

Abstract
Calcium is a critical mediator of many intracellular processes in eukaryotic cells. In the obligate intracellular parasite Toxoplasma gondii, for example, a rise in [Ca2+] is associated with significant morphological changes and rapid egress from host cells. To understand the mechanisms behind such dramatic effects, we isolated a mutant that is altered in its responses to the Ca2+ ionophore A23187 and found the affected gene encodes a homologue of Na+/H+ exchangers (NHEs) located on the parasite's plasma membrane. We show that in the absence of TgNHE1, Toxoplasma is resistant to ionophore-induced egress and extracellular death and amiloride-induced proton efflux inhibition. In addition, the mutant has increased levels of intracellular Ca2+, which explains its decreased sensitivity to A23187. These results provide direct genetic evidence of a role for NHE1 in Ca2+ homeostasis and important insight into how this ubiquitous pathogen senses and responds to changes in its environment.
Keywords: Toxoplasma; ionophore; egress; calcium; NHE
Introduction
Toxoplasma gondii is an obligate intracellular parasite with the ability to infect virtually any nucleated cell from a wide range of mammalian and avian species (Joiner and Dubremetz, 1993). In humans, Toxoplasma infections are widespread and can lead to severe disease in individuals with an immature or suppressed immune system. Consequently, Toxoplasmosis has become one of the main opportunistic infections in AIDS patients (Luft and Remington, 1992). A devastating consequence of the uncontrolled growth of Toxoplasma, and a cause of much of its associated disease, is the lethal lysis of the host cell as the parasite exits the parasitophorous vacuole in which it replicates. This process of egress is thought to share many of the molecular mechanisms used during the active invasion of host cells, allowing the parasite to quickly move from the lysed cell to a new one with little exposure to the extracellular environment (Hoff and Carruthers, 2002).
One of the aspects functionally connecting invasion and egress is the dependence of both events on Ca2+ signaling (Pezzella et al., 1997; Black and Boothroyd, 2000; Moudy et al., 2001). The relation between Ca2+ fluxes and egress is particularly evident in experiments with the calcium ionophore A23187, which induces the parasite to quickly exit its host cell (Endo et al., 1982). This ionophore-induced egress (IIE) will stimulate a population of intracellular parasites to rapidly exit their parasitophorous vacuole and the host cell in a manner similar to natural egress except that IIE can be induced at any stage during the lytic cycle, whereas in vitro, at least, natural egress generally occurs only when there are 64 or more parasites inside the host cell. As in natural egress, parasites that undergo IIE become motile and change their morphology before they exit, suggesting that this is an active process requiring the cytoskeleton and motility machinery (Black et al., 2000; Black and Boothroyd, 2000; Hoff and Carruthers, 2002). Similarly, when exposed to the ionophore A23187, extracellular parasites activate the secretory and cytoskeletal events required for invasion (Mondragon and Frixione, 1996; Carruthers et al., 1999). Prolonged exposure to the ionophore while extracellular causes Toxoplasma to irreversibly lose its ability to invade host cells, presumably due to the exhaustion of required invasion factors. The result of such treatment for this obligate intracellular parasite is death, and thus this phenomenon is known as ionophore-induced death (IID) (Mondragon and Frixione, 1996).
We have previously reported the isolation and characterization of chemically induced mutants deficient in IIE (Iie− mutants) (Black et al., 2000). From those analyses, a stepwise model for IIE was proposed. First, the ionophore induces the release of intracellular Ca2+ stores (either within the host or the parasite). The parasites respond to these changes in Ca2+ concentrations by first extending their conoid, an anterior cytoskeletal structure involved in invasion, and then permeabilizing the host plasma membrane and the parasitophorous vacuole by an unknown mechanism. This allows a proposed final egress signal to reach the parasite, inducing it to leave. In addition, Iie− mutants are also defective in the earliest stages of the lytic cycle (i.e., invasion) and some of them exhibit a resistance to IID (Iie−Iid− mutants), confirming commonality between IIE, IID, and normal processes of the parasite (Black et al., 2000).
The permeabilization of the host cell has also been reported to occur before natural egress (Moudy et al., 2001). While it is unclear whether that permeabilization event is parasite driven, Moudy and colleagues report that it is the subsequent loss of K+ in the host cell that initiates the Ca2+-dependent egress process. The involvement of changes in both [K+] and [Ca2+] during egress highlights the interplay and importance of ions in mediating crucial functions in Toxoplasma.
While the study of chemically induced IIE mutants yielded much information about Toxoplasma egress, identifying the disrupted genes has proven elusive, rendering the interpretation of the mutant phenotype and the study of egress genes difficult (Black et al., 2000). Random insertional mutagenesis is an alternative and previously successful method of mutating Toxoplasma, which simplifies the identification of the affected gene (Donald and Roos, 1995). We have now isolated and characterized an insertional mutant with a delayed IIE phenotype. This mutant was isolated using a selection based on the observations that most mutants selected for resistance to IID also show a defect in IIE (Black et al., 2000). Identification of the disrupted gene in the isolated Iid−Iie− mutant revealed an insertion within a previously undescribed Toxoplasma Na+/H+ exchanger (TgNHE1). In this report, we provide evidence that TgNHE1 is in the plasma membrane, where it is active in proton transport, and that its disruption results in elevated intraparasitic [Ca2+]. These results are used to formulate a model for the role of the Na+/H+ exchanger (NHE) in the Ca2+-dependent IIE and IID processes.
Results
Isolation of insertional mutants defective in IID
Previously, a genetic selection was employed to isolate chemically induced mutants of the RHΔhpt strain that were resistant to IID (Black et al., 2000). Several of these Iid− mutants were found to also have a delay in IIE (Iid−Iie− mutants). Due to the ease of the IID selection and its success at identifying mutants in the IIE process, we repeated the selection scheme using insertional mutants, which should simplify the identification of the disrupted genes. For this purpose, two independent populations of insertional mutants were created by transforming extracellular parasites of the RHΔhpt strain with a vector carrying HPT, as previously described (Knoll et al., 2001; see Materials and methods). Parasites that carried a random insertion of the plasmid were selected for the resistance to mycophenolic acid and xanthine conferred by the HPT marker, and the stably transformed populations were used in the subsequent selection for Iid− mutants. In brief, extracellular parasites from two mutant populations (GAD1 and GAD2) and nonmutagenized RHΔhpt parasites were exposed to 1 μM A23187 for 30–60 min before allowing them to infect fibroblasts to recover the survivors (see Materials and methods). After the third round of selection, no survivors from either the GAD2 population or the nonmutagenized sample were recovered. After six rounds of selection, the surviving parasites from the GAD1 population were plated at a limiting dilution, and 30 clones were isolated. The Iid− phenotype of two clones (GAD1.7 and GAD1.15) is shown in Fig. 1. Both strains showed a 10% survival rate after 60 min, at which time point no survivors were ever detected with the parental strain.
Figure 1.

IID phenotype of the parental strain RHΔhpt and two Iid − mutants, GAD1.7 and GAD1.15. Extracellular parasites were exposed to A23187 and then assayed for the ability to form plaques. Efficiency of plating (EOP) was defined as the percentage of plaque-forming units arising from parasites incubated with A23187 versus the DMSO-treated control. Each data point represents the average of three experiments and the error bars represent the standard deviation.
GAD1.7, but not GAD1.15, exhibits a delay in IIE
To investigate whether any of the clones obtained in the IID selection were also defective in IIE, as expected from previous results (Black et al., 2000), intracellular parasites from all 30 clones were treated with 1 μM A23187 for 3 min, and the levels of egress were assessed visually as compared with the parental strain (see Materials and methods). In this manner, five clones (GAD1.3, GAD1.7, GAD1.17, GAD1.24, and GAD1.26) were identified as having 10–30% of vacuoles intact after the incubation in three replicate experiments (Iie− clones), while all other clones and the parental strain exhibited <1% intact vacuoles by this time point (Iie+). The detailed IIE phenotypes of the parental strain and clones GAD1.7 (Iid−Iie−) and GAD1.15 (Iid−Iie+) are shown in Fig. 2. At 2 min, 100% of the parental strain parasites are out of their vacuoles as previously reported (Black et al., 2000). Clone GAD1.15 behaves as wild type, while clone GAD1.7 parasites exhibit a consistently delayed reaction to the ionophore with only 61% of parasites having exited their host cells by 2 min. Nevertheless, with longer incubations, the mutants do eventually respond such that by 5 min, ∼95% of vacuoles are lysed (Fig. 2). Since clones with and without an Iie− phenotype were isolated from the same insertional mutant population, the delay in IIE is most likely due to the disruption of a gene and not a general effect of the vector used. Finally, this selection confirms that selecting for Iid− mutants results in parasites that also have an Iie− phenotype and provides further evidence that these two processes are genetically linked (Black et al., 2000).
Figure 2.

IIE phenotype of GAD and knockout mutants. Percentage of vacuoles lysed at specific time points after induction with 1 μM A23187 in DMEi was measured for the parental strain RHΔhpt, the two Iid− mutants GAD1.7 and GAD1.15, the NHE1 knockout strain RHΔnhe1, and the HXGPRT-expressing strain RHΔhpt+HPT. Each data point represents the average of three experiments, and the error bars represent the standard deviation.
The mutagenic vector in GAD1.7 is inserted near a region of homology to sodium hydrogen exchangers
The most likely explanation for the Iie− defect of GAD1.7 is that the gene disrupted by the random insertion is involved in this process and in IID. Hence, the site of insertion was identified for this mutant. A molecular cloning approach was used to recover the inserted plasmid along with one of the flanking genomic DNA fragments (see Materials and methods). This flanking fragment was sequenced and determined to be within a 10-kb genomic region from the Toxoplasma genome project (http://ToxoDB.org). PCR analysis was used to determine that all five Iie− clones (see above) had the same insertion, while none of the 10 Iie+ clones tested showed this disruption (unpublished data). These five clones, therefore, are most likely the result of a single insertion event and subsequent expansion of the resulting mutant during the six rounds of selection. Sequence analysis of the targeted region revealed a segment ∼2,000 base pairs away from the insertion site with high homology to NHEs from various organisms. Given that this mutant was selected for a delay in responding to Ca2+ fluxes, an ion exchange protein fits the profile of a candidate gene whose disruption might lead to such a result. While intriguing, the homology to NHE was not conclusive about the gene disrupted however, because the insertion was not within the NHE homology region and therefore could be in an adjacent gene.
While only GAD1.7 (Iid−Iie−) is discussed here in detail, the insertion for GAD1.15 (Iid−Iie+) has also been determined. The disturbed region shows no homology to known genes, no EST lie close to the insertion site, and no long open reading frame is affected. The basis for the resulting phenotype, therefore, is unknown.
TgNHE1 encodes an unusually large protein with 12 TM domains
To further characterize this previously undescribed Toxoplasma NHE and confirm that the insertion was indeed within its gene, we cloned and sequenced the entire cDNA (Fig. 3 A). We call this gene and its predicted product TgNHE1, since it is the first of its class to be described in Toxoplasma and analysis of sequence databases suggests the presence of three additional NHE homologues in this parasite (see below). TgNHE1 is encoded by an 8,997-base transcript and, as Fig. 3 A depicts, the gene has 12 exons and 11 introns. The complete cloning of TgNHE1 along with PCR products from GAD1.7 genomic DNA (unpublished data) confirmed that TgNHE1 was indeed the gene into which the mutagenic vector had inserted; specifically it was within the sixth exon (Fig. 3 A, arrow).
Figure 3.

Genomic arrangement and protein domains of NHE1 . The Toxoplasma NHE1 gene encodes a protein 2,097 amino acids long with 12 TM domains. (A) The distribution of exons (dark lines), introns (broken lines), and coding sequences (white boxes) along the genomic region encoding NHE1 are shown. Base 1 in the gDNA and RNA graph represents the transcription start site as determined by 5′ RACE (nucleotide 19397 in TGG_10551, ToxoDB v.2.1). Also shown are the genomic regions (black boxes) used as flanking fragments in the generation of the NHE1 knockout construct. The bracket in the gDNA drawing indicates the region that is deleted and replaced by the HPT gene in the RHΔnhe1 knockout strain, while the arrow shows the site of the original insertion in the GAD1.7 mutant. The arrowheads in the RNA indicate the position of the two putative translation initiation sites. (B) Shown is the schematic of the TgNHE1 protein, with its signal sequence (black box) and the predicted 12 TM domains (gray boxes). Below and in the same scale is the Kyte-Doolitle hydropathy plot for the entire protein (Kyte and Doolittle, 1982). (C) Sequence comparison of the two highly conserved TM domains in Toxoplasma (NHE1, TM domains indicated by * in B), human (NHE1) (Sardet et al., 1988), P. falciparum (putative NHE) (PlasmoDB.org), and Arabadopsis (NHE/SOS1) (Shi et al., 2000) is shown. The conserved polar amino acids proposed to be involved in cation binding and translocation are marked by X (Wiebe et al., 2001). The amino acid number of the first residue in each line is shown. Complete sequence of TgNHE1 is available from GenBank/EMBL/DDBJ, accession no. AY485268.
Conceptual translation of the TgNHE1 cDNA reveals two potential start methionine (Met) residues, 190 amino acids apart. While no direct data exist to determine the actual start codon of TgNHE1, two lines of reasoning favor the second AUG as the start point of translation: (1) the second codon has a better context for a Toxoplasma translational initiation site with an adenine at the −3 position from the AUG (Seeber, 1997), and (2) only the translated product from the second Met has a predicted signal peptide within the first 65 amino acids as expected for a protein of this type. Hence, Fig. 3 shows a schematic of the TgNHE1 protein as starting from the second potential initiator methionine (base 1161 of the transcript) and extending for 2,097 amino acids. Computer algorithms predict TgNHE1 to have a signal peptide with a putative cleavage site between residues 62 and 63 (SignalP, http://www.cbs.dtu.dk/services/SignalP-2.0/), and 12 transmembrane (TM) domains (TMpred, http://www.ch.embnet.org/software/TMPRED_form.html) within its NH2-terminal region as expected for an NHE (Fig. 3 B; Putney et al., 2002).
TgNHE1 shows homology within the 12 TMs to NHEs from a variety of organisms, including bacteria, other apicomplexan parasites, mammals, and plants. The two highly conserved TMs thought to be involved in ion translocation (Wiebe et al., 2001) correspond to TM domains V and VI respectively in Toxoplasma, which has ∼72% identity to human NHE1 in this region (Fig. 3 C). The TM domains of TgNHE1 exhibit homology to a plethora of other NHEs but do not show significantly higher homology to any particular isoform from the best-studied eukaryotes, which typically encode three to six NHE homologues. Hence, we have chosen TgNHE1 as a name simply to denote that it is the first of its class in T. gondii. The COOH-terminal region shows little similarity to other NHEs, even from the related apicomplexan Plasmodium falciparum, but this is typical for these exchangers. While the COOH-terminal tail for most NHEs ranges up to 500 amino acids, this domain is particularly long in TgNHE1 with ∼1,800 residues. Only the predicted Plasmodium NHE also exhibits this unusually long COOH-terminal tail.
The Iie− phenotype is recapitulated in a directed knockout of TgNHE1
The methods used to develop random insertional mutants here do not preclude the possibility of multiple insertions or sporadic point mutations in other genes. Therefore, further experiments are necessary to unequivocally show that it is the insertion within the TgNHE1 gene, and not a separate mutation, that is responsible for the phenotype observed. For this purpose, we set out to disrupt the NHE1 locus in the parental strain using techniques developed for targeting specific genes in Toxoplasma (Kim et al., 1993; Donald and Roos, 1995). This de novo nhe1 − mutant will not share any other mutation with GAD1.7, and thus, any phenotype shared by the two strains should be a consequence of the disruption described. To produce such a strain, a construct was designed using segments of the TgNHE1 genomic region flanking the Toxoplasma selectable marker HPT (pKONHE1). Positive selection for HPT guaranteed the presence of the pKONHE1 construct in transformed parasites, while negative selection for a downstream marker, GFP, increased the chance that the vector had inserted by double homologous recombination. The insertion replaces 1,274 bases from TgNHE1 with HPT (Fig. 3 A), eliminating an entire exon and introducing a complete gene, including a polyA addition site, into its middle. This will almost certainly result in no functional TgNHE1 protein being produced. In addition, the knockout was designed so that the HPT marker is transcribed in the opposite direction from the HPT in the original mutant, GAD1.7. This serves as a control for at least some of the potential polar effects caused by the expression of this marker within this specific region of the genome.
A PCR-based approach was used to identify a clone (called hereafter RHΔnhe1) that was gfp− and had the desired disruption in TgNHE1. A separate gfp− clone expressing HPT, which in the PCR test did not carry the TgNHE1 disruption, was also maintained for use as a control for the effects of HPT expression and called RHΔhpt+HPT. The disruption of TgNHE1 in both the GAD1.7 and RHΔnhe1 strains was confirmed by Southern blot analysis probing for a TgNHE1 fragment that was predicted to change sizes in the mutants (unpublished data). As expected, the control strain, RHΔhpt+HPT, appeared as wild type in the Southern blot.
The IIE phenotype of the established nhe1 knockout was tested in parallel with the parental strain, the original GAD1.7 mutant, and the RHΔhpt+HPT strain. The two lines carrying an NHE1 disruption exhibit the same level of delay in IIE with ∼60% egress by 2 min of ionophore incubation, whereas the RHΔhpt+HPT strain, which carries a heterologous insertion of the pKONHE1 vector but not a disruption in NHE1, has a normal IIE curve with 100% egress by 2 min (Fig. 2). RHΔnhe1also exhibits resistance to IID, with 13% of parasites surviving a 60-min ionophore treatment compared with <1% survival with wild-type or the RHΔhpt+HPT strains (not depicted). The fact that the targeted disruption of TgNHE1 results in the same phenotypes as seen for GAD1.7 mutant proves that insertion into the TgNHE1 locus is responsible for the Iie− and Iid− phenotypes seen in this mutant.
An inhibitor of NHE function causes a delay in IIE
The above results strongly suggest a direct role for TgNHE1 in IIE. The possibility exists however that the phenotype is instead the consequence of a nearby gene being affected by the insertion or a polar effect caused by the expression of HPT within this specific region of the genome. To independently test the hypothesis that eliminating NHE function causes an Iie− phenotype, therefore, we investigated the effect on IIE of pharmacologically inhibiting NHE function. Amiloride and its derivatives are known to be powerful inhibitors of NHEs, acting by competitively inhibiting Na+ binding (Paris and Pouyssegur, 1983) and by noncompetitively inhibiting ion translocation (Ives et al., 1983). Hence, intracellular parasites were preincubated with dimethylamiloride (DMA) before inducing egress with the ionophore (see Materials and methods). As can be seen in Fig. 4, preincubation with DMA does indeed retard IIE: only 67% of DMA-treated parasites are out of their host cells by 2 min, whereas 100% of nontreated parasites have emerged by the same time point. This level of reduction is essentially the same as that seen with the genetic disruption of NHE1 in either the knockout or the original mutant (Fig. 4). The specificity of the drug is confirmed by the fact that DMA treatment of nhe1 − parasites gives no further delay in IIE (Fig. 4). These results confirm that the effect seen in the genetic mutant is caused by the loss of TgNHE1 function and is not an indirect consequence of the insertion.
Figure 4.

Effect of pretreating parasites with an NHE inhibitor on IIE. Percentage of vacuoles lysed at specific time points after induction with 1 μM A23187 in DMEi was measured for RHΔhpt, GAD1.7, and RHΔnhe1 parasites that were pretreated with either 100 μM DMA dissolved in DMSO or an equivalent amount of DMSO alone for 10 min. The data are pooled from three independent trials, and the error bars represent the standard deviation.
TgNHE1 is localized in the plasma membrane
To understand how TgNHE1 might affect IIE and other events, it is critical to determine its localization within the parasite. For this purpose, we created a polyclonal antibody against a bacterially expressed fragment of TgNHE1 (see Materials and methods). The fragment expressed was chosen from the COOH-terminal domain of the protein to avoid cross-reactivity with other NHEs, which usually show homology only in the TM domains. Specificity of the antibodies to this segment was also predicted by the fact that sequence analysis did not identify any other regions in the Toxoplasma genome with significant similarity to this fragment.
The mouse antibody produced was used to stain intracellular parasites by immunofluorescence. This approach yielded a clear staining at the extreme periphery of wild-type parasites (Fig. 5 A), which was absent when preimmune serum was used (not depicted). The specificity of this antibody for TgNHE1 was confirmed by the absence of staining in the GAD1.7 mutant and the nhe1 knockout strain (Fig. 5, B and C). Some faint, nondistinct intraparasitic staining can be seen in the nhe1 − mutants. This is presumably the result of weak cross-reactivity to another NHE or an unrelated protein, despite the absence of detectable similarity to the region used for immunization in any hypothetical Toxoplasma protein. The fluorescence pattern seen with wild-type parasites does not change during different stages of intracellular growth, indicating that the signal is from protein within the parasite plasma membrane rather than the inner membrane complex just below, which forms within dividing parasites and exhibits an immunofluorescence pattern distinct from plasma membrane (Hu et al., 2002). The fact that this antibody does not stain unpermeabilized parasites (unpublished data) indicates that the COOH-terminal region of TgNHE1 is not exposed to the outside, as predicted from results with other NHE proteins (Putney et al., 2002).
Figure 5.
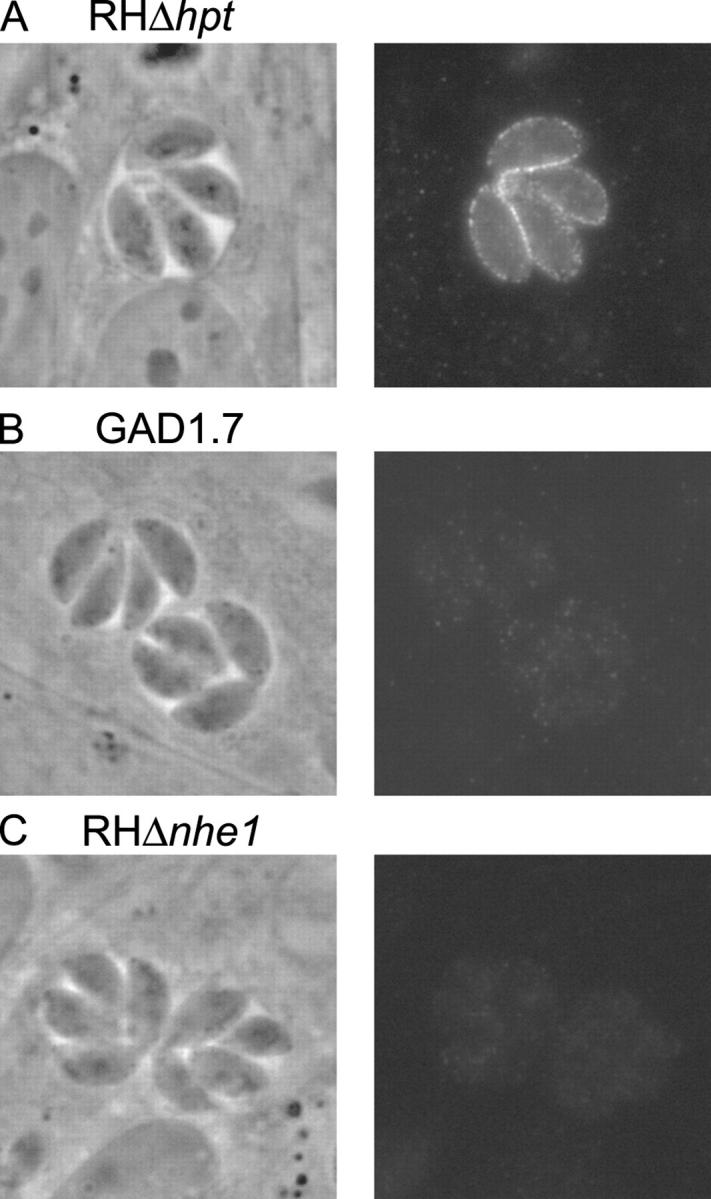
Intracellular localization of TgNHE1. Intracellular parasites of either (A) the wild-type RHΔhpt, (B) the mutant GAD1.7, or (C) the NHE knockout RHΔnhe1 were stained with antibodies raised in mouse against amino acids 716–1073 of the TgNHE1 protein. An Alexa fluor 488–conjugated goat anti–mouse antibody was used to visualize the TgNHE1 signal. The phase contrast and fluorescence images of the same vacuole are shown for each strain.
Proton efflux and calcium homeostasis in the TgNHE1 mutants
Plasma membrane NHEs in other systems exchange Na+ for H+, linking the electrochemical potential of sodium and hydrogen ions maintained across the membrane. In animal cells, NHEs have been implicated in a variety of functions, including the regulation of cytoplasmic pH and Na+ concentration and the control of cell volume (Putney et al., 2002). Assuming TgNHE1 has a similar function, its disruption should have an effect on the proper balance of intracellular ions. To investigate this possibility, and to further explore its role in IIE and IID, several physiological assays were performed with RHΔhpt, GAD1.7, and RHΔnhe1 parasites that are purified away from host cells. First, the intraparasitic pH (pHi) was measured for the three strains under conditions where the extracellular pH (pHe) ranged pHe 6–8. The mean baseline pHi for all three strains was not significantly different from each other under any of the conditions tested (unpublished data). While this result might suggest a lack of a role for NHE1 in pHi homeostasis, it is possible that either the pHi readings obtained in our experiments are not representative of what occurs while the parasites are within host cells or that other proton pumps present in the parasites' membranes overcome any defects in H+ exchange. To test the latter possibility, the effect of the NHE inhibitor amiloride on the efflux of protons was tested (Fig. 6). Whereas in a standard buffer, wild type and the two NHE1 mutant parasites show the same level of proton efflux, addition of amiloride reduces proton efflux by half in RHΔhpt cells but does not affect either GAD1.7 or RHΔnhe1 parasites. The resistance to the reduction of proton efflux exhibited by both TgNHE1 mutant strains is specific to the NHE inhibitor because the V-H+-ATPase inhibitor bafilomycin A1 affects proton efflux to the same level in wild-type and mutant parasites (Fig. 6). Addition of bafilomycin A1 after amiloride had an additive effect on inhibition of proton efflux over that produced by amiloride alone in the RHΔhpt cells, while this effect was not observed with the GAD1.7 and RHΔnhe1 parasites (not depicted). The effects of bafilomycin A1 on proton efflux, and on recovery of pHi after an acid load reported previously (Moreno et al., 1998), are in agreement with a significant role of the V-H+-ATPase in pH homeostasis. The present results indicate that TgNHE1 can contribute to proton transport in extracellular, wild-type parasites, although this does not establish its natural function when the parasites reside within a host cell.
Figure 6.

Effect of NHE inhibition on proton efflux. The level of proton efflux was measured for wild-type parasites (RHΔhpt) or either of the NHE1 mutants GAD1.7 and RHΔnhe1 by incubating the parasites in a weakly buffered solution in the presence of BCECF. The effect of the NHE inhibitor amiloride and the V-H+-ATPase inhibitor bafilomycin A1 on proton efflux for each strain is shown. Each bar represents the average of six independent experiments, and the error bars represent the standard deviation. Values that are statistically different from the RHΔhpt control as assessed by a t test are marked by * (P < 0.05).
Given the effect of disrupting TgNHE1 on a calcium-dependent process such as IIE, we next investigated the regulation of the intracellular concentration of calcium [Ca2+]i in extracellular parasites. [Ca2+]i measurements taken over a 20-min period in either the nominal absence of extracellular Ca2+ (EGTA present; Fig. 7, A and B) or in the presence of CaCl2 (Fig. 7, C and D) revealed that both NHE1 mutant strains were less able to regulate their [Ca2+]i and exhibited an elevated [Ca2+]i as compared with the wild-type strain (Fig. 7 E). In the absence of extracellular Ca2+, the [Ca2+]i for RHΔhpt parasites was 173 ± 28 (n = 7) at the 10-min time point, while the [Ca2+]i at the same time point for both the mutant GAD1.7 and the knockout strain RHΔnhe1 was significantly higher (550 ± 132 [n = 3] and 399 ± 127 [n = 4], respectively). This twofold increase in [Ca2+]i in either of the NHE1 mutants at 10 min is also observed when CaCl2 is present (Fig. 7 E; 224 ± 59 [n = 4] for RHΔhpt, 763 ± 373 [n = 4] for GAD1.7, and 450 ± 100 [n = 3] for RHΔnhe1). Taken together, these results indicate that the disruption of TgNHE1 can result in a deficient ability to regulate [Ca2+]i levels and subsequent accumulation of Ca2+ in the cytosol of the parasites. As described further below, this is most likely due to a coupling of TgNHE1 and one or more Ca2+/H+ exchange functions such that loss of TgNHE1 results in an inability to modulate [Ca2+]i in the normal way.
Figure 7.

Ca 2+ homeostasis in the parental strain RHΔhpt and the nhe − mutants RHΔnhe1 and GAD1.7. Intracellular Ca2+ concentration of Fura-2AM–loaded parasites was measured in the presence of either EGTA (A and B) or CaCl2 (C and D) by loading the parasites with Fura-2AM. Traces shown are from representative experiments. (E) The average [Ca2+]i for each of the three strains at 10 min in the presence of either EGTA or CaCl2 is shown. Each bar represents the average of at least three independent experiments, and the error bars represent the standard deviation. Values that are statistically different from the RHΔhpt control as assessed by a t test are marked by * (P < 0.015).
Discussion
In the studies described here, an insertional mutant resistant to the effects of the Ca2+ ionophore A23187 was isolated and characterized to identify the genes and pathways involved in IIE. The mutants obtained from one population selected for resistance to IID fall into two distinct classes: Iid−Iie− (e.g., GAD1.7) and Iid−Iie+ (e.g., GAD1.15). A genetic link between IIE and IID had been proposed previously (Black et al., 2000) and is further corroborated by the current report.
The Iid− phenotype of both mutant classes is not complete, but it was enough to provide a selective advantage over wild-type parasites. Interestingly, the level of IID resistance of GAD1.7 and GAD1.15 is considerably lower than the one reported for Iid− mutants generated by chemical mutagenesis (80–100% resistance at 60 min; Black et al., 2000), and this difference has been confirmed by parallel IID assays of the insertional and chemical mutants (unpublished data). This is also seen with the Iie− defect of GAD1.7: in parallel assays, wild-type parasites exhibited 100% egress after 2 min of ionophore treatment, compared with 61% for GAD1.7 and 16% for MBE1.1 (a chemical Iie−Iid− mutant; Black et al., 2000) (unpublished data). The weaker phenotype of GAD1.7 is interesting given that the insertional mutagenesis produced a complete loss of TgNHE1 function. This suggests that TgNHE1 is probably not the gene disturbed in the chemically treated mutant strains. Chemically induced point mutations, however, can result in a stronger phenotype than a complete knockout if the change produces a gain-of-function allele (so-called dominant negatives in diploid systems). This possibility remains to be explored for the many Iie− chemical mutants whose causal mutations have yet to be identified.
Our analysis of the Iie− mutant GAD1.7 has not revealed a detectable defect in natural egress. This is not surprising given that the difference in IIE between the mutant and wild type is on the scale of minutes. Such a difference would be undetectable during natural egress, which is not synchronized and occurs ∼48 h after infection with a variability of hours from vacuole to vacuole.
NHEs are ubiquitous membrane proteins involved in the exchange of H+ for Na+. The direction of this exchange is solely determined by the ions' electrogradient (Wiebe et al., 2001). In many eukaryotes, NHEs function to remove excess intracellular H+ by the uptake of extracellular Na+ in an electroneutral fashion, thus regulating pHi and cell volume (Putney et al., 2002). In bacteria, yeast, and some plants, the exchange is in the opposite direction, which in plants serves to confer salt tolerance (Shi et al., 2000). We have been unable to observe any differences in pHi between the mutant and wild-type strains under steady-state conditions in extracellular parasites. Nevertheless, our experiments (Fig. 6) provide pharmacological and genetic evidence that TgNHE1 can function in the efflux of protons at least under the conditions of these experiments. Under physiological conditions, however, TgNHE1 could function in efflux or in the opposite direction (i.e., sodium extrusion) as has been proposed for Plasmodium (Saliba and Kirk, 1999).
No other NHE has been previously described in Toxoplasma. Nevertheless, in the closely related parasite Plasmodium, the presence of a membrane Na+/H+ antiport activity has been suggested (Bosia et al., 1993), and the annotated genomic sequence predicts the presence of an NHE with high homology to TgNHE1. Similarly, in both Leishmania donovani and in Trypanosma brucei, kinetic and pharmacological studies argue for the presence of an NHE within the acidocalcisome (Vercesi and Docampo, 1996; Vercesi et al., 2000). Interestingly, those studies also suggested that the activation of the acidocalcisome's NHEs lead to an increase in cytosolic Ca2+ concentration by facilitating Ca2+ release from this organelle. This connection between an NHE and Ca2+ release is consistent with our results that the elimination of TgNHE1 affects processes dependent on Ca2+ fluxes. Immunofluorescence studies indicate that TgNHE1 is predominantly, if not exclusively, in the plasma membrane and not in an intracellular compartment. Sequence analysis of the Toxoplasma genome and a collection of ESTs reveal the existence of three other NHE homologues in this parasite. It is likely that one of these is present in the Toxoplasma acidocalcisome, and further studies will be needed to understand the role of these NHEs in ion homeostasis.
We had previously hypothesized that the Ca2+ detected by the parasites before egress was of extra-parasitic origin (Black et al., 2000). This was mostly based on the observation that BAPTA-AM, which should not enter intracellular parasites, blocked the effects of the ionophore in IIE. Nevertheless, we could not exclude the possibility that BAPTA was in fact entering the parasite and that therefore the Ca2+ signal was coming from intracellular compartments within the parasites. Additional studies on natural egress clearly argue for this latter possibility (Moudy et al., 2001). Regardless, egress in wild-type parasites appears to be the result of parasites sensing an increase in [Ca2+]i to ∼400 nM (Moudy et al., 2001). The delay in IIE caused by the disruption of TgNHE1 therefore could be related to the fact that in the nhe1 − mutant, the resting [Ca2+]i may already be elevated because of an inability to appropriately regulate [Ca2+]i. Accordingly, addition of an ionophore and induction of Ca2+ fluxes might produce less of an initial percentage change in [Ca2+]i and hence a delay in IIE and IID.
While a role for an NHE in Ca2+ homeostasis has been suggested in human platelets (Siffert and Akkerman, 1987), this interpretation has been disputed based on the lack of specificity of the pharmacological inhibitors used (Hunyady et al., 1987). Our studies using an organism with a genetic elimination of a specific NHE unequivocally show a role for this exchanger in the regulation of Ca2+ levels. While the mutant parasites are clearly able to adapt to defects in Ca2+ regulation, they exhibit a defect in responding to changes in local ion concentrations as seen by the delay in IIE. The exact mechanism responsible for the altered ability to maintain calcium levels under our experimental conditions in the nhe1 − mutants is not known. While we cannot exclude the possibility that TgNHE1 serves to directly transport Ca2+, Ca2+ extrusion from the cytosol is thought to be catalyzed by Ca2+/H+ countertransporting ATPases located in the plasma membrane, endoplasmic reticulum, and/or acidocalcisomes (Moreno and Docampo, 2003). Coupling of the plasma membrane Ca2+-ATPase with TgNHE1, therefore, could be involved in Ca2+ efflux and Na+ entry so that disruption of the TgNHE1 would result in higher [Ca2+]i. Such coupling between a Ca2+/H+ ATPase and an NHE has been suggested in intracellular Ca2+ regulation in stimulated Jurkat T cells (Berthe et al., 1991). It is also possible that TgNHE1 is not involved in pH homeostasis under physiological conditions, and that the effect on Ca2+ regulation is a consequence of some unidentified function of TgNHE1. Since our experiments suggest that TgNHE1 can influence proton fluxes, an alternative explanation for our results is that, under physiological conditions, TgNHE1 is linked to inward proton movement and functions in the extrusion of Na+. The absence of the TgNHE could lead to cytosolic Na+ increase, which has been shown to stimulate Ca2+ release from the acidocalcisomes of L. donovani (Vercesi et al., 2000) and T. brucei (Vercesi and Docampo, 1996).
The results reported here show that efficient IIE is dependent on the function of an NHE homologue. Furthermore, our work has revealed that the maintenance of Ca2+ homeostasis in Toxoplasma is dependent on this same protein. This provides strong evidence for a connection between Na+ and Ca2+ exchange and for a specific role in these processes for TgNHE1. This discovery also validates the use of forward genetics in the study of IIE since, a priori, this specific plasma membrane NHE would not have been predicted to be involved in Ca2+ homeostasis and the parasite functions regulated by Ca2+.
Materials and methods
Parasite cultivation and reagents
The tachyzoite stage of the RHΔHXGPRT strain (RHΔhpt; Donald et al., 1996) was the parent strain used for the mutagenesis and phenotypic analysis. Parasites of the Prugniaud strain (Zenner et al., 1993) were used to extract RNA for cloning and sequencing of the NHE1 gene. Strains were maintained in vitro by serial passage on monolayers of human foreskin fibroblasts (HFFs) at 37°C in 0.5% CO2 as previously described (Roos et al., 1994). DME (GIBCO BRL) was modified for use in three different forms: (1) DMEc for normal propagation was supplemented with 10% NuSerum (Collaborative Biomedical Products), 2 mM glutamine, and 20 μg/ml gentamycin; (2) DMEm/x consisted of DMEc with 50 μg/ml mycophenolic acid (MPA) and 50 μg/ml xanthine; (3) DMEi was same as DMEc without serum.
The calcium ionophore A23187 (Sigma-Aldrich) was dissolved in DMSO at 1 mM. DMA (Sigma-Aldrich) was dissolved in DMSO at 10 mM. MPA and xanthine (Sigma-Aldrich) were both dissolved in 0.3 N NaOH at 25 mg/ml.
Insertional mutagenesis and selection of Iid− mutants
The previously described plasmid, pHANA, containing the hypoxanthine-xanthine-guanine phosphoribosyl transferase (HPT) gene under the dihyrofolate reductase-thymidylate synthase (DHFR) promoter, was used to generate random insertional mutants (Black and Boothroyd, 1998). Insertional mutagenesis was performed by transfecting extracellular RHΔhpt parasites by electroporation (Soldati and Boothroyd, 1993) with 50 μg of pHANA linearized with NotI. In addition, a mock transfection without DNA was performed as a control. Parasites were then allowed to expand in a monolayer of HFFs in a T75 flask. After 24 h, the medium of the mutagenized parasites was changed to DMEm/x. Parasites were allowed to lyse and a third of the parasites were passed to a new T75 continuing the selection with DMEm/x. After 48 h, the mutagenized and nonmutagenized populations were syringed with a 27-gauge needle and pelleted at 1,000 g for 10 min. The parasites were resuspended in warm DMEi that contained 1 μM A23187 and incubated for 30 min at 37°C followed by pelleting for 10 min at 1,000 g. The entire pellet of parasites was resuspended in DMEc plated onto a fresh HFF monolayer in a T75 flask to recover the survivors. After 48 h, a second round of selection was performed by incubating syringe-lysed extracellular parasites in 1 μM A23187 for 45 min at 37°C. Surviving parasites were recovered by adding them to HFFs in T25 flasks in DMEc followed by four additional rounds of selection using 60 min of incubation in 1 μM A23187 for each round. Only parasites from the mutagenized GAD1 population survived this series of selections. These were plated out on monolayers in a 96-well plate at a limiting dilution for isolating clones. 30 clones were isolated from the GAD1 population and maintained in 24-well plates with DMEm/x for further analysis. Consistent with nomenclature previously used (Black et al., 2000), clones were labeled GADx.y, where GA indicate the initials of the researcher performing the selection, “D” indicates that the mutant came from a death selection, x represents the experiment number, and y is the clone number in that experiment.
Screening for Iie− mutants
30 μl of lysed parasites from each of the 30 clones and the parental strain were passed to HFFs in a 24-well plate with DMEc. Parasites were allowed to replicate for 30 h at 37°C. The growth medium was washed off the monolayer with PBS and replaced with warm DMEi containing 1 μM A23187 for 3 min at 37°C, at which point wild-type parasites show 100% egress (Black et al., 2000). The ionophore was removed and the monolayers were fixed with 100% methanol and stained with Difquick (Dade-Behring). Vacuoles were examined by light microscopy to determine if egress had occurred as previously described (Black et al., 2000). Those clones with >10% intact vacuoles were selected for confirmation of an Iie− phenotype.
Quantitation of IID and IIE
The exact quantification of the IID and IIE phenotype was performed as previously described with the following changes (Black et al., 2000). The ionophore incubations were performed in warm DMEi containing 1 μM A23187 in DMSO. For the IIE assays, the monolayers were fixed with 100% methanol and stained with Difquick (Dade-Behring). Vacuoles were then examined by light microscopy counting no fewer than five fields from each well (≥100 vacuoles) at a magnification of 400×.
The same methods were used to analyze the effect of DMA on IIE, incubating the parasites with 100 μM DMA in DMSO or a comparable amount of DMSO alone in DMEi at 37°C for 10 min before the addition of an equal volume of DMEi + 2 μM A23187. The statistical significance of the differences between the mutants and the wild-type strains was assessed by t test.
Plasmid rescue of GAD1.7 mutant
Genomic Toxoplasma DNA was purified from freshly lysed tachyzoites using standard methods (Black and Boothroyd, 1998). To rescue fragments flanking the inserted pHANA vector, 1 μg of genomic DNA was cut with AgeI for 12 h, followed by heat inactivation. 20 ng of digested DNA was incubated with T4 DNA ligase at a concentration of 2 ng/μl to favor intramolecular ligation and subsequently transformed into DH5α chemically competent cells. Plasmids were isolated from ampicillin-resistant colonies and analyzed by restriction digest and sequencing to confirm the presence and identity of flanking genomic sequences.
Cloning of NHE1
A genomic contig (TGG_10551) from version 2.1 of the Toxoplasma genome project (http://ToxoDB.org) was identified as the region of insertion in mutant GAD1.7 as described above. The internal sequence of the NHE1 cDNA was identified from a directional cDNA library (from day 3 in vitro bradyzoites from pH stressed Prugniaud parasites; a gift from M. Cleary [Stanford University] and Compligen) using primers designed against regions of TgNHE1 homology within the Toxoplasma genomic sequence and library vector primers. The primers used were 5′-aaaggcgagaagagagcaatc-3′ in combination with M13forward in a primary reaction and the nested primer 5′-gtaagtcacaatgccgctgat-3′ used with T7 in a secondary reaction. In addition, 5′-ccgtcatcacagtctgctaca-3′ was used with M13reverse and the nested primer 5′-gctacatcgcctactttgtcg-3′ was used with T3 in a secondary reaction. The ends of the cDNA were identified using 5′RACE and 3′RACE systems (Invitrogen) with RNA isolated from Prugniaud parasites as described before (Singh et al., 2002) and using primers 5′-gccgctgatgatgaaaatagc-3′, 5′-gcattgcgtcatgggatattt-3′, 5′-cgtgcacagtttgtctgaagc-3′, 5′-acacgatctccggttttgtc-3′, and 5′-aaagatgtcgggagacgatgg-3′.
Generation of NHE1-specific knockout
The parental vector pMini-GFP.ht was generated by inserting a Klenow-blunted HindIII to BamHI fragment from pgraGFP (Kim et al., 2001) with GFP under the GRA2 promoter into the HPT-containing vector pMiniHXGPRT-I (Donald et al., 1996) digested with SacI and blunted with T4 polymerase. This approach generated pMini-GFP.ht with GFP in the 5′ to 3′ end direction in respect to HPT. To design the knockout construct, first a PCR fragment from bases 6463–8676 (flank 2 in Fig. 3 A) from NHE1 gDNA was digested with HindIII and KpnI and cloned into pMini-GFP.ht digested with the same restriction enzymes. The resulting plasmid was digested with NotI and XbaI and ligated to a PCR fragment from genomic bases 2292–5189 (flank 1 in Fig. 3 A) also cut with NotI and XbaI. The primers used to expand the flanking regions were 5′-ggggcgaagcttagaaacgcgaagacgaaaaa-3′ and 5′-ggggggtaccccatcgtctccgacatcttt-3′ for flank 1 and 5′-gggaaagggaaagcggccgcgattggtggatgcgtgcgtc-3′ and 5′-gctaatcacgaagttctagagacgg-3′ for flank 2.
The resulting vector, pKONHE1, was linearized with NotI, and 50 μg of DNA was introduced into RHΔhpt strain parasites by standard methods (Soldati and Boothroyd, 1993). Six independent transformations were performed, and parasites were then grown in HFFs in T25 flasks for 24 h, at which point the media was changed to DMEm/x. A fifth of the surviving parasites from each population were passed to new cells when lysed and kept under the MPA/xanthine selection. 10 d after the transformation, genomic DNA was obtained from the selected population and used in PCR to confirm the knockout event using primers that yielded a product only if the knockout had occurred (5′-gtggcgattctcatcgactt-3′ and 5′-aacgtaatctcgtggcttcg-3′). Parasites from the one population with the knockout were grown in 24-well plates in pools of 50 parasites per well. Parasites from each of 24 wells were tested again for the knockout parasite by PCR, and parasites from one of the positive wells were cloned by limiting dilution. 40 clones that did not express GFP were tested by PCR, and one clone was determined to have the desired knockout. Besides the knockout clone RHΔnhe1, a clone with a heterologous insertion of the knockout construct was maintained and dubbed RHΔhpt+HPT.
Generation of TgNHE1 antibodies and immunofluorescence analysis
Coding sequences corresponding to amino acids 716–1073 of TgNHE1 were amplified from cDNA and subcloned into the T7 expression vector pET28a(+) (Novagen) that had been digested with NdeI and EcoRI. Primers used in this step were 5′-gggcagcatatgaccgtcgtgaagcgcatggcgacg-3′ and 5′-ctgtgtgcgcatgctggtcaggcgat-3′. The resulting vector was transformed into Escherichia coli strain BL21(DE3) (Novagen), and expression of the recombinant protein, which encoded 6X-His tags at both ends, was induced with IPTG. TgNHE1(716–1073) was purified using affinity chromatography with Ni-NTA agarose from QIAGEN and following the manufacturer's instructions. 100 μg of purified recombinant protein was injected into Balb/C mice (Teconic) along with 100 μl of RIBI adjuvant (Corixa). Three boost injections were performed, at 3-wk intervals. Mouse serum was tested for specific immunoreactivity 4 d after the second and third boost by immunofluorescence staining of wild-type and NHE1 knockout parasites.
For immunofluorescence analysis, parasites were grown for 24 h in HFFs on glass coverslips. Staining of intracellular parasites was performed as described before (Singh et al., 2002). For primary staining, serum from the TgNHE1(716–1073) immunized mouse was used at 1:1,000 in PBS/0.2% Triton X-100/3% BSA for 1 h at room temperature. The secondary antibody, Alexa fluor 488–conjugated goat anti–mouse (Molecular Probes), was used at 1:2,000 antibody in PBS with 3% BSA for 1 h. Coverslips were mounted on slides with vectashield (Vector Laboratories) and examined using a 100× lens of an Olympus BX60 fluorescence microscope, and images were captured using a Hamamatsu digital camera (model C4742-95) and the Image Pro Plus software.
Spectrofluorometric determinations
Spectrofluorometric determinations were performed as described previously (Moreno and Zhong, 1996). In summary, for the proton extrusion experiments, freshly lysed and filtered tachyzoites were washed twice and resuspended in buffer A (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 5.5 mM d-glucose, and 50 mM Hepes, pH 7.4) at 109 parasites/ml and kept in ice until use. 108 parasites were centrifuged and resuspended in 100 μl of a weakly buffered (100 μM Hepes-Tris, pH 7.4) standard solution and then mixed in with 2.4 ml of standard solution containing 0.38 μM of free acid BCECF (Molecular Probes) and either 100 μM amiloride (Sigma-Aldrich) or 1 μM bafilomycin (Kamiya Biomedical). Fluorescence measurements started after 1 min of stirring in a cuvette. For Ca2+ measurements, freshly lysed and filtered parasites were washed three times with buffer A and preincubated at 25°C for 30 min in buffer A with 1% sucrose, and 4.5 μM Fura 2-AM (Molecular Probes) at 109 parasites/ml. Parasites were then washed twice and resuspended in buffer A at the same concentration. For each measurement, 50 μl of parasite suspension was diluted in a cuvette with 2.5 ml of a standard buffer, with either 1 mM EGTA or 1mM CaCl2, and allowed to stir for 2 min before recording [Ca2+].
Acknowledgments
The host strain RHΔhpt was obtained through the AIDS Research and Reference Program, Division of AIDS, NIAID, NIH from Dr. David Roos. We would also like to thank Mike Black and members of the Boothroyd and Moreno labs for critical input into this work, and Linda Brown for technical assistance.
This work was supported in part by the National Institutes of Health (AI45051, AI07328-12, GM20872-02, and AI43614).
Abbreviations used in this paper: DMA, dimethylamiloride; HFF, human foreskin fibroblast; IID, ionophore-induced death; IIE, ionophore-induced egress; MPA, mycophenolic acid; NHE, Na+/H+ exchanger; pHe, extracellular pH; pHi, intraparasitic pH; TM, transmembrane.
References
- Berthe, P., J.L. Cousin, and J.P. Breittmayer. 1991. Intracellular Ca2+ regulation in CD3 stimulated Jurkat T cells involves H+ fluxes. Cell. Signal. 3:453–459. [DOI] [PubMed] [Google Scholar]
- Black, M.W., and J.C. Boothroyd. 1998. Development of a stable episomal shuttle vector for Toxoplasma gondii. J. Biol. Chem. 273:3972–3979. [DOI] [PubMed] [Google Scholar]
- Black, M.W., and J.C. Boothroyd. 2000. Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 64:607–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, M.W., G. Arrizabalaga, and J.C. Boothroyd. 2000. Ionophore-resistant mutants of Toxoplasma gondii reveal host cell permeabilization as an early event in egress. Mol. Cell. Biol. 20:9399–9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosia, A., D. Ghigo, F. Turrini, E. Nissani, G.P. Pescarmona, and H. Ginsburg. 1993. Kinetic characterization of Na+/H+ antiport of Plasmodium falciparum membrane. J. Cell. Physiol. 154:527–534. [DOI] [PubMed] [Google Scholar]
- Carruthers, V.B., O.K. Giddings, and L.D. Sibley. 1999. Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cell. Microbiol. 1:225–235. [DOI] [PubMed] [Google Scholar]
- Donald, R.G., and D.S. Roos. 1995. Insertional mutagenesis and marker rescue in a protozoan parasite: cloning of the uracil phosphoribosyltransferase locus from Toxoplasma gondii. Proc. Natl. Acad. Sci. USA. 92:5749–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald, R.G., D. Carter, B. Ullman, and D.S. Roos. 1996. Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine-xanthine-guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. J. Biol. Chem. 271:14010–14019. [DOI] [PubMed] [Google Scholar]
- Endo, T., K.K. Sethi, and G. Piekarski. 1982. Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp. Parasitol. 53:179–188. [DOI] [PubMed] [Google Scholar]
- Hoff, E.F., and V.B. Carruthers. 2002. Is Toxoplasma egress the first step in invasion? Trends Parasitol. 18:251–255. [DOI] [PubMed] [Google Scholar]
- Hu, K., T. Mann, B. Striepen, C.J. Beckers, D.S. Roos, and J.M. Murray. 2002. Daughter cell assembly in the protozoan parasite Toxoplasma gondii. Mol. Biol. Cell. 13:593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunyady, L., B. Sarkadi, E.J. Cragoe Jr., A. Spat, and G. Gardos. 1987. Activation of sodium-proton exchange is not a prerequisite for Ca2+ mobilization and aggregation in human platelets. FEBS Lett. 225:72–76. [DOI] [PubMed] [Google Scholar]
- Ives, H.E., V.J. Yee, and D.G. Warnock. 1983. Mixed type inhibition of the renal Na+/H+ antiporter by Li+ and amiloride. Evidence for a modifier site. J. Biol. Chem. 258:9710–9716. [PubMed] [Google Scholar]
- Joiner, K.A., and J.F. Dubremetz. 1993. Toxoplasma gondii: a protozoan for the nineties. Infect. Immun. 61:1169–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K., D. Soldati, and J.C. Boothroyd. 1993. Gene replacement in Toxoplasma gondii with chloramphenicol acetyltransferase as selectable marker. Science. 262:911–914. [DOI] [PubMed] [Google Scholar]
- Kim, K., M.S. Eaton, W. Schubert, S. Wu, and J. Tang. 2001. Optimized expression of green fluorescent protein in Toxoplasma gondii using thermostable green fluorescent protein mutants. Mol. Biochem. Parasitol. 113:309–313. [DOI] [PubMed] [Google Scholar]
- Knoll, L.J., G.L. Furie, and J.C. Boothroyd. 2001. Adaptation of signature-tagged mutagenesis for Toxoplasma gondii: a negative screening strategy to isolate genes that are essential in restrictive growth conditions. Mol. Biochem. Parasitol. 116:11–16. [DOI] [PubMed] [Google Scholar]
- Kyte, J., and R.F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132. [DOI] [PubMed] [Google Scholar]
- Luft, B.J., and J.S. Remington. 1992. Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis. 15:211–222. [DOI] [PubMed] [Google Scholar]
- Mondragon, R., and E. Frixione. 1996. Ca2+-dependence of conoid extrusion in Toxoplasma gondii tachyzoites. J. Eukaryot. Microbiol. 43:120–127. [DOI] [PubMed] [Google Scholar]
- Moreno, S.N., and R. Docampo. 2003. Calcium regulation in protozoan parasites. Curr. Opin. Microbiol. 6:359–364. [DOI] [PubMed] [Google Scholar]
- Moreno, S.N., and L. Zhong. 1996. Acidocalcisomes in Toxoplasma gondii tachyzoites. Biochem. J. 313:655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno, S.N.J., L. Zhong, H.G. Lu, W. de Souza, and M. Benchimol. 1998. A vacuolar type H+-ATPase regulates cytoplasmic pH in Toxoplasma gondii tachyzoites. Biochem. J. 330:853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudy, R., T.J. Manning, and C.J. Beckers. 2001. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. J. Biol. Chem. 276:41492–41501. [DOI] [PubMed] [Google Scholar]
- Paris, S., and J. Pouyssegur. 1983. Biochemical characterization of the amiloride-sensitive Na+/H+ antiport in Chinese hamster lung fibroblasts. J. Biol. Chem. 258:3503–3508. [PubMed] [Google Scholar]
- Pezzella, N., A. Bouchot, A. Bonhomme, L. Pingret, C. Klein, H. Burlet, G. Balossier, P. Bonhomme, and J.M. Pinon. 1997. Involvement of calcium and calmodulin in Toxoplasma gondii tachyzoite invasion. Eur. J. Cell Biol. 74:92–101. [PubMed] [Google Scholar]
- Putney, L.K., S.P. Denker, and D.L. Barber. 2002. The changing face of the Na+/H+ exchanger, NHE1: structure, regulation, and cellular actions. Annu. Rev. Pharmacol. Toxicol. 42:527–552. [DOI] [PubMed] [Google Scholar]
- Roos, D.S., R.G. Donald, N.S. Morrissette, and A.L. Moulton. 1994. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 45:27–63. [DOI] [PubMed] [Google Scholar]
- Saliba, K.J., and K. Kirk. 1999. pH regulation in the intracellular malaria parasite, Plasmodium falciparum. H(+) extrusion via a v-type h(+)-atpase. J. Biol. Chem. 274:33213–33219. [DOI] [PubMed] [Google Scholar]
- Sardet, C., A. Franchi, and J. Pouyssegur. 1988. Molecular cloning of the growth-factor-activatable human Na+/H+ antiporter. Cold Spring Harb. Symp. Quant. Biol. 53:1011–1018. [DOI] [PubMed] [Google Scholar]
- Seeber, F. 1997. Consensus sequence of translational initiation sites from Toxoplasma gondii genes. Parasitol. Res. 83:309–311. [DOI] [PubMed] [Google Scholar]
- Shi, H., M. Ishitani, C. Kim, and J.K. Zhu. 2000. The Arabidopsis thaliana salt tolerance gene SOS1 encodes a putative Na+/H+ antiporter. Proc. Natl. Acad. Sci. USA. 97:6896–6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siffert, W., and J.W. Akkerman. 1987. Activation of sodium-proton exchange is a prerequisite for Ca2+ mobilization in human platelets. Nature. 325:456–458. [DOI] [PubMed] [Google Scholar]
- Singh, U., J.L. Brewer, and J.C. Boothroyd. 2002. Genetic analysis of tachyzoite to bradyzoite differentiation mutants in Toxoplasma gondii reveals a hierarchy of gene induction. Mol. Microbiol. 44:721–733. [DOI] [PubMed] [Google Scholar]
- Soldati, D., and J.C. Boothroyd. 1993. Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science. 260:349–352. [DOI] [PubMed] [Google Scholar]
- Vercesi, A.E., and R. Docampo. 1996. Sodium-proton exchange stimulates Ca2+ release from acidocalcisomes of Trypanosoma brucei. Biochem. J. 315:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercesi, A.E., C.O. Rodrigues, R. Catisti, and R. Docampo. 2000. Presence of a Na+/H+ exchanger in acidocalcisomes of Leishmania donovani and their alkalization by anti-leishmanial drugs. FEBS Lett. 473:203–206. [DOI] [PubMed] [Google Scholar]
- Wiebe, C.A., E.R. Dibattista, and L. Fliegel. 2001. Functional role of polar amino acid residues in Na+/H+ exchangers. Biochem. J. 357:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenner, L., F. Darcy, M.F. Cesbron-Delauw, and A. Capron. 1993. Rat model of congenital toxoplasmosis: rate of transmission of three Toxoplasma gondii strains to fetuses and protective effect of a chronic infection. Infect. Immun. 61:360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]