

Abstract
Bloom's syndrome (BS), a disorder associated with genomic instability and cancer predisposition, results from defects in the Bloom's helicase (BLM) protein. In BS cells, chromosomal abnormalities such as sister chromatid exchanges occur at highly elevated rates. Using Xenopus egg extracts, we have studied Xenopus BLM (Xblm) during both unperturbed and disrupted DNA replication cycles. Xblm binds to replicating chromatin and becomes highly phosphorylated in the presence of DNA replication blocks. This phosphorylation depends on Xenopus ATR (Xatr) and Xenopus Rad17 (Xrad17), but not Claspin. Xblm and Xenopus topoisomerase IIIα (Xtop3α) interact in a regulated manner and associate with replicating chromatin interdependently. Immunodepletion of Xblm from egg extracts results in accumulation of chromosomal DNA breaks during both normal and perturbed DNA replication cycles. Disruption of the interaction between Xblm and Xtop3α has similar effects. The occurrence of DNA damage in the absence of Xblm, even without any exogenous insult to the DNA, may help to explain the genesis of chromosomal defects in BS cells.
Keywords: RecQ helicase; ATR; Rad17; DNA replication; DNA damage
Introduction
RecQ helicases comprise a highly conserved family of DNA-unwinding enzymes that are essential for maintenance of genomic stability in a wide variety of organisms (Bachrati and Hickson, 2003). Mutations in three of the five human RecQ homologues (e.g., the Bloom's, Werner's, and RECQL4 helicases) result in diseases associated with genomic instability and predisposition to cancer. Bloom's syndrome (BS) is a rare genetic disorder that arises from mutations in the Bloom's helicase (BLM). Affected individuals display sensitivity to sunlight, immunodeficiency, dwarfism, and elevated incidence of cancer. Cultured cells from BS patients are highly prone to acquire numerous chromosomal aberrations, notably sister chromatid exchanges (SCEs).
In mammals, BLM is a constituent of the BRCA1-associated genome surveillance complex, which contains other tumor suppressors such as BRCA1, MLH1, MSH2, MSH3, ATM, and the MRE11–RAD50–NBS1 complex (Wang et al., 2000). BLM also interacts with topoisomerase IIIα and both proteins colocalize in nuclear promyelocytic leukemia bodies (Johnson et al., 2000; Wu et al., 2000; Hu et al., 2001; Bachrati and Hickson, 2003). Topoisomerase IIIα cannot be localized properly in BS cells (Johnson et al., 2000; Wu et al., 2000). A fragment of BLM containing its NH2-terminal 133 amino acids appears to be necessary and sufficient for binding topoisomerase IIIα (Hu et al., 2001). The expression of full-length BLM, but not BLMΔ1-133, can restore focal staining of topoisomerase IIIα in BS cells. Furthermore, expression of BLMΔ1-133 does not rescue the elevated SCE phenotype of BS cells (Hu et al., 2001).
For maintenance of genomic integrity, cells have developed checkpoint control mechanisms that coordinate DNA repair with cell cycle progression (Melo and Toczyski, 2002; Osborn et al., 2002). In vertebrates, ATM and ATR, members of the phosphatidylinositol 3-kinase–related family of proteins, are critical checkpoint regulators (Abraham, 2001). ATM, which responds to DNA damage caused by ionizing irradiation, phosphorylates and activates the downstream effector kinase Chk2 (Bartek et al., 2001). On the other hand, an ATR-dependent pathway detects incompletely replicated and UV-damaged DNA and promotes activation of the effector kinase Chk1 (Guo et al., 2000; Hekmat-Nejad et al., 2000; Liu et al., 2000). During this process, ATR collaborates with multiple other checkpoint proteins, including Claspin and Rad17 (Melo and Toczyski, 2002; Osborn et al., 2002). Claspin may act as a mediator or adaptor protein for the phosphorylation of Chk1 by ATR (Kumagai and Dunphy, 2000). Rad17 is necessary for loading of the Rad9–Rad1–Hus1 (9-1-1) checkpoint sliding clamp complex onto DNA. Significantly, ATR, Claspin, and Rad17 appear to recognize different features of chromatin containing incompletely replicated/UV-damaged DNA (Melo and Toczyski, 2002; Osborn et al., 2002; Lee et al., 2003). These observations suggest that the cell detects multiple aspects of chromosomal DNA during checkpoint responses. Once activated, Chk1 inhibits the entry into mitosis by modulating Cdc25 and Wee1, which coordinately regulate Cdc2–cyclin B, a critical inducer of mitosis (Melo and Toczyski, 2002).
It has been shown that ATM associates with and phosphorylates BLM after γ-irradiation (Ababou et al., 2000; Beamish et al., 2002). Treatment with hydroxyurea also induces the phosphorylation of BLM, and this phosphorylation is compromised by expression of kinase-defective ATR (Franchitto and Pichierri, 2002). More recently, it has been shown that ATR binds to human BLM and phosphorylates both threonine-99 and threonine-122 (Davies et al., 2004). Cells harboring a mutant of BLM that cannot be phosphorylated on these two sites display a defect in recovery from S phase arrest.
Although many experiments have been performed on human BLM, its exact biological function and the rationale for how mutations in this protein result in tumorigenesis in BS patients are still unknown. In this paper, we have investigated the physiological function of the Xenopus homologue of BLM (Xblm) using the Xenopus egg extract system. These experiments indicate that BLM needs to function during a normal S phase to forestall DNA damage.
Results
Xblm associates with chromatin and undergoes checkpoint-dependent phosphorylation
First, we examined the localization of Xblm under both normal and replication checkpoint–inducing conditions in Xenopus egg extracts. We incubated demembranated Xenopus sperm chromatin in extracts to form reconstituted nuclei. To induce formation of DNA replication blocks, we added the DNA polymerase inhibitor aphidicolin. In parallel, we also added sperm chromatin that had been damaged with UV light. UV-treated chromatin also accumulates DNA replication blocks. After 90 min, the reconstituted nuclei were isolated and separated into both nuclear soluble and chromatin fractions. We found that Xblm mainly binds to chromatin in untreated extracts, aphidicolin-treated extracts, and extracts containing UV-damaged DNA (Fig. 1 A). There was an undetectable amount of Xblm protein in the nuclear soluble fractions.
Figure 1.
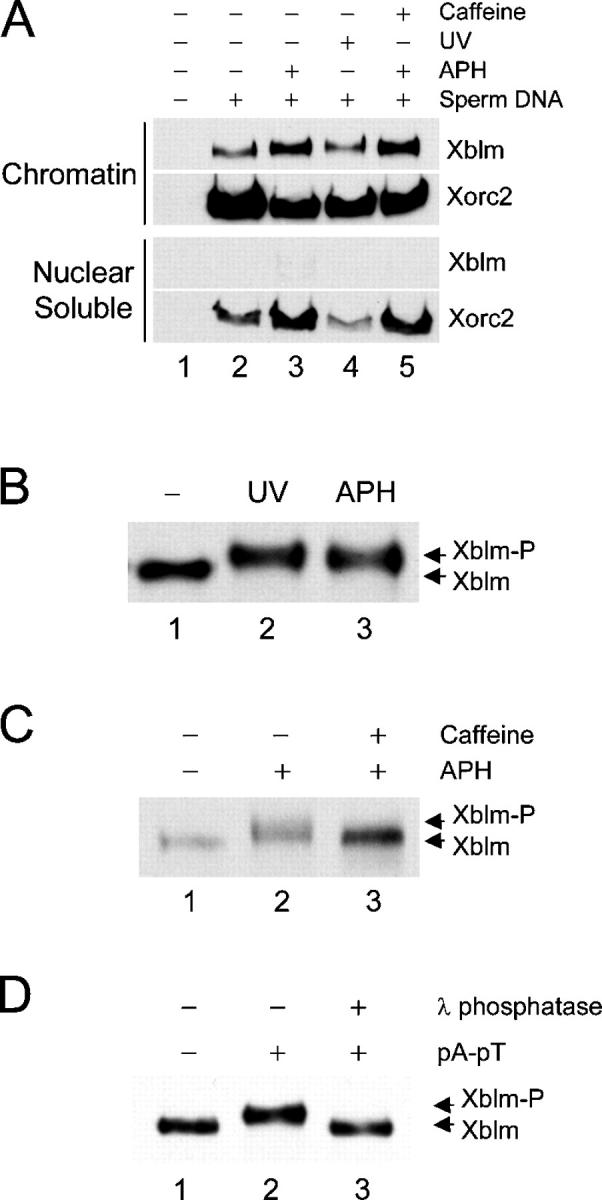
Xblm binds to chromatin and becomes phosphorylated in a checkpoint-dependent manner. (A) Extracts were incubated with no sperm chromatin (lane 1), untreated sperm chromatin (lane 2), sperm chromatin plus 100 μg/ml aphidicolin (APH; lane 3), UV-damaged sperm chromatin (lane 4), or sperm chromatin plus aphidicolin and caffeine (lane 5). After 90 min, nuclei were isolated and separated into nuclear soluble and chromatin fractions. The fractions were immunoblotted for Xblm and Xorc2 (as a loading control). (B) Extracts were incubated for 90 min with untreated sperm chromatin (lane 1), UV-damaged sperm chromatin (lane 2), or sperm chromatin plus aphidicolin (lane 3). Whole nuclear fractions were immunoblotted for Xblm. (C) Extracts were incubated with sperm chromatin alone (lane 1), sperm chromatin plus aphidicolin (lane 2), or sperm chromatin plus aphidicolin and caffeine (lane 3). Nuclear fractions were immunoblotted for Xblm. (D) Anti-Xblm immunoprecipitates from extracts containing no DNA (lane 1) or pA-pT (lanes 2 and 3) were incubated without (lanes 1 and 2) or with (lane 3) λ phosphatase and were immunoblotted for Xblm.
Next, we investigated whether incompletely replicated or UV-damaged DNA could induce any modification of Xblm. We observed that, upon aphidicolin or UV treatment, chromatin-bound Xblm migrated at a reduced mobility during SDS-PAGE (Fig. 1 B). Treatment with caffeine abolished the aphidicolin-induced modification of Xblm (Fig. 1 C). Caffeine overrides checkpoint responses by inhibiting ATR and ATM (Abraham, 2001). We also examined the effect of annealed oligomers of poly(dA)70 and poly(dT)70 (which we will refer to as pA-pT) on Xblm. This DNA template can induce the phosphorylation of both Xchk1 and Xchk2 under the appropriate conditions (Guo and Dunphy, 2000; Kumagai and Dunphy, 2000). As shown in Fig. 1 D, pA-pT induced a clear upward mobility shift of Xblm. This shift was largely abolished by λ phosphatase, which indicates that the modification corresponds to phosphorylation.
Phosphorylation of Xblm depends on Xatr and Xrad17
Incompletely replicated and UV-damaged DNA activate the Xenopus ATR (Xatr)–dependent checkpoint pathway (Guo et al., 2000; Hekmat-Nejad et al., 2000). The observation that Xblm undergoes caffeine-sensitive phosphorylation in the presence of aphidicolin-treated or UV-damaged chromatin raised the possibility that Xatr controls the phosphorylation of Xblm. To test this hypothesis, we immunodepleted Xatr from egg extracts and examined phosphorylation of Xblm in nuclear fractions from these extracts. We observed that the phosphorylation of Xblm in the presence of aphidicolin was largely abolished in Xatr-depleted extracts in comparison with mock-depleted extracts (Fig. 2 A). Claspin is necessary for the Xatr-dependent activation of Xchk1 during a replication checkpoint response (Kumagai and Dunphy, 2000). Therefore, we investigated whether Claspin is also required for the phosphorylation of Xblm. Interestingly, depletion of Claspin had no discernible effect on the phosphorylation of Xblm (Fig. 2 A). As positive controls, we showed that depletion of either Claspin or Xatr completely abolished the aphidicolin-induced phosphorylation of Xchk1 on serine-344 under the same conditions (Fig. 2 A). Therefore, in contrast to Xchk1, the Xatr-dependent phosphorylation of Xblm appears not to require Claspin.
Figure 2.

Xatr and Xrad17, but not Claspin, are required for phosphorylation of Xblm. (A) Extracts were treated with control (lanes 1 and 2), anti-Xatr (lanes 3 and 4), or anti-Claspin (lanes 5 and 6) antibodies. Sperm chromatin was incubated in extracts for 90 min in the absence (lanes 1, 3, and 5) or presence (lanes 2, 4, and 6) of aphidicolin. Nuclear fractions were isolated and immunoblotted for Xatr, Claspin, Xblm, Xchk1, and P-Ser344 of Xchk1. (B) Extracts were treated with control (lanes 1, 2, 5, and 6) or anti-Xrad17 (lanes 3, 4, 7, and 8) antibodies. Sperm chromatin was incubated in extracts for 90 min in the absence (lanes 1 and 5) or presence (lanes 2–4 and 6–8) of aphidicolin. His6-Xrad17 was added back to some extracts (lanes 4 and 8). Chromatin fractions and whole-extract aliquots were immunoblotted for Xatr, Xblm, and Xrad17.
In parallel, we examined the role of Xenopus Rad17 (Xrad17) in the phosphorylation of Xblm. We immunodepleted Xrad17 from control and aphidicolin-treated extracts and examined the phosphorylation of Xblm on chromatin. We found that the aphidicolin-induced phosphorylation of Xblm was abolished in the absence of Xrad17 (Fig. 2 B). Consistent with previous results, Xatr bound in elevated amounts to chromatin in aphidicolin-treated extracts lacking Xrad17 (Lee et al., 2003). As a control, we showed that addition of recombinant His6-Xrad17 to the Xrad17-depleted extracts restored the phosphorylation of Xblm (Fig. 2 B). The addition of recombinant Xrad17 also decreased the binding of Xatr to normal levels. These results indicate that both Xatr and Xrad17 are necessary for the phosphorylation of Xblm.
Xblm is dispensable for phosphorylation of Xchk1 and Xchk2
Because Xblm is phosphorylated in a checkpoint-dependent manner, we examined whether Xblm might be required for triggering a checkpoint response. For these experiments, we tested if Xblm is necessary for activation of the checkpoint effector kinases Xchk1 or Xchk2 in egg extracts. First, we immunodepleted Xblm from egg extracts and then monitored whether Xchk1 could be phosphorylated upon formation of DNA replication blocks. As shown in Fig. 3 A, depletion of Xblm did not inhibit the aphidicolin-induced phosphorylation of Xchk1. In parallel, we also treated extracts with pA-pT, a double-stranded DNA template that induces phosphorylation of Xchk2 very effectively (Guo and Dunphy, 2000). We found that depletion of Xblm does not abolish the phosphorylation of Xchk2 elicited by pA-pT (Fig. 3 B).
Figure 3.
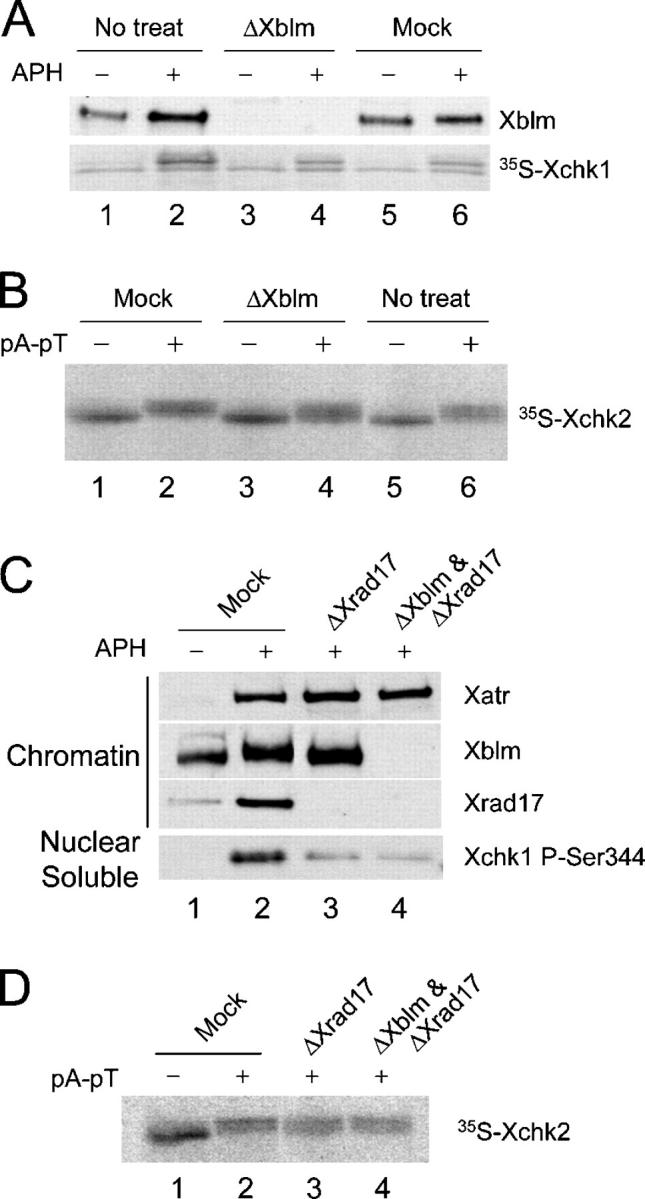
Xblm is not required for the checkpoint-dependent phosphorylation of Xchk1 or Xchk2. (A) Untreated (lanes 1 and 2), Xblm-depleted (lanes 3 and 4), and mock-depleted (lanes 5 and 6) egg extracts containing 35S-labeled Xchk1 were incubated for 90 min in the absence or presence of aphidicolin. Nuclear fractions were isolated and immunoblotted for Xblm or were examined for phosphorylation of 35S-labeled Xchk1 by SDS-PAGE and phosphorimaging. (B) Extracts were treated with control antibodies (lanes 1 and 2), anti-Xblm antibodies (lanes 3 and 4), or no antibodies (lanes 5 and 6). 35S-labeled Xchk2 was incubated in these depleted extracts for 90 min in the absence or presence of pA-pT. 35S-labeled Xchk2 in the whole-egg extracts was examined by phosphorimaging. (C) Extracts were treated with control antibodies (lanes 1 and 2), anti-Xrad17 antibodies (lane 3), or both anti-Xrad17 and anti-Xblm antibodies (lane 4). Extracts were incubated with sperm chromatin in the absence (lane 1) or presence (lanes 2–4) of aphidicolin. Chromatin fractions were immunoblotted for Xatr, Xblm, and Xrad17. Phosphorylation of Xchk1 in the soluble nuclear fraction was detected with anti-P-Ser344 antibodies. (D) Extracts were treated with control (lanes 1 and 2), anti-Xrad17 (lane 3), or both anti-Xrad17 and anti-Xblm (lane 4) antibodies. Phosphorylation of 35S-labeled Xchk2 after incubation in the absence (lane 1) or presence (lanes 2–4) of pA-pT in egg extracts was examined.
In budding yeast, Sgs1 is involved in phosphorylation of the checkpoint effector kinase Rad53 in a manner partially redundant with Rad24, the homologue of fission yeast Rad17 (Frei and Gasser, 2000; Myung and Kolodner, 2002). To test if Xblm is involved in the activation of Xchk1 or Xchk2 through a pathway redundant with one containing Xrad17, we depleted both Xblm and Xrad17 from egg extracts. As reported (Jones et al., 2003; Lee et al., 2003), depletion of Xrad17 greatly reduces the phosphorylation of Xchk1 after treatment with aphidicolin (Fig. 3 C). However, depletion of both Xrad17 and Xblm did not further diminish the phosphorylation of Xchk1 (Fig. 3 C). Consistent with previous results (Jones et al., 2003), depletion of Xrad17 did not block the phosphorylation of Xchk2 upon addition of pA-pT (Fig. 3 D). Furthermore, phosphorylation of Xchk2 occurred to a very similar extent in extracts lacking both Xrad17 and Xblm. Together, these results indicate that Xblm is not required for activation of Xchk1 or Xchk2.
Xblm interacts with Xenopus topoisomerase IIIα in a regulated manner
It has been reported that human BLM interacts with topoisomerase IIIα (Johnson et al., 2000; Wu et al., 2000; Hu et al., 2001). To examine whether this interaction is conserved in the Xenopus system, we immunoblotted anti-Xblm immunoprecipitates from egg extracts with antibodies against Xenopus topoisomerase IIIα (Xtop3α; Fig. 4, A and B). We incubated the extracts in the absence or presence of the DNA template pA-pT. Using two different anti-Xblm antibodies, we observed that binding of Xtop3α to Xblm was strongly stimulated in the presence of pA-pT.
Figure 4.

Xblm interacts with Xtop3α in a regulated manner. (A) Control (lane 1) and anti-Xblm (lanes 2 and 3) antibodies on protein A beads were incubated in extracts in the absence (lanes 1 and 2) or presence (lane 3) of pA-pT. The beads were collected and immunoblotted for Xblm and Xtop3α. (B) Control antibodies (lanes 1 and 4) and two different anti-Xblm antibodies (#870, lanes 2 and 5; #868, lanes 3 and 6) on protein A beads were incubated in extracts in the absence (lanes1–3) or presence (lanes 4–6) of pA-pT. The beads were collected and immunoblotted for Xtop3α. (C) His6-tagged Xblm-N122 on nickel beads and blank nickel beads were incubated in interphase egg extracts. The beads were collected and immunoblotted for Xtop3α. (D) Xblm-N122 on nickel beads was incubated in extracts containing no DNA (lane 1), pA-pT (lane 2), or pA-pT plus caffeine (lane 3). The beads were collected and immunoblotted with anti-Xtop3α and anti-His6 antibodies (to detect Xblm-N122). (E) Egg extracts were incubated with no DNA (lane 1), pA-pT (lane 2), or pA-pT plus caffeine (lane 3) and were immunoblotted for Xblm. (F) Anti-Xblm antibodies on protein A beads were incubated in extracts in the presence of different oligonucleotides. In some cases, either purified Xblm-N122 (lanes 6–10) or caffeine (lanes 11–14) was also added. After 2 h, the beads were collected and immunoblotted for Xblm and Xtop3α.
The NH2-terminal 133 amino acids of human BLM mediate binding to human topoisomerase IIIα (Hu et al., 2001; Bachrati and Hickson, 2003). We prepared a His6-tagged version of a comparable fragment from Xblm containing its NH2-terminal 122 residues (Xblm-N122). We observed that Xblm-N122 on nickel beads could be used to pull down Xtop3α efficiently from egg extracts (Fig. 4 C). There was no binding of Xtop3α to blank nickel beads. Interestingly, unlike the results for full-length Xblm, Xtop3α bound equally well to Xblm-N122 in the absence and presence of pA-pT (Fig. 4 D). Furthermore, this interaction was not affected by caffeine, indicating that it does not depend on ATR/ATM. Under these conditions, pA-pT does induce a caffeine-sensitive modification of Xblm (Fig. 4 E). An explanation for these results is that, upon removal of the COOH-terminal region of Xblm, the NH2-terminal end of Xblm can bind constitutively to Xtop3α. Consistent with this interpretation, analyses in budding yeast have indicated that COOH-terminally truncated (but not full-length) Sgs1 interacts strongly with Top3 (Duno et al., 2000).
We also examined the abilities of other DNA molecules besides pA-pT to promote the binding of Xtop3α to Xblm. We could observe a strong interaction between Xtop3α and Xblm in the presence of annealed poly(dC)70 and poly(dG)70 (i.e., pC-pG; Fig. 4 F). Furthermore, both Y-shaped and fork-structured DNA molecules also stimulated this interaction. By contrast, there was only a small amount of Xtop3α that bound to Xblm in the presence of a single-stranded DNA template such as poly(dA)70 (i.e., pA).
To evaluate whether Xtop3α associates with Xblm protein through direct protein–protein interactions rather than by being bridged by DNA, we examined whether Xblm-N122 could compete away the interaction of Xblm with Xtop3α in the presence of the various oligonucleotides. As shown in Fig. 4 E, we observed that addition of Xblm-N122 to egg extracts completely abolished the DNA-induced binding of full-length Xblm to Xtop3α, suggesting that this binding involves direct protein–protein interactions. Xblm-N122 itself cannot associate with DNA (unpublished data). Finally, we examined whether the binding of Xblm to Xtop3α is checkpoint regulated. For this purpose, we added caffeine to extracts containing the various oligonucleotides and then immunoprecipitated Xblm. Significantly, caffeine had little (if any) effect on the binding of Xtop3α to Xblm (Fig. 4 E), which indicates that the checkpoint-dependent phosphorylation of Xblm is not required for this interaction.
One explanation for these results would be that, in the presence of DNA containing double-stranded regions, Xblm undergoes a conformational change that exposes its NH2-terminal end. Consequently, Xblm can interact with Xtop3α with higher affinity in the presence of double-stranded DNA. Another possibility is that double-stranded DNA promotes the assembly of protein networks containing Xblm and Xtop3α.
Xblm is required for the binding of Xtop3α to chromatin in the absence, but not the presence, of DNA replication blocks
To investigate further the interaction between Xblm and Xtop3α, we examined if Xblm and Xtop3α could associate with one another on chromatin in reconstituted nuclei. First, we characterized the chromatin-binding properties of Xtop3α. We observed that Xtop3α could bind to chromatin in both the absence and presence of aphidicolin (Fig. 5 A). Treatment with both aphidicolin and caffeine resulted in somewhat elevated binding of Xtop3α to chromatin.
Figure 5.

The NH2-terminal 122 amino acids of Xblm are involved in the interaction with Xtop3α on chromatin. (A) Extracts were treated with no antibodies (lanes 1–3), anti-Xblm antibodies (lanes 4 and 5), or control antibodies (lanes 6 and 7) and then incubated with sperm chromatin alone (lanes 1, 4, and 6), sperm chromatin plus aphidicolin (lanes 2, 5, and 7), or sperm chromatin plus aphidicolin and caffeine (lane 3). After 90 min, nuclear fractions were isolated and separated into chromatin and nuclear soluble fractions. These fractions were immunoblotted for Xblm, Xtop3α, and Xorc2. (B) The NH2-terminal 122 amino acids of Xblm are required for interaction with Xtop3α on chromatin. Egg extracts containing sperm chromatin were incubated in the absence (lanes 1–4) or presence (lanes 5–8) of aphidicolin. The extracts were also treated without (lanes 1 and 5) or with (10–40 μg/μl; lanes 2–4 and 6–8) Xblm-N122. After 90 min, chromatin fractions were immunoblotted for Xblm, Xtop3α, and Xorc2. (C) Extracts were treated with no antibodies (lane 1), control antibodies (lane 2), or anti-Xtop3α antibodies (lane 3) and were immunoblotted for Xblm (top) and Xtop3α (bottom). (D) Mock-depleted extracts (lanes 1 and 2), Xtop3α- depleted extracts (lanes 3 and 4), and extracts treated with no antibodies (lanes 5 and 6) from C were incubated with sperm chromatin in the absence (lanes 1, 3, and 5) or presence (lanes 2, 4, and 6) of aphidicolin. Chromatin fractions were isolated and immunoblotted for Xblm, Xtop3α, and Xorc2.
Next, we immunodepleted Xblm from the extracts and examined the chromatin-binding properties of Xtop3α. In the absence of aphidicolin, there was no binding of Xtop3α to chromatin in Xblm-depleted extracts (Fig. 5 A). In the same experiment, Xtop3α bound very well to chromatin in mock-depleted extracts lacking aphidicolin. Interestingly, when we added aphidicolin to the Xblm-depleted extracts, we observed that Xtop3α could then bind well to chromatin. In these experiments, we found that depletion of Xblm did not affect the amount of Xtop3α in the nuclear soluble fractions, which eliminates the possibility that Xblm and Xtop3α might be codepleted under some conditions.
To further characterize the interaction of Xblm and Xtop3α with chromatin, we used the Xblm-N122 fragment. We added increasing amounts of Xblm-N122 to extracts lacking or containing aphidicolin and subsequently immunoblotted chromatin fractions for Xblm and Xtop3α. In the absence of aphidicolin, we observed that high concentrations of Xblm-N122 abolished the binding of both Xblm and Xtop3α to chromatin (Fig. 5 B). One interpretation is that Xblm-N122 blocks the binding of full-length Xblm to Xtop3α. Because Xblm is required for stable association of Xtop3α with chromatin in the absence of aphidicolin, Xblm-N122 thereby inhibits the binding of Xtop3α to chromatin. In the presence of aphidicolin, Xblm-N122 likewise prevented the binding of full-length Xblm to chromatin. However, there was still a significant amount of Xtop3α on chromatin in aphidicolin-treated extracts containing the highest concentration of Xblm-N122. This observation is consistent with the fact that Xtop3α can still bind to chromatin in Xblm-depleted extracts that contain aphidicolin, albeit at reduced levels.
As another method to evaluate the interaction between Xblm and Xtop3α on chromatin, we immunodepleted Xtop3α from egg extracts and then examined the binding of Xblm to chromatin. We observed that no Xblm binds to chromatin when Xtop3α was depleted either in the absence or presence of aphidicolin (Fig. 5, C and D). Overall, these results suggest that interaction between Xblm and Xtop3α stabilizes the binding of both proteins to chromatin under normal conditions. However, in the presence of DNA replication blocks, some Xtop3α remains tethered to chromatin independently of Xblm.
Phosphorylation of Xblm depends on association with Xtop3α
As described above, Xblm on chromatin containing incompletely replicated DNA has become phosphorylated in a checkpoint-dependent manner. Furthermore, interaction with Xtop3α promotes binding of Xblm to chromatin. We investigated the relationship between interaction with Xtop3α, association with chromatin, and phosphorylation of Xblm. Taking advantage of the finding that Xblm-N122 can disrupt the binding of Xblm to chromatin in aphidicolin-treated extracts, we tested whether Xblm could still be phosphorylated under these conditions. For this purpose, we added Xblm-N122 to egg extracts in the absence or presence of aphidicolin. After 90 min, we isolated nuclear fractions from the extracts and examined the phosphorylation of Xblm. As shown in Fig. 6 A, we found that the addition of Xblm-N122 largely abolished the aphidicolin-induced phosphorylation of Xblm. A potential concern is that Xblm-N122 could affect phosphorylation of full-length Xblm by inhibiting operation of the DNA replication checkpoint response. We ruled out this possibility by showing that Xblm-N122 did not affect the phosphorylation of Xchk1 on serine-344 in the presence of aphidicolin (Fig. 6 A).
Figure 6.

Phosphorylation of Xblm depends on association with Xtop3α. (A) Buffer alone (lanes 1 and 2) or Xblm-N122 (100 μg/μl; lanes 3 and 4) was added to extracts lacking (lanes 1 and 3) or containing (lanes 2 and 4) aphidicolin. After 90 min, nuclear fractions were isolated and immunoblotted with antibodies against Xblm and P-Ser344 of Xchk1. (B) Mock-depleted (lanes 1 and 2) and Xtop3α-depleted (lanes 3 and 4) extracts were incubated with sperm chromatin in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of aphidicolin. After 90 min, nuclear fractions were isolated and immunoblotted with antibodies against Xblm, Xtop3α, and the whole Xchk1 protein.
As another approach, we immunodepleted Xtop3α from egg extracts and then examined the aphidicolin-induced phosphorylation of Xblm (Fig. 6 B). As described above, Xblm cannot associate stably with chromatin in the absence of Xtop3α. Under these conditions, we observed that Xblm in the nuclear fraction did not become phosphorylated upon the addition of aphidicolin. By contrast, immunodepletion of Xtop3α had no effect on the checkpoint-dependent phosphorylation of Xchk1 (Fig. 6 B). Together, these results suggest that Xblm must associate stably with Xtop3α in order to undergo phosphorylation in extracts containing DNA replication blocks. Because this binding also promotes association with chromatin, this result might imply that phosphorylation of Xblm occurs on chromatin. However, it is also possible that binding of Xtop3α is required for phosphorylation of Xblm irrespective of association with chromatin.
Xblm binds to chromatin after origin unwinding
Human BLM has been reported to accumulate during S phase (Bachrati and Hickson, 2003), but the exact roles that it might play during S phase are not clear. To investigate this issue, we characterized Xblm in S phase Xenopus egg extracts. First, we examined the time course for binding of Xblm to chromatin in the absence and presence of aphidicolin. Nuclear envelope assembly around exogenously added chromatin in egg extracts occurs in ∼30–40 min. When the nuclear membrane seals off the nucleoplasm from the cytoplasm, the initiation of DNA replication takes place shortly thereafter. In the absence of aphidicolin, Xblm bound to chromatin in a time-dependent manner (Fig. 7 A). Binding of Xblm to the chromatin began at 30 min, peaked at ∼60 min, and then declined by 90 min, as DNA replication typically approaches completion. By contrast, in the presence of aphidicolin, the amount of Xblm increased steadily after 30 min and reached high levels by 90 min. The phosphorylation-dependent mobility shift of Xblm was very evident at this time. The binding profile of Xblm was very similar to that of RPA70, which is known to accumulate on aphidicolin-treated chromatin (Walter and Newport, 2000).
Figure 7.

Xblm associates with replicating chromatin, but is not essential for DNA replication. (A) Sperm chromatin was incubated in egg extracts in the absence (lanes 1–5) or presence (lanes 6–10) of aphidicolin. At various times, chromatin fractions were isolated and immunoblotted for Xblm, RPA70, and Xorc2. (B) Xblm-depleted (lanes 1–4) and mock-depleted (lanes 5–8) egg extracts were incubated with sperm chromatin. Chromosomal DNA replication was assayed as described in the Materials and methods. The two bands in each lane depict 32P incorporation into chromosomal DNA. (C) Xblm-depleted (lanes 1 and 2) and mock-depleted (lanes 3 and 4) extracts were incubated with sperm chromatin in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of aphidicolin. Chromatin fractions were isolated and immunoblotted for Xblm, RPA70, and Xorc2. (D) Mock-depleted (lanes 1 and 2) and RPA70-depleted (lanes 3 and 4) extracts were incubated with sperm chromatin in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of aphidicolin. Chromatin fractions were isolated and immunoblotted for Xblm, RPA70, and Xorc2. (E) Extracts were left untreated (lane 1) or were treated with either geminin (lane 2) or p27 (lane 3) as described previously (Lee et al., 2003), and sperm chromatin was added. After 90 min, chromatin fractions were immunoblotted for Xblm (top) and Xorc2 (bottom).
BLM can unwind many different DNA structures (Bachrati and Hickson, 2003). A significant question is whether this helicase has some direct role in DNA replication. To assess this matter, we immunodepleted Xblm from egg extracts and then assayed chromosomal DNA replication by incorporation of 32P from α[32P]dATP. We observed that removal of Xblm did not have any noticeable effect on either the timing or overall extent of DNA replication (Fig. 7 B). These results contradict those of a previous report (Liao et al., 2000), in which it was observed that immunodepletion of Xblm severely inhibits DNA replication in egg extracts (see Discussion).
To further assess whether there is any relationship between Xblm and the process of DNA replication, we examined the interdependencies between binding of Xblm to chromatin and known steps in DNA replication. In one approach, we examined the relationship between Xblm and RPA for association with chromatin. First, we prepared Xblm-depleted extracts and examined the binding of RPA70 to chromatin in the absence and presence of aphidicolin. As shown in Fig. 7 C, the chromatin-binding properties of RPA70 in extracts lacking or containing aphidicolin were unaffected by the immunodepletion of Xblm. Likewise, the binding of Pol α to chromatin in these extracts was not altered by the removal of Xblm (unpublished data). Because RPA is required to stabilize unwound DNA for the initiation of replication, these observations are consistent with the idea that Xblm is not required for the replicative unwinding of DNA.
In another experiment, we immunodepleted RPA with anti-RPA70 antibodies and examined the binding of Xblm to chromatin in extracts either lacking or containing aphidicolin. We found that binding of Xblm to chromatin was largely abolished in RPA-depleted extracts in both the absence and presence of aphidicolin (Fig. 7 D). There is also no binding of Xtop3α to RPA-depleted chromatin (unpublished data). Therefore, RPA is necessary for the association of Xblm as well as Xtop3α with chromatin. To examine explicitly whether binding of Xblm occurs during the process of DNA replication, we treated interphase extracts with geminin or p27, both of which abolish replication origin firing. Geminin blocks loading of the minichromosome maintenance complex onto the DNA, and p27 inhibits the kinase activity of Cdk2 (McGarry and Kirschner, 1998; Lee et al., 2003). As shown in Fig. 7 E, there is no binding of Xblm to interphase chromatin in extracts treated with either geminin or p27. Together, these observations suggest that normal unwinding of the DNA at replication forks is a prerequisite for the binding of Xblm to chromatin during S phase.
Xblm is required for preventing accumulation of chromosomal breaks during both unperturbed and disrupted S phases
We asked whether any problems might arise during the course of DNA replication due to the absence of Xblm. In particular, we examined whether absence of Xblm might result in DNA damage during S phase. For this purpose, we performed TUNEL assays to detect DNA broken ends. We used conditions that were used previously to detect double-stranded DNA breaks in egg extracts (Costanzo et al., 2001). Initially, we prepared Xblm- and mock-depleted extracts, incubated these extracts with untreated sperm chromatin, and then performed TUNEL assays. We detected a strong signal for incorporation of 32P into the DNA from Xblm-depleted extracts, but not from mock-depleted extracts (Fig. 8). For comparison, we deliberately damaged the sperm chromatin by adding the restriction endonuclease EcoRI to egg extracts (Kobayashi et al., 2002). As expected, treatment with EcoRI resulted in a strong positive signal in the TUNEL assay, but interestingly, the signal was only about twofold greater than for Xblm-depleted extracts containing no DNA-damaging agent. As a control, we showed that the incorporation of 32P into DNA depended on exogenously added TdT. Finally, we asked whether the DNA damage in the Xblm-depleted extracts depended on the occurrence of DNA replication. To address this issue, we treated Xblm-depleted extracts with geminin. Significantly, treatment with geminin completely abolished the positive signal in the TUNEL assay. By contrast, geminin did not suppress the signal arising from DNA damage induced by EcoRI. These results indicate that the chromosomal breaks that occur in Xblm-depleted extracts arise from the process of DNA replication.
Figure 8.
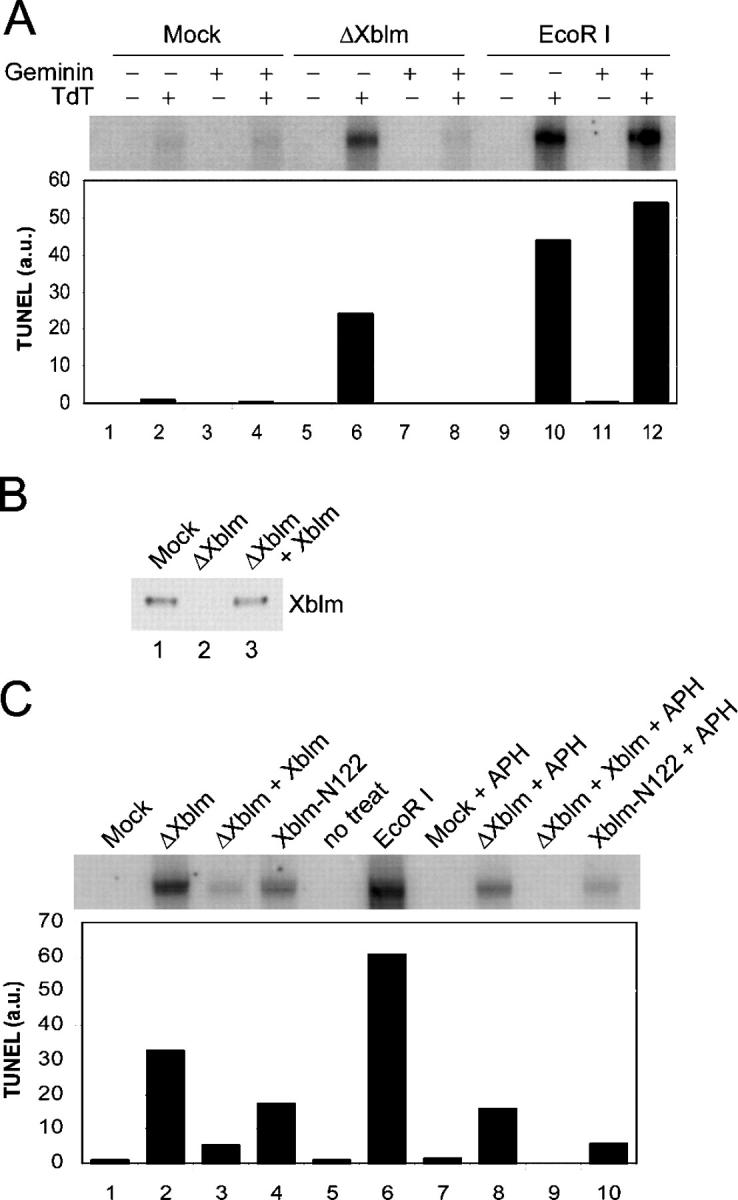
Xblm is required for preventing accumulation of chromosomal breaks during normal and disrupted S phases. (A) Extracts were treated with control antibodies (lanes 1–4), anti-Xblm antibodies (lanes 5–8), or no antibodies (lanes 9–12) and were incubated with sperm chromatin. Some extracts were treated with 0.1 U/μl EcoRI (lanes 9–12) and geminin (lanes 3, 4, 7, 8, 11, and 12). After 2 h, TUNEL assays were performed in the presence (lanes 2, 4, 6, 8, 10, and 12) or absence (lanes 1, 3, 5, 7, 9, and 11) of TdT. Samples were subjected to agarose gel electrophoresis and phosphorimaging (top). The radioactivity in each band was quantitated (bottom). (B) Mock-depleted (lane 1) and Xblm-depleted (lanes 2 and 3) extracts were prepared. Recombinant Xblm was made in the TnT® system and added back to an Xblm-depleted extract (lane 3). Samples were immunoblotted for Xblm. (C) Extracts were treated with control antibodies (lanes 1 and 7), anti-Xblm antibodies (lanes 2, 3, 8, and 9), or no antibodies (lanes 4, 5, 6, and 10) and were incubated with sperm chromatin. The reactions also contained 100 μg/ml Xblm-N122 (lanes 4 and 10), 0.1 U/μl EcoRI (lane 6), and aphidicolin (lanes 7–10). Full-length recombinant Xblm was added to some samples (lanes 3 and 9). After 2 h, TUNEL assays were performed as in A.
To determine whether the chromosomal breaks in the Xblm-depleted extracts were due specifically to the absence of Xblm, we performed rescue experiments. We observed that addition of recombinant Xblm that had been produced in the TnT® expression system greatly reduced the TUNEL assay signals in Xblm-depleted extracts (Fig. 8, B and C). By contrast, mock TnT® lysates lacking Xblm did not suppress DNA damage in Xblm-depleted extracts (unpublished data).
As described above, the Xblm-N122 fragment can disrupt the binding of Xblm to chromatin. Therefore, we examined whether the addition of Xblm-N122 to egg extracts would also cause the occurrence of chromosomal breaks. As shown in Fig. 8 C, Xblm-N122 elicited a positive signal in the TUNEL assay that was roughly one half of that observed in Xblm-depleted extracts. This experiment suggests that damage arises when Xblm, even though present in egg extracts, cannot bind to chromatin. It should also be noted that addition of Xblm-N122 and immunodepletion of Xblm both compromise the binding of Xtop3α to chromatin (see Discussion).
It has been proposed that human BLM protein is involved in resolving deleterious structures that arise as a consequence of blocks in DNA replication (Enomoto, 2001; Imamura et al., 2001; Franchitto and Pichierri, 2002). Therefore, we also examined the effect of a DNA replication inhibitor on the formation of chromosomal breaks in the absence and presence of Xblm. For this experiment, we incubated Xblm- and mock-depleted extracts with aphidicolin. We also observed a positive TUNEL assay signal from Xblm-depleted extracts containing aphidicolin (Fig. 8 C). Significantly, we could not detect any TUNEL assay signal in mock-depleted extracts containing aphidicolin. Together, these observations indicate that chromosomal breaks clearly occur in Xblm-depleted extracts containing aphidicolin. Overall, we conclude that if Xblm is either absent or unable to bind to chromatin, chromosomal breaks accumulate in egg extracts in both the absence and presence of a DNA replication inhibitor.
Discussion
Xblm binds to chromatin in S phase egg extracts and becomes phosphorylated in response to incompletely replicated or UV-damaged DNA. This phosphorylation is sensitive to caffeine and dependent on Xatr. Because Xatr responds to the presence of incompletely replicated or UV-damaged DNA, these observations suggest that Xblm functions in some Xatr-regulated pathway. Recent reports have indicated that phosphorylation of human BLM by ATR on threonine-99 and threonine-122 is involved in recovery from S phase arrest (Davies et al., 2004). Interestingly, these phosphorylation sites appear not to be conserved in Xblm. In humans, BLM is also phosphorylated by ATM in response to ionizing radiation (Beamish et al., 2002). Our results do not exclude the possibility that Xblm is also a target of Xenopus ATM. Overall, BLM appears to be a target of ATR/ATM-dependent pathways in a number of different checkpoint responses in vertebrates.
It is well established that ATR collaborates with other proteins to regulate the phosphorylation of Chk1 during checkpoint responses (O'Connell et al., 2000; Melo and Toczyski, 2002; Osborn et al., 2002). These proteins include a checkpoint clamp loader that contains Rad17 and the four small RFC subunits (RFC2–5). In addition, there is a checkpoint-sliding clamp that consists of the Rad9–Rad1–Hus1 (9-1-1) complex. Rad17-(RFC2–5) is responsible for loading the 9-1-1 complex onto various checkpoint-inducing DNA structures. This pathway has been well characterized in Xenopus egg extracts (Lupardus et al., 2002; Stokes et al., 2002; You et al., 2002; Jones et al., 2003; Lee et al., 2003). Moreover, the Xatr-dependent phosphorylation of Xchk1 also depends on the checkpoint mediator/adaptor protein called Claspin (Kumagai and Dunphy, 2000). Interestingly, we have found that the Xatr-dependent phosphorylation of Xblm requires Xrad17 and presumably the 9-1-1 complex, but not Claspin. The implication is that this phosphorylation requires a distinct mediator protein or perhaps can occur without the assistance of such a protein.
Although Xblm becomes phosphorylated in a checkpoint-dependent manner, it appears not to be necessary for the activation of the two major checkpoint-signaling pathways that have been characterized in the Xenopus egg extract system. Immunodepletion of Xblm does not compromise the phosphorylation of Xchk1 and Xchk2 in response to DNA replication blocks and double-stranded DNA ends, respectively. Furthermore, the cell cycle delay induced by aphidicolin is not diminished by removal of Xblm (unpublished data). In budding yeast, Sgs1 is involved in the phosphorylation of Rad53 in a Rad24-redundant manner (Frei and Gasser, 2000; Myung and Kolodner, 2002), but we did not observe any additional effect on phosphorylation of either Xchk1 or Xchk2 upon removal of both Xblm and Xrad17. These results suggest that Xblm functions in some other process besides imposition of cell cycle arrest.
Human BLM is known to interact with topoisomerase IIIα (Johnson et al., 2000; Wu et al., 2000; Hu et al., 2001). This interaction appears to be conserved in both budding and fission yeast (Bachrati and Hickson, 2003). We have likewise observed a physical association between Xblm and Xtop3α in the Xenopus system. In addition, this interaction is stimulated by the presence of DNA molecules with double-stranded regions. This increased binding is possibly due to a conformational change in Xblm that exposes its NH2-terminal, Xtop3α-interacting region. Although both human BLM and topoisomerase IIIα were shown to associate with DNA, the issue of how these proteins interact on chromatin has not been addressed explicitly (Wu and Hickson, 2002). Xblm and Xtop3α bind to normally replicating chromatin interdependently. Thus, when either Xblm or Xtop3α is removed from the extracts, the other protein is unable to associate stably with chromatin, at least in the absence of aphidicolin. Furthermore, treatment with Xblm-N122, which competes with full-length Xblm for the binding to Xtop3α, abrogates binding of both Xblm and Xtop3α to chromatin. These observations argue that Xblm and Xtop3α form a specific complex on chromatin to function in a coordinated manner. Notably, in the presence of aphidicolin-induced DNA replication blocks, some Xtop3α remains associated with chromatin in the absence of Xblm. In this case, Xtop3α may be tethered to chromatin through binding to an unknown protein and/or unique DNA structure without Xblm.
Xblm binds to chromatin specifically during S phase, but appears not to be essential for DNA replication. Upon complete removal of Xblm from egg extracts, we did not observe any obvious decrease in either the rate or overall extent of DNA replication. Likewise, immunodepletion of Xtop3α, which binds to chromatin interdependently with Xblm, does not inhibit DNA replication (unpublished data). Additionally, Xblm can bind to chromatin only after RPA has loaded onto unwound single-stranded DNA, which indicates that Xblm is not required for at least initial DNA unwinding at origins. Our results are different from those of an earlier work in which it was observed that depletion of Xblm from egg extracts led to a large reduction in DNA synthesis (Liao et al., 2000). The reason for the discrepancy is unclear, but it is notable that BLM and its homologues appear not to be absolutely essential for DNA replication in any other organism for which this question has been addressed (Bachrati and Hickson, 2003). In particular, human BS cells are viable and thus able to complete DNA replication.
Despite the fact that Xblm is dispensable for DNA replication, we did observe that immunodepletion of Xblm or addition of Xblm-N122 results in accumulation of chromosomal breaks during S phase. Because these treatments also disrupt binding of Xtop3α to chromatin, damage could arise from lack of Xblm, Xtop3α, or both at replication forks. Significantly, when we added aphidicolin to control egg extracts, we could not detect the formation of chromosomal breaks. This observation suggests that egg extracts normally have the capacity to avoid accumulation of damage at stalled replication forks. However, when Xblm is absent from the system, damage occurs readily in aphidicolin-blocked chromatin. In a recent complementary report, it was found that untreated and aphidicolin-treated human BS cells acquire DNA damage in late S phase (Rassool et al., 2003).
There are various models that could explain the accumulation of chromosomal breaks in the absence of Xblm. Even during apparently normal DNA replication, the replication fork can undergo pausing or stalling. As one example, so-called “fragile sites” in human cells may correspond to chromosomal regions that undergo inefficient replication (Casper et al., 2002). One model has been that BLM, and therefore Xblm, could function as a “roadblock remover” in advancing forks (Bachrati and Hickson, 2003). Xblm could disrupt DNA secondary structures or other obstacles to fork progression. Ineffective removal of such structures could lead to fork stalling and damage. Another possibility is that Xblm prevents the collapse or demise of replication forks that have undergone stalling, as is the case for budding yeast Sgs1 (Cobb et al., 2003). Furthermore, Xblm could be involved in repairing forks that have already undergone spontaneous damage. Recent evidence has indicated a role for BLM in DNA repair in the Drosophila system (Adams et al., 2003). Finally, it should be noted that these models are not mutually exclusive.
A related model is that lack of BLM leads to abnormal processing of replication intermediates. BLM interacts functionally with flap endonuclease 1, which is involved in Okazaki fragment processing (Imamura and Campbell, 2003; Sharma et al., 2004). Aberrant processing of Okazaki fragments, perhaps selectively in slowly replicating zones, may result in damage. Whatever the mechanism, the implication is that advancing replication forks encounter problems with some frequency and that Xblm must cope continuously with such events. Inaccurate repair of this damage could lead to the chromosomal abnormalities like those found in BS cells (e.g., SCEs). Curiously, the damage that results from absence of Xblm in egg extracts lacking aphidicolin does not trigger detectable activation of either Xchk1 or Xchk2. Perhaps this damage is simply below a threshold for detection, but this observation also raises the possibility that this sort of damage is not readily detected by at least these checkpoint systems during S phase. This property could enhance the likelihood that DNA damage would be propagated to daughter cells in intact cells lacking BLM.
In conclusion, we have found that Xblm is necessary for guarding against accumulation of DNA damage during both normal and perturbed S phases. Further analysis of how Xblm exerts this effect may help to reveal mechanisms for preservation of genomic integrity during S phase in vertebrates.
Materials and methods
Production of in vitro–translated proteins
The sequence of Xblm (Liao et al., 2000) was cloned into pCRII-TOPO (Invitrogen) after the T7 promoter. This plasmid was used for in vitro translation of Xblm in the TnT® system (Promega) with nonradioactive amino acids. Full-length Xchk1 was labeled with [35S]methionine as before (Kumagai et al., 1998). Xchk2 and His6-Xrad17 were produced using pET30a constructs (Novagen) with the TnT® system.
Antibodies
Anti-Xblm antibodies were raised against two His6 fusions containing aa 364–614 (antibody #870) and aa 1150–1360 (antibody #868). Fragments were expressed in bacteria and purified as described previously (Kumagai and Dunphy, 2003). Anti-Xtop3α antibodies were raised against a COOH-terminal fragment of Xtop3α. Encoding DNA was amplified by PCR with primers based on Xenopus EST sequences (AW64024 and TC21034) and cloned into pET3-His6X for protein and antibody production. Antibodies against Claspin, Xchk1, P-Ser344 of Xchk1, Xatr, RPA70, and Xorc2 were described previously (Lee et al., 2003).
Immunodepletions
For immunodepletions, anti-Xblm (#870) or anti-Xtop3α antibodies on Affiprep protein A beads (Bio-Rad Laboratories) were incubated in egg extracts for two rounds of 40 min at 4°C.
Production of the His6-tagged, NH2-terminal fragment of Xblm
A DNA fragment encoding the NH2-terminal 122 amino acids of Xblm was produced by PCR and cloned into pET3-His6X. The His6 fusion polypeptide was expressed in bacteria and purified as described above.
Immunoprecipitation of Xblm
Anti-Xblm antibodies on Affiprep protein A beads were incubated in egg extracts for 100 min at 4°C and centrifuged at 570 g for 1 min. Supernatants were removed and the beads were washed four times with Hepes-buffered saline containing 2.5 mM EGTA and 20 mM β-glycerolphosphate. Samples from caffeine-treated extracts were washed with caffeine-containing buffer.
Oligonucleotides
Oligonucleotides poly(dA)70 (referred to as pA) and annealed poly(dA)70-poly(dT)70 (referred to as pA-pT) were prepared as described previously (Kumagai and Dunphy, 2000). Oligonucleotides 5′-(dC)40(dA)30-3′ and 5′-(dA)30(dG)40-3′ were annealed to form Y-shape structured DNA. The oligonucleotide (dT)30 was annealed with this Y-shaped DNA to yield fork-structured DNA.
Isolation of nuclear and chromatin fractions
Egg extracts containing 3,000 sperm nuclei/μl were overlaid on a 1-M sucrose cushion in chromatin isolation buffer (CIB; 20 mM Hepes-KOH, pH 7.6, 80 mM KCl, 2.5 mM potassium gluconate, and 10 mM magnesium gluconate) and centrifuged at 6,100 g for 5 min. Nuclear pellets were washed twice with CIB. For isolation of chromatin, CIB plus 0.5% NP-40 was added to the washed nuclear pellets. Samples were incubated on ice for 5 min before centrifugation at 6,100 g for 5 min. The supernatant corresponds to the soluble nuclear fraction. The pellet (chromatin fraction) was washed with CIB three times.
Replication assays
DNA replication assays were performed by labeling with α[32P]dATP as described previously (Yanow et al., 2003).
TUNEL assays
Egg extracts were incubated with 10,000 sperm nuclei/μl for 120 min at RT and processed for TUNEL assays with terminal transferase (TdT) in the presence of α[32P]dATP essentially as described previously (Costanzo et al., 2001). Radiolabeled bands were quantitated with ImageQuant® software (Molecular Dynamics).
Acknowledgments
We thank our colleagues in the lab for helpful comments on the manuscript.
S.-M. Kim and J. Lee are associates and W.G. Dunphy is an investigator in the Howard Hughes Medical Institute.
Abbreviations used in this paper: BLM, Bloom's helicase; BS, Bloom's syndrome; SCE, sister chromatid exchange; Xatr, Xenopus ATR; Xblm, Xenopus BLM; Xrad17, Xenopus Rad17; Xtop3α, Xenopus topoisomerase IIIα.
References
- Ababou, M., S. Dutertre, Y. Lecluse, R. Onclercq, B. Chatton, and M. Amor-Gueret. 2000. ATM-dependent phosphorylation and accumulation of endogenous BLM protein in response to ionizing radiation. Oncogene. 19:5955–5963. [DOI] [PubMed] [Google Scholar]
- Abraham, R.T. 2001. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196. [DOI] [PubMed] [Google Scholar]
- Adams, M.D., M. McVey, and J.J. Sekelsky. 2003. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science. 299:265–267. [DOI] [PubMed] [Google Scholar]
- Bachrati, C.Z., and I.D. Hickson. 2003. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem. J. 374:577–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek, J., J. Falck, and J. Lukas. 2001. CHK2 kinase–a busy messenger. Nat. Rev. Mol. Cell Biol. 2:877–886. [DOI] [PubMed] [Google Scholar]
- Beamish, H., P. Kedar, H. Kaneko, P. Chen, T. Fukao, C. Peng, S. Beresten, N. Gueven, D. Purdie, S. Lees-Miller, et al. 2002. Functional link between BLM defective in Bloom's syndrome and the ataxia-telangiectasia-mutated protein, ATM. J. Biol. Chem. 277:30515–30523. [DOI] [PubMed] [Google Scholar]
- Casper, A.M., P. Nghiem, M.F. Arlt, and T.W. Glover. 2002. ATR regulates fragile site stability. Cell. 111:779–789. [DOI] [PubMed] [Google Scholar]
- Cobb, J.A., L. Bjergbaek, K. Shimada, C. Frei, and S.M. Gasser. 2003. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 22:4325–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo, V., K. Robertson, M. Bibikova, E. Kim, D. Grieco, M. Gottesman, D. Carroll, and J. Gautier. 2001. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol. Cell. 8:137–147. [DOI] [PubMed] [Google Scholar]
- Davies, S.L., P.S. North, A. Dart, N.D. Lakin, and I.D. Hickson. 2004. Phosphorylation of the Bloom's syndrome helicase and its role in recovery from S-phase arrest. Mol. Cell. Biol. 24:1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duno, M., B. Thomsen, O. Westergaard, L. Krejci, and C. Bendixen. 2000. Genetic analysis of the Saccharomyces cerevisiae Sgs1 helicase defines an essential function for the Sgs1-Top3 complex in the absence of SRS2 or TOP1. Mol. Gen. Genet. 264:89–97. [DOI] [PubMed] [Google Scholar]
- Enomoto, T. 2001. Functions of RecQ family helicases: possible involvement of Bloom's and Werner's syndrome gene products in guarding genome integrity during DNA replication. J. Biochem. (Tokyo). 129:501–507. [DOI] [PubMed] [Google Scholar]
- Franchitto, A., and P. Pichierri. 2002. Bloom's syndrome protein is required for correct relocalization of RAD50/MRE11/NBS1 complex after replication fork arrest. J. Cell Biol. 157:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei, C., and S.M. Gasser. 2000. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 14:81–96. [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., and W.G. Dunphy. 2000. Response of Xenopus Cds1 in cell-free extracts to DNA templates with double-stranded ends. Mol. Biol. Cell. 11:1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., A. Kumagai, S.X. Wang, and W.G. Dunphy. 2000. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 14:2745–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Nejad, M., Z. You, M.C. Yee, J.W. Newport, and K.A. Cimprich. 2000. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr. Biol. 10:1565–1573. [DOI] [PubMed] [Google Scholar]
- Hu, P., S.F. Beresten, A.J. van Brabant, T.Z. Ye, P.P. Pandolfi, F.B. Johnson, L. Guarente, and N.A. Ellis. 2001. Evidence for BLM and topoisomerase IIIα interaction in genomic stability. Hum. Mol. Genet. 10:1287–1298. [DOI] [PubMed] [Google Scholar]
- Imamura, O., and J.L. Campbell. 2003. The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast dna2 mutants. Proc. Natl. Acad. Sci. USA. 100:8193–8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura, O., K. Fujita, A. Shimamoto, H. Tanabe, S. Takeda, Y. Furuichi, and T. Matsumoto. 2001. Bloom helicase is involved in DNA surveillance in early S phase in vertebrate cells. Oncogene. 20:1143–1151. [DOI] [PubMed] [Google Scholar]
- Johnson, F.B., D.B. Lombard, N.F. Neff, M.A. Mastrangelo, W. Dewolf, N.A. Ellis, R.A. Marciniak, Y. Yin, R. Jaenisch, and L. Guarente. 2000. Association of the Bloom syndrome protein with topoisomerase IIIα in somatic and meiotic cells. Cancer Res. 60:1162–1167. [PubMed] [Google Scholar]
- Jones, R.E., J.R. Chapman, C. Puligilla, J.M. Murray, A.M. Car, C.C. Ford, and H.D. Lindsay. 2003. XRad17 is required for the activation of XChk1 but not XCds1 during checkpoint signaling in Xenopus. Mol. Biol. Cell. 14:3898–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T., S. Tada, T. Tsuyama, H. Murofushi, M. Seki, and T. Enomoto. 2002. Focus-formation of replication protein A, activation of checkpoint system and DNA repair synthesis induced by DNA double-strand breaks in Xenopus egg extract. J. Cell Sci. 115:3159–3169. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., and W.G. Dunphy. 2000. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell. 6:839–849. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., and W.G. Dunphy. 2003. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat. Cell Biol. 5:161–165. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., Z. Guo, K.H. Emami, S.X. Wang, and W.G. Dunphy. 1998. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J. Cell Biol. 142:1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J., A. Kumagai, and W.G. Dunphy. 2003. Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol. Cell. 11:329–340. [DOI] [PubMed] [Google Scholar]
- Liao, S., J. Graham, and H. Yan. 2000. The function of Xenopus Bloom's syndrome protein homolog (xBLM) in DNA replication. Genes Dev. 14:2570–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q., S. Guntuku, X.S. Cui, S. Matsuoka, D. Cortez, K. Tamai, G. Luo, S. Carattini-Rivera, F. DeMayo, A. Bradley, et al. 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Lupardus, P.J., T. Byun, M.C. Yee, M. Hekmat-Nejad, and K.A. Cimprich. 2002. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 16:2327–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry, T.J., and M.W. Kirschner. 1998. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 93:1043–1053. [DOI] [PubMed] [Google Scholar]
- Melo, J., and D. Toczyski. 2002. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 14:237–245. [DOI] [PubMed] [Google Scholar]
- Myung, K., and R.D. Kolodner. 2002. Suppression of genome instability by redundant S-phase checkpoint pathways in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 99:4500–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell, M.J., N.C. Walworth, and A.M. Carr. 2000. The G2-phase DNA-damage checkpoint. Trends Cell Biol. 10:296–303. [DOI] [PubMed] [Google Scholar]
- Osborn, A.J., S.J. Elledge, and L. Zou. 2002. Checking on the fork: the DNA-replication stress-response pathway. Trends Cell Biol. 12:509–516. [DOI] [PubMed] [Google Scholar]
- Rassool, F.V., P.S. North, G.J. Mufti, and I.D. Hickson. 2003. Constitutive DNA damage is linked to DNA replication abnormalities in Bloom's syndrome cells. Oncogene. 22:8749–8757. [DOI] [PubMed] [Google Scholar]
- Sharma, S., J.A. Sommers, L. Wu, V.A. Bohr, I.D. Hickson, and R.M. Brosh, Jr. 2004. Stimulation of flap endonuclease-1 by the Bloom's syndrome protein. J. Biol. Chem. 279:9847–9856. [DOI] [PubMed] [Google Scholar]
- Stokes, M.P., R. Van Hatten, H.D. Lindsay, and W.M. Michael. 2002. DNA replication is required for the checkpoint response to damaged DNA in Xenopus egg extracts. J. Cell Biol. 158:863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, J., and J. Newport. 2000. Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase α. Mol. Cell. 5:617–627. [DOI] [PubMed] [Google Scholar]
- Wang, Y., D. Cortez, P. Yazdi, N. Neff, S.J. Elledge, and J. Qin. 2000. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 14:927–939. [PMC free article] [PubMed] [Google Scholar]
- Wu, L., and I.D. Hickson. 2002. The Bloom's syndrome helicase stimulates the activity of human topoisomerase IIIα. Nucleic Acids Res. 30:4823–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, L., S.L. Davies, P.S. North, H. Goulaouic, J.F. Riou, H. Turley, K.C. Gatter, and I.D. Hickson. 2000. The Bloom's syndrome gene product interacts with topoisomerase III. J. Biol. Chem. 275:9636–9644. [DOI] [PubMed] [Google Scholar]
- Yanow, S.K., D.A. Gold, H.Y. Yoo, and W.G. Dunphy. 2003. Xenopus Drf1, a regulator of Cdc7, displays checkpoint-dependent accumulation on chromatin during an S-phase arrest. J. Biol. Chem. 278:41083–41092. [DOI] [PubMed] [Google Scholar]
- You, Z., L. Kong, and J. Newport. 2002. The role of single-stranded DNA and polymerase α in establishing the ATR, Hus1 DNA replication checkpoint. J. Biol. Chem. 277:27088–27093. [DOI] [PubMed] [Google Scholar]