

Abstract
The steroid hormone ecdysone regulates moulting, cell death, and differentiation during insect development. Ecdysone mediates its biological effects by either direct activation of gene transcription after binding to its receptor EcR–Usp or via hierarchical transcriptional regulation of several primary transcription factors. In turn, these transcription factors regulate the expression of several downstream genes responsible for specific biological outcomes. DRONC, the Drosophila initiator caspase, is transcriptionally regulated by ecdysone during development. We demonstrate here that the dronc promoter directly binds EcR–Usp. We further show that mutation of the EcR–Usp binding element (EcRBE) reduces transcription of a reporter and abolishes transactivation by an EcR isoform. We demonstrate that EcRBE is required for temporal regulation of dronc expression in response to ecdysone in specific tissues. We also uncover the participation of a putative repressor whose function appears to be coupled with EcR–Usp. These results indicate that direct binding of EcR–Usp is crucial for controlling the timing of dronc expression in specific tissues.
Keywords: nuclear hormone receptors; gene transcription; steroid hormones; apoptosis; transcriptional regulation
Introduction
Programmed cell death (PCD) is an essential biological process required for the sculpturing of various tissues and removal of unwanted cells during development. PCD is mainly executed by the process of apoptosis and involves a highly conserved machinery (for review see Baehrecke, 2002; Adams, 2003). Various signals such as cytotoxic insults, hormones, and growth factors regulate the activation of PCD by controlling the balance between pro- and anti-death factors of the cell death machinery (Baehrecke, 2002; Adams, 2003). Although the composition of the cell death effector machinery is now largely understood, how the upstream signals communicate with the core components of the machinery remains poorly defined. Many recent studies suggest that transcription plays a key role in the control of the cell death machinery by regulating the intracellular levels of the pro- and anti-death factors (for review see Kumar and Cakouros, 2004).
In Drosophila melanogaster a single steroid hormone 20-hydroxyecdysone (ecdysone) regulates PCD to remove obsolete larval tissues (for review see Riddiford, 1993; Thummel, 1996; Baehrecke, 2000, 2002; Truman and Riddiford, 2002). Pulses of ecdysone are produced at various times during fly development and regulate cell proliferation, differentiation, and death in a temporally and spatially controlled manner. An ecdysone pulse toward the end of the larval stage signals puparium formation and histolysis of the larval midgut. A second pulse ∼12 h later initiates head eversion and histolysis of the larval salivary glands. These events are followed by progenitor cells giving rise to adult tissues (Thummel, 1996; Baehrecke, 2000, 2002; Truman and Riddiford, 2002). Ecdysone binds to its heterodimeric receptor, EcR–Usp (ecdysone receptor–ultraspiracle), and transcriptionally regulates several primary response genes. There are three EcR isoforms in Drosophila, EcR-A, B1, and B2 (Yao et al., 1993). These isoforms are highly homologous in the DNA and ligand binding domains but differ in their amino terminal transactivation domain. The EcR-B1 isoform is predominantly expressed in tissues destined to undergo PCD, whereas the EcR-A isoform is expressed in tissues that differentiate in response to ecdysone (Talbot et al., 1993; Yao et al., 1993).
In the larval salivary glands and midgut, ecdysone controls the expression of several transcription factors, which in turn regulate several secondary response genes (Jiang et al., 1997; Baehrecke, 2000, 2002). EcR–Usp and ecdysone-induced transcription factors βFTZ-F1, BR-C, E74, E75, and E93 have been shown to play a role in ecdysone-mediated cell death in larval salivary gland and midgut. For example, EcR–Usp directly regulates rpr transcription in salivary glands and BR-C is required for maximal rpr and dronc expression (Jiang et al., 2000, Cakouros et al., 2002). BR-C and E74A are also required for the optimal induction of hid in salivary glands (Jiang et al., 2000). In salivary glands of the E93 mutants rpr, hid, ark, and dronc mRNA levels are severely reduced (Lee et al., 2000). These results suggest that ecdysone-mediated up-regulation of death initiators such as rpr, hid, dark, and dronc is crucial for PCD in salivary glands and midgut during Drosophila metamorphosis.
Of the seven caspases in Drosophila, DRONC is the CED3/caspase-9-like apical caspase (Dorstyn et al., 1999a; for review see Kumar and Doumanis, 2000; Richardson and Kumar, 2002). DRONC is the only fly caspase containing a caspase recruitment domain, and its function is essential for PCD in Drosophila (Hawkins et al., 2000; Meier et al., 2000; Quinn et al., 2000; Dorstyn et al., 2002). Interestingly, dronc is transcriptionally regulated by ecdysone in salivary glands and midgut during larval-pupal metamorphosis, and dronc overexpression is sufficient to mediate ecdysone-induced PCD (Dorstyn et al., 1999a,b; Cakouros et al., 2002). These studies underscore the importance of direct regulation of caspase levels in initiating PCD in vivo. Furthermore, they provide evidence that developmental PCD is controlled at the level of transcription, rather than activation, of a preexisting cell death machinery alone. Given this evidence, it is essential to understand the transcriptional control of the caspase activation machinery. As ecdysone-mediated dronc regulation in Drosophila provides a convenient model to study transcriptional regulation of the core PCD machinery we have been dissecting out the dronc promoter for important transcriptional regulatory elements. Previous work has shown that BR-C and E93 are important for dronc expression by ecdysone (Lee et al., 2000; Cakouros et al., 2002). Preliminary promoter deletion studies suggest that dronc transcriptional regulation is complex and involves both temporal and spatial control (Daish et al., 2003). In the present paper, we demonstrate that EcR-B1 directly binds and transactivates the dronc promoter and that this binding is necessary for the correct timing of expression of dronc in specific tissues. These studies provide a basis for investigating both temporal and spatial regulation of gene transcription of a caspase during development by a steroid hormone receptor.
Results
A specific region of the dronc promoter is essential for ecdysone-mediated transcription
In an effort to identify regions of the dronc promoter that are essential for ecdysone-mediated transcription, a series of deletion constructs containing 2.8, 1.1, and 0.54 kb of the dronc promoter, cloned in front of the luciferase reporter gene, were generated and analyzed for their ability to drive reporter expression by ecdysone. The Drosophila cell line l(2)mbn, which undergoes ecdysone-induced and dronc-dependent cell death, was used in this paper (Ress et al., 2000; Cakouros et al., 2002). Transient transfections with the reporter constructs showed that the 2.8-kb promoter was highly responsive to ecdysone, whereas 1.1 or 0.54 kb promoters were unable to drive reporter expression after ecdysone treatment (Fig. 1 A). These experiments also revealed that deletion of the 2.8-kb promoter to 1.1 kb resulted in a dramatic increase in basal promoter activity, suggesting the possible recruitment of a repressor to this region (Fig. 1 A). Given that most repressors recruit histone deacetylases to repress transcription, we used the dronc promoter-reporter constructs in transient transfections to determine if they were responsive to the histone deacetylase inhibitor Trichostatin A (TSA). TSA treatment of l(2)mbn cells transfected with the dronc-reporter constructs revealed that the 2.8-kb promoter was activated by TSA, whereas 1.1 and 0.54 kb promoters had lost the ability to respond to TSA (Fig. 1 B). To further test that dronc repression can be alleviated by TSA, we analyzed the endogenous dronc transcript and protein levels (Fig. 1 C). In l(2)mbn cells treated with TSA for 0–16 h, endogenous DRONC precursor increased at 4 h, and then quickly stabilized as it was processed to its active form (Fig. 1 C). It should be noted that TSA treatment of l(2)mbn cells for 16 h results in some apoptosis. Northern blot analysis showed that TSA treatment increased the levels of dronc up to 8 h, which then stabilized (Fig. 1 C). These data suggest that alleviation of repression is required for dronc expression. Overall, these experiments demonstrate that the region between 2.8 and 1.1 kb of the dronc promoter is essential for ecdysone-induced dronc transcription and that this region also harbors a putative repressor element, which presumably acts by recruiting a histone deacetylase.
Figure 1.
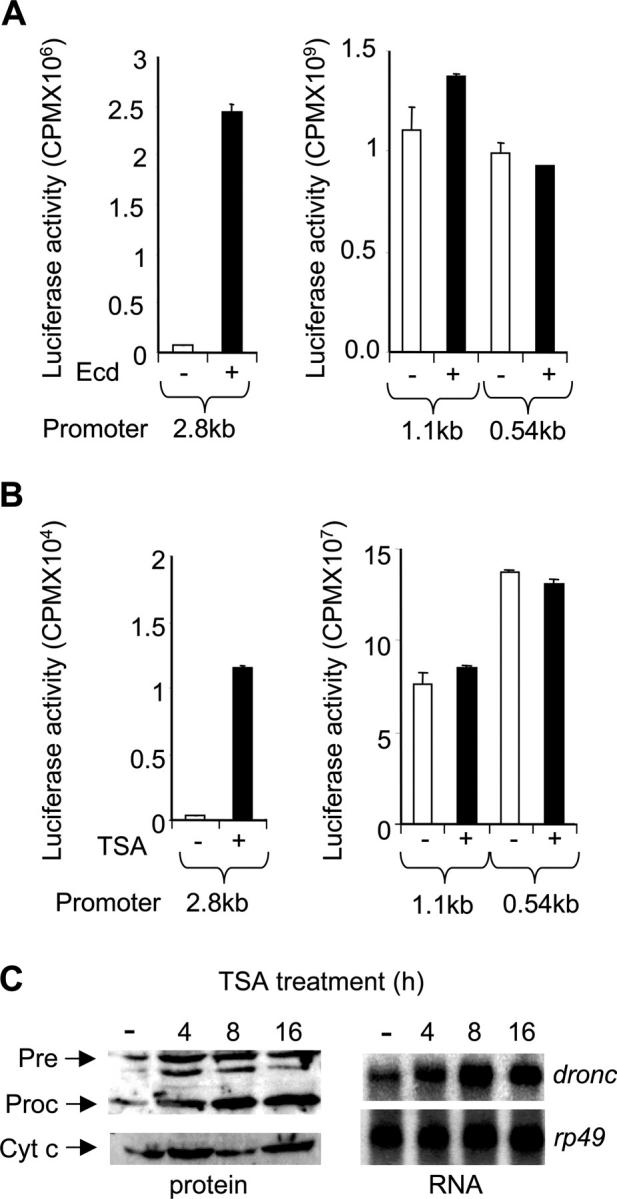
Upstream dronc promoter is responsive to ecdysone and TSA. (A) 2.5 × 106 l(2)mbn cells were transfected in triplicate with 2 μg of dronc luciferase reporter, pxpDR2.8kbLuc, pxpDR1.1kbLuc, or pxpDR0.54kbLuc. After 24 h, cells were treated with 10 μM ecdy-sone (Ecd) for 24 h where indicated (+). Cell extracts were prepared and assayed in triplicate for luciferase activity. Background luciferase activity obtained from empty luciferase vector transfections was subtracted from values shown. Error bars represent SD. (B) Experiment was conducted as in A except trichostatin A (TSA) was used at 1 μM where indicated (+). (C) l(2)mbn cells were treated with ethanol for 16 h (−) or with TSA (1 μM) for the indicated time. All cells were harvested at the same time. 107 cells were used for immunoblotting using a DRONC antibody. Full-length precursor (Pre) and processed (Proc) DRONC species are indicated. For Northern blot analysis, 15 μg of total RNA was electrophoresed, transferred onto nitrocellulose membrane, and probed with dronc probe or a control rp49 probe.
Cycloheximide partially inhibits dronc transcription
Although dronc has been shown to be regulated by the ecdysone-induced transcription factors BR-C and E93 (Lee et al., 2000; Cakouros et al., 2002), two observations suggest the possibility of EcR–Usp directly binding and activating the dronc upstream promoter. First, previous work has shown that ecdysone can regulate dronc transcription in l(2)mbn very early (∼2 h), whereas other transcription factors such as BR-C bind to the promoter after 6 h of ecdysone exposure (Cakouros et al., 2002). Second, the upstream promoter region seems to harbor sites for both repressors and activators (Daish et al., 2003). Nuclear hormone receptors tend to recruit corepressors in the absence of ligand to repress transcription and coactivators in the presence of ligand to activate transcription (Kumar and Thompson, 2003). To examine this possibility, l(2)mbn cells were treated with ecdysone for various times in the presence or absence of the protein translation inhibitor cycloheximide. If ecdysone-induced transcription factors are solely responsible for dronc transcription, then cycloheximide should inhibit the ecdysone-mediated dronc increase. RT-PCR analysis showed that dronc transcript levels increased with ecdysone treatment from 0–12 h (Fig. 2 A). In the presence of cycloheximide, dronc levels at 0 and 6 h were unaffected but were reduced at 12 h (Fig. 2 A). These results were confirmed by Northern blotting (Fig. 2 B) using earlier time points to demonstrate the early onset of dronc transcription, which is insensitive to cycloheximide treatment. Under these conditions, the control rp49 levels were not significantly affected. These data support the possibility that the dronc promoter is directly activated by the preexisting ecdysone receptor, which acts in tandem with other ecdysone responsive transcription factors to activate dronc transcription.
Figure 2.

Ecdysone-mediated dronc transcription is partially cycloheximide sensitive. (A) 107 l(2)mbn cells were treated with 10 μM ecdysone (Ecd) for the indicated time in the presence or absence (Control) of 10 μg/ml cycloheximide (CHX). RNA extracted from cells was analyzed by RT-PCR. (B) Northern blot analysis was performed on RNA samples from cells treated as in A. Where indicated, cells were treated with cycloheximide (CHX) for 2 h.
dronc promoter contains an EcR–Usp binding element (EcRBE)
To screen for possible binding of EcR–Usp to the dronc promoter, we performed electrophoretic mobility shift analysis (EMSA) and competition experiments. EMSA experiments were performed using the hsp70 EcRBE as a probe and in vitro translated EcR–Usp proteins (Fig. 3 A). The EcR–Usp complex was completely abolished when competed with the cold hspEcRBE oligonucleotide, illustrating the specificity of the complex. 400 bp PCR products spanning the upstream dronc promoter region were used as competitors in an attempt to locate the EcRBE. Competitors spanning the region between 2.8 to 1.42 kb had no significant effect on the EcR–Usp complex, however the region between 1.42–1.0 kb clearly competed out most of the EcR–Usp complex (Fig. 3 A). Further experiments mapped the potential EcRBE to the region between 1.42 to 1.2 kb (Fig. 3 B). The ∼200-bp region was analyzed in more detail using 60 bp overlapping oligonucleotides spanning the region. As shown in Fig. 3 C, only oligonucleotide 2 successfully competed out the EcR–Usp complex in EMSA, suggesting that the potential EcRBE in the dronc promoter resides within this sequence.
Figure 3.
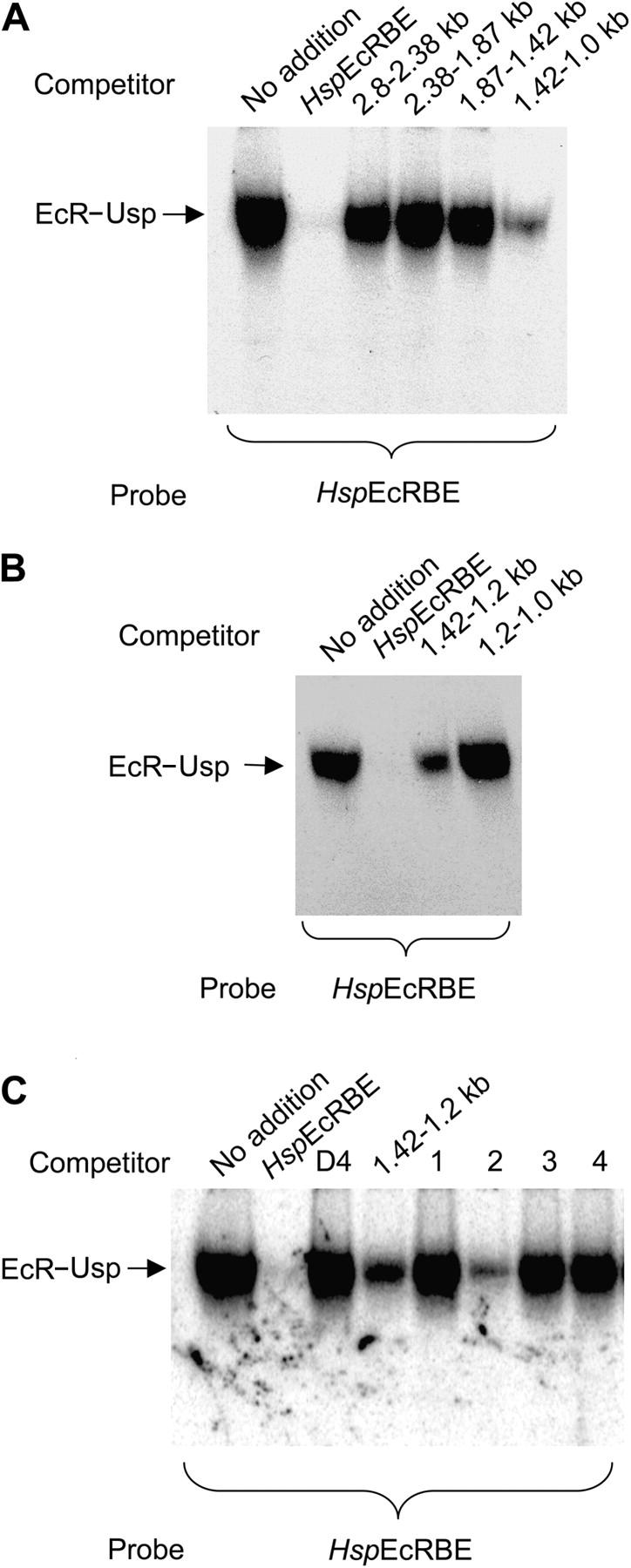
dronc upstream promoter harbors a putative EcRBE. (A) In vitro translated EcR and Usp proteins were incubated with the EcR consensus probe hspEcRBE for 20 min in the presence of dronc promoter fragments. 400 bp PCR fragments spanning the dronc promoter region from 2.8 to 1.1 kb were gel purified and 400 ng was used in each reaction. Positive control (hspEcRBE) was used at 40 ng (equimolar). Complexes were resolved on an acrylamide/TBE gel, dried on 3 MM Whatmann paper, and exposed to Kodak film overnight. EcR–UsP complex is indicated. (B) EMSA experiment was performed as in A except PCR fragments used as competitors spanned the regions from 1.42 to 1.0 kb. hspEcRBE was used as the probe. (C) EMSA was performed as in A. Negative control competitor (D4) corresponds to the dronc promoter region between 67 to 7 bp upstream of the transcription start site. Oligonucleotide competitors correspond to the dronc promoter region 1.44 to 1.2 kb upstream of the transcription start site. HspEcRBE was used as the probe.
An EcR–Usp binding site in the dronc promoter
Analysis of the 60 bp region revealed a potential EcR–Usp binding site that has a 10 out of 13 bp match to the consensus EcRBE (Fig. 4 A). An oligonucleotide containing this potential EcRBE was used as a competitor as well as a mutant oligonucleotide that had specific mutations in the binding site (Fig. 4 A). As shown in Fig. 4 B, the EcR–Usp complex with the hspEcRBE could be competed out with the hspEcRBE. Increasing amounts of the droncEcRBE oligonucleotide also competed the EcR–Usp complex, however higher amounts were needed to abolish binding of EcR–Usp to hspEcRBE. The mutant oligonucleotide failed to compete for the EcRBE (Fig. 4 B). The ability of the droncEcRBE to bind EcR–Usp was determined by EMSA, and as shown in Fig. 4 C, EcR–Usp formed a specific complex that was abolished when competed with the hspEcRBE oligonucleotide. Mutation of the droncEcRBE rendered it incapable of binding EcR–Usp (Fig. 4 C).
Figure 4.
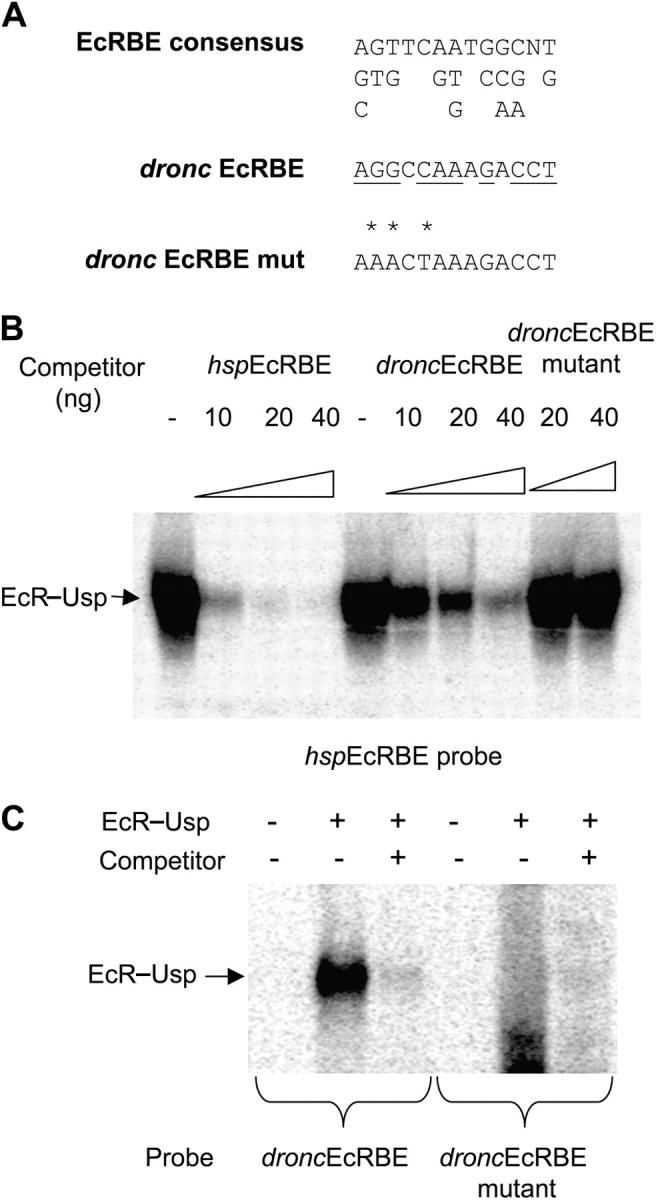
dronc EcRBE binds EcR–Usp. (A) The EcR consensus binding site, its variations, and the putative droncEcRBE sequences are shown. The conserved residues in the droncEcRBE are underlined. The asterisks represent the residues that have been mutated in a mutant droncEcRBE, which was used in the following studies. These sequences correspond to the oligonucleotides used in EMSA. (B) In vitro translated EcR and Usp proteins were incubated with the EcR consensus probe (hspEcRBE) for 20 min in the presence of increasing amounts of hspEcRBE competitor, droncEcRBE competitor, or droncEcRBE mutant competitor oligonucleotides. Complexes were resolved on an acrylamide/TBE gel, dried on 3 MM Whatmann paper, and exposed to Kodak film overnight. EcR–UsP complex is indicated. (C) EMSA was performed as in B. Probes used were the droncEcRBE and droncEcRBE mutant oligonucleotides.
The EcR-B1 isoform specifically binds to the dronc promoter
In Drosophila, the EcR-B1 isoform is predominantly expressed in tissues that are destined to undergo PCD, whereas the EcR-A isoform is predominant in tissues which undergo morphogenesis and form adult structures (Talbot et al., 1993). Based on these observations, we predicted that dronc, which is expressed in tissues undergoing PCD, is likely to be regulated by the EcR-B1 isoform. Therefore, we analyzed the expression of EcR isoforms in l(2)mbn cells that also undergo PCD in response to ecdysone. Using primers specific for each isoform in RT-PCR analysis, we found that the EcR-B1 and EcR-B2 isoforms were highly abundant in l(2)mbn cells, whereas EcR-A was not detectable (Fig. 5 A). In cDNA prepared from total larvae, EcR-A was clearly evident. To determine which isoform is recruited to the dronc promoter, EMSA analysis was performed using isoform-specific EcR antibodies. Nuclear extracts from l(2)mbn cells revealed a single complex binding to the droncEcRBE probe, which was entirely supershifted by an EcR antibody that recognizes all EcR isoforms (Fig. 5 B). The EcR-B1 isoform antibody also supershifted the complex, whereas the EcR-A isoform-specific antibody had no effect. As expected, the hspEcRBE oligonucleotide competed most of the complex. Mutation of the droncEcRBE completely inhibited binding of the EcR–Usp complex, and competitor oligonucleotide or EcR common antibody had no effect as no complex bound to this probe (Fig. 5 B). These results clearly demonstrate that l(2)mbn cells express EcR-B1, and this isoform is specifically recruited to the dronc promoter. As specific EcR-B2 antibodies are unavailable, we were unable to test if this isoform also binds to the droncEcRBE. However, as EcR-B1 specific antibody supershifts most of the complex, it is apparent that most of the binding in the l(2)mbn nuclear extracts is due to the EcR-B1 isoform.
Figure 5.

l(2)mbn cells express the EcR-B1 isoform, which binds to the dronc promoter. (A) 107 l(2)mbn cells were treated with 10 μM ecdysone for the indicated time. RNA was extracted from cells and analyzed by RT-PCR to detect specific EcR isoforms. The last lane of the gel shows EcR isoforms expressed in late third instar larvae/prepupae (120 h after egg laying). (B) 9 μg of nuclear extracts prepared from l(2)mbn cells treated with ecdysone for 6 h was incubated with the droncEcRBE or the EcRBE mutant probe for 20 min in the presence of 2 μl of EcR common, EcR-B1, or EcR-A antibody. A mouse control antibody was also used. Complexes were resolved on an acrylamide/TBE gel, dried on 3 MM Whatmann paper, and exposed to Kodak film overnight. EcR–Usp complex (EcR) and supershifted EcR–Usp complex (ss) are indicated.
EcR-B1 is predominantly expressed in salivary glands and midgut and binds to the dronc promoter in these tissues
Because dronc is predominantly expressed in midgut and salivary glands from early (2 h after puparium formation in the midgut) and late prepupae (12 h after puparium formation in salivary glands), these tissues were analyzed for EcR expression. RT-PCR analysis showed the expression of EcR-B1 but not EcR-B2 or EcR-A in both salivary glands and midgut (Fig. 6 A). Increasing the number of PCR cycles did also reveal the presence of EcR-B2 (unpublished data). Nuclear extracts prepared from staged animals revealed binding of the EcR–Usp to the droncEcRBE probe from early to late prepupae, which correlates with the stages of midgut and salivary gland cell death (Fig. 6 B). Binding of the EcR complex was abolished upon mutation of the droncEcRBE (Fig. 6 B). Antibody supershift experiments using nuclear extracts from total larvae at the mid prepupal stage showed the binding of EcR-B1 isoform to the EcRBE (Fig. 6 C). Interestingly, EcR-A isoform was also capable of binding to the EcRBE, but given its lack of expression in salivary gland and midgut, this is likely due to the contribution of other larval tissues in the extracts prepared from whole prepupae (Fig. 6 C). When analyzing nuclear extracts from specific tissues, it was evident that EcR-B1 in prepupal salivary gland (12 h) and midgut (2 h) binds to the dronc promoter, however no supershift was seen with the EcR-A antibody (Fig. 6 D). Cold EcRBE (droncEcRBE) competitor eliminated binding of the EcR–Usp complex as expected. These results clearly demonstrate that EcR-B1 predominantly binds the dronc promoter at least in these two tissues.
Figure 6.
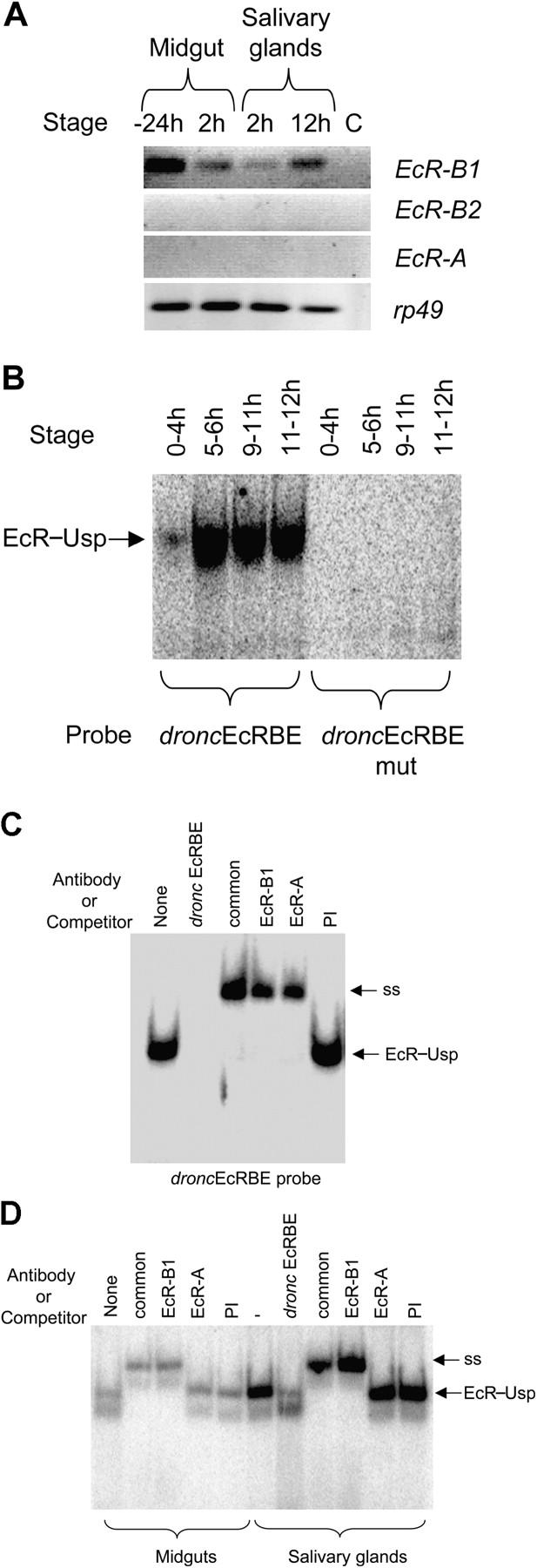
Salivary glands and midgut express EcR-B1, which binds to dronc promoter. (A) Salivary glands and midgut were dissected from animals at −24, 2, or 12 h relative to puparium formation. RNA was analyzed by RT-PCR to detect EcR isoform expression. Rp49 was used as a control. (B) 9 μg of nuclear extracts prepared from various staged animals were incubated with the droncEcRBE or the EcRBE mutant probe for 20 min. Complexes were analyzed as in Figs. 4 and 5. EcR–Usp complex and supershift (ss) are indicated. Developmental stages are shown as hours relative to puparium formation. These stages represent early (0–4 h), mid (5–6 h), and late (9–11 h) prepupae and early (11–12 h) pupae. (C) EMSA was performed as in B in the presence of droncEcRBE cold competitor, 2 μl of EcR common antibody, EcR-B1, or EcR-A. EcR–Usp (EcR) and supershifted EcR–Usp (ss) complexes are indicated. (D) EMSA was performed as in C with 6 μg on nuclear extract from salivary glands or midguts from 12 h (salivary gland) and 2 h (midgut) staged prepupae. EcR–Usp and supershifted (ss) complexes are shown. 40 ng of cold competitor (droncEcRBE) was also added where indicated.
droncEcRBE is important for ecdysone-mediated dronc transcription
Having established the binding of EcR–Usp to the dronc promoter, we determined the significance of direct EcR–Usp binding in ecdysone-mediated dronc transcription of this caspase. To assess this, the dronc promoter-luciferase reporter constructs with or without EcRBE mutations were introduced into l(2)mbn cells, and ecdysone-mediated reporter expression was analyzed. The wild-type promoter was up-regulated in response to ecdysone treatment, and cotransfection of EcR-B1 isoform enhanced ecdysone-mediated transcription (Fig. 7 A). However, mutation of the EcR binding site in the promoter abolished reporter expression in response to ecdysone treatment, and cotransfection of increasing amounts of EcR-B1 had no enhancing effect (Fig. 7 A). Because the region between 2.8–1.1 kb of the promoter is sensitive to TSA treatment (Fig. 1 B), we wished to determine if the EcR mutation inhibits the effects of TSA to determine if the response to TSA is due to the EcRBE or another region of the upstream promoter. The wild-type dronc promoter-reporter was activated by TSA treatment, whereas the EcR mutation significantly inhibited this activation (Fig. 7 B). These results demonstrate that the EcR binding site possibly recruits a histone deacetylase for repression of transcription in the absence of ecdysone and is important for ecdysone-mediated dronc transcription.
Figure 7.

Mutation of droncEcRBE reduces transcription. (A) 2.5 × 106 l(2)mbn cells were transfected in triplicate with 2 μg of dronc luciferase reporter, under the control of the 2.8-kb dronc promoter (Wt) or the promoter with the EcRBE mutated (EcRBE mutant). Where indicated, cells were also cotransfected with 1 or 5 μg of the EcR-B1 expression vector. After 24 h, cells were treated with 10 μM ecdysone for 24 h where indicated (+). Cell extracts were prepared and 100 μg of protein assayed in triplicate for luciferase activity. Luciferase activity was subtracted from values obtained from empty luciferase vector transfections alone. (B) Transfections were performed as in A except trichostatin A (TSA) was added at 1 μM as indicated. Error bars represent SD.
droncEcRBE is important for dronc expression in specific tissues
We have recently reported that the 2.8-kb dronc promoter region contains most necessary elements required for correct spatial and temporal expression (Daish et al., 2003). Interestingly, we also found that the deletion of the 2.8-kb promoter down to 1.1 kb abolished expression in larval/prepupal salivary glands and brain lobes, but not in midgut, suggesting that an element located between 2.8 and 1.1 kb promoter region is necessary for tissue-specific regulation of dronc (Daish et al., 2003). One possibility is that the EcRBE described in this work is responsible for this regulation of dronc. To test this possibility, we created additional promoter deletions and generated transgenic flies carrying the dronc promoter driving expression of the LacZ gene. A transgenic construct containing the 1.64-kb dronc promoter and including the droncEcRBE was able to drive reporter LacZ gene expression in salivary glands, midgut (Fig. 8 A), and brain lobes (not depicted) as assessed by the β-galactoside activity staining of dissected tissues. It should be noted that some premature β-galactoside activity was seen in salivary glands of the 1.64-kb dronc promoter-LacZ transgenic flies (unpublished data). This finding could imply that the 1.64-kb promoter lacks some control elements that govern precise timing of expression. A 1.33-kb promoter construct that deletes the region just past the EcRBE was unable to drive LacZ gene expression in salivary glands (Fig. 8 A) and brain lobes (not depicted). As expected, 1.33 kb of the promoter could efficiently drive reporter expression in the midgut (Fig. 8 A). These data suggested that the region between 1.64 and 1.33 kb, which harbors the EcRBE, is required for dronc expression in specific tissues.
Figure 8.
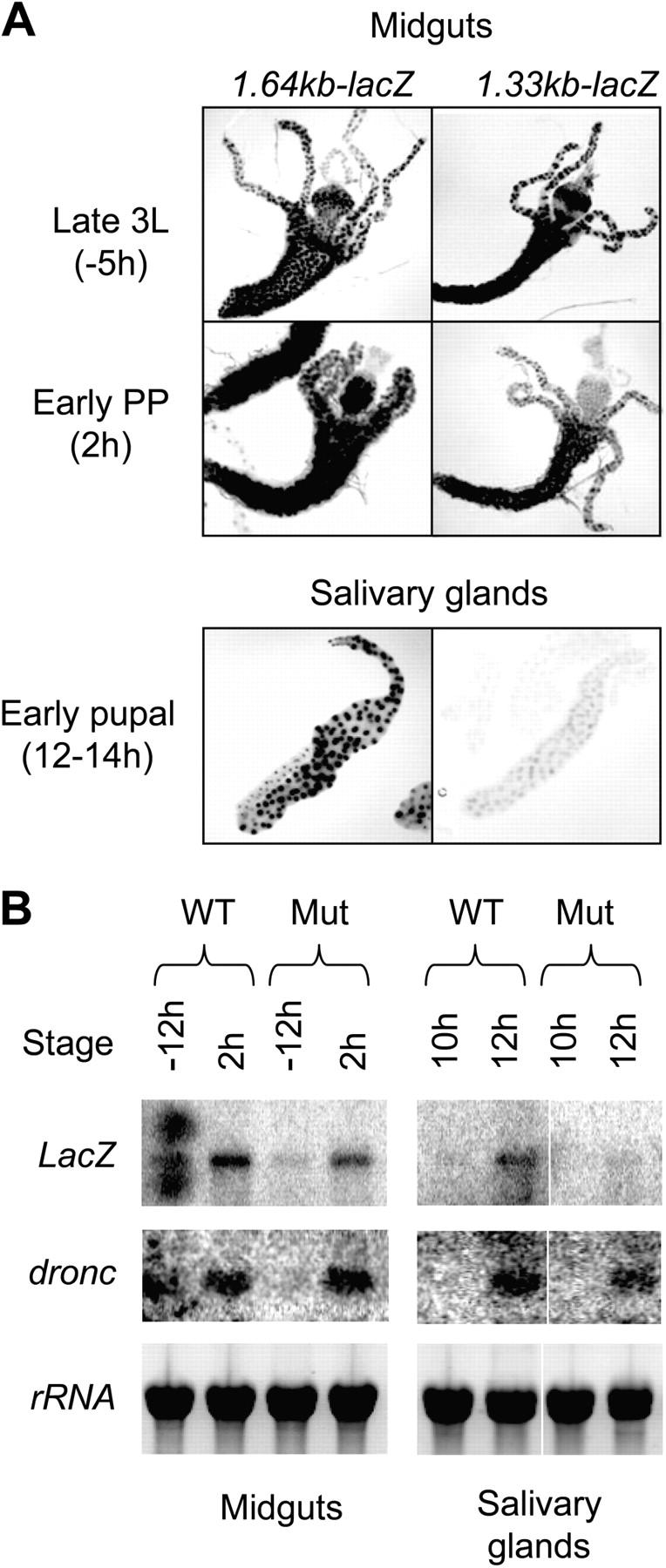
A region of the dronc promoter is required for spatial regulation of expression. β-Galactoside staining of midguts and salivary glands from dronc promoter-reporter transgenic Drosophila is shown. Tissues were dissected out at indicated developmental stages and stained with X-gal. (A, top) Qualitatively similar staining patterns for 1.64 kb and 1.33 kb transgenes with expression throughout midgut and gastric cecae from late third instar larvae (3L) and early prepupae (PP). (bottom) Tissue-specific expression between 1.64 and 1.33 kb transgenes in salivary glands from early pupae demonstrating a requirement for the EcRBE for salivary gland-specific expression. Images are representative of multiple lines analyzed. Slight adjustments of brightness and contrast were applied to all images to enhance quality of the presentation. However, this did not obscure or eliminate any information. These images were originally in color. (B) RNA was collected from staged animals carrying the wild-type dronc 2.8 kb promoter-LacZ transgene (WT) or EcRBE mutant 2.8 kb dronc promoter-LacZ transgene (Mut) at various times (in hours) relative to puparium formation from midguts (−12 and 2 h) and salivary glands (10 and 12 h) and subjected to Northern blot analysis. Filters were probed with LacZ and dronc. Slight adjustments of brightness and contrast were applied to images to enhance quality of the presentation. (bottom) A picture of the gel before blotting to demonstrate that approximately equal amounts of intact RNA were present in all lanes. White lines indicate that intervening lanes have been spliced out.
The role of direct EcR–Usp–mediated dronc regulation was analyzed further in transgenic flies carrying mutations in the droncEcRBE (Fig. 4 A) that abrogate EcR–Usp binding. Transgenic flies carrying mutant EcRBE (in the 2.8 kb dronc promoter LacZ transgene) were carefully staged together with transgenic flies carrying the wild-type 2.8-kb dronc promoter-reporter, and Northern blot analysis was performed comparing LacZ and endogenous dronc expression. In lines where LacZ expression was driven by the wild-type dronc promoter, LacZ and endogenous dronc show low to undetectable levels of expression in midgut and salivary gland at −12 and 10 h (relative to puparium formation), respectively. Both LacZ and endogenous dronc transcription was up-regulated in the midgut and salivary gland at 2 and 12 h, respectively (Fig. 8 B), at a time when PCD occurs in these tissues. Mutation of the EcRBE did not significantly affect LacZ expression in midgut, whereas expression in salivary gland was compromised at 12 h (Fig. 8 B) but detected at later stages (not depicted). As expected, endogenous dronc was transcriptionally up-regulated in both tissues (Fig. 8 B). These results demonstrate that the droncEcRBE is required for proper timing of dronc expression in the salivary gland.
Discussion
One of the major questions associated with the biological actions of nuclear hormones is how a single hormone can control differentiation, PCD, and proliferation in different tissues. Although useful information has been obtained from mammalian systems, problems of redundancy, different nuclear receptor isoforms, and developmental defects has hampered the ability to decipher the precise mechanisms of nuclear receptor actions. Drosophila provides an excellent system to address some fundamental questions associated with nuclear hormone actions. The steroid hormone ecdysone specifically mediates the removal of larval tissues, such as salivary glands and midgut at specific times during development that are then replaced by adult tissues from differentiating progenitor cells (Jiang et al., 1997; Baehrecke, 2000, 2002). We have previously discovered that ecdysone transcriptionally up-regulates dronc expression in salivary glands and midgut before PCD in these tissues (Dorstyn et al., 1999a; Daish et al., 2003). The understanding of ecdysone-mediated spatial and temporal regulation of dronc expression will greatly assist in deciphering how nuclear hormones control PCD of specific tissues at precise stages of development.
With the use of luciferase reporter constructs and an ecdysone responsive cell line in this paper, we have established that the promoter region spanning 2.8 to 1.1 kb contains important elements for ecdysone-mediated transcription. We have further shown that this region harbors a putative repressor that acts by recruiting a histone deacetylase as TSA treatment alleviates the repression of transcription. However, TSA has no effect on the 1.1-kb promoter, implying a histone deacetylase is recruited specifically to the 2.8–1.1 kb region. It was further shown that an EcR binding site was present at 1.36 kb from the transcription start site that specifically binds the EcR-B1 isoform in l(2)mbn cells and Drosophila prepupal salivary gland/midgut nuclear extracts. Functional experiments in l(2)mbn cells have established that this site is vital for dronc transcription as mutation reduces ecdysone-mediated activation and transactivation by EcR-B1. Due to the anomalies associated with cell lines, the importance of the EcRBE was also assessed in transgenic flies. The results are supported by our recent findings that the 2.8-kb promoter contains all necessary elements for correct spatial regulation in Drosophila and deleting the promoter to 1.1 kb (eliminating the EcR binding site) renders it inactive in salivary glands and brain lobes (Daish et al., 2003). This finding was further supported by two in vivo approaches. First, the importance of droncEcRBE was demonstrated by deletion just before the EcR binding site, which had no abrogating effect on expression, whereas deletion of the EcR binding site eliminated transcription in both salivary glands and brain lobe without any effect in the midgut. In addition, specific mutation of the EcRBE delayed expression in salivary glands without affecting midgut expression.
The lack of effect of EcRBE mutation in the midgut can be explained in many ways. For example, a different chromatin structure along the dronc promoter in this tissue may allow other factors to play a dominant role for dronc expression. The chromatin structure surrounding the EcR binding site in the midgut may also preclude EcR–Usp from binding to EcRBE (chromatin effects are not taken into consideration in EMSA experiments). Coactivators play a key role in modifying chromatin in nuclear hormone-mediated transcription. For example, CARMER, a Drosophila histone methyl transferase, required for ecdysone-induced expression of cell death genes in l(2)mbn cells (Cakouros et al., 2004), may have some role in tissue-specific gene expression. Alternatively, a midgut-specific transcription factor may inhibit binding of EcR–Usp to the dronc promoter. Our results show that the droncEcRBE has a lower affinity for the EcR–Usp than the consensus site (Fig. 4 B), and expression analysis shows a decrease in EcR-B1 expression at the time of dronc expression in the midgut but an increase in expression in the salivary gland (Fig. 6 A). The lower affinity and lower EcR expression suggests that insufficient amounts of EcR bind to the promoter in the midgut, whereas the increase in EcR-B1 expression in salivary glands overcome this problem. In fact, this result is observed in EMSA performed with extracts from tissues (Fig. 6 D) as identical levels of nuclear extracts show better binding from salivary gland extracts when compared with midgut. In any case, our results show that the EcRBE is important for expression of dronc specifically in salivary gland and brain lobes, but not in midgut (Fig. 9).
Figure 9.

A model of ecdysone-mediated regulation of dronc expression. Ecdysone binds its heterodimeric receptor EcR–Usp and activates dronc expression in specific tissues by directly interacting with an EcRBE in the promoter. Recruitment of a potential repressor to EcRBE region is likely to be required for the correct temporal regulation of dronc expression. EcR–Usp also regulates dronc transcription via ecdysone-induced transcription factors BR-C and E93, which may regulate temporal expression. Because E93 is required for expression of dronc in both larval salivary gland and midgut (Lee et al., 2000), it may also play a role in the spatial regulation of dronc expression.
Although the EcRBE is important for proper salivary gland dronc transcription, it is possible that it does not function alone but in cooperation with other factors that might govern tissue-specific expression. Numerous examples of this already exist such as the dGATAb transcription factor which binds to three binding sites flanking a EcR–Usp binding site of the Fbp1 promoter directing fat body-specific transcription (Brodu et al., 1999). Studies on Sgs4 gene transcription, which is specifically expressed in larval salivary glands, have revealed that its tissue-specific expression is governed by the Forkhead transcription factor in this tissue (Lehmann and Korge, 1996). In light of this, we have previously shown that BR-C binding sites exist in the dronc promoter (Cakouros et al., 2002), and given their similarity to Forkhead binding sites, it is possible that in salivary glands these sites also bind Forkhead. However, this possibility remains to be tested.
Our data show that both EcR-B1 and EcR-A isoforms are able to bind the dronc promoter EcRBE. Because EcR-A isoform is not expressed in l(2)mbn cells or larval/pupal midgut and salivary glands (or expressed at levels below detection), it is unlikely to play an important role in ecdysone-mediated dronc transcription and PCD. Consistent with this, previous works have shown that ectopic expression of the EcR-A isoform in EcR-B1–deficient salivary glands does not restore their ability to respond to ecdysone (Bender et al., 1997).
In mammals it is well documented that nuclear receptors recruit corepressors in the absence of ligand and specific coactivators in the presence of ligands (Shibata et al., 1997). These coactivators can be tissue and promoter specific, and bind to selected receptor isoforms. In this work, we have provided evidence of the possible recruitment of a repressor to the upstream dronc promoter region. This repressor seems to recruit a histone deacetylase as assessed by sensitivity to TSA. However, TSA sensitivity is reduced when the EcRBE is mutated, indicating that the EcR–Usp binding is partly responsible for recruiting this repressor. If a repressor was recruited by EcR–Usp, then the mutation of EcRBE site should show increased basal expression. However, this increase was not observed. One possible reason for this lack of increased activity is because increased promoter activity is likely to be seen when the corepressor is inactivated or eliminated, enabling coactivators to bind to the EcR–Usp, increasing basal activity. However, in the absence of EcR binding to the promoter, coactivators are unable to be recruited to the promoter, and there will therefore be no increase in basal activity. Given that these results are suggestive of the actions of a corepressor, more detailed experiments are being undertaken to identify the recruitment of such a repressor.
Overall, we have demonstrated that the EcR-B1 isoform is directly recruited to the dronc promoter and is required for proper temporal dronc transcription in specific tissues. The data presented here forms the foundation of future work to address important questions associated with spatio-temporal gene expression and PCD and the role of nuclear hormones in these processes. We believe that the dronc promoter provides an important tool for such studies.
Materials and methods
Cell culture
l(2)mbn cells (a gift from A. Dorn, Johannes Gutenberg University, Mainz, Germany; Ress et al., 2000) were grown in Schneider's medium supplemented with 10% FBS. 2.5 × 106 per well cells were seeded in 6-well plates in triplicate. Where necessary, 10 μM ecdysone (Sigma-Aldrich) was added for the desired time. Cycloheximide was used at 10 μg/ml. TSA (Sigma-Aldrich) was used at 1 μM. Cell viability was assessed by trypan blue exclusion.
Northern blotting and RT-PCR
Total RNA was extracted using Trizol reagent (Life Technologies), and 15–20 μg was analyzed by Northern blotting using 32P-labeled probes as described previously (Colussi et al., 2000; Cakouros et al., 2002). For RT-PCR, 2 μg RNA was used to generate cDNA using Superscript II reverse transcriptase (Invitrogen). cDNA was diluted 1:10 and 1 μl was used in a standard PCR reaction. PCR conditions have been described previously (Daish et al., 2003).
Immunoblotting
Cell lysates were electrophoresed on a 10% SDS PAGE, transferred onto PVDF membrane (Schleicher & Schuell), and blocked for 4 h in 5% skim milk. Affinity-purified DRONC antibody (Quinn et al., 2000; Dorstyn et al., 2002) was used at a 1:300 dilution. Purified antibodies to EcR common, EcR-B1, and EcR-A from the hybridoma bank were used at a 1:2,000 dilution. Secondary HRP-conjugated anti–mouse antibody (Amersham Biosciences) was used at a 1:2,000 dilution. Signals were detected using the ECL system (Amersham Biosciences).
Expression constructs
The luciferase reporter pxpGDR2.8kbLuc (generated by P. Colussi, Hanson Institute, Adelaide, Australia) contains a 2.8-kb region of the dronc promoter upstream of the transcription start site up to the ATG cloned into the luciferase reporter vector pxpG (provided by P. Cockerill, Hanson Institute, Adelaide, Australia). Deletions of the promoter were made by PCR amplification from 1.1 kb and 0.54 kb relative to transcription start site up to the ATG site and cloned into the pxpG luciferase reporter vector. EcR-B1 expression construct was provided by M. Bender (University of Georgia, Athens, GA).
Preparation of nuclear extracts and EMSA
7.5 × 106 l(2)mbn cells were pelleted, washed once in PBS, resuspended in 800 μl of buffer A (10 mM Hepes, pH 7.6, 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.1 mM EGTA, and Complete™ protease inhibitors from Roche), and placed on ice for 15 min. 0.1% NP-40 was added, and cell suspension was vortexed for 30 s and centrifuged at 13 K for 30 s at 4°C. Nuclear pellets were resuspended in 80 μl of buffer C (10 mM Hepes, pH 7.6, 400 mM NaCl, 7.5 mM MgCl2, 0.2 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, and Complete™) and incubated on ice for 40 min while shaking. Extracts were centrifuged for 5 min at 13 K, and supernatant were aliquoted and frozen at −70°C. Nuclear extracts were prepared from staged larvae ranging from 120–132 h AEL by homogenizing 50 larvae in 300 μl of buffer A and removed to a fresh tube in a total of 600 μl devoid of larval debri. After incubation on ice for 15 min, 10 μl of 10% NP-40 was added, lysates were vortexed for 30 s, and spun at 13 K for 30 s. Nuclear pellets were resuspended in 60–100 μl of buffer C and incubated on ice with shaking for 1 h. Lysates were centrifuged for 5 min at 13 K at 4°C and supernatant frozen as aforementioned. EMSA was performed by incubating 7–12 μg (4 μl) of nuclear extracts in binding buffer (20 mM Hepes, pH 7.9, 80 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF, and 10% glycerol) containing 1mg/ml BSA and 1 μg of poly dI/dC for 5 min on ice. Where needed, 2 μl of murine EcR-B1, EcR common, EcR-A antibody, or a control antibody was also added to the nuclear extracts and incubated on ice before the addition of labeled probe. Where indicated, 40 ng of cold competitor oligonucleotide was also included. 0.2 ng of labeled probe was added and incubated on ice for a further 20 min. 5 μl of 5× loading buffer was added, and samples were electrophoresed on a 5% polyacrylamide/0.5×TBE gel. The gel was dried down onto 3 MM Whatmann paper and exposed to Kodak X-ray film.
Transfection and luciferase assay
2 μg of pxpGDR2.8kbLuc, pxpGDR2.8kbEcRmutLuc, pxpGDR1.1kbLuc, or pxpGDR0.54kbLuc was transfected using Cellfectin alone or with 1–5 μg of EcR-B1 expression constructs. Equal amounts of DNA were used with the pIE1-4 expression vector. DNA in a total volume of 100 μl in Schneider media (without FBS) was added to Cellfectin (2:9 ratio) in 100 μl of total media devoid of FBS and incubated at RT for 15 min. 800 μl of serum-free medium was added and overlaid onto 2.5 × 106 cells in 6-well plates. Cells were incubated with the DNA/Cellfectin mixture for 5 h. The medium was replaced by 3 ml of Schneider media supplemented with 10% FBS, and cells were allowed to recover for 24 h. Where needed, 10 μM ecdysone was added for 24 h. Cells were harvested 48 h after transfection, resuspended in lysis buffer (100 mM phosphate buffer, pH 7.8, 10 μM EDTA, and 2 mM DTT), and frozen three times in liquid nitrogen. After centrifugation for 5 min at 13 K, the supernatant was analyzed for luciferase activity. 60–100 μg of protein was assayed in 200 μl of assay buffer (100 mM phosphate buffer, 8 mM MgSO4, 2 mM DTT, 0.75 mM ATP, and 0.175 mM coenzyme A) using an illuminometer (Packard Instrument Co.).
Generation and analysis of dronc reporter lacZ lines
1.64 kb and 1.33 kb of the dronc promoter were PCR amplified from Drosophila genomic DNA using Expand High Fidelity PCR System (Roche) with the primer sets DrPrF3cBglII (5′ CCG AGA TCT ATG TAC GTT ATG TTA TAG TAA GTG TA 3′); DrPrR1BglII (5′ CGG AGA TCT CCG GAT ATG GCT TCC ACG CGT 3′) and DrPrF3eBglII (5′ CGA AGA TCT AAT TGT GTA CAA CTA AAG GAA 3′); DrPrR1BglII, respectively. PCR products were cloned into pGem-T easy (Promega), and then subcloned into the BglII site of the p-element transformation lacZ reporter vector pCaSpeR-NLSlacZ (provided by Carl Thummel, Howard Hughes Medical Institute, University of Utah, Salt Lake City, UT) after BglII digestion. The 2.8-kb dronc-LacZ reporter construct has been described previously (Daish et al., 2003). The EcRBE mutations were introduced by PCR using standard techniques. Clones were sequenced for correct orientation, and transgenic flies were generated and transgenes mapped by established techniques. Animals were staged and tissues stained for β-galactosidase activity as described previously (Daish et al., 2003). In brief, third instar larval stages were determined by the gut clearance technique after growth of animals on bromophenol blue–supplemented food. Prepupal and pupal stages were attained by collecting newly pupariated animals from clear gutted third instar larvae populations every 30 min and ageing at 25°C to desired stages before collection and analysis. Images were acquired using a microscope (model SZ40; Olympus) set at 3–4× objective with a 2× adaptor lens (110AL2x), fitted with a digital camera (model DP11; Olympus). Images were cropped and processed using Adobe Photoshop software.
Acknowledgments
We thank Drs. Carl Thummel, Peter Cockerill, Paul Colussi, Augustus Dorn, and Michael Bender for reagents and cell lines.
This work was supported by the National Health and Medical Research Council of Australia. T. Daish is the holder of a Dawes Postgraduate Scholarship.
Abbreviations used in this paper: EcRBE, EcR–Usp binding element; EMSA, electrophoretic mobility shift analysis; PCD, programmed cell death; TSA, Trichostatin A.
References
- Adams, J.M. 2003. Ways of dying: multiple pathways to apoptosis. Genes Dev. 17:2481–2495. [DOI] [PubMed] [Google Scholar]
- Brodu, V., B. Mugat, J. Roignant, J.-A. Lepesant, and C. Antoniewski. 1999. Dual requirement for the EcR/USP nuclear receptor and the dGATAb factor in an ecdysone response in Drosophila melanogaster. Mol. Cell. Biol. 19:5732–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehrecke, E.H. 2000. Steroid regulation of programmed cell death during Drosophila development. Cell Death Differ. 7:1057–1062. [DOI] [PubMed] [Google Scholar]
- Baehrecke, E.H. 2002. How death shapes life during development. Nat. Rev. Mol. Cell Biol. 3:779–787. [DOI] [PubMed] [Google Scholar]
- Bender, M., F.B. Imam, W.S. Talbot, B. Ganetzky, and D.S. Hogness. 1997. Drosophila ecdysone receptor mutations reveal functional differences among receptor isoforms. Cell. 91:777–788. [DOI] [PubMed] [Google Scholar]
- Cakouros, D., T. Daish, D. Martin, E.H. Baehrecke, and S. Kumar. 2002. Ecdysone-induced expression of the caspase DRONC during hormone-dependent programmed cell death in Drosophila is regulated by Broad-Complex. J. Cell Biol. 157:985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakouros, D., T. Daish, K. Mills, and S. Kumar. 2004. An arginine-histone methyltransferase, CARMER, coordinates ecdysone-mediated apoptosis in Drosophila cells. J. Biol. Chem. 279:18467–18471. [DOI] [PubMed] [Google Scholar]
- Colussi, P.A., L.M. Quinn, D.C.S. Huang, M. Coombe, S.H. Read, H. Richardson, and S. Kumar. 2000. Debcl, a proapoptotic Bcl-2 homologue, is a component of the Drosophila melanogaster cell death machinery. J. Cell Biol. 148:703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daish, T.J., D. Cakouros, and S. Kumar. 2003. Distinct promoter regions regulate spatial and temporal expression of the Drosophila caspase dronc. Cell Death Differ. 10:1348–1356. [DOI] [PubMed] [Google Scholar]
- Dorstyn, J., P.A. Colussi, L.M. Quinn, H. Richardson, and S. Kumar. 1999. a. DRONC, an ecdysone-inducible Drosophila caspase. Proc. Natl. Acad. Sci. USA. 96:4307–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorstyn, L., S.H. Read, L.M. Quinn, H. Richardson, and S. Kumar. 1999. b. DECAY, a novel Drosophila caspase related to mammalian caspase-3 and caspase-7. J. Biol. Chem. 274:30778–30783. [DOI] [PubMed] [Google Scholar]
- Dorstyn, L., S. Read, D. Cakouros, J.R. Huh, B.A. Hay, and S. Kumar. 2002. The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J. Cell Biol. 156:1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins, C.J., S.J. Yoo, E.P. Peterson, S.L. Wang, S.Y. Vernooy, and B.A. Hay. 2000. The Drosophila caspase DRONC is a glutamate/aspartate protease whose activity is regulated by DIAP1, HID and GRIM. J. Biol. Chem. 275:27084–27093. [DOI] [PubMed] [Google Scholar]
- Jiang, C., E.H. Baehrecke, and C.S. Thummel. 1997. Steroid regulated programmed cell death during Drosophila metamorphosis. Development. 124:4673–4683. [DOI] [PubMed] [Google Scholar]
- Jiang, C., A.J. Lamblin, H. Steller, and C.S. Thummel. 2000. A steroid triggered hierarchy controls salivary gland cell death during Drosophila metamorphosis. Mol. Cell. 5:445–455. [DOI] [PubMed] [Google Scholar]
- Kumar, R., and E.B. Thompson. 2003. Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol. Endocrinol. 17:1–10. [DOI] [PubMed] [Google Scholar]
- Kumar, S., and J. Doumanis. 2000. The fly caspases. Cell Death Differ. 7:1039–1044. [DOI] [PubMed] [Google Scholar]
- Kumar, S., and D. Cakouros. 2004. Transcriptional control of the core cell-death machinery. Trends Biochem. Sci. 29:193–199. [DOI] [PubMed] [Google Scholar]
- Lee, C.Y., D.P. Wendel, P. Reid, G. Lam, C.S. Thummel, and E.H. Baehrecke. 2000. E93 directs steroid-triggered programmed cell death in Drosophila. Mol. Cell. 6:433–443. [DOI] [PubMed] [Google Scholar]
- Lehmann, M., and G. Korge. 1996. The fork head product directly specifies the tissue specific hormone responsiveness of the Drosophila Sgs-4 gene. EMBO J. 15:4825–4834. [PMC free article] [PubMed] [Google Scholar]
- Meier, P., J. Silke, S.J. Leevers, and G.I. Evan. 2000. The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 19:598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn, L.M., L. Dorstyn, P.A. Colussi, P. Chen, M. Coombe, J. Abrams, S. Kumar, and H. Richardson. 2000. An essential role for the caspase Dronc in developmentally programmed cell death in Drosophila. J. Biol. Chem. 275:40416–40424. [DOI] [PubMed] [Google Scholar]
- Ress, C., W. Holtmann, U. Maas, J. Sofsky, and A. Dorn. 2000. 20-Hydroxyecdysone-induced differentiation and apoptosis in the Drosophila cell line, l(2)mbn. Tissue Cell. 32:464–477. [DOI] [PubMed] [Google Scholar]
- Richardson, H., and S. Kumar. 2002. Death to flies: Drosophila as a model system to study programmed cell death. J. Immunol. Methods. 265:21–38. [DOI] [PubMed] [Google Scholar]
- Riddiford, L.M. 1993. Hormones and Drosophila development. The Development of Drosophila melanogaster. M. Bate and A. Martinez-Arias, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 899–939.
- Shibata, H., T.E. Spencer, S.A. Onate, G. Jenster, S.Y. Tsai, M.J. Tsai, and B.W. O'Malley. 1997. Role of co-activators and co-repressors in the mechanism of steroid/thyroid receptor action. Recent Prog. Horm. Res. 52:141–164. [PubMed] [Google Scholar]
- Talbot, W.S., E.A. Swyryd, and D.S. Hogness. 1993. Drosophila tissues with different metamorphic responses to ecdysone express different ecdysone receptor isoforms. Cell. 73:1323–1337. [DOI] [PubMed] [Google Scholar]
- Thummel, C.S. 1996. Files on steroids—Drosophila metamorphosis and the mechanisms of steroid hormone action. Trends Genet. 12:306–310. [DOI] [PubMed] [Google Scholar]
- Truman, J.W., and L.M. Riddiford. 2002. Endocrine insights into the evolution of metamorphosis in insects. Annu. Rev. Entomol. 47:467–500. [DOI] [PubMed] [Google Scholar]
- Yao, T.P., B.M. Forman, Z. Jiang, L. Cherbas, J.D. Chen, M. McKeown, P. Cherbas, and R.M. Evans. 1993. Functional ecdysone receptor is the product of EcR and Ultraspiracle genes. Nature. 366:476–479. [DOI] [PubMed] [Google Scholar]