

Abstract
Thymocyte and thymic epithelial cell (TEC) development are interdependent processes. Although lineage relationships among progressively maturing thymocyte subsets have been characterized, the developmental relationships among TEC subsets are obscure. Because epithelial cells express distinct keratin (K) species as a function of differentiation stage and proliferative status, we used K expression patterns to identify mouse TEC subsets and determine their lineage relationships. As expected, cortical and medullary TEC subsets express distinct K expression patterns in the normal thymus. However, we detected two distinct cortical TEC subsets, a major K8+K5− subset and a minor K8+K5+ subset, which is highly represented at the cortico-medullary junction. Both cortical TEC subsets are also present in recombination activating gene 1 (RAG-1−/−) and TCRβxδ−/− thymi in which T-cell development is blocked at the CD4−CD8−CD25+CD44− pre-T cell stage. In contrast, K8+K5+ TECs predominate in the thymi of human CD3ɛ transgenic mice in which thymocyte development is blocked at an earlier CD4−CD8−CD25−CD44+ stage. Transplantation of newborn human CD3ɛ transgenic thymi under the kidney capsule of RAG-1−/− mice results in the emergence of K8+K5− TECs concomitant with the appearance of CD25+ thymocytes. Together, the data suggest that cortical TEC development proceeds from a K8+K5+ precursor subset to a K8+K5− stage in a differentiation process concomitant with T-cell lineage commitment.
The intrathymic developmental pathways that generate mature CD4 and CD8 single positive T cells from immature CD4−CD8− double-negative (DN) thymocyte precursors have been extensively investigated (reviewed in refs. 1 and 2). During the T-lineage commitment process, multipotent CD44+CD25− progenitors in the DN compartment up-regulate CD25, down-regulate CD44, and initiate T cell antigen receptor (TCR)γ-, δ-, and β-gene rearrangements. CD44−CD25+ pre-T cells that productively rearrange the TCRβ locus and express pre-TCR/CD3 complexes proliferate and differentiate to the CD4+CD8+ double-positive (DP) stage. The DN to DP transition is accompanied by loss of CD25 expression, prohibition of TCRβ locus rearrangements, and induction of TCRα locus rearrangements. Signaling through the αβTCR/CD3 complexes on DP thymocytes mediates the positive and negative selection processes that shape the T-cell repertoire. DP thymocytes that are positively selected by self-peptide/major histocompatibility complex molecules presented on cortical epithelial cells terminate CD8 or CD4 expression and migrate to the thymic medulla.
Thymic epithelial cells (TECs) are not only involved in the selection of DP cells, but also promote differentiation of early DN thymocyte precursors. Nude mice that are unable to generate a normal thymic epithelial compartment because of an inactivating whn gene mutation have a primitive thymic anlage devoid of T cells (3). Furthermore, recent studies have shown that the maturation of DN precursors in thymic organ culture requires the presence of major histocompatibility complex class II+ TECs (4–6). Although TECs are known to play a critical role in T-cell maturation, the factors that govern TEC development are incompletely understood, particularly in comparison to the well-characterized T-cell developmental process. Nevertheless, it is clear that the establishment of normal thymic architecture and thus, TEC differentiation, depends on thymocyte/TEC interactions (7, 8). This interdependence is apparent in mice that express a human CD3ɛ (hCD3ɛ) transgene, which prevents T-cell maturation beyond the primitive CD44+CD25− DN stage (9). As a consequence of the early T-cell developmental block, the thymus is extremely hypoplastic, contains atypically arranged cortical TECs, and lacks an organized medulla (9, 10). In contrast, an organized cortex is present in recombination activating gene (RAG)-1−/− or RAG-2−/− deficient and severe combined immunodeficiency (SCID) mice, which sustain a later developmental block at the CD44−CD25+ differentiation stage (9, 11, 12). Because DN thymocytes that have progressed to the CD44−CD25+ stage are developmentally committed to the T lineage, it appears that normal cortical organization accompanies the T-lineage commitment process. However, the TEC precursors with which DN thymocytes presumably interact to generate an organized cortical epithelial compartment have not been previously identified.
Various approaches have been taken to identify TEC subsets and determine their lineage relationships. Morphological studies using electron microscopy identified subcapsular, cortical, and two distinct medullary TEC subsets (13, 14). Similar TEC subsets were identified in immunohistochemical studies using a panel of clusters of thymic epithelial staining mAbs (15, 16). In a related approach, two medullary epithelial subsets were defined by differential reactivity with the lectin UEA-1 and mAbs that recognize classical versus nonpolymorphic major histocompatibility complex molecules (17, 18). Although it is well established that morphologically and antigenically unique subsets of TECs occupy distinct microenvironmental niches, the developmental regulation imposed by early DN thymocytes on cortical TEC maturation and the lineage relationships involved in this process are not well defined. We have explored these issues in normal mice and in mice that contain specific blocks in T-cell development by characterizing TEC subsets based on keratin expression patterns. Keratins are intermediate filament proteins that are expressed in all epithelial cells as heterodimers of acidic type I and basic type II polypeptides (19). Keratins comprise a multigene family that consists of approximately 20 members. The expression of different keratin species is regulated in a tissue-specific manner and also depends on epithelial cell differentiation stage and proliferative activity. Thus, keratin expression patterns have been used as markers to identify epithelial cell subsets and determine their lineage relationships in a variety of tissues (19–21).
By using a panel of antibodies specific for mouse keratins, we show that the normal thymic cortex does not consist of a homogenous population, but rather contains two distinct TEC subsets. The major cortical population expresses keratin 8 (K8), but not K5, whereas a minor subset scattered throughout the cortex, but highly enriched at the cortico-medullary border, expresses both K8 and K5. A similar analysis in hCD3ɛ transgenic, RAG-1−/− and TCRβxδ−/− mice revealed that blocks at specific stages of thymocyte development differentially impact the two cortical TEC subsets. Furthermore, the lineage relationship between K8+K5+ and K8+K5− subsets was established by engrafting newborn hCD3ɛ transgenic thymi under the kidney capsule of hCD3ɛ transgenic versus RAG-1−/− recipients. These experiments demonstrate that K8+K5+ epithelial cells contain precursors that give rise to the major cortical K8+K5− subset, a TEC differentiation process that depends on interactions between TEC progenitors and T-lineage committed thymocytes.
MATERIALS AND METHODS
Mice.
C57BL/6J, hCD3ɛ transgenic, RAG-1−/− and TCRβxδ−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME).
Antibodies.
Rabbit antisera specific for mouse K5 or K14 were developed as described by Roop et al. (22). Troma-1, a mAb that recognizes K8, and LE61, a mAb that recognizes the K8/18 complex, were kindly provided by Rolf Kemler (Max-Planck-Institut fur Immunbiologie, Freiburg, Germany) (23) and Birgitte Lane (The University of Dundee, Scotland) (24), respectively. Fluorochrome-conjugated or biotinylated anti-Ig second-step reagents and MTS10 mAb were purchased from Jackson ImmunoResearch and PharMingen, respectively. Anti-CD25 (clone 3C7) was a hybridoma supernatant. Biotinylated UEA-1 and fluorescein-isothiocynate-conjugated streptavidin were obtained from Vector Laboratories.
Immunohistology.
Five-micrometer serial frozen sections were air-dried for 30 min before acetone fixation. Alternatively, 5-μm serial sections were obtained from 70% ethanol-fixed, paraffin-embedded thymi. Thin sections were incubated with normal serum or with avidin-biotin blocking kit (Vector Laboratories) and were incubated with optimal dilutions of antibodies, nonimmune rabbit IgG, or isotype-matched mouse Ig controls for at least 1 hr at 25°C. For double-staining, primary antibodies from different species were added simultaneously and after incubation at 25°C, the slides were washed and incubated with the secondary reagents described above. For immunohistochemistry, primary antibody binding was detected with biotinylated secondary reagents followed by incubation with avidin-biotin-horseradish peroxidase complexes (ABC Vectastain; Vector Laboratories), and the sections were developed with 3,3′-diaminobenzidine.
Thymic Grafts.
Thymi from newborn hCD3ɛ transgenic mice were grafted under the kidney capsule of anesthetized adult hCD3ɛ transgenic or RAG-1−/− recipients. A small incision was made in the peritoneal cavity, and the left kidney was exposed. By using an intravenous cannula, two thymic lobes from individual hCD3ɛ transgenic newborn mice were positioned under the kidney capsule after which the capsule defect was plugged with gel foam and the wound closed by using surgical sutures.
RESULTS
Keratin Expression Patterns Define Two Cortical And Two Medullary TEC Subsets In The Normal Thymus.
Cortical and medullary TEC subsets were characterized by differential expression of four keratin species: K8, K18, K5, and K14. K8 and its heterodimerization partner, K18, are the first intermediate filament proteins expressed during mouse embryogenesis and are characteristic of simple epithelia (25). K5 and its heterodimerization partner, K14, are expressed in the proliferating compartment of stratified squamous epithelia, but are down-regulated during epithelial differentiation as other keratin species are up-regulated (20, 21). Fig. 1 shows that in the young adult C57BL/6J thymus, K8 and K18 are expressed by the vast majority, if not all, cortical and subcapsular stellate TECs. In addition, K8 and K18 are expressed by a subset of TECs localized in the medulla, which have a globular rather than stellate morphology (Fig. 1 and data not shown). In contrast, K5 is expressed by the predominant medullary TEC subset, which has a distinctive stellate appearance. Note that K5-expressing TECs are not restricted to the inner medullary region, but extend well into the cortico-medullary junction. Furthermore, scattered TECs in the cortex and subcapsular region also express K5, demonstrating that the cortex does not contain a homogenous population of TECs as previously described (13, 14, 26). Interestingly, the K5 and K14 expression pattern is not identical. Although K5 and K14 are both expressed in stellate medullary TECs, K14 is not found in association with K5+ TECs in the cortex or at the cortico-medullary junction, suggesting that another keratin species heterodimerizes with K5 in these regions. A likely candidate is K15, which was demonstrated to be an alternative K5 partner in K14 null mice (27).
Figure 1.

Keratin expression patterns in normal adult C57BL6/J thymus. Serial 5-μm cryostat sections of a normal adult thymus were stained as indicated with antibodies specific for K8, K5, K18, or K14. K8 and K18 expression were detected with an anti-Ig fluorescein isothiocynate reagent (green), and K5 or K14 expression was detected with an anti-Ig-Texas Red reagent (red). Single filter and computer-generated image overlays are shown. Cells costaining for K8 and K5 appear yellow. The original magnification was ×100.
Double-staining confirmed that K5+ TECs in the cortex and cortico-medullary junction coexpress K8. Moreover, although K8+K5+ TECs are found infrequently in the cortex, they are highly represented at the cortico-medullary junction. In contrast, the predominant medullary subset expresses a K5+K8− phenotype. Double-staining for K8 and K14 confirmed that K14 expression is restricted to the inner medullary region and does not extend to the K8+K5+ TECs demarcating the cortico-medullary junction. The failure of K14+ TECs to coexpress K8 delineates two distinct medullary subsets, namely a K8−K14+ stellate subset and a K8+K14− globular subset.
To integrate keratin expression patterns with earlier reports describing TEC subsets, TECs were costained with antikeratin antibodies and either MTS10, a cluster of thymic epithelial staining II mAb, or the UEA-1 lectin. In agreement with previous studies, cortical TECs were generally negative for MTS10 and UEA-1, whereas medullary TECs gave strongly positive staining patterns (11, 16–18). The results in Fig. 2 show that the stellate TECs in the medulla coexpress K5 and the MTS10 ligand. However, it is notable that the K5+ TECs at the cortico-medullary junction and extending out into the cortex fail to stain with the MTS10 mAb. In contrast, UEA-1 reacts with a distinct K5− medullary TEC subset that has a globular morphology suggestive of the K8+ medullary subset. Indeed, UEA-1 and K8 double-staining confirms the coexpression of these two markers (data not shown).
Figure 2.

TEC subsets characterized by costaining for expression of K5, MTS10, and UEA-1 ligand. Cryosections of a normal adult thymus were costained for expression of K5 (red) and MTS10 (green) by using fluorescein isothiocynate (FITC)- or Texas red-conjugated anti-Ig second steps. Alternatively, the slides were costained for K5 (red) and biotyinylated UEA-1 followed by FITC-conjugated streptavidin (green). The microscopic fields shown include medullary and cortico-medullary junction TECs. Single filter and computer-generated image overlays are shown. The original magnification was ×300. Although K5+MTS10+ stellate cells are present in the inner medulla, K5+ TECs at the cortico-medullary junction do not coexpress MTS10 ligand. Distinct populations of medullary TECs express K5 and the ligand for UEA-1.
Table 1 synthesizes the data from the keratin, MTS10, and UEA-1 expression profiles into a summary of normal murine TEC subsets defined by these parameters. The majority of subcapsular and cortical TECs have a K8+K18+K5−K14− MTS10−UEA-1− phenotype, whereas a less prominent K8+ K18+K5+K14−MTS10−UEA-1− subset is highly enriched at the cortico-medullary junction, as well as scattered throughout the subcapsular and cortical regions. The medulla contains both a stellate K8−K18−K5+K14+MTS10+UEA-1− subset localized to the inner medullary region and a globular K8+ K18+K5−K14−MTS10−UEA-1+ subset that tends to be concentrated toward the outer region of the medullary compartment.
Table 1.
TEC subsets in the normal adult murine thymus
| Medullary | Major cortical subset | Minor cortical subset | Major medullary subset | Minor medullary subset |
|---|---|---|---|---|
| K8 | + | + | − | + |
| K18 | + | + | − | + |
| K5 | − | + | + | − |
| K14 | − | − | + | − |
| MTS10 | − | − | + | − |
| UEA-1 | − | − | − | + |
| Relative location | Cortical and subcapsular | Concentrated at CMJ*; infrequent in cortex | Restricted to medulla | Restricted to medulla |
Corticomedullary junction.
Disruption of Thymocyte Differentiation at Specific Early Maturation Stages Affects Cortical TEC Development.
T-cell maturation is blocked at early, but distinct, developmental control points in hCD3ɛ transgenic compared with RAG-1−/− or TCRβxδ−/− mice (9, 28). Therefore, these model systems provide an opportunity to assess the influence of early thymocyte subsets on TEC differentiation and organization. The hCD3ɛ transgenic mice sustain a block at the DN CD44+CD25− stage and have severely hypoplastic thymi that contain large cysts and aberrantly arranged cortical TECs (9). In RAG-1−/− mice, T-cell development proceeds through T-lineage commitment, but is arrested at the CD44−CD25+ stage. In contrast to the hCD3ɛ transgenic model, the RAG-1−/− thymic cortex has a relatively normal histological appearance (9, 11, 12). T-cell development is arrested at a similar stage in TCRβxδ−/− mice, although in contrast to RAG-1−/− mice, B-cell development is intact (28).
Fig. 3 shows a comparison of keratin expression patterns in TECs from representative hCD3ɛ transgenic, RAG-1−/−, and TCRβxδ−/− mice. As expected, very few K14+ or UEA-1+ TECs were observed in any of these models in the absence of an organized medulla (Fig. 3 and data not shown). Surprisingly, however, the vast majority of hCD3ɛ transgenic TECs coexpress K8 and K5 similar to TECs that are concentrated in the normal thymus at the cortico-medullary junction and sparsely scattered throughout the cortex. In contrast, the TECs in RAG-1−/− and TCRβxδ−/− mice contain not only K8+K5+ cells, but also a prominent K8+K5− subset. The latter subset expresses the same keratin expression pattern as the major cortical/subcapsular population in the normal thymus. Thus, these data confirm the existence of two cortical subsets and further suggest that development of the K8+K5− TEC subset depends on advancement of thymocyte maturation to the CD44+25+ or CD44−25+ stage. The fact that both cortical TEC subsets are present in RAG-1−/− as well as in TCRβxδ−/− mice indicates that B-cell development is not necessary to induce development of the K8+K5− TEC subset. Taken together the results suggest that during the process of T-lineage commitment, thymocytes acquire the ability to induce the differentiation of K8+K5+ TEC precursors to the major K8+K5− cortical subset.
Figure 3.
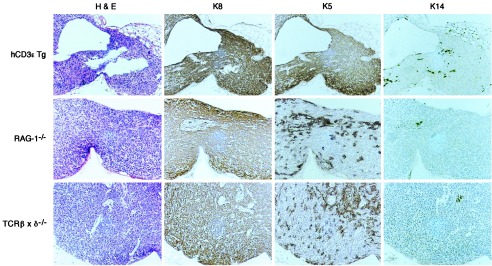
TEC subsets in hCD3ɛ transgenic, RAG-1−/− and TCRβxδ−/− mice. Serial paraffin-embedded sections were stained with hematoxylin and eosin, or antibodies to K8, K5, or K14 as indicated. Antibody binding was detected by secondary reagents conjugated with horseradish peroxidase, and the reaction was developed with diaminobenzidine. The slides were lightly counterstained with hematoxylin. The original magnification was ×200.
The K8+K5+ TEC Subset Contains Precursors that Generate the Major Cortical TEC K8+K5− Subset.
To directly examine the lineage relationship between the K8+K5+ and K8+K5− cortical TEC subsets, hCD3ɛ transgenic thymi were reconstituted with hematopoietic precursors that could differentiate beyond the block imposed by the hCD3ɛ transgene. Previous reports demonstrated that reconstitution of hCD3ɛ transgenic thymi by nontransgenic hematopoietic precursors can be achieved only within a restricted developmental window occurring up to 6 days after birth (29). Therefore, thymi were obtained from newborn hCD3ɛ transgenic mice for engraftment under the kidney capsule of either adult hCD3ɛ transgenic or RAG-1−/− mice. The thymic grafts were harvested 1 month after transplantation. Fig. 4 shows that hCD3ɛ transgenic thymi recovered from hCD3ɛ recipients were devoid of CD25+ thymocytes and contained a TEC population that maintained the K8+K5+ profile originally observed. In sharp contrast, hCD3ɛ transgenic thymi recovered from RAG-1−/− recipients not only were reconstituted with CD25+ thymocytes, but also contained both K8+K5+ and K8+K5− cortical TEC subsets. Thus, down-regulation of K5 in the cortical TEC compartment was achieved in conjunction with reconstitution by hematopoietic precursors that could advance to the DN CD44−25+ stage. These data are therefore consistent with the notion of a direct lineage relationship between K8+K5+ TEC precursors and K8+K5− TEC progeny, a developmental process regulated by early thymocyte subsets at or just before the DN CD44−25+ stage.
Figure 4.

TEC subsets in hCD3ɛ transgenic thymi transplanted under the kidney capsule of hCD3ɛ transgenic or RAG-1−/− recipients. Newborn hCD3ɛ transgenic thymi were transplanted under the kidney capsule of adult hCD3ɛ transgenic or RAG-1−/− mice as described in Materials and Methods. Four weeks later the transplanted thymi were recovered and double-stained for K8 (green) and K5 (red) or single-stained for CD25. The original magnification was ×200 except for the inset, which was ×600. The thymi recovered from the hCD3ɛ transgenic retained a K8+K5+ phenotype, whereas thymi recovered from RAG-1−/− recipients contained both K8+K5+ and K8+K5− subsets as well as CD25+ thymocytes.
DISCUSSION
Thymic organogenesis is a complex process that depends on mutually inductive thymocyte/TEC interactions to generate a functional microenvironment that serves as the major site of T-cell differentiation (1, 2). Considerable attention has been focused on the cellular and molecular events that control thymocyte maturation as well as on the lineage relationships among thymocyte subsets. In contrast, the lineage relationships among TEC subsets and the thymocyte interactions that regulate their development are not as well characterized. In the present study, TECs subsets were distinguished by differential expression of members of the keratin family, an approach that has been used successfully to characterize epithelial cells from other organs. Two findings emerged from this analysis. First, the thymic cortex contains at least two distinct TEC subsets: a K8+K18+K5−K14− subset (referred to as K8+K5−) that predominates in the cortex and a K8+K18+K5+K14− subset (referred to as K8+K5+) that is infrequently scattered throughout the cortex, but concentrated at the cortico-medullary junction. Second, the results obtained with hCD3ɛ thymic grafts demonstrate that K8+K5+ precursors give rise to K8+K5− progeny in the cortex, a developmental relationship that depends on inductive interactions between TEC progenitors and early DN thymocyte subsets at or just before the DN CD44−CD25+ stage.
Previous studies have reported the presence of two distinct medullary TEC subsets (13, 14, 17, 18), whereas the epithelial cells in the cortex generally have been considered to be a relatively homogeneous population (13, 14, 26). However, although earlier investigations showed that K8 and K18 were uniformly expressed in the cortex, neither K5 nor K14 expression was evaluated (30, 31). In the present study, the existence of two cortical TEC subsets in the normal adult thymus was confirmed by analysis of TECs in hCD3ɛ transgenic and RAG-1−/− mice. Although the K8+K5+ subset is a minor constituent of the cortical epithelial compartment in the normal adult thymus, it is highly represented in hCD3ɛ transgenic and RAG-1−/− thymi. Indeed, the TECs in hCD3ɛ thymi are uniformly K8+K5+, whereas both K8+K5+ and K8+K5− subsets are present in RAG-1−/− and TCRβxδ−/− thymi. The presence of both cortical TEC subsets in RAG-1−/− and TCRβxδ−/− thymi demonstrates that mature T cells are not required for the generation of K8+K5− TECs. However, the fact that K8+K5+ TECs comprise a greater proportion of TECs in the cortex of RAG-1−/− as compared with normal C57BL/6J mice suggests that thymocyte differentiation beyond the DN CD44−CD25+ stage promotes the development and/or expansion of the K8+K5− cortical subset.
The notable differences in thymic architecture and TEC subsets found in hCD3ɛ transgenic and RAG-1−/− mice correspond to blocks at distinct stages of T-cell development. Thus, although the medullary region fails to develop in RAG-1−/− mice because of the absence of mature T cells, the cortical epithelial compartment appears to be normally organized and contains both K8+K5+ and K8+K5− subsets in intimate contact with DN CD44−CD25+ thymocytes. The hypoplastic thymus containing exclusively K8+K5+ TEC in hCD3ɛ transgenic mice suggests that the K8+K5+ subset is a primitive epithelial population containing precursors that generate K8+K5− progeny in a differentiation process dependent on interactions with thymocytes that have matured beyond the block imposed by the hCD3ɛ transgene. This hypothesis predicts that the failure of hCD3ɛ transgenic K8+K5+ precursors to differentiate to K8+K5− cells is correctable by reconstituting hCD3ɛ transgenic thymi with hematopoietic progenitors that can mature to the DN CD44−CD25+ stage. The results from the thymic graft experiments strongly support this hypothesis. Thus, the K8+K5− subset developed in newborn hCD3ɛ transgenic thymi that were grafted under the kidney capsule of RAG-1−/− mice. In contrast, the K8+K5− subset failed to develop and only K8+K5+ TECs were found in newborn hCD3ɛ transgenic thymi recovered from hCD3ɛ transgenic recipients. Because T-lineage commitment takes place during the transition of DN CD44+25− progenitors to the DN CD44−CD25+ pre-T cell stage (2), it is possible that TEC maturation depends on inductive interactions with thymocyte precursors that are developmentally restricted to the T lineage.
Keratin expression patterns also provided additional information on the composition of the medullary TEC compartment. Thus, the medulla contains a K8+K18+K5−K14− subset that has a globular morphology and tends to localize to the outer portion of the medullary region, near the cortico-medullary junction. Another subset that expresses a K8−K18−K5+K14+ phenotype has a stellate morphology and is situated more centrally in the medullary region. Simultaneous analysis of UEA-1 reactivity and keratin expression patterns revealed that the globular K8+K18+K5−K14− subset is highly reactive with UEA-1, in contrast to the stellate K8−K18−K5+K14+ medullary TEC subset, which fails to bind this lectin. MTS10 displays the opposite binding profile in that only the K8−K18−K5+K14+ medullary TECs react with this reagent. These results unify the present characterization of medullary subsets with previous studies and expand the parameters that identify the two medullary subsets to include distinct keratin expression patterns.
There are conflicting views as to whether cortical and medullary TECs develop from a common epithelial stem cell. Because the thymus forms from a fusion of the third pharyngeal cleft ectoderm with endoderm from the third pharyngeal pouch, it has been suggested that the cortex develops from an ectodermal stem cell, whereas the medulla develops from an endodermal stem cell (32). In contrast, other studies suggest that TECs are derived primarily from endodermal epithelium (33, 34). A recent report described the presence of rare isolated TECs that express both cortical and medullary clusters of thymic epithelial staining antigens, suggesting the existence of a common epithelial stem cell, although the ectodermal versus endodermal origin of this putative progenitor population was not determined (35). It is not known whether the K8+K5+ TEC subset that contains precursors of the major K8+K5− cortical subset also gives rise to the two medullary subsets described in this and previous investigations (13, 14, 17, 18). We find that both hCD3ɛ transgenic and RAG-1−/− thymi contain small clusters of K14+ as well as UEA-1+ cells that may contain precursors that are induced to proliferate by mature T cells to form an organized medulla. Alternatively, mature T cells may enhance differentiation of K14+ and/or UEA-1+ TECs from the precursor K8+K5+ TEC subset. Penit et al. (11) recently demonstrated that in RAG-2−/− mice, the MTS10+ TECs (which we demonstrated are equivalent to the K5+K14+ subset) failed to incorporate bromodeoxyuridine during bone marrow reconstitution experiments, suggesting that this medullary TEC subset arises postmitotically from a precursor population. However, in the absence of direct evidence for precursor-progeny relationships in the medulla, the issue of whether both cortical and medullary TEC subsets arise from a common epithelial subset requires further investigation.
Acknowledgments
We are grateful to Dr. Lezlee Coghlan for performing the thymic transplants and to Dale Weiss, Pam Kille, and Donna Schutz of the Science Park Animal Facility for their excellent support. We also thank Carrie McKinley for help in manuscript preparation. This research was supported by National Institute on Environmental Health Sciences Center Grant ES07784, National Institutes of Health Training Grant HDO7296, and a grant from the Bruce McMillan, Jr. Foundation.
ABBREVIATIONS
- TEC
thymic epithelial cell
- K
keratin
- hCD3ɛ
human CD3ɛ
- RAG
recombination activating gene
- DN
double negative
- TCR
T cell antigen receptor
- DP
double positive
References
- 1. Robey E, Fowlkes B J. Annu Rev Immunol. 1994;12:675–705. doi: 10.1146/annurev.iy.12.040194.003331. [DOI] [PubMed] [Google Scholar]
- 2.Shortman K, Wu L. Annu Rev Immunol. 1996;14:29–47. doi: 10.1146/annurev.immunol.14.1.29. [DOI] [PubMed] [Google Scholar]
- 3.Nehls M, Pfiefer D, Schorpp M, Hedrich H, Boehm T. Nature (London) 1994;372:103–105. doi: 10.1038/372103a0. [DOI] [PubMed] [Google Scholar]
- 4.Anderson G, Jenkinson E J, Moore N C, Owen J J T. Nature (London) 1993;362:70–73. doi: 10.1038/362070a0. [DOI] [PubMed] [Google Scholar]
- 5.Anderson G, Anderson K L, Tchilian E Z, Owen J J T, Jenkinson E J. Eur J Immunol. 1997;27:1200–1206. doi: 10.1002/eji.1830270522. [DOI] [PubMed] [Google Scholar]
- 6.Oosterwegel M A, Haks M C, Jeffry U, Murray R, Kruisbeek A M. Immunity. 1997;6:351–360. doi: 10.1016/s1074-7613(00)80337-4. [DOI] [PubMed] [Google Scholar]
- 7.van Ewijk W, Shores E W, Singer A. Immunol Today. 1994;15:214–217. doi: 10.1016/0167-5699(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 8.Boyd R L, Tucek C L, Godfrey D I, Izon D J, Wilson T J, Davidson N J, Bean A G D, Ladyman H M, Ritter M A, Hugo P. Immunol Today. 1993;14:445–459. doi: 10.1016/0167-5699(93)90248-J. [DOI] [PubMed] [Google Scholar]
- 9.Hollander G A, Wang B, Nichogiannopoulou A, Platenburg P P, van Ewijk W, Burakoff S J, Gutierrez-Ramos J-C, Terhorst C. Nature (London) 1995;373:350–353. doi: 10.1038/373350a0. [DOI] [PubMed] [Google Scholar]
- 10.Wang N, van Ewijk W, Hollander G, Terhorst C, Wang B. FASEB J. 1996;10:A1047. , Abstr. [Google Scholar]
- 11.Penit C, Lucas B, Vasseur F, Rieker T, Boyd R L. Dev Immunol. 1996;5:25–36. doi: 10.1155/1996/61035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shores E W, VanEwijk W, Singer A. Int Immunol. 1994;6:1393–1402. doi: 10.1093/intimm/6.9.1393. [DOI] [PubMed] [Google Scholar]
- 13.Moraux-Moyson A, Scheiff J-M, Haumont S. Thymus. 1988;12:89–109. [PubMed] [Google Scholar]
- 14.Nabarra B, Andrianarison I. Thymus. 1987;9:95–121. [PubMed] [Google Scholar]
- 15.Kampinga J, Berges S, Boyd R L, Brekelmans P, Colic M, VanEwijk W, Kendall M D, Ladyman H, Nieuwenhuis P, Ritter M A, et al. Thymus. 1989;13:165–173. [PubMed] [Google Scholar]
- 16.Godfrey D I, Izon D J, Tucek C L, Wilson T J, Boyd R L. Immunology. 1990;70:66–74. [PMC free article] [PubMed] [Google Scholar]
- 17.Farr A G, Anderson S K. J Immunol. 1985;134:2971–2977. [PubMed] [Google Scholar]
- 18.Surh C D, Gao E-K, Kosaka H, Lo D, Ahn C, Murphy D B, Karlsson L, Peterson P, Sprent J. J Exp Med. 1992;176:495–505. doi: 10.1084/jem.176.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuchs E, Weber K. Annu Rev Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- 20.Franke W W, Schiller D L, Moll R, Winger S, Schmid E, Engelbrecht I, Denk H, Krepler R, Platzer B. J Mol Biol. 1991;153:933–959. doi: 10.1016/0022-2836(81)90460-5. [DOI] [PubMed] [Google Scholar]
- 21.Huitfeldt H S, Heyden A, Clausen O P F, Thrane E V, Roop D, Yuspa S H. Carcinogenesis. 1991;12:2063–2067. doi: 10.1093/carcin/12.11.2063. [DOI] [PubMed] [Google Scholar]
- 22.Roop D R, Cheng C K, Titterington L, Meyers C A, Stanley J R, Steinert P M, Yuspa S H. J Biol Chem. 1984;259:8037–8040. [PubMed] [Google Scholar]
- 23.Kemler R, Brulet P, Schnebelen M-T, Gaillard J, Jacob F. J Embryol Exp Morphol. 1981;64:45–60. [PubMed] [Google Scholar]
- 24.Lane E B. J Cell Biol. 1982;92:665–673. doi: 10.1083/jcb.92.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baribault H, Price J, Miyai K, Oshima R G. Genes Dev. 1993;7:1191–1202. doi: 10.1101/gad.7.7a.1191. [DOI] [PubMed] [Google Scholar]
- 26.Van Vliet E, Melis M, VanEwijk W. Eur J Immunol. 1984;14:524–529. doi: 10.1002/eji.1830140608. [DOI] [PubMed] [Google Scholar]
- 27.Lloyd C, Qian-Chun Y, Cheng J, Turksen K, Degenstein L, Hutton E, Fuchs E. J Cell Biol. 1995;129:1329–1344. doi: 10.1083/jcb.129.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mombaerts P, Clarke A R, Rudnicki M A, Iacomini J, Itohara S, Lafaille J J, Wang L, Ichikawa Y, Jaenisch R, Hooper M L, Tonegawa S. Nature (London) 1992;360:225–231. doi: 10.1038/360225a0. [DOI] [PubMed] [Google Scholar]
- 29.Wang B, Simpson S J, Hollander G A, Terhorst C. Immunol Rev. 1997;157:53–60. doi: 10.1111/j.1600-065x.1997.tb00973.x. [DOI] [PubMed] [Google Scholar]
- 30.Colic M, Dratojevic-Simic V, Gasic S, Dujic A. Dev Comp Immunol. 1990;14:347–354. doi: 10.1016/0145-305x(90)90025-a. [DOI] [PubMed] [Google Scholar]
- 31.Savino W, Dardenne M. J Histochem Cytochem. 1988;36:1123–1129. doi: 10.1177/36.9.2457046. [DOI] [PubMed] [Google Scholar]
- 32.Cordier A C, Haumont S M. Am J Anat. 1980;157:227–263. doi: 10.1002/aja.1001570303. [DOI] [PubMed] [Google Scholar]
- 33.Wallin J, Eibel H, Neubuser A, Wilting J, Koseki H, Balling R. Development (Cambridge, UK) 1996;22:23–30. doi: 10.1242/dev.122.1.23. [DOI] [PubMed] [Google Scholar]
- 34.LeDouarin N, Bussonnet C, Chaumont F. Ann Embryol Morphol. 1968;1:29–37. [Google Scholar]
- 35.Ropke C, VanSoest P, Platenburg P P, VanEwijk W. Dev Immunol. 1995;4:149–156. doi: 10.1155/1995/23168. [DOI] [PMC free article] [PubMed] [Google Scholar]