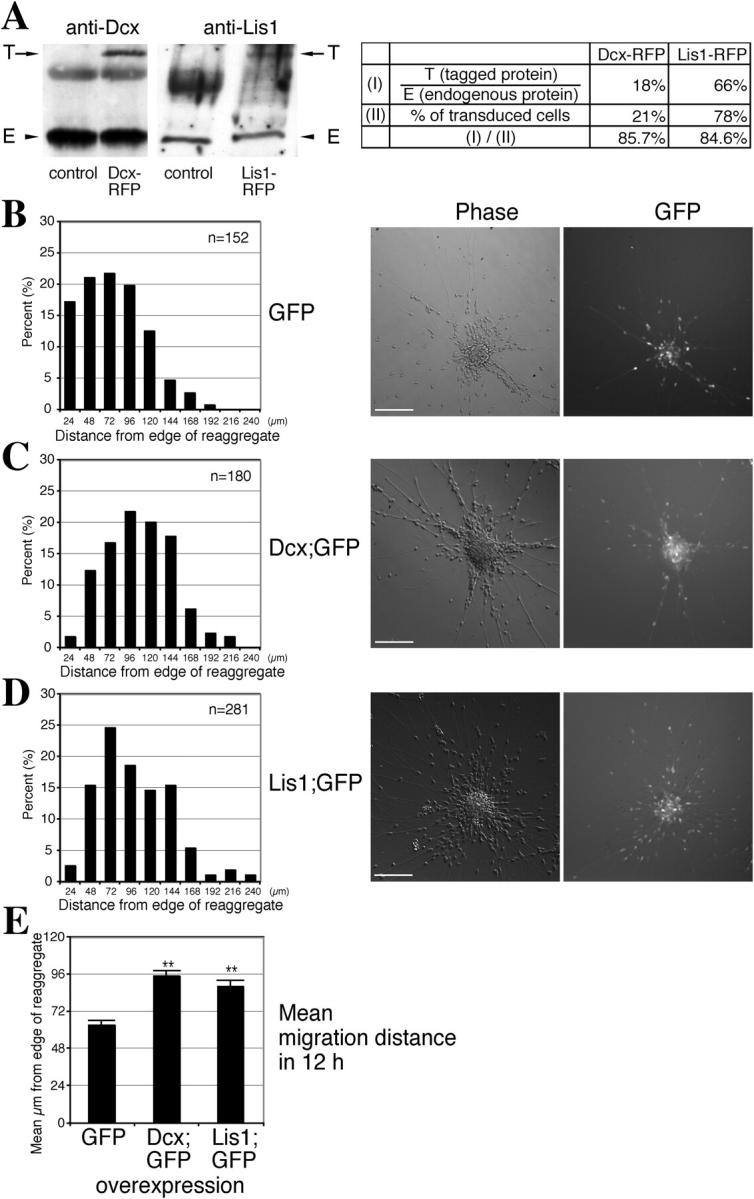
Dcx or Lis1 overexpression increases neuronal migration. (A) Retroviral transduction nearly doubles the amount of Dcx or Lis1. Lysates from neurons transduced with RFP-tagged Dcx or Lis1 show bands corresponding to endogenous protein (E) and transduced tagged protein (T). From the ratio of band intensity (T/E) and percentage of transduced cells, transduced protein was ∼85% of the endogenous level. (B–D) Transduction of either Dcx or Lis1 into WT neurons results in an increase in net neuronal migration. The migration distance of each GFP-positive neuron in 12 h is binned. Neurons transduced with either Dcx;GFP or Lis1;GFP display a shift in the bin distribution toward the right. Migration distances are represented in μm along the x axis, and percentage of total neuron number in each bin is represented along the y axis. n = number of neurons measured for each variable. Representative images for each condition are shown. (E) Mean migration distance for neurons overexpressing either Dcx or Lis1 is increased 40–50% compared with GFP alone. **, differs from GFP control; P < 0.01, SNK test. Similar results were obtained on three separate experimental trials. Error bars represent SEM. Bars, 100 μm.
Wild-type (WT) granule neurons were transduced with either retrovirus encoding GFP alone, Dcx in the first cistron and GFP in the second cistron (herein referred to as Dcx;GFP), or Lis1 in the first cistron and GFP in the second cistron (Lis1;GFP). Migration distances in 12 h were binned. Neurons transduced with GFP alone (Fig. 2 B) were positioned indistinguishably from untransduced neurons, suggesting that retroviral transduction itself had no effect on migration. Dcx- or Lis1-overexpressing neurons displayed a rightward shift in bin distribution (Fig. 2, B–D). Mean migration distance for each increased by ∼50% compared with GFP control (Fig. 2 E). These data suggest that neuronal migration can be positively regulated by Dcx or Lis1 expression.
Patient mutations abrogate the effect of overexpression on migration
Intragenic LIS1 or DCX mutations in patients with lissencephaly include missense amino acid changes and truncations (Cardoso et al., 2000, 2002; Gleeson et al., 2000). The mutant proteins are either incorrectly folded (Sapir et al., 1999; Caspi et al., 2003) and fail to associate with binding partners (Feng et al., 2000) or fail to bind MTs (Taylor et al., 2000), but no data exist on the effect of these mutations on migration. To assess the function of the mutant proteins quantitatively, patient-related mutant Dcx (R89G, T203R, 303stop; Gleeson et al., 1998) or Lis1 (H149R, S169R, D317H; Lo Nigro et al., 1997; Pilz et al., 1999) was overexpressed. Neurons transduced with single amino acid substitution mutations in either Dcx or Lis1 displayed a mean migration slightly above GFP control, but were not statistically different (Fig. 3). There were slight differences in the bin distribution for each mutation, but the overall migration distances were not significantly different from one another. Truncated Dcx showed no increase in migration over the control level. Thus, patient mutations abrogate the effect of overexpression of Dcx and Lis1 on migration and appear to function as null or hypomorphic alleles.
Figure 3.

Patient mutations of Dcx or Lis1 abrogate the effect of overexpression on migration. WT neurons transduced with Dcx or Lis1 harboring missense mutations (C, D, and G–I) display only a slight rightward shift in migration bin distribution and a corresponding slight increase in the mean migration distance from GFP control, whereas the truncated mutant Dcx (E) showed no increase over control level. Similar results were obtained on two separate experimental trials. Error bars represent SEM.
Dcx overexpression as well as Lis1-forced expression in Lis1-deficient neurons largely restore neuronal migration
We tested whether Dcx overexpression was sufficient to rescue the migration defect in Lis1-deficient neurons. Lis1 +/− cerebellar granule neurons were previously shown to have a defect in migration compared with WT (Hirotsune et al., 1998; Gambello et al., 2003). We confirmed in our assay that Lis1 +/− neurons displayed a leftward shift of the bin distribution of migration distance, and the mean distance decreased by ∼50% from the WT level (Fig. 4, B and E). Lis1 +/− neurons transduced with Lis1;GFP displayed a shift in migration distance bin back toward the right, similar to that of Lis1+/+ neurons (Fig. 4 C). The mean distance was restored to slightly above WT level (Fig. 4 E). These data suggest that Lis1-forced expression likely restored migration to WT level in Lis1 +/− neurons through correction of the Lis1 deficiency.
Figure 4.
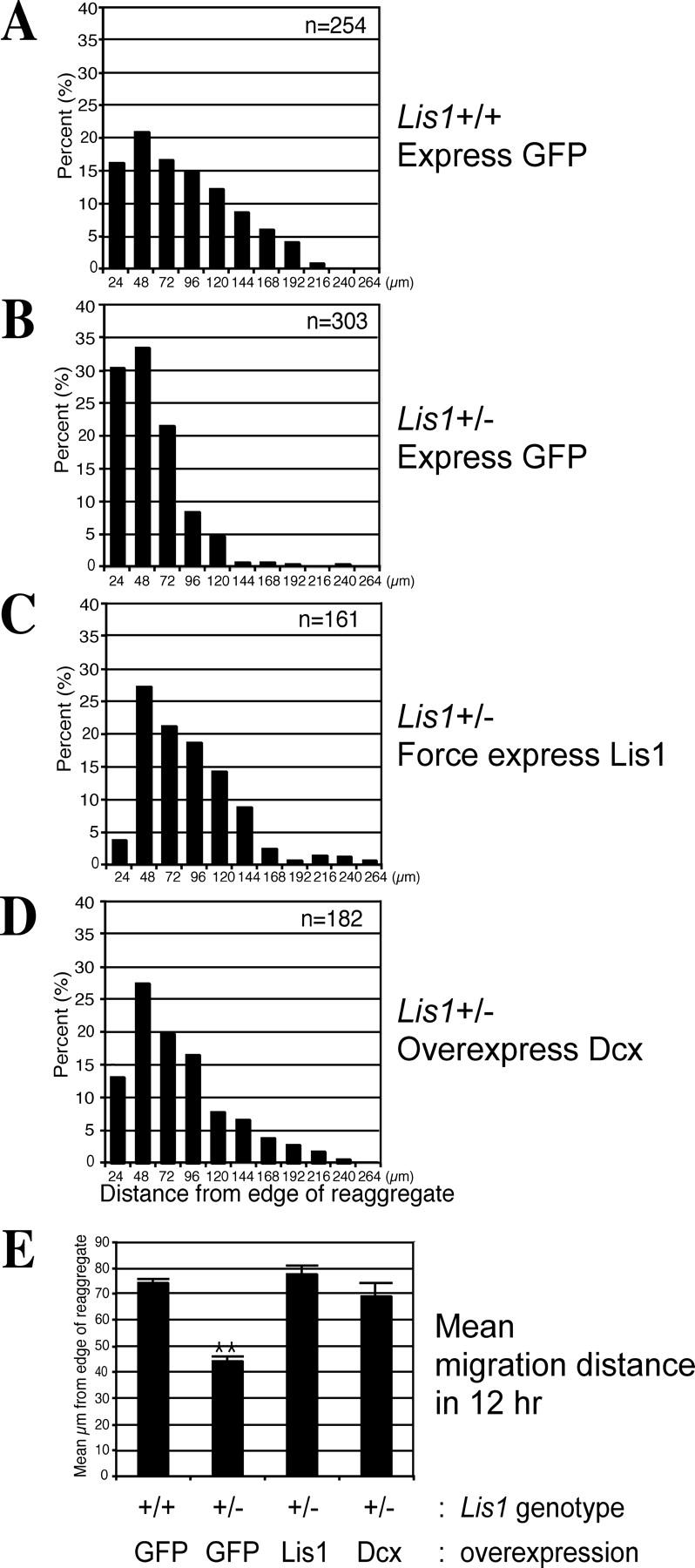
Lis1-forced expression or Dcx overexpression largely corrects the migration defect associated with a Lis1 deficiency. Lis1 +/− neurons (B) display a leftward shift of migration bin distribution from Lis1 +/+ neurons (A). With Lis1-forced expression (C) or Dcx overexpression (D), there is a correction in bin distribution back toward the right. (E) The correction in the mean migration distance for Lis1 +/− neurons transduced with either Lis1 or Dcx. **, differs from Lis1 +/+ neurons transduced with GFP; P < 0.01, SNK test. Similar results were obtained on three separate experimental trials. Error bars represent SEM. n = number of neurons for each variable.
Dcx overexpression largely rescued the migration defect in Lis1 +/− neurons. There was a shift in the migration bin distribution back to the right in Lis1 +/− neurons transduced with Dcx;GFP, and mean migration distance was restored to 94% of WT level (Fig. 4 E). There was no statistical difference in distance between Lis1 +/+ neurons transduced with GFP, or Lis1 +/− transduced with Dcx;GFP or Lis1;GFP (P > 0.05 for both comparisons; Student-Newman-Kleus [SNK] test). These data suggest that Dcx and Lis1 may function on a common pathway to regulate neuronal migration.
Lis1 is localized at the centrosome in migrating neurons
In fixed cells of various types, Lis1 has been reported to overlap in localization with several structures. To test the localization in live migrating neurons, RFP-tagged Lis1 (Lis1-RFP) was transduced in granule neurons. Localization of Lis1-RFP was nearly identical with native Lis1 visualized by immunostaining (Fig. 5 A), suggesting that it reports the localization of the native protein.
Figure 5.
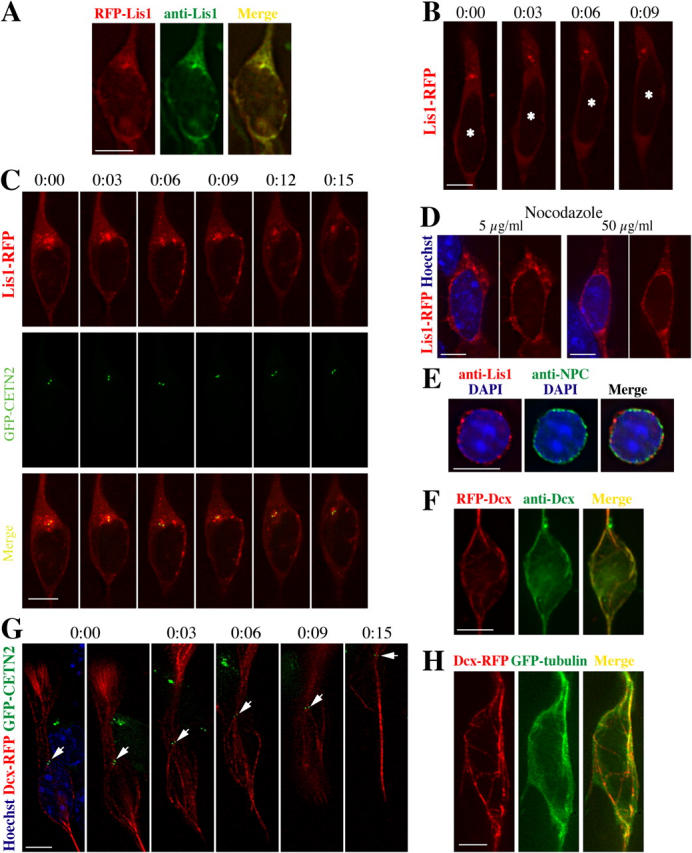
Lis1 is localized at the centrosome, whereas Dcx outlines MTs extending from a perinuclear cage to the centrosome. (A) Localization of Lis1-RFP is nearly identical with native Lis1. Cerebellar granule neuron transduced with Lis1-RFP is immunostained with anti-Lis1 antibody. (B) Concentrated Lis1-RFP signal is maintained ahead of the nucleus in the direction of migration. The nucleus is indicated by the asterisk. (C) Concentrated Lis1 represents the location of the centrosome in live neurons. Time-lapse images of WT neuron transduced with Lis1-RFP and GFP-CETN2 show the major consistent focus of Lis1 localizes with the CETN2 signal. (D) Redistribution of Lis1 to the perinuclear region by disruption of MTs. Live images show perinuclear redistribution of Lis1-RFP after 2-h treatment of nocodazole. (E) Lis1 localizes on the nuclear membrane after nocodazole treatment. Isolated nucleus immunostained with anti-Lis1 and anti-NPC (nucleoporin) antibodies shows Lis1 localization on the nuclear membrane. (F) Localization of Dcx-RFP is nearly identical with native Dcx. Dcx-RFP–transduced neuron is immunostained with anti-Dcx antibody. (G) Dcx outlines a cage-like fibrillar bundle encapsulating the nucleus and converging at the centrosome (arrow) ahead of the nucleus. Time-lapse images of migrating neuron transduced with Dcx-RFP and GFP-CETN2. (H) Cotransduction with Dcx-RFP and GFP-tubulin shows Dcx localizes on the MTs. Bars, 5 μm.
Using time-lapse deconvolution microscopy, neurons transduced with Lis1-RFP displayed a concentrated RFP signal just ahead of the nucleus in the direction of migration (Fig. 5 B). There were occasionally less intense, additional foci or spread of Lis1, but a major focus remained throughout each recording. This focus suggested the likely position of the centrosome, based on its reported location in neurons (Rakic, 1971; Gregory et al., 1988; Rivas and Hatten, 1995). To test this, a GFP-tagged constitutive centrosomal protein, centrin 2 (CETN2; White et al., 2000), was retrovirally transduced, producing intense GFP signal at the pair of centrioles. Cotransduction of Lis1-RFP and GFP-CETN2 revealed that the focus of Lis1 localized at the GFP-CETN2 signal (Fig. 5 C). There were occasional frames with more than one Lis1 focus that were less or similarly intense; in all of these a major focus remained throughout each recording and was consistently colocalized with GFP-CETN2 signal. These data suggest that Lis1 predominantly localizes to centrosomal or pericentrosomal structures during migration.
Lis1 localizes prominently to the nuclear membrane after MT disruption
A previous report has found that treatment with the MT-depolymerizing drug nocodazole led to some Lis1 localized to the nuclear membrane in fixed nonneuronal cells (Smith et al., 2000), suggesting that some fraction of Lis1 may be localized to the nuclear membrane and that an intact MT cytoskeleton is required for Lis1 localization to the centrosome. We tested this in migrating neurons using nocodazole. In approximately half of the cells analyzed at 5 and 50 μg/ml nocodazole concentration, there was redistribution of Lis1 to the perinuclear region (Fig. 5 D). There continued to be some localization of Lis1 to the region of the presumed centrosome, although it was less intense than in untreated cells. The half of the cells not showing perinuclear distribution also displayed less intense centrosomal Lis1 localization. Therefore, a fraction of Lis1 is redistributed to the nuclear membrane after MT disruption.
Because there is very little perinuclear cytoplasm in neurons, the previous experiment could not easily distinguish between perinuclear versus cytoplasmic localization of Lis1. Therefore, nuclei were isolated from granule neurons in the presence of nocodazole and immunostained for Lis1. Nucleoporin, a constitutive nuclear pore complex protein (Davis and Blobel, 1986), outlined the nuclear membrane, whereas Lis1 was detected as punctate signals (Fig. 5 E). Together, these data suggest that some Lis1 is associated with the nuclear membrane and that intact MT structures may be required in localizing Lis1 to the centrosome.
Dcx outlines MTs extending from the perinuclear “cage” to the centrosome
To study the localization of Dcx during migration, RFP-tagged Dcx (Dcx-RFP) was transduced in granule neurons. The localization of Dcx-RFP was nearly identical to endogenous Dcx (Fig. 5 F). Dual transduction with Dcx-RFP and GFP-CETN2 displayed a bright fibrillar bundle-like signal of Dcx encapsulating the nucleus and converging to the centrosome ahead of the nucleus throughout migration (Fig. 5 G). Cotransduction with Dcx-RFP and GFP-tubulin indicated that the fibrillar signal represented MTs (Fig. 5 H), thus Dcx outlined the perinuclear cage-like MT structures previously described in migrating neurons (Rivas and Hatten, 1995). Together, these data suggest that Lis1 and Dcx display distinct but adjacent localization during migration.
Coupling of the nucleus to the centrosome is defective in Lis1-deficient neurons
Our time-lapse recording of migrating neurons using GFP-CETN2 demonstrated that in essentially every cell that was observed moving, the centrosome precedes the nucleus in the direction of migration. According to the reported MT organization in neurons (Aumais et al., 2001; Hatten, 2002), MTs would couple (1) the leading process to the centrosome and (2) the centrosome to the nucleus, in order to translocate the nucleus (Fig. 6 A). Our data on subcellular localization in migrating neurons, together with the known role of Lis1 in mediating dynein motor function, suggests that Lis1 and Dcx might be involved in coupling the nucleus to the preceding centrosome (Fig. 6 B).
Figure 6.

Schematic cytoarchitecture of migrating neuron using MT structures to translocate nucleus. (A) The centrosome is positioned ahead of the nucleus, with MTs forming a perinuclear cage-like structure converging into the centrosome and projecting into the leading process from the centrosome. MT structures couple (1) the leading process to the centrosome and (2) the centrosome to the nucleus to translocate the nucleus. (B) Model for the role of Lis1 and Dcx in neuronal migration. Dcx is distributed to the perinuclear MT structure, and the Lis1–dynein complexes move in an MT minus end direction, attached to the nuclear membrane, to displace the nucleus toward the centrosome. Schematic modified from Hatten (2002).
To test this coupling and its dependence on Lis1 levels, the distances between the nucleus and the preceding centrosome during migration were analyzed. WT and Lis1 +/− neurons were transduced with GFP-CETN2 and labeled with Hoechst nuclear stain. After time-lapse deconvolved images were collected, the distance between the anterior edge of the nucleus marked by Hoechst and the centrosome, projected on the horizontal plane, was measured (Fig. 7 A). To be sure that Hoechst signal reported the edge of the nucleus accurately, cells were fixed and stained with nucleoporin, and the Hoechst and nucleoporin signals were contiguous in the two genotypes (unpublished data). There was a significant difference in the N-C distance between WT and Lis1 +/− neurons. Lis1 +/− neurons displayed a 70% increase in the mean N-C distance with greater variability compared with WT, suggesting defective N-C coupling (Fig. 7 B; mean 0.96 μm, SD 0.78 μm, n = 54, WT vs. mean 1.64 μm, SD 1.21 μm, n = 76, Lis1 +/−; P < 0.01, t test).
Figure 7.

Both Lis1 deficiency and dynein inhibition cause defective N-C coupling, and are rescued by Dcx overexpression. (A) Lis1-deficient neurons display increased separation between the nucleus and preceding centrosome. Deconvolved images of Lis1 +/+ and Lis1 +/− migrating neurons transduced with GFP-CETN2. In the Lis1 +/− neuron, the nucleus is positioned further behind the centrosome (a caliper indicates N-C distance). Direction of migration indicated by black arrow. (B) The average N-C distance is increased by 70% in Lis1 +/− neurons compared with Lis1 +/+. (C) Increase in the N-C distance in Lis1 +/− neurons is rescued by Lis1 or Dcx overexpression. Similar results were obtained on three separate experimental trials. (D) Deconvolved images of migrating neurons transduced with CETN2-RFP only (left) or together with GFP-dynamitin (right), demonstrating GFP-dynamitin transduction results in an increase in N-C distance. Centrosome position indicated by arrows and N-C distance indicated by a caliper. Exposure time for GFP was calculated so that signal was just above background, to minimize phototoxicity. (E) A 63% increase in the mean N-C distance is seen in dynamitin-overexpressing neurons. (F) Dynein inhibition by dynamitin overexpression results in a neuronal migration defect, shown by a leftward shift in the bin distribution of migration distance similar to Lis1 +/− neurons. (G) Representative images for the rescue of dynamitin-induced coupling defect by Dcx-RFP transduction. (Left) WT neuron transduced with CETN2-RFP and GFP-dynamitin. (Right) WT neuron transduced with CETN2-RFP, GFP-dynamitin, and Dcx-RFP. Centrosome position indicated by arrows, Dcx-RFP indicated by arrowheads, and N-C distances by calipers. (H) Correction of the dynamitin-induced N-C coupling defect after Dcx-RFP transduction. (I) Disruption of MTs by nocodazole leads to N-C coupling defects. GFP-CETN2–transduced neurons treated with nocodazole display a graded increase in the N-C distance according to the concentrations of nocodazole. **, differs from control at P < 0.01, error bars represent SEM. Bars, 5 μm.
N-C coupling defect in Lis1-deficient neurons is rescued by Dcx overexpression
To determine the relevance of the increased N-C distance in Lis1 +/− neurons during migration, we tested whether this could be corrected by Lis1-forced expression or Dcx overexpression, which rescued the migration defect in these neurons. Lis1 +/− neurons were transduced with GFP-CETN2 and either Lis1-RFP or Dcx-RFP, and the N-C distances in dual-transduced neurons were analyzed likewise. The increased N-C distance in Lis1 +/− neurons was normalized after Lis1-RFP transduction (Fig. 7 C; mean 2.14 μm, SD 1.41 μm, n = 36, Lis1 +/− vs. mean 1.14 μm, SD 0.93 μm, n = 51, Lis1+/− with Lis1-RFP; P < 0.01). Similarly, overexpression of Dcx-RFP resulted in normalization of this N-C distance in Lis1 +/− neurons (mean 1.11 μm, SD 0.84 μm, n = 35, Lis1+/− with Dcx-RFP; P < 0.01, SNK test). Thus, Dcx overexpression rescued the subcellular defect in N-C coupling as well as the cellular defect in migration due to Lis1 deficiency. These results suggest that this N-C coupling defect may underlie the migration defect in Lis1-deficient neurons.
Dynein inhibition results in defects in N-C coupling and neuronal migration
Previous works demonstrated the direct and functional interaction between Lis1 and cytoplasmic dynein (Faulkner et al., 2000; Sasaki et al., 2000; Smith et al., 2000), so we reasoned that inhibiting dynein function may recapitulate the defects due to Lis1 deficiency. To test this, dynamitin overexpression, which disassembles the cytoplasmic dynein component dynactin (Echeverri et al., 1996), was used to disrupt dynein function in migrating neurons.
WT neurons were transduced with RFP-tagged CETN2 (CETN2-RFP) and GFP-dynamitin, nuclei were labeled with Hoechst stain, and the N-C distance was measured in each dual-transduced neuron (Fig. 7 D). Dynamitin overexpression led to a 63% increase in the mean N-C distance with greater variability (Fig. 7 E; mean 1.09 μm, SD 0.77 μm, n = 46, control vs. mean 1.78 μm, SD 1.33 μm, n = 21, GFP-dynamitin; P < 0.01). Thus, N-C coupling in migrating neurons is defective with disruption of dynein function.
To test the effect of dynein inhibition on neuronal migration, the migration assay was performed on granule neurons overexpressing GFP-dynamitin. This resulted in a shift in a migration bin distribution similar to Lis1 +/− neurons (Fig. 7 F), and led to a 30% decrease in mean migration distance (47 μm, n = 356, GFP-dynamitin vs. 65 μm, n = 704, GFP control; P < 0.01, t test). Thus, dynein motor function is required for proper N-C coupling and neuronal migration. These data indicate that recapitulation of the N-C coupling defect using a different method also leads to defective migration.
Dcx rescues the impaired N-C coupling caused by dynein inhibition
As Dcx overexpression rescued the N-C coupling defect in Lis1 +/− neurons, we examined whether Dcx overexpression rescued the coupling defect seen with dynamitin overexpression. WT neurons were transduced with CETN2-RFP and GFP-dynamitin, and some were additionally transduced with Dcx-RFP (Fig. 7 G). Although both CETN2 and Dcx are labeled with the same fluorophore, localization of these two is distinct, and dual-transduced cells were easily identifiable. Dcx overexpression corrected the N-C coupling defect caused by dynamitin overexpression back to a WT level (Fig. 7 H; mean 1.48 μm, SD 1.34 μm, n = 60, GFP-dynamitin vs. mean 0.84 μm, SD 0.77 μm, n = 36, GFP-dynamitin with Dcx-RFP; P < 0.01). These data suggest that the mechanism by which Dcx overexpression rescues Lis1 deficiency is through restoration of dynein function. Together, our data suggest that Lis1 and Dcx function with dynein to couple the nucleus to the centrosome in neuronal migration.
Disruption of MTs leads to N-C coupling defects
The N-C coupling model suggests that intact MT structures are required. To test this, MTs were disrupted by nocodazole in WT neurons transduced with GFP-CETN2, and N-C distances during migration were analyzed. Mean N-C distances were increased significantly by treatment with nocodazole (Fig. 7 I; mean 0.92 μm, SD 0.70 μm, n = 60, control vs. mean 1.54 μm, SD 1.43 μm, n = 52, 100 nM nocodazole, P < 0.01; mean 2.41 μm, SD 1.42 μm, n = 35, 1 μM nocodazole, P < 0.01, SNK test). These data indicate correct N-C coupling requires MT integrity, and is consistent with the dynein-based coupling mechanism.
Dcx is part of the cytoplasmic dynein complex
The data mentioned in the previous paragraph suggest that Dcx may interact with dynein. We tested whether Dcx, like Lis1 (Faulkner et al., 2000; Sasaki et al., 2000; Smith et al., 2000), is part of the dynein motor complex. Protein complexes were immunoprecipitated using specific antisera. Before immunoprecipitation, whole brain lysates from postnatal d 5 mice were incubated at 4°C in order to depolymerize MTs, and were then centrifuged to remove MT-bound proteins. In this way, proteins complexed through MT bridges alone should be largely removed. Dcx immunoprecipitated with both dynein heavy chain (DHC) and dynein intermediate chain (DIC) antibodies in a fashion similar to Lis1 (Fig. 8). This suggests that Dcx is part of a complex with dynein.
Figure 8.

Dcx is part of the dynein motor complex. Coimmunoprecipitation of Dcx and Lis1 with components of dynein motor complex. Whole brain lysate (WBL) was immunoprecipitated with anti-Dcx, anti-DIC, no antibody (control), or anti-DHC. An additional control with no brain lysate but with anti-Dcx antibody is included. S, 1% of supernatant; P, 10% of pellet. WBL reveals Dcx running as a doublet (double arrowhead). Dcx immunoprecipitates with its own antibody, and with DIC and DHC antibody. Lis1 is included as a positive control, as it was previously shown to coimmunoprecipitate with both Dcx and dynein subunits.
Discussion
Dosage-sensitive roles of Dcx and Lis1 regulating neuronal migration
We demonstrated that overexpression of Dcx or Lis1 led to enhanced neuronal migration. Together with the loss-of-function phenotypes, these data suggest that both Dcx and Lis1 play dosage-sensitive roles in migration.
Previous reports showed that LIS1 overexpression in COS-7 or MDCK cells resulted in altered organization and distribution of MTs, cytoplasmic dynein, dynactin, and the Golgi complex (Smith et al., 2000), increase in mitotic index, a variety of spindle defects, unattached chromosomes, and irregular cell shapes (Faulkner et al., 2000). These analyses were performed in nonneuronal cells in which the physiological LIS1 levels were lower than neuronal cells (Smith et al., 2000), and used adenoviral or cytomegalovirus promoters in which expression levels are much higher than with the retroviral 5′ long terminal repeats promoter used here. The resultant nonphysiological changes in LIS1 dosage might be responsible for the toxic effect on these cells. In our work, the level of Lis1 or Dcx after retroviral transduction was a little less than twice the WT level. In this range, overexpression of Dcx or Lis1 appears to be capable of increasing neuronal migration without obvious detrimental or toxic effects. It remains to be seen if even higher levels of expression would produce yet further enhancement in migration.
Patient-related mutations of Dcx or Lis1 abrogate the effects on migration
Overexpression of patient-related mutant proteins had little to no obvious effects on migration. This is the first demonstration of a system that can be used to test the effect of patient mutations on neuronal migration. Previous reports have demonstrated that Lis1 with introduced patient mutations is either unstable (Sapir et al., 1999), displays abnormal folding (Caspi et al., 2003), or cannot properly bind to ligands such as mNudE (Feng et al., 2000). Dcx with introduced patient mutations leads to impaired MT polymerizing ability (Sapir et al., 2000; Taylor et al., 2000) or cannot bind to the FIGQY-phosphotyrosine motif in neurofascin (Kizhatil et al., 2002). Our data clearly indicate these patient-related mutations abrogate their ability to positively regulate migration rate, suggesting the mutant proteins are non- or hypo-functional, and lead to the migration defects that result in the lissencephaly phenotype in humans.
N-C coupling requires normal levels of Lis1 and is regulated by Dcx and dynein
We confirmed that in live migrating neurons the centrosome is nearly uniformly positioned ahead of the nucleus, as previously reported in fixed cells (Rakic, 1971; Gregory et al., 1988; Rivas and Hatten, 1995). In migrating neurons, Lis1 localized at the centrosome and after MT disruption redistributed to the perinuclear region, suggesting that Lis1 translocates from the perinuclear region toward the centrosome via MT structures in physiological conditions, or that at least its localization elsewhere requires MTs. Dcx localized to a perinuclear cage-like MT structure intersecting the centrosome. In Lis1-deficient neurons, we identified defective N-C coupling. Based on our evidence, the known effects of Dcx on MT polymerization, the minus end of MTs being located at the centrosome, and association of Lis1 with the dynein motor complex, we speculate that Dcx is required for proper formation of the perinuclear MT structure, and that the Lis1–dynein complex may move in an MT minus end direction, attached to the nuclear membrane, to displace the nucleus toward the centrosome (Fig. 6 B). Our preliminary data suggest a possible defect in perinuclear MT structure in Dcx null migrating neurons, but more work is needed.
There is almost nothing known about N-C coupling during neuronal migration. We demonstrated that in WT migrating neurons, the N-C distance measured 0.8–1.1 μm on average, and that manipulation of Lis1, Dcx, dynein, or MTs significantly changed this distance. We measured the distance projected in a single horizontal plane in which migration occurred. Any differences in the z axis should have been randomly distributed and should not interfere with these analyses. All our analyses were done on deconvolved images that had clear contours, assuring accuracy in measurement. Our findings are consistent: (1) the increase in N-C distance by Lis1 deficiency was rescued by Lis1-forced expression or Dcx overexpression, which also rescued migration defects; (2) N-C distance was increased by disruption of MTs as well as dynein inhibition; and (3) the differences detected between experimental conditions were highly reproducible, and although this distance was dynamic as neurons move, measurements were collected from several hundred neurons and so these internal variations due to the phases of migration should be captured in our dataset. Therefore, we conclude that the N-C distances collectively measured in live neurons reflect the N-C coupling states and pertain to migration.
Dcx, Lis1, and dynein pathways in nuclear translocation
Previous data indicated Lis1-deficient cells display a defect of dynein function (Sasaki et al., 2000; Smith et al., 2000). Our data show that Lis1 deficiency and dynein inhibition display the same effect on N-C coupling and neuronal migration, suggesting that Lis1 deficiency affects N-C coupling by impairment of dynein function. Dcx overexpression rescued the coupling defects caused by Lis1 deficiency and dynein inhibition. Additionally, we showed coimmunoprecipitation between Dcx and dynein subunits, which may be mediated by Lis1. These data suggest that the mechanism by which Dcx overexpression rescues Lis1 deficiency might be through restoration of dynein function. However, other possible mechanisms of rescue exist, including a modulation of the MT cytoskeleton by Dcx, which might compensate for less efficient Lis1-mediated N-C coupling. Further genetic and cell biological evidence for this rescue will be necessary.
How these proteins function to mediate N-C coupling remains to be determined. There is a growing body of evidence that the dynein–dynactin complex associates with nuclear membranes in many cell types, and these complexes coupled to the nucleus may move in a minus end–directed fashion toward the centrosome. Synthetic nuclei assembled from Xenopus egg extracts move along MTs toward their minus ends, the movement required both dynein and the nuclear membrane, and female pronuclear movement is suggested to use this mechanism (Reinsch and Karsenti, 1997). Dynein is required for positioning or nuclear attachment of centrosomes in C. elegans (Gönczy et al., 1999) and Drosophila (Robinson et al., 1999), and localizes to the nuclear envelope of mammalian cells and C. elegans embryos before nuclear envelope breakdown (Busson et al., 1998; Gönczy et al., 1999; Salina et al., 2002). Recently, compelling evidence of a dynein-dependent N-C coupling mechanism has emerged from the study of the Hook protein ZYG-12 in C. elegans (Malone et al., 2003), which localizes to both the nuclear envelope and the centrosome, binds dynein light intermediate chain, and is proposed to move the centrosome toward the nucleus.
Together, these data suggest an evolutionarily conserved pathway using dynein in centrosome–nuclear interactions. Mammalian neurons may use this mechanism to translocate the nucleus during migration, where we propose the centrosome is fixed in location by MT structures so that the nucleus moves toward the centrosome. Molecular mechanisms of capture and translocation of the nucleus toward the centrosome and the role of dynein should be further investigated.
Materials and methods
Retroviral vector constructs
NH2-terminal tagged DCX (GenBank/EMBL/DDBJ accession no. 2792349) and LIS1 (GenBank/EMBL/DDBJ accession no. 6031206) (Gleeson et al., 1999a) were ligated into the first cistronic position of pCX-IEGFP (Akagi et al., 2000) to create pCX-DCX-IEGFP and pCX-LIS1-IEGFP constructs. Dcx-RFP and Lis1-RFP were ligated into pCX-IAP to create pCXDCX-RFP-IAP and pCXLIS1-RFP-IAP, respectively. GFP-CETN2 (provided by J. Salisbury, Mayo Clinic College of Medicine, Rochester, NY) was shuttled into pCXbsr, from which IRES-bsr had been removed to increase the retrovirus titer, to create pCX-GFP-CETN2. CETN2-RFP, which was created by ligation of CENT2 into the multiple cloning site of pDsRed1-N1, was ligated into pCXbsr to create pCX-CETN2-RFP. N-myc GFP-tagged (provided by T. Schroer, The Johns Hopkins University, Baltimore, MD) dynamitin was shuttled into pCXbsr, from which IRES-bsr had been removed, to create pCX-GFP-dynamitin.
Production of recombinant retroviruses
Recombinant ecotropic replication-incompetent Moloney murine leukemia retrovirus (Naviaux et al., 1996; Tomoda et al., 1999) or pseudotyped retrovirus (Yee et al., 1994) was produced according to standard protocols.
Reaggregate neuronal migration assay
Cerebellar granule cells were isolated as described previously (Hatten, 1985) and cultured at 106 cells/ml with retrovirus for 12 h, resulting in uniform-sized reaggregates (100–150 μm in diameter), which were then transferred to poly-d-lysine– (Sigma-Aldrich) and laminin (Sigma-Aldrich)-treated slides (Bix and Clark, 1998). Imaging was commenced after an additional 12 h using a 20× objective lens and images were analyzed using MetaMorph® v.4.5 (Universal Imaging Corp.).
Isolation of nuclei from cerebellar granule neurons
Nuclei were isolated from cerebellar granule neurons according to the protocols for isolation of mammalian cerebellar nuclei with modifications (Blobel and Potter, 1966; Thompson, 1973; Thomas and Thompson, 1977). Neurons were treated with 10 μg/ml nocodazole and 10 μg/ml latrunculin B for 2 h, centrifuged at 2,000 rpm for 10 min, and the pellet was resuspended in 2.0 M sucrose in TKM (50 mM Tris-HCl, pH 7.5, 25 mM KCl, 5 mM MgCl2, 1 mM PMSF, 1 mM DTT) and homogenized in an Aldrich-type homogenizer. The homogenate diluted with 2.0 M sucrose/TKM was centrifuged at 64,000 g for 30 min. The pellet was resuspended in 2.4 M sucrose/TKM, overlaid with 1.8 M sucrose/TKM, and recentrifuged at 85,000 g for 30 min. The pellet at the interface of two sucrose layers was collected, resuspended in 0.25 M sucrose/TKM, and centrifuged for 10 min at 1,000 g to remove remaining contaminants.
Immunofluorescence
Neurons were fixed in 4% PFA for 10 min, blocked in 4% normal donkey serum in PBS, and immunostained with anti-Lis1 (provided by L.-H. Tsai, Harvard Medical School, Boston, MA, and D. Smith, University of South Carolina, Columbia, SC; 1:250; Smith et al., 2000), anti-Dcx (1:300; Gleeson et al., 1999a), and anti-nucleoporin (1:100, Mab414; Covance) antibodies controlled by omitting primary antibodies. Cells were washed and stained with Alexa Fluor® secondary antibodies (1:600; Molecular Probes, Inc.).
Image acquisition
Images in Fig. 5 (A–F and H) and Fig. 7 were collected using a DeltaVision® deconvolution imaging system (Applied Precision) on a microscope (model IX-70; Olympus) with an oil immersion 100× (NA 1.4) or 60× (NA 1.4) lens at 37°C in a 5% CO2 chamber, using 0.28–0.35-μm Z-steps, with 10 cycles of deconvolution by softWoRx v.3.2.3. Images in Fig. 5 G were collected by a DeltaVision® system on a microscope (model TE200; Nikon) with a 100× lens (NA 1.4) at 37°C, using 0.3-μm Z-steps, with 10 cycles of deconvolution by softWoRx v.2.5.
Western analysis and immunoprecipitation
Cells were lysed with modified RIPA buffer with protease inhibitors (PMSF, aprotinin, leupeptin), incubated at 4°C for 30 min, calf intestinal alkaline phosphatase was added, and cells were further incubated at 37°C for 1 h. For immunoprecipitation, whole brains from postnatal d 5 mice were homogenized in 1 ml modified RIPA buffer, incubated at 4°C for 30 min, centrifuged at 14,000 rpm, and the supernatant was collected. To 250 μl brain lysate, 10 μg antibody, anti-Dcx (Gleeson et al., 1999a), anti-DIC (CHEMICON International), anti-DHC (CHEMICON International), or control antibody was added and incubated for 1 h, followed by addition of 30 μl protein A–Sepharose beads (Amersham Biosciences) for 30 min. Beads were washed with PBS four times, boiled in sample buffer, analyzed by Western blotting as previously described (Taylor et al., 2000) with anti-Dcx (1:2,000) or anti-Lis1 (1:200, N-19; Santa Cruz Biotechnology, Inc.), and were developed using chemiluminescence. Band intensities were quantitated in ImageQuant v.1.1 (Molecular Dynamics).
Statistical analysis
For single pairwise comparisons, a two-tailed t test was used. For multiple pairwise comparisons, the SNK test was used, which corrects the P value for multiple comparisons (Glantz, 1996).
Animal experiments
All animal work was performed at the animal facility at the University of California, San Diego School of Medicine under approved animal protocols and in accordance with institutional guidelines.
Acknowledgments
The authors wish to thank Don Cleveland, Larry Goldstein, Karen Oegema, Arshad Desai, and members of the Gleeson laboratory for helpful comments and suggestions; Gary Clark, Kelli Mullen, and Mary Hatten for sharing a detailed cerebellar granule neuron isolation protocol; and Toshifumi Tomoda for retroviral protocols.
This work was supported by a Post-doctoral Research Training Fellowship (T. Tanaka) and the Junior Investigator Research Grant from the Epilepsy Foundation of America, by the National Institute of Neurological Disorders and Stroke (K12NS01701, R01 NS41537), the University of California, San Diego Neuroscience Microscopy Shared Facility (NS047101), the John Merck Award in the Developmental Disabilities in Childhood, the Searle Scholars Program, and the Klingenstein Foundation.
Abbreviations used in this paper: CETN2, centrin 2; DCX, doublecortin; DHC, dynein heavy chain; DIC, dynein intermediate chain; MT, microtubule; N-C, nucleus–centrosome; SNK, Student-Newman-Kleus; WT, wild type.
References
- Akagi, T., T. Shishido, K. Murata, and H. Hanafusa. 2000. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc. Natl. Acad. Sci. USA. 97:7290–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumais, J.P., J.R. Tunstead, R.S. McNeil, B.T. Schaar, S.K. McConnell, S.H. Lin, G.D. Clark, and L.Y. Yu-Lee. 2001. NudC associates with Lis1 and the dynein motor at the leading pole of neurons. J. Neurosci. 21:RC187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, J., R.L. Ramos, J.B. Ackman, A.M. Thomas, R.V. Lee, and J.J. LoTurco. 2003. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 6:1277–1283. [DOI] [PubMed] [Google Scholar]
- Berg, M.J., G. Schifitto, J.M. Powers, C. Martinez-Capolino, C.T. Fong, G.J. Myers, L.G. Epstein, and C.A. Walsh. 1998. X-linked female band heterotopia-male lissencephaly syndrome. Neurology. 50:1143–1146. [DOI] [PubMed] [Google Scholar]
- Bix, G.J., and G.D. Clark. 1998. Platelet-activating factor receptor stimulation disrupts neuronal migration In vitro. J. Neurosci. 18:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel, G., and V.R. Potter. 1966. Nuclei from rat liver: isolation method that combines purity with high yield. Science. 154:1662–1665. [DOI] [PubMed] [Google Scholar]
- Busson, S., D. Dujardin, A. Moreau, J. Dompierre, and J.R. De Mey. 1998. Dynein and dynactin are localized to astral microtubules and at cortical sites in mitotic epithelial cells. Curr. Biol. 8:541–544. [DOI] [PubMed] [Google Scholar]
- Cardoso, C., R.J. Leventer, N. Matsumoto, J.A. Kuc, M.B. Ramocki, S.K. Mewborn, L.L. Dudlicek, L.F. May, P.L. Mills, S. Das, et al. 2000. The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene. Hum. Mol. Genet. 9:3019–3028. [DOI] [PubMed] [Google Scholar]
- Cardoso, C., R.J. Leventer, J.J. Dowling, H.L. Ward, J. Chung, K.S. Petras, J.A. Roseberry, A.M. Weiss, S. Das, C.L. Martin, et al. 2002. Clinical and molecular basis of classical lissencephaly: Mutations in the LIS1 gene (PAFAH1B1). Hum. Mutat. 19:4–15. [DOI] [PubMed] [Google Scholar]
- Caspi, M., R. Atlas, A. Kantor, T. Sapir, and O. Reiner. 2000. Interaction between LIS1 and doublecortin, two lissencephaly gene products. Hum. Mol. Genet. 9:2205–2213. [DOI] [PubMed] [Google Scholar]
- Caspi, M., F.M. Coquelle, C. Koifman, T. Levy, H. Arai, J. Aoki, J.R. De Mey, and O. Reiner. 2003. LIS1 missense mutations: variable phenotypes result from unpredictable alterations in biochemical and cellular properties. J. Biol. Chem. 278:38740–38748. [DOI] [PubMed] [Google Scholar]
- Coquelle, F.M., M. Caspi, F.P. Cordelieres, J.P. Dompierre, D.L. Dujardin, C. Koifman, P. Martin, C.C. Hoogenraad, A. Akhmanova, N. Galjart, et al. 2002. LIS1, CLIP-170's key to the dynein/dynactin pathway. Mol. Cell. Biol. 22:3089–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo, J.C., T.A. Deuel, J.M. Long, P. LaPorte, E. Tsai, A. Wynshaw-Boris, and C.A. Walsh. 2002. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J. Neurosci. 22:7548–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, L.I., and G. Blobel. 1986. Identification and characterization of a nuclear pore complex protein. Cell. 45:699–709. [DOI] [PubMed] [Google Scholar]
- Dobyns, W.B., and C.L. Truwit. 1995. Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics. 26:132–147. [DOI] [PubMed] [Google Scholar]
- Dobyns, W.B., E. Andermann, F. Andermann, D. Czapansky-Beilman, F. Dubeau, O. Dulac, R. Guerrini, B. Hirsch, D.H. Ledbetter, N.S. Lee, et al. 1996. X-linked malformations of neuronal migration. Neurology. 47:331–339. [DOI] [PubMed] [Google Scholar]
- Dobyns, W.B., C.L. Truwit, M.E. Ross, N. Matsumoto, D.T. Pilz, D.H. Ledbetter, J.G. Gleeson, C.A. Walsh, and A.J. Barkovich. 1999. Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephaly. Neurology. 53:270–277. [DOI] [PubMed] [Google Scholar]
- Dujardin, D.L., L.E. Barnhart, S.A. Stehman, E.R. Gomes, G.G. Gundersen, and R.B. Vallee. 2003. A role for cytoplasmic dynein and LIS1 in directed cell movement. J. Cell Biol. 163:1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverri, C.J., B.M. Paschal, K.T. Vaughan, and R.B. Vallee. 1996. Molecular characterization of the 50-kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J. Cell Biol. 132:617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner, N.E., D.L. Dujardin, C.Y. Tai, K.T. Vaughan, C.B. O'Connell, Y. Wang, and R.B. Vallee. 2000. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2:784–791. [DOI] [PubMed] [Google Scholar]
- Feng, Y., E.C. Olson, P.T. Stukenberg, L.A. Flanagan, M.W. Kirschner, and C.A. Walsh. 2000. LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron. 28:665–679. [DOI] [PubMed] [Google Scholar]
- Francis, F., A. Koulakoff, D. Boucher, P. Chafey, B. Schaar, M.C. Vinet, G. Friocourt, N. McDonnell, O. Reiner, A. Kahn, et al. 1999. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 23:247–256. [DOI] [PubMed] [Google Scholar]
- Friocourt, G., A. Koulakoff, P. Chafey, D. Boucher, F. Fauchereau, J. Chelly, and F. Francis. 2003. Doublecortin functions at the extremities of growing neuronal processes. Cereb. Cortex. 13:620–626. [DOI] [PubMed] [Google Scholar]
- Gambello, M.J., D.L. Darling, J. Yingling, T. Tanaka, J.G. Gleeson, and A. Wynshaw-Boris. 2003. Multiple dose-dependent effects of Lis1 on cerebral cortical development. J. Neurosci. 23:1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz, S.A. 1996. Primer of Biostatistics. 4th ed. McGraw-Hill Inc., New York. 473 pp.
- Gleeson, J.G., K.M. Allen, J.W. Fox, E.D. Lamperti, S. Berkovic, I. Scheffer, E.C. Cooper, W.B. Dobyns, S.R. Minnerath, M.E. Ross, and C.A. Walsh. 1998. doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 92:63–72. [DOI] [PubMed] [Google Scholar]
- Gleeson, J.G., P.T. Lin, L.A. Flanagan, and C.A. Walsh. 1999. a. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 23:257–271. [DOI] [PubMed] [Google Scholar]
- Gleeson, J.G., S.R. Minnerath, J.W. Fox, K.M. Allen, R.F. Luo, S.E. Hong, M.J. Berg, R. Kuzniecky, P.J. Reitnauer, R. Borgatti, et al. 1999. b. Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann. Neurol. 45:146–153. [DOI] [PubMed] [Google Scholar]
- Gleeson, J.G., S. Minnerath, R.I. Kuzniecky, W.B. Dobyns, I.D. Young, M.E. Ross, and C.A. Walsh. 2000. Somatic and germline mosaic mutations in the doublecortin gene are associated with variable phenotypes. Am. J. Hum. Genet. 67:574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gönczy, P., S. Pichler, M. Kirkham, and A.A. Hyman. 1999. Cytoplasmic dynein is required for distinct aspects of MTOC positioning, including centrosome separation, in the one cell stage Caenorhabditis elegans embryo. J. Cell Biol. 147:135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gönczy, P., J.-M. Bellanger, M. Kirkham, A. Pozniakowski, K. Baumer, J.B. Phillips, and A.A. Hyman. 2001. zyg-8, a gene required for spindle positioning in C. elegans, encodes a doublecortin-like kinase. Dev. Cell. 1:363–375. [DOI] [PubMed] [Google Scholar]
- Gregory, W.A., J.C. Edmondson, M.E. Hatten, and C.A. Mason. 1988. Cytology and neuron-glial apposition of migrating cerebellar granule cells in vitro. J. Neurosci. 8:1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten, M.E. 1985. Neuronal regulation of astroglial morphology and proliferation in vitro. J. Cell Biol. 100:384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten, M.E. 2002. New directions in neuronal migration. Science. 297:1660–1663. [DOI] [PubMed] [Google Scholar]
- Hirotsune, S., M.W. Fleck, M.J. Gambello, G.J. Bix, A. Chen, G.D. Clark, D.H. Ledbetter, C.J. McBain, and A. Wynshaw-Boris. 1998. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat. Genet. 19:333–339. [DOI] [PubMed] [Google Scholar]
- Horesh, D., T. Sapir, F. Francis, S.G. Wolf, M. Caspi, M. Elbaum, J. Chelly, and O. Reiner. 1999. Doublecortin, a stabilizer of microtubules. Hum. Mol. Genet. 8:1599–1610. [DOI] [PubMed] [Google Scholar]
- Kizhatil, K., Y.X. Wu, A. Sen, and V. Bennett. 2002. A new activity of doublecortin in recognition of the phospho-FIGQY tyrosine in the cytoplasmic domain of neurofascin. J. Neurosci. 22:7948–7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W.L., J.R. Oberle, and J.A. Cooper. 2003. The role of the lissencephaly protein Pac1 during nuclear migration in budding yeast. J. Cell Biol. 160:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, S., and K.A. Crutcher. 1992. Neuronal migration on laminin in vitro. Brain Res. Dev. Brain Res. 66:127–132. [DOI] [PubMed] [Google Scholar]
- Lo Nigro, C., C.S. Chong, A.C. Smith, W.B. Dobyns, R. Carrozzo, and D.H. Ledbetter. 1997. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum. Mol. Genet. 6:157–164. [DOI] [PubMed] [Google Scholar]
- Lois, C., J.M. Garcia-Verdugo, and A. Alvarez-Buylla. 1996. Chain migration of neuronal precursors. Science. 271:978–981. [DOI] [PubMed] [Google Scholar]
- Malone, C.J., L. Misner, N. Le Bot, M.C. Tsai, J.M. Campbell, J. Ahringer, and J.G. White. 2003. The C. elegans hook protein, ZYG-12, mediates the essential attachment between the centrosome and nucleus. Cell. 115:825–836. [DOI] [PubMed] [Google Scholar]
- Matsumoto, N., R.J. Leventer, J.A. Kuc, S.K. Mewborn, L.L. Dudlicek, M.B. Ramocki, D.T. Pilz, P.L. Mills, S. Das, M.E. Ross, et al. 2001. Mutation analysis of the DCX gene and genotype/phenotype correlation in subcortical band heterotopia. Eur. J. Hum. Genet. 9:5–12. [DOI] [PubMed] [Google Scholar]
- Naviaux, R.K., E. Costanzi, M. Haas, and I.M. Verma. 1996. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70:5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer, M., D.S. Smith, R. Ayala, J. Peng, J. Ko, M.S. Lee, M. Morabito, and L.H. Tsai. 2000. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 28:697–711. [DOI] [PubMed] [Google Scholar]
- Pilz, D.T., J. Kuc, N. Matsumoto, J. Bodurtha, B. Bernadi, C.A. Tassinari, W.B. Dobyns, and D.H. Ledbetter. 1999. Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum. Mol. Genet. 8:1757–1760. [DOI] [PubMed] [Google Scholar]
- Rakic, P. 1971. Guidance of neurons migrating to the fetal monkey neocortex. Brain Res. 33:471–476. [DOI] [PubMed] [Google Scholar]
- Reinsch, S., and E. Karsenti. 1997. Movement of nuclei along microtubules in Xenopus egg extracts. Curr. Biol. 7:211–214. [DOI] [PubMed] [Google Scholar]
- Rivas, R.J., and M.E. Hatten. 1995. Motility and cytoskeletal organization of migrating cerebellar granule neurons. J. Neurosci. 15:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J.T., E.J. Wojcik, M.A. Sanders, M. McGrail, and T.S. Hays. 1999. Cytoplasmic dynein is required for the nuclear attachment and migration of centrosomes during mitosis in Drosophila. J. Cell Biol. 146:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, M.E., K. Swanson, and W.B. Dobyns. 2001. Lissencephaly with cerebellar hypoplasia (LCH): a heterogeneous group of cortical malformations. Neuropediatrics. 32:256–263. [DOI] [PubMed] [Google Scholar]
- Salina, D., K. Bodoor, D.M. Eckley, T.A. Schroer, J.B. Rattner, and B. Burke. 2002. Cytoplasmic dynein as a facilitator of nuclear envelope breakdown. Cell. 108:97–107. [DOI] [PubMed] [Google Scholar]
- Sapir, T., M. Eisenstein, H.A. Burgess, D. Horesh, A. Cahana, J. Aoki, M. Hattori, H. Arai, K. Inoue, and O. Reiner. 1999. Analysis of lissencephaly-causing LIS1 mutations. Eur. J. Biochem. 266:1011–1020. [DOI] [PubMed] [Google Scholar]
- Sapir, T., D. Horesh, M. Caspi, R. Atlas, H.A. Burgess, S.G. Wolf, F. Francis, J. Chelly, M. Elbaum, S. Pietrokovski, and O. Reiner. 2000. Doublecortin mutations cluster in evolutionarily conserved functional domains. Hum. Mol. Genet. 9:703–712. [DOI] [PubMed] [Google Scholar]
- Sasaki, S., A. Shionoya, M. Ishida, M.J. Gambello, J. Yingling, A. Wynshaw-Boris, and S. Hirotsue. 2000. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 28:681–696. [DOI] [PubMed] [Google Scholar]
- Smith, D.S., M. Niethammer, R. Ayala, Y. Zhou, M.J. Gambello, A. Wynshaw-Boris, and L.H. Tsai. 2000. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol. 2:767–775. [DOI] [PubMed] [Google Scholar]
- Swan, A., T. Nguyen, and B. Suter. 1999. Drosophila Lissencephaly-1 functions with Bic-D and dynein in oocyte determination and nuclear positioning. Nat. Cell Biol. 1:444–449. [DOI] [PubMed] [Google Scholar]
- Taylor, K.R., A.K. Holzer, J.F. Bazan, C.A. Walsh, and J.G. Gleeson. 2000. Patient mutations in doublecortin define a repeated tubulin-binding domain. J. Biol. Chem. 275:34442–34450. [DOI] [PubMed] [Google Scholar]
- Thomas, J.O., and R.J. Thompson. 1977. Variation in chromatin structure in two cell types from the same tissue: a short DNA repeat length in cerebral cortex neurons. Cell. 10:633–640. [DOI] [PubMed] [Google Scholar]
- Thompson, R.J. 1973. Studies on RNA synthesis in two populations of nuclei from the mammalian cerebral cortex. J. Neurochem. 21:19–40. [DOI] [PubMed] [Google Scholar]
- Tomoda, T., R.S. Bhatt, H. Kuroyanagi, T. Shirasawa, and M.E. Hatten. 1999. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 24:833–846. [DOI] [PubMed] [Google Scholar]
- White, R.A., Z. Pan, and J.L. Salisbury. 2000. GFP-centrin as a marker for centriole dynamics in living cells. Microsc. Res. Tech. 49:451–457. [DOI] [PubMed] [Google Scholar]
- Xiang, X. 2003. LIS1 at the microtubule plus end and its role in dynein-mediated nuclear migration. J. Cell Biol. 160:289–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, X., S.M. Beckwith, and N.R. Morris. 1994. Cytoplasmic dynein is involved in nuclear migration in Aspergillus nidulans. Proc. Natl. Acad. Sci. USA. 91:2100–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, X., A.H. Osmani, S.A. Osmani, M. Xin, and N.R. Morris. 1995. NudF, a nuclear migration gene in Aspergillus nidulans, is similar to the human LIS-1 gene required for neuronal migration. Mol. Biol. Cell. 6:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, X., W. Zuo, V.P. Efimov, and N.R. Morris. 1999. Isolation of a new set of Aspergillus nidulans mutants defective in nuclear migration. Curr. Genet. 35:626–630. [DOI] [PubMed] [Google Scholar]
- Yee, J.K., T. Friedmann, and J.C. Burns. 1994. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 43:99–112. [DOI] [PubMed] [Google Scholar]