

Abstract
Listeria monocytogenes uptake by nonphagocytic cells is promoted by the bacterial invasion proteins internalin and InlB, which bind to their host receptors E-cadherin and hepatocyte growth factor receptor (HGF-R)/Met, respectively. Here, we present evidence that plasma membrane organization in lipid domains is critical for Listeria uptake. Cholesterol depletion by methyl-β-cyclodextrin reversibly inhibited Listeria entry. Lipid raft markers, such as glycosylphosphatidylinositol-linked proteins, a myristoylated and palmitoylated peptide and the ganglioside GM1 were recruited at the bacterial entry site. We analyzed which molecular events require membrane cholesterol and found that the presence of E-cadherin in lipid domains was necessary for initial interaction with internalin to promote bacterial entry. In contrast, the initial interaction of InlB with HGF-R did not require membrane cholesterol, whereas downstream signaling leading to F-actin polymerization was cholesterol dependent. Our work, in addition to documenting for the first time the role of lipid rafts in Listeria entry, provides the first evidence that E-cadherin and HGF-R require lipid domain integrity for their full activity.
Keywords: Listeria; phagocytosis; rafts; E-cadherin; HGF-R/c-Met
Introduction
Listeria monocytogenes is a Gram-positive pathogenic bacterium which causes severe infections such as septicaemias, meningitis, and abortions (Vazquez-Boland et al., 2001). This food-born pathogen has evolved strategies to cross the intestinal, blood-brain, and placental barriers. In cultured cells, L. monocytogenes enters nonprofessional phagocytic cells by a receptor-mediated zipper mechanism (Dramsi and Cossart, 1998; Cossart and Sansonetti, 2004). After phagocytosis, the vacuolar membrane is disrupted by the bacterial toxin listeriolysin O (LLO) and the secreted phospholipases PlcA and PlcB (Cossart et al., 1989; Camilli et al., 1991). An actin-driven mechanism allows the bacterium to disseminate from cell to cell, avoiding its exposure to the extracellular environment (Tilney and Portnoy, 1989). Our laboratory has identified two major bacterial proteins, internalin and InlB, that mediate L. monocytogenes entry into cells (Cossart et al., 2003). Internalin binds to the cell adhesion molecule E-cadherin, and InlB binds to the hepatocyte growth factor (HGF) receptor (HGF-R or Met; Mengaud et al., 1996b; Shen et al., 2000). Other molecules, such as gC1qR, may contribute in entry (Cossart et al., 2003). The HGF-R is predominantly expressed in cells of epithelial or endothelial origins, whereas E-cadherin is expressed in epithelial cells. Hence, depending on the target cell, L. monocytogenes entry can involve one or both bacterial invasion proteins. Although some components of the signaling pathways implicated in entry have been identified, the spatio-temporal regulations of the interactions between bacterial and host molecular effectors within the plasma membrane remain elusive (Ireton et al., 1999; Bierne et al., 2000, 2001; Lecuit et al., 2000; Sousa et al., 2004).
Cellular invasion by viruses or parasites and the uptake of some intracellular bacteria, such as FimH-expressing enterobacteria, mycobacteria, Salmonella, Chlamydia, and Shigella into professional or nonprofessional phagocytic cells, has been shown to require a specialized host membrane microenvironment organized into lipid domains (Gatfield and Pieters, 2000; Shin et al., 2000; Garner et al., 2002; Lafont et al., 2002; Jutras et al., 2003). In addition, bacterial toxins such as cholera toxin, LLO, and anthrax toxin target lipid domains (Orlandi and Fishman, 1998; Coconnier et al., 2000; Abrami et al., 2003).
Formation of membrane lipid domains is thought to be based on the segregation of different lipid phases within the lipid bilayer. A liquid-ordered phase (raft-like domains) and a liquid-disordered phase (more fluid regions of the membrane) might coexist in biological membranes (Brown and Rose, 1992; Brown and London, 1998; Rietveld and Simons, 1998). The first type of membrane domain, often called lipid rafts, has been extensively studied. A biochemical approach for their analysis is the isolation of detergent-resistant membranes (DRMs) after cellular extraction with cold nonionic detergents, and successive flotation on density gradients (London and Brown, 2000; Giurisato et al., 2003; Schuck et al., 2003). Although this methodology might not preserve the native structural organization of lipid rafts, it has been used to study their composition. DRMs are characterized by a high content of cholesterol and glycosphingolipids compared with the average membrane content. Cholesterol, the major lipid of the membrane, is a critical component that controls lipid phase separation and stabilizes the liquid-ordered phase (London and Brown, 2000). Drugs that modify membrane cholesterol concentration disrupt membrane domain organization, and serve as tools to study lipid domain functions in cellular processes (Kilsdonk et al., 1995). Raft domains comprise different classes of proteins such as glycosylphosphatidylinositol (GPI)-anchored proteins, myristoylated or palmitoylated proteins, cholesterol-linked and some transmembrane proteins (Harder and Simons, 1997; Zacharias et al., 2002; Edidin, 2003). Different raft domains might coexist and be present in distinct areas of the membranes, as for example caveolar (caveolin-containing) rafts and noncaveolar rafts. Functionally, membrane domain segregation provides a mechanism for the selection of molecular effectors into functional units for efficient signaling and sorting processes. Cellular events such as cell trafficking, cell migration, and phagocytosis require membrane domain segregation (Simons and Ikonen, 1997; Seveau et al., 2001; Manes et al., 2003), and key components of their molecular machineries have been isolated in DRMs.
In the present work, we addressed the role of membrane lipid domains in L. monocytogenes entry and focused our work on the study of the interactions between the bacterial invasion proteins internalin and InlB, and their receptors E-cadherin and HGF-R. Whether E-cadherin and the HGF-R requires lipid assemblies to function properly was previously unknown. Our work provides the first demonstration that both E-cadherin and HGF-R require lipid domains to mediate L. monocytogenes entry. Interestingly, depending on the receptor involved, lipid domains are important for different steps of the internalization process.
Results
Listeria monocytogenes entry into mammalian cells requires plasma membrane cholesterol
We investigated if cholesterol-enriched membrane microdomains could be implicated in L. monocytogenes entry into nonphagocytic mammalian cells. We used methyl-β-cyclodextrin (MβCD), a water-soluble cyclic oligosaccharide (Kilsdonk et al., 1995) to deplete selectively membrane cholesterol and disrupt lipid domains in the fibroblastic cell line L2071hEcad, and in the Vero epithelial cell line (two cell lines that we routinely use in our invasion studies). The L2071hEcad cells express E-cadherin and HGF-R and consequently allow internalin- and InlB-mediated entry. The Vero cell line expresses HGF-R but not E-cadherin limiting entry to InlB-mediated mechanism. Cells were depleted with 10 mM MβCD for 1 h (L2071hEcad) or for 30 min (Vero cells) and were assayed for bacterial entry by the gentamicin assay. As shown in Fig. 1, for wild-type L. monocytogenes (EGD strain), internalin- and InlB-mediated entry into L2071hEcad cells and InlB-mediated entry into Vero cells were strongly inhibited after cholesterol depletion. After cholesterol depletion cellular viability, permeability to gentamicin, and cellular adherence were not affected (unpublished data). The use of lower concentration of MβCD (5 mM) also decreased bacterial entry by 50% in both cell lines (unpublished data). To further demonstrate that inhibition of bacterial entry was specifically due to cholesterol depletion and not to a secondary effect of the MβCD, cells were first depleted with MβCD and cholesterol was then added back to the membrane with cholesterol-chelated MβCD. As shown in Fig. 1, cholesterol repletion restored bacterial uptake.
Figure 1.

Cholesterol-dependent entry of L. monocytogenes in mammalian cells. Control, cholesterol-depleted, and cholesterol-repleted L2071hEcad or Vero cells were incubated at 37°C for 30 min (L2071hEcad) or 1 h (Vero) with bacteria at a multiplicity of infection = 50. The number of intracellular bacteria (EGD strain) per cell was quantified by the gentamicin assay and results were expressed relative to control nondepleted cells (mean of at least three independent experiments).
Internalin- and InlB-mediated entry require plasma membrane cholesterol independently of the presence of the bacterial toxin LLO
To analyze further the effect of cholesterol depletion on internalin- and InlB-dependent uptake, we used two isogenic mutant strains, EGDΔInlB and EGDΔInlA that only express internalin or InlB, respectively. As shown in Fig. 2 A, internalin- and InlB-mediated entry into L2071hEcad cells were cholesterol dependent.
Figure 2.

Internalin- and InlB-mediated entry require membrane cholesterol independently of the presence of the cholesterol-binding toxin LLO. (A) Control (−) and cholesterol-depleted (+) L2071hEcad or Vero cells were incubated at 37°C with the EGD isogenic mutant strains (EGDΔInlB, EGDΔInlA, or EGDhly::Tn917) for 30 min (L2071hEcad) or 1 h (Vero). The number of intracellular bacteria (EGD strain) per cell was quantified by the gentamicin assay and results were expressed relative to control nondepleted cells (mean of at least three independent experiments). (B) Control (−) and cholesterol-depleted (+) cells were incubated for 30 min at 37°C with InlB- or internalin-coated latex beads. After three washes, cells were fixed and extracellular beads were fluorescently labeled. The percent of intracellular beads per cell was quantified by microscopy. Results of at least three independent experiments were expressed relative to control nondepleted cells.
The secreted LLO, a major virulence factor, is known to bind to plasma membrane cholesterol and trigger several signaling events (Coconnier et al., 2000; Dramsi and Cossart, 2003). Cholesterol-dependent entry of L. monocytogenes could thus be attributed to a decreased binding of LLO to the plasma membrane after MβCD treatment. The use of a L. monocytogenes mutant defective in LLO expression (EGDhly::Tn917) demonstrated that, independently of the presence of the toxin LLO, L. monocytogenes entry into L2071hEcad and Vero cells required membrane cholesterol (Fig. 2 A). Furthermore, entry of the bacterial species L. innocua expressing internalin or InlB, but not LLO was also cholesterol dependent (not depicted).
To definitively establish if the specific interactions of internalin or InlB with their respective receptors required plasma membrane cholesterol, we used latex beads coated with the purified recombinant bacterial invasion proteins. As shown previously (Lecuit et al., 1997; Braun et al., 1998), internalin- and InlB-coated beads are able to induce their specific entry into mammalian cell lines expressing their receptors. As shown in Fig. 2 B, in the absence of any other bacterial factors, internalin- and InlB-mediated uptake also required plasma membrane cholesterol.
Recruitment of raft markers at the internalin and InlB entry sites
To further demonstrate the implication of lipid domains in L. monocytogenes entry, we performed colocalization studies of raft and nonraft markers with bacteria or beads bound to the cell surface. For this purpose, cells were transiently transfected with the Aequorea GFP fused to GPI moiety (GFP-GPI), or with the cyan variant of Aequorea GFP fused to short peptide sequences which are the site for posttranslational modifications: myristoylation and palmitoylation (CFP-MyrPalm); and geranylgeranylation (CFP-GerGer). These chimeras are preferentially targeted to lipid domains at the outer leaflet (GFP-GPI) or at the inner leaflet (CFP-MyrPalm), whereas CFP-GerGer is present at the inner leaflet of the plasma membrane, does not display any preferential association with lipid domains, and was used as a negative control (Zacharias et al., 2002). In addition, we performed ganglioside 1 (GM1) labeling by using the Alexa Fluor 488–conjugated B subunit of cholera toxin after cellular fixation (see Materials and methods). Prion protein (PrP) was also used as a raft marker (Naslavsky et al., 1997) and the transferrin receptor as a nonraft marker.
To study the internalin pathway, we infected Lovo epithelial cells with L. innocua, which expresses internalin. Lovo cells are permissive for both internalin- and InlB-mediated entry (Pizarro-Cerda et al., 2004), are more easily transfected, and more efficiently labeled by the B subunit of cholera toxin than the L2071hEcad cells. Our results show that L. innocua expressing internalin recruited at the plasma membrane the raft markers GM1 and CFP-MyrPalm, but not the nonraft marker CFP-GerGer (Fig. 3). The raft markers GFP-GPI, and PrP were also recruited at the bacterial entry site (Fig. 3).
Figure 3.

Recruitment of raft markers during internalin-dependent entry. Non-transfected Lovo cells (A), Lovo cells transfected with CFP-MyrPalm (B), with CFP-GerGer (C), or with GFP-GPI (D), and epithelial Rov9 cells stably expressing the PrP (E), were incubated with L. innocua expressing internalin (BUG 1489) for 10 min, washed and fixed. The images B–D represent the CFP or GFP fluorescence, and the images A′, B′, C′, D′′, and E′′ correspond to phase contrast images. GM1 was labeled with fluorescent B subunit of cholera toxin (A). F-actin was labeled with Alexa Fluor 546 (D′). PrP (E) and internalin (E′) were labeled with mAbs (clone L7.7 and clone 4F2) and secondary fluorescent antibodies. Note that L. innocua expressing internalin, in some cases, appears as chains. Bars, 10 μm.
To study the InlB pathway, we infected Vero epithelial cells with L. monocytogenes overexpressing an InlB protein covalently anchored at the bacterial surface or incubated the cells with InlB-coated beads. As presented in Fig. 4 A, L. monocytogenes corecruited F-actin and the lipid raft marker GM1. The CFP-MyrPalm, but not the CFP-GerGer, was recruited at the bacterial entry site. We repeated these experiments with InlB-coated beads, and as shown in Fig. 4 B, GM1 and HGF-R were corecruited, whereas the transferrin receptor was not. HGF-R colocalized with the CFP-MyrPalm but not with CFP-GerGer, and finally the GFP-GPI chimera was corecruited with the HGF-R and F-actin. These results clearly showed that lipid raft markers are recruited at both internalin and InlB entry site.
Figure 4.
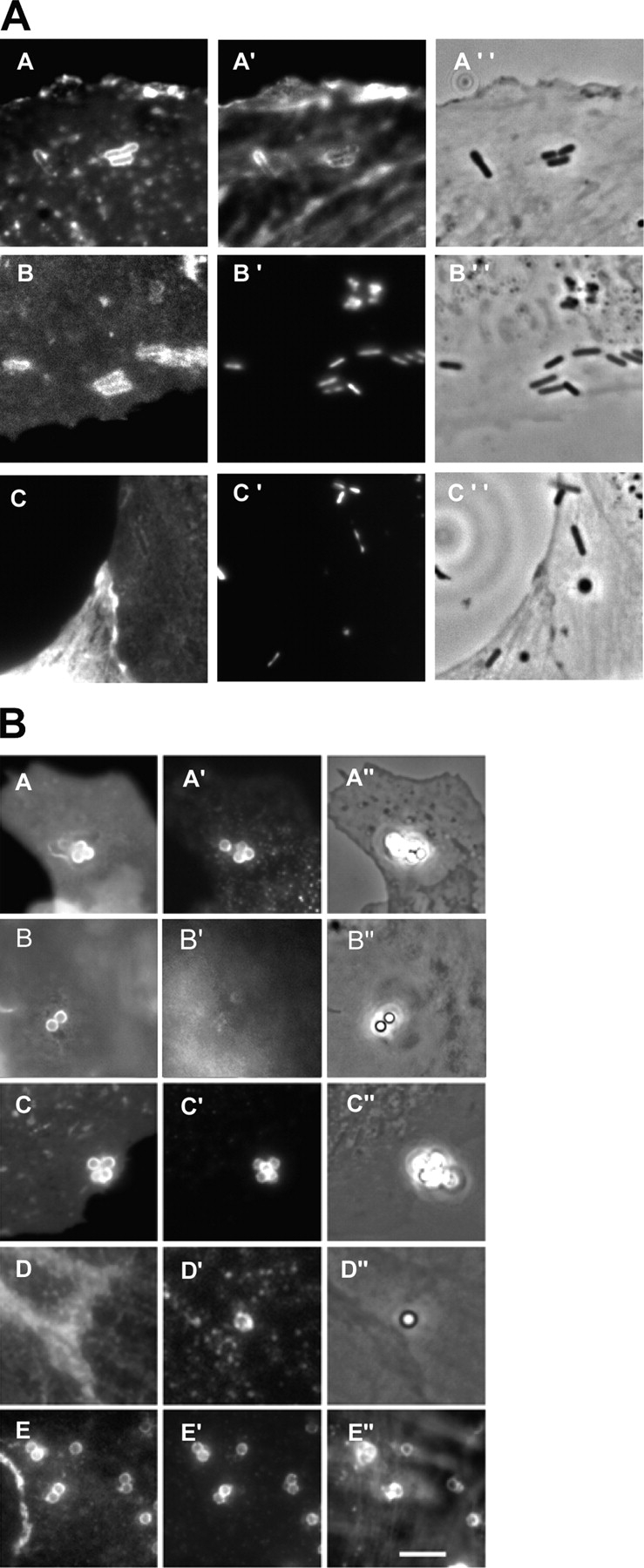
Recruitment of raft markers during InlB-dependent entry. (A) Non-transfected Vero cells (A) and Vero cells transfected with CFP-MyrPalm (B) or with CFP-GerGer (C), were incubated with L. monocytogenes (BUG 1641) for 10 min, washed, and fixed. GM1 was labeled with the fluorescent B subunit of cholera toxin (A) and F-actin was labeled with Alexa Fluor 647 (A′). The images B and C represent the CFP fluorescence, and A′′, B′′, and C′′ correspond to the phase contrast images. Bacteria were labeled with DAPI (B′ and C′). (B) Non-transfected Vero cells (A and B), Vero cells transfected with CFP-MyrPalm (C), with CFP-GerGer (D), or with GFP-GPI (E), were incubated with InlB-coated beads for 10 min, washed, and fixed. Cells were then labeled for GM1 (A and B), for HGF-R (A′, C′, D′, and E′), for transferrin receptor (B′), and for F-actin (E′′). A′′, B′′, C′′, and D′′ correspond to phase contrast images. Bar, 10 μm.
Cholesterol depletion has a different effect on internalin–E-cadherin and InlB–HGF-R interactions
We analyzed which steps (adherence and/or internalization) of internalin–E-cadherin- and InlB–HGF-R–mediated uptake were affected by cholesterol depletion. To study the internalin- and InlB-mediated entry pathways independently and specifically, control and cholesterol-depleted cells were incubated with internalin- or InlB-coated beads for 30 min and then fixed. The total number of beads associated with the cells (adherent extracellular beads plus intracellular beads) was quantified by phase-contrast microscopy and the number of extracellular and intracellular beads was assessed by fluorescence microscopy (see Materials and methods). As seen in Fig. 5, after cholesterol depletion the total number of internalin-coated beads associated with L2071hEcad cells decreased, whereas the total number of InlB-coated beads associated with L2071hEcad or with Vero cells was not significantly affected. The number of extracellular internalin-coated beads interacting with L2071hEcad cells was reduced after cholesterol depletion, whereas cholesterol depletion of Vero cells had no effect on InlB-coated bead adherence. In conclusion, initial internalin-mediated attachment of the beads to the cell surface was strongly affected after cholesterol depletion. In contrast, InlB-mediated attachment at the cell surface did not significantly decrease after cholesterol depletion, but downstream events necessary for the phagocytic uptake were abrogated.
Figure 5.

Cholesterol depletion affects differently the internalin–E-cadherin and the InlB–HGF-R interactions. Control (−) and cholesterol-depleted (+) cells were incubated for 30 min at 37°C with internalin- or with InlB-coated beads. After three washes cells were fixed and extracellular beads were immunolabeled. The total number of beads (extracellular plus intracellular) associated with the cells, and the number of extracellular beads associated to the cell surface, were assessed by microscopy. Results of at least three independent experiments were expressed relative to control nondepleted cells.
Cholesterol depletion affects internalin–E-cadherin interaction and E-cadherin recruitment at the entry site
To explain the decreased adherence of internalin-coated beads to L2071hEcad cells, we investigated if cholesterol depletion affected the amount of E-cadherin molecules present at the plasma membrane. By fluorescence microscopy, we quantified the level of E-cadherin expression at the plasma membrane (see Materials and methods). As shown in Fig. 6 A, similar amounts of E-cadherin were present at the surface of control and cholesterol-depleted L2071hEcad cells. This result demonstrated that the diminution in internalin-coated bead adherence to the plasma membrane was not due to a lower expression of E-cadherin but rather to a decrease in receptor affinity or avidity. We then assessed the ability of internalin-coated beads to recruit E-cadherin. Control and cholesterol-depleted cells were incubated for 30 min with internalin-coated beads, cells were then fixed and E-cadherin was labeled with fluorescent antibodies. As shown in control cells (Fig. 6 B), E-cadherin molecules were massively recruited at the entry site forming a fluorescent ring around the beads, whereas after cholesterol depletion E-cadherin was poorly recruited. We confirmed this result by showing that the percentage of beads associated to the cell surface and surrounded by a ring of fluorescent E-cadherin, severely decreased in cholesterol depleted cells (Fig. 6 C). In conclusion, cholesterol depletion affected the recruitment of E-cadherin at the cell surface. This result prompted us to analyze if E-cadherin had to be present in DRMs to promote internalin-dependent entry.
Figure 6.

E-cadherin recruitment at the entry site is cholesterol dependent. (A) Control and cholesterol-depleted L2071hEcad cells were fixed and immunofluorescently labeled for E-cadherin (clone HECD1; Alexa Fluor 546–conjugated secondary antibodies). Images were acquired with a 40× objective, and were processed for quantitative analysis using the Metamorph software. Results of three independent experiments were expressed relative to control cells. (B and C) Control and cholesterol-depleted L2071hEcad cells were incubated for 30 min with internalin-coated beads. Cells were washed three times, fixed, permeabilized, and labeled for E-cadherin. (B) Phase-contrast and fluorescent images were acquired with a 100× objective. Two different plans acquired at different focus points were shown for the fluorescent images. (C) Quantitative analysis of the percentage of internalin-coated beads which recruited E-cadherin. Bars, 10 μm. Arrows indicate the internalin-coated beads, which recruited E-cadherin.
E-cadherin is present in DRMs
We performed cold detergent extraction of the L2071hEcad cells, using the nonionic detergent Brij 58. Cell lysates were fractionated according to density criteria by ultracentrifugation on sucrose gradients. Fractions were collected and subjected to Western blotting analysis. We used caveolin-2 as a control of our fractionation because it is well known that caveolin-2 is present within the low density DRM fractions (Mora et al., 1999). Interestingly, a significant proportion of E-cadherin molecules was present in DRMs suggesting an association of E-cadherin with lipid microdomains (Fig. 7 A, left). To further demonstrate this result, cholesterol was depleted before cell lysis. After cholesterol depletion E-cadherin molecules were not present in the DRMs confirming that a significant proportion of E-cadherin molecules was present in DRMs in a cholesterol-dependent manner. α-Catenin, the cytoskeletal molecule which links E-cadherin to the F-actin cytoskeleton was, similarly to E-cadherin, present in DRMs. We also performed cold Triton X-100 extraction of L2071hEcad cells (Fig. 7 B) and of Lovo cells (Fig. 7 C) and found that E-cadherin, as well as HGF-R, were present in the low density DRM fraction. Cells were then incubated with soluble internalin to determine if internalin would associate preferentially with the population of E-cadherin present in DRMs. Internalin was exclusively present to the low density fractions (Fig. 7 A, right). In contrast, after cholesterol depletion, internalin was associated to higher density fractions in which E-cadherin was present. These results show that internalin interacts preferentially with E-cadherin present within DRMs and that incubation of cells with soluble internalin does not affect the amount of E-cadherin present in the DRM fractions.
Figure 7.

E-cadherin and HGF-R are present in DRMs. (A) Control and cholesterol-depleted L2071hEcad cells, preincubated or not with soluble internalin (InlA), were extracted with 0.5% Brij58. (B) L2071hEcad cells were extracted with 0.1% Triton X-100. (C) Lovo cells were extracted with 0.5% Triton X-100. After detergent extraction, cell lysates were subjected to ultracentrifugation on sucrose gradients. Fractions were collected from top to bottom and were processed for Western blotting analysis. Fractions 2–4/low density fractions (LD), and fractions 9 and 10/high density fractions (HD).
Cholesterol depletion does not affect HGF-R recruitment and initial membrane extensions at the entry site
It has been shown that InlB-coated beads or bacteria induce HGF-R recruitment at the entry site, tyrosine phosphorylation of HGF-R, and the activation of signaling cascades leading to F-actin polymerization (Bierne and Cossart, 2002). We first analyzed the ability of InlB-coated beads to trigger HGF-R recruitment after cholesterol depletion. Control and cholesterol-depleted Vero cells were incubated for 30 min with InlB-coated beads, fixed, permeabilized, and labeled for HGF-R. As shown in Fig. 8 A, HGF-R recruitment around the InlB-coated beads could be observed for control as well as for cholesterol-depleted cells. We quantified this result by calculating the percentage of InlB-coated beads which had recruited detectable amounts of HGF-R. No significant difference between control and cholesterol-depleted cells was observed (Fig. 8 B).
Figure 8.

Cholesterol depletion does not affect HGF-R recruitment or initial membrane recruitment at the InlB entry site but subsequent closure of the phagocytic cup. (A) Control and cholesterol-depleted Vero cells were incubated for 30 min with InlB-coated beads. Cells were washed three times, fixed, permeabilized, and labeled for HGF-R (clone DL21; Alexa Fluor 546–conjugated secondary antibodies) and for F-actin (Alexa Fluor 488 phalloidin). Phase-contrast and fluorescent images were acquired with a 100× objective. Arrows indicate the InlB-coated beads that recruited HGF-R. Arrowheads indicate the InlB-coated beads that recruited HGF-R and F-actin. Bar, 10 μm. (B) Percentage of InlB-coated beads which have recruited HGF-R. (C) Control and cholesterol-depleted Vero cells were incubated for 5, 15, and 30 min with InlB-coated beads. Cells were washed three times and processed for scanning EM as described in Materials and methods. Extracellular portion of the beads were colorized using the Adobe photoshop software. Bar, 1 μm.
We then examined, by scanning EM, the membrane rearrangements during phagocytic uptake. As shown in Fig. 8 C, initial membrane attachment to the beads was not inhibited in depleted cells and beads were surrounded by adherent membranes. At later time points, membrane extension and closure were impaired, suggesting that cholesterol depletion affects late stage of the uptake process.
HGF-R phosphorylation and PI 3 kinase recruitment are not affected by cholesterol depletion
Some of the InlB- and HGF-signaling events occurring downstream from the initial interaction with HGF-R have been identified, including HGF-R tyrosine phosphorylation, recruitment and activation of Gab1, Cbl and Shc, and formation of signaling complexes containing these adapters and the p85 subunit of PI 3-kinase (Ireton et al., 1999). We investigated whether InlB could still induce HGF-R tyrosine phosphorylation and PI 3-kinase recruitment after cholesterol depletion. Vero cells incubated at 37°C for 1 min with soluble InlB (8 nM) were lysed and cell lysates were immunoprecipitated with anti–HGF-R or with anti-phosphotyrosine antibodies as described previously (Ireton et al., 1999). As shown in Fig. 9, MβCD treatment did not induce HGF-R phosphorylation or p85α recruitment in Vero cells. This control was important as it has been reported that MβCD treatment could induce by itself PI 3-Kinase activation and the phosphorylation of the epidermal growth factor (Chen and Resh, 2002). As previously reported, in control cells (not incubated with MβCD), soluble InlB induced HGF-R tyrosine phosphorylation and coimmunoprecipitation of the p85α subunit of PI 3-kinase with phosphotyrosine proteins (Ireton et al., 1999). After cholesterol depletion, soluble InlB and HGF still induced tyrosine phosphorylation of HGF-R (Fig. 9 A) as well as the coimmunoprecipitation of the p85α subunit of PI 3-kinase with phosphotyrosine proteins (Fig. 9 B).
Figure 9.

Cholesterol depletion does not affect HGF-R phosphorylation and association of PI 3-kinase with phosphotyrosine proteins. (A) Control (−) and cholesterol-depleted (+) cells were stimulated or not (NS) with soluble InlB (8 nM) for 1 min at 37°C. Cells were lysed and lysates were immunoprecipitated with anti–HGF-R antibodies. Immunoprecipitated proteins were analyzed by Western blotting with anti-phosphotyrosine and anti–HGF-R. (B) Control (−) and cholesterol-depleted (+) cells were stimulated or not (NS) with soluble InlB (8 nM) or HGF (0.6 nM) for 1 min at 37°C. Cells were lysed and lysates were immunoprecipitated with anti-phosphotyrosine antibodies. Total cell lysates and immunoprecipitated proteins were analyzed by Western blotting with anti–p85-α antibodies.
Cholesterol depletion impairs actin polymerization and membrane ruffles
We then analyzed if cholesterol depletion could affect the formation of actin-rich phagocytic cups at the entry site. As shown in Fig. 8 A, in contrast to control cells, after cholesterol depletion F-actin cups were rarely visualized around the InlB-coated beads which recruited HGF-R. Furthermore, cholesterol depletion partially impaired the formation of F-actin-rich membrane ruffles normally induced by soluble InlB (8 nM) or HGF (0.6 nM; Fig. 10).
Figure 10.
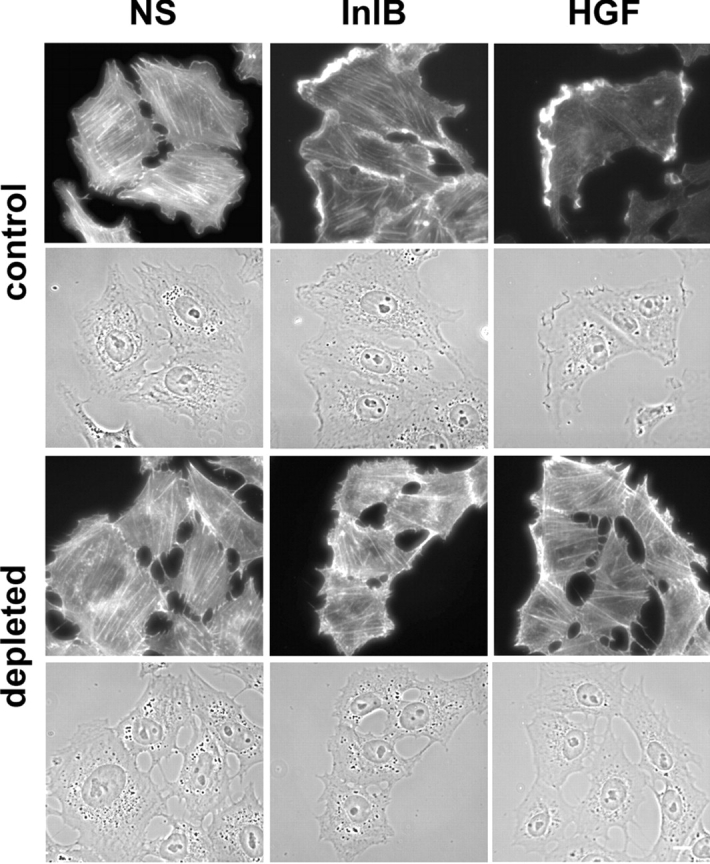
Cholesterol depletion abrogates partially the formation of actin-rich membrane ruffles. Control and cholesterol-depleted Vero cells were stimulated or not (NS) for 2 min with soluble InlB or HGF. Cells were then fixed and labeled for F-actin (Alexa Fluor 488 phalloidin). Phase contrast and fluorescent images were acquired with a 40× objective. Bar, 10 μm.
Together, the results indicate that initial steps of InlB-mediated entry, i.e., HGF-R recruitment, and initial membrane recruitment were not affected by cholesterol depletion, but that the downstream events leading to the massive F-actin polymerization and cup closure were abrogated.
Discussion
In the present work, we have demonstrated structural and functional implications of membrane microdomains during L. monocytogenes entry into nonphagocytic epithelial and fibroblastic cells. As the two bacterial factors involved in entry and their cognate host receptors are known and well documented, L. monocytogenes provides an exceptional model to address the role of lipid domains during bacterial entry. We have shown that internalin–E-cadherin– and InlB–HGF-R–mediated entry processes both required lipid domain integrity, although at different stages of the entry process. Furthermore, aside from having important role in bacterial entry, E-cadherin and HGF-R are implicated in various cellular processes such as tissue morphogenesis, cell migration, and metastasis. It was previously not known if these molecules were interacting with raft domains. Thus, our work also documents the role of membrane domains in their respective functions.
E-cadherin present within DRMs acts as a receptor for the invasion protein internalin
We conducted functional and biochemical studies to address the role of lipid rafts in internalin-mediated entry. We showed that internalin-dependent adherence and internalin-dependent recruitment of E-cadherin at the entry site were inhibited after cholesterol depletion by MβCD. E-cadherin, α-catenin- and E-cadherin–associated internalin were present in the low density DRMs in a cholesterol-dependent manner. In addition, lipid raft markers are recruited at internalin-dependent entry site. Altogether, our results indicate that E-cadherin engagement into lipid domains is critical for internalin-dependent entry of L. monocytogenes. We favor the hypothesis that lipid rafts are important for the initial clustering of E-cadherin molecules during multivalent binding to internalin present on bacteria or beads. This conclusion is based on several observations: (a) for control and MβCD-treated cells, the same amount of E-cadherin is present at the plasma membrane (Fig. 6); (b) soluble internalin associates with E-cadherin present in DRM fractions (Fig. 7); and (c) the internalin-coated beads failed to recruit E-cadherin at the plasma membrane of cholesterol-depleted cells.
It has been reported that in polarized epithelial cells, adherens junctions were not disassembled after cholesterol depletion whereas tight junctions were disrupted (Nusrat et al., 2000). Thus, cholesterol would be important for tight junction but not for adherens junction maintenance. An alternative possibility is that both junctions are raft dependent but that in adherens junction, cholesterol is protected from cholesterol extraction. We favor the hypothesis that similarities exist between adherens junction formation and internalin-mediated phagocytosis and that lipid rafts would be necessary for adherens junction establishment.
HGF-R signaling pathway involves membrane cholesterol
In contrast to what was observed for the E-cadherin–internalin interactions, cholesterol depletion did not affect InlB binding to HGF-R nor HGF-R recruitment at the plasma membrane. However, downstream signaling events required membrane cholesterol because InlB-induced membrane ruffles and actin-rich phagocytic cups were not observed after cholesterol depletion. HGF, as observed with soluble InlB, could not either induce the formation of actin-rich membrane ruffles after cholesterol depletion. Some events involved in the InlB- and HGF-mediated signaling have been described previously (Ireton et al., 1999; Shen et al., 2000; Bierne et al., 2001), among which tyrosine phosphorylation of the HGF-R, activation of PI 3-kinase, production of phosphoinositides, and Rac activation. In this work, we report that after cholesterol depletion, tyrosine phosphorylation of the HGF-R as well as PI 3-kinase recruitment to phosphotyrosine proteins were still occurring, indicating that the signaling cascade was interrupted further downstream, in agreement with our finding that HGF-R is present in DRMs. It remains to determine which of the downstream events of InlB- and HGF-mediated signaling pathways, between PI 3-kinase recruitment and F-actin polymerization, is cholesterol dependent.
Cross-talk between HGF-R and E-cadherin
Several reports point to a cross-talk between HGF-R and E-cadherin. The activation of epithelial cells by HGF results in the disruption of E-cadherin–dependent adherens junctions and in the coendocytosis of E-cadherin and HGF-R (Kamei et al., 1999). HGF stimulation induces MAPK activation and phosphorylation of β-catenin (Liou et al., 2002). In the case of L. monocytogenes, depending on the target cell, the bacterium exploits HGF-R and E-cadherin independently or in conjunction, to trigger its entry. In Lovo cells (Pizarro-Cerda et al., 2004) and in L2071hEcad (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200406078/DC1), E-cadherin and HGF-R can be corecruited at the phagosomal membrane bacterial entry site. However, the molecular basis of the possible synergy between the two receptors during L. monocytogenes entry has not been elucidated. Our work shows that lipid domain integrity is a common requirement for both receptors to allow bacterial uptake. Altogether, these observations lead to the hypothesis that upon InlB or HGF stimulation, both receptors could be localized into lipid rafts and be cointernalized through these microdomains. Whether this coendocytosis has a synergistic effect on bacterial entry has still to be demonstrated.
Exploitation of lipid rafts in bacterial entry
Over the past years lipid rafts have been reported as implicated in the entry of various bacterial pathogens into professional and nonprofessional phagocytic cells. However, the integrity of lipid domains is not necessarily required for all phagocytic events. Raft integrity is required for nonopsonic uptake of mycobacteria and Brucella by macrophages, whereas after opsonization by antibodies, these pathogens could still infect macrophages despite cholesterol depletion (Peyron et al., 2000). To our knowledge, the only example of cholesterol-independent entry into nonphagocytic cells is that of Yersinia via the invasion protein invasin, which binds to β1 integrins (Lafont et al., 2002). These results indicate that depleting plasma membrane cholesterol has no general effect and does not always inhibit receptor activity, signaling events, or cytoskeletal rearrangements.
Interestingly, it was proposed that entry via raft microdomains determines the fate of some intracellular pathogens. For example, lipid raft-mediated entry of Brucella and FimH-expressing E. coli, prevents phago–lysosomal fusion and permits intracellular survival of the pathogens (Gatfield and Pieters, 2000; Shin et al., 2000; Naroeni and Porte, 2002). Our present work demonstrates that in the case of L. monocytogenes, entry occurs in raft containing microdomains. Whether this determines the fate of the bacteria is unknown. Quite strikingly, the secreted bacterial toxin, LLO, which allows escape from the phagocytic vacuole and also induces signaling pathways, binds to cholesterol in mammalian membranes. It will be of particular interest to study the spatiotemporal interactions of LLO with plasma membrane during L. monocytogenes internalization, and the possible synergy between LLO and bacterial invasion proteins internalin and InlB.
Materials and methods
Reagents and antibodies
Alexa Fluor 488 cholera toxin subunit B, Alexa Fluor 488–, 546–, and 647–conjugated phalloidin, Alexa Fluor 488– and 546–conjugated goat anti–rabbit and goat anti–mouse antibodies were purchased from Molecular Probes. mAbs anti–E-cadherin (clone HECD-1) was purchased from R&D systems, anti–α-catenin (clone 5) was purchased from Transduction Laboratories. Mouse monoclonal anti-internalin and anti-InlB have been described previously (Braun et al., 1999; Mengaud et al., 1996a). Mouse mAbs anti–human Met (clone DL-21) and anti-phosphotyrosine (clone 4G10) were purchased from Upstate Biotechnology. Rabbit antisera against human Met (C-28) was purchased from Santa Cruz Biotechnology, Inc., against caveolin-2 was purchased from BD Transduction Laboratories, against PI 3-kinase p85α subunit was purchased from Upstates Biotechnology, and against human transferrin receptor (H-300) was purchased from Santa Cruz Biotechnology, Inc. Mouse monoclonal anti-Prp, clone 4F2 (Krasemann et al., 1996), was provided by D. Vilette and J. Grassi (Institut National de la Recherche Agronomique [INRA], Jouy-en-Josas, France). Surfact-Amps58 (Brij 58) and Triton X-100, 10% solutions, were purchased from Pierce Chemical Co. MβCD and cholesterol-water soluble were purchased from Sigma-Aldrich. 1-μm-diam latex beads were coated by J. Pizarro-Cerdá (Institut Pasteur), following the manufacturer's instructions (Molecular Probes), with purified InlB (Braun et al., 1997; Ireton et al., 1999) or internalin (Mengaud et al., 1996b). HGF and DAPI were purchased from Sigma-Aldrich.
Mammalian cells and bacterial strains
African green monkey kidney epithelial Vero cells (American Type Culture Collection; CCL-81), L2071 mouse fibroblasts (American Type Culture Collection; CCL1.1), were cultured in DME glutamax (GIBCO BRL) supplemented with 10% FBS. L2071 cells transfected with the human E-cadherin cDNA (L2071-hEcad; Lecuit et al., 2000) were cultured in the presence of 800 μg/ml of G418 (GIBCO BRL). Human epithelial Lovo cells (American Type Culture Collection; CCL-229) were cultured with F-12K nutrient mixture (GIBCO BRL) supplemented with 10% FBS and 2 mM glutamine. Rabbit kidney epithelial Rov9 cells, stably expressing the PrP protein (Vilette et al., 2001) were cultured with MEM glutamax (GIBCO BRL) supplemented with 10% FBS.
Wild-type L. monocytogenes EGD (BUG600) and its isogenic deletion mutants, ΔinlA (BUG947) and ΔinlB (BUG1047; Dramsi et al., 1995), EGDhly::Tn917 mutant strain obtained by signature-tagged mutagenesis, L. monocytogenes variant EGDΔinlB (LRRs-IR-SPA) (BUG 1641; Bierne et al., 2001), and L. innocua transformed with pRB474 harboring the inlA gene (BUG 1489) were grown overnight at 37°C in brain–heart infusion agar (Difco), diluted 10 times in brain–heart infusion, and cultured until OD600nm = 0.8. Bacteria were washed in culture medium three times before assayed.
The DNA encoding monomeric CFP fused onto short peptides containing consensus sequences for acylation (MyrPalm-CFP) or prenylation (GerGer-CFP) in vector were provided by R.Y. Tsien (University of California, San Diego, La Jolla, CA) and have been described in Zacharias et al. (2002). The GPI moiety of the folate receptor cloned as a GFP-GPI chimera in pJB20 vector was provided by C. Zurzolo and S. Paladino (Institut Pasteur). Cells were transfected with Lipofectin (GIBCO BRL) following the manufacturer's instructions.
Cholesterol depletion and repletion
For cholesterol depletion, cells were washed twice with DME (without serum) and incubated at 37°C for 1 h (L2071hEcad) or 30 min (Vero cells) with MβCD 10 mM in DME. Cells were then washed twice and assayed as described. For cholesterol repletion, L2071hEcad and Vero cells were incubated with a solution of 2.5 mM and 5 mM cholesterol-MβCD for 2 h and 15 min, respectively, then washed and assayed as described.
Gentamicin survival assay
Gentamicin assays were performed in 24-well tissue culture plates (Costar), three wells for each condition. Cells (±105 cell/well) were washed twice with DME without antibiotic, and bacterial suspensions were added to mammalian cells at multiplicity of infection of 50. Cells were incubated with the bacterial suspension at 37°C for 30 min (L2071hEcad) or 1 h (Vero), then washed three times with DME, and overlaid with DME containing gentamicin (10 μg/ml) for 1 h at 37°C. Cells were washed three times in DME and were lysed by adding 0.2% Triton X-100 in PBS. The number of viable bacteria released from the cells was assessed by titration on agar plates. The results were expressed as the number of intracellular bacteria per cell relative to control nondepleted cells. Results are the mean value of at least three independent experiments.
Internalin- and InlB-coated beads adhesion and entry assay
Cells were plated onto glass coverslips in 24-well tissue culture plates. Control and cholesterol-depleted cells were incubated 30 min with internalin- or InlB-coated latex beads at 37°C. Cells were washed three times with DME and fixed with paraformaldehyde solution (3.5% in PBS) for 30 min. Cells were then washed twice with a 0.1 M glycine in PBS solution and incubated in blocking solution (PBS; 10% BSA) at RT for 30 min. Extracellular beads were labeled with anti-internalin or anti-InlB antibodies and with Alexa Fluor 546–conjugated goat anti–mouse secondary antibodies. Total number of beads associated with cells (extracellular plus intracellular beads) was determined by phase-contrast microscopy. The number of extracellular beads was quantified by fluorescence microscopy. Results were the mean of at least three independent experiments and were expressed relative to control nondepleted cells.
Immunolabeling, wide-field microscopy, and fluorescence quantification of E-cadherin expression
For immunolabeling, cells were fixed with a PFA solution (3.5% in PBS) for 30 min, then permeabilized (0.2% Triton X-100 for 5 min in PBS) or not. Cells were then washed twice with a 0.1 M glycine in PBS solution and incubated in blocking solution (PBS; 10% BSA) at RT for 30 min. Antibodies were incubated for 30 min at RT in blocking solution.
For GM1 labeling, cells were fixed with cold PFA for 30 min and labeled with a 40-μg/ml cold solution of Alexa Fluor 488 cholera toxin subunit B for 30 min.
Images were acquired on a fluorescence inverted microscope (Axiovert 135; Carl Zeiss MicroImaging, Inc.) equipped with a cooled charge-coupled device camera (MicroMax 5 MHz; Princetown Instruments) driven by Metamorph Imaging System software (Universal Imaging Corp). For quantification of fluorescence, images were acquired under the same condition (40× oil immersion objective, acquisition time, and microscope setting), were background corrected, and a mask was applied to consider only the fluorescence associated with cells. Fluorescence intensity per cell was calculated by the ratio of the total fluorescence intensity per field over the cell number within the field. The results were expressed relative to control cells. Three independent experiments were performed, ∼500 cells were analyzed for each experiment.
Scanning EM
For scanning EM analysis, cells were washed in PBS and fixed in 2.5% (vol/vol) glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2, overnight at 4°C. Cells were washed three times for 5 min (each time) in 0.2 M cacodylate buffer, pH 7.2, fixed after for 1 h in 1% (wt/vol) osmium tetroxide in 0.2 M cacodylate buffer, pH 7.2, and then rinsed with distilled water. Cells were dehydrated through a graded series of 25, 50, 75, and 95% ethanol solutions for 10 min (each time) and washed two times for 10 min (each time) in 100% ethanol. Dehydrated cells were consecutively immersed in 25, 50, and 75% (vol/vol) hexamethyldisilazane in ethanol for 5 min (each time), immersed twice in 100% hexamethyldisilazane for 5 min (each time), and quickly air dried. Coverslips were sputter coated twice with carbon with a BALTEC MED010 evaporator. Samples were examined and photographed with a JEOL JSM 6700F field emission scanning electron microscope operating at 5 Kv.
Phosphotyrosine and HGF-R immunoprecipitations
Vero cells were cultured in 75-cm2 tissue culture flasks (∼106 cell/flask), serum starved for 5 h, then depleted or not for 30 min with 10 mM MβCD. After two washes, cells were stimulated with 8 nM soluble InlB or with 0.6 nM HGF for 1 min at 37°C. Cell were washed with cold PBS and solubilized by 1 ml of ice-cold immunoprecipitation buffer (1% NP-40, 50 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 3 mM sodium orthovanadate, 1 mM PMSF, and 10 μg/ml aprotinin, leupeptin, pepstatin, and chymostatin) according to Ireton et al. (1999). Cell lysates were precleared with protein A–Sepharose beads for 1 h at 4°C. After protein quantification, cell lysates were immunoprecipitated with 1 μg anti-phosphotyrosine (clone 4G10) or anti HGF-R antibodies (C-28) overnight. Protein A Sepharose was added for 2 h. After washes by centrifugation, samples were boiled for 2 min in buffer containing 50 mM Tris-HCl, pH 7.5, 0.5% SDS, and 5 mM DTT. Protein A–Sepharose beads were then removed by centrifugation. Samples were analyzed by SDS-PAGE and by Western blotting for PI 3-kinase p85α subunit, phosphotyrosine or HGF-R.
Equilibrium density gradient centrifugation
Cells were cultured on 60-mm Petri dishes (Costar), treated as described were washed twice with cold TKM buffer (50 mM Tris-HCl, ph 7.4, 25 mM KCl, 5 mM MgCl2, and 1 mM EGTA), then were lysed with 500 μl TKM buffer containing, Brij58 (0.5%) or Triton X-100 (0.5% or 0.1%) and protease inhibitor cocktail (1:50), for 30 min on ice according to Ilangumaran (Ilangumaran et al., 1998). Aliquots of cell lysates were used to assess protein concentration. Cell lysates were mixed with an equal volume of 80% sucrose containing 0.5% Brij58, or 0.5% Triton X-100. Centrifuge tubes (Beckman Coulter) were filled successively with 1 ml of 80% sucrose in TKM, 1 ml of 40% sucrose containing the cell lysate, 6 ml of 36% sucrose, and 3.5 ml of 5% sucrose. The gradients were centrifuged in an ultracentrifuge (rotor SW41; Beckman Coulter) at 4°C for 18 h at 38,000 rpm. Fractions (1 ml) were collected from top to bottom, and proteins were precipitated with TCA. Samples corresponding to each fraction were analyzed by SDS-PAGE and Western blotting, using antibodies against E-cadherin, caveolin-2, α-catenin, internalin, transferring receptor, and HGF-R/Met.
Online supplemental material
In Fig. S1, Lovo cells were incubated with L. monocytogenes (BUG 1641) for 30 min at 37°C, cells were then washed, fixed, and labeled for E-cadherin and HGF-R/Met. Images were acquired with a 100× objective. Bar, 10 μm. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200406078/DC1.
Acknowledgments
We thank Dr. Joel Swanson, Dr. Javier Pizarro-Cerdá, Dr. Juan Jose Martinez, and Pierre Mandin for critical reading of the manuscript. We thank Dr. H. Fsihi (Institut Pasteur) for providing the strain EGDhly::Tn917. We are grateful to Dr. R.Y. Tsien for providing the CFP-MyrPalm and the CFP-GerGer constructs, and to Dr. C. Zurzolo for her advices and for providing the GFP-GPI construct.
This work was supported by the Pasteur Institute, INSERM, Association pour la Recherche contre le Cancer (ARC), and INRA, and by a fellowship (to S. Seveau) from the ARC, Paris, France. P. Cossart is an international research scholar from the Howard Hughes Medical Institute.
Abbreviations used in this paper: DRM, detergent-resistant membrane; Ger Ger, geranylgeranylation; GM1, ganglioside 1; GPI, glycosylphosphatidylinositol; HGF, hepatocyte growth factor; HGF-R; HGF receptor; LLO, listeriolysin O; MβCD, methyl-β-cyclodextrin; MyrPalm, myristoylation and palmitoylation; Prp, Prion protein.
References
- Abrami, L., S. Liu, P. Cosson, S.H. Leppla, and F.G. van der Goot. 2003. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 160:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne, H., and P. Cossart. 2002. InlB, a surface protein of Listeria monocytogenes that behaves as an invasin and a growth factor. J. Cell Sci. 115:3357–3367. [DOI] [PubMed] [Google Scholar]
- Bierne, H., S. Dramsi, M.P. Gratacap, C. Randriamampita, G. Carpenter, B. Payrastre, and P. Cossart. 2000. The invasion protein InIB from Listeria monocytogenes activates PLC-gamma1 downstream from PI 3-kinase. Cell. Microbiol. 2:465–476. [DOI] [PubMed] [Google Scholar]
- Bierne, H., E. Gouin, P. Roux, P. Caroni, H.L. Yin, and P. Cossart. 2001. A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J. Cell Biol. 155:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, L., S. Dramsi, P. Dehoux, H. Bierne, G. Lindahl, and P. Cossart. 1997. InlB: an invasion protein of Listeria monocytogenes with a novel type of surface association. Mol. Microbiol. 25:285–294. [DOI] [PubMed] [Google Scholar]
- Braun, L., F. Nato, B. Payrastre, J.C. Mazie, and P. Cossart. 1999. The 213-amino-acid leucine-rich repeat region of the Listeria monocytogenes InlB protein is sufficient for entry into mammalian cells, stimulation of PI 3-kinase and membrane ruffling. Mol. Microbiol. 34:10–23. [DOI] [PubMed] [Google Scholar]
- Braun, L., H. Ohayon, and P. Cossart. 1998. The InIB protein of Listeria monocytogenes is sufficient to promote entry into mammalian cells. Mol. Microbiol. 27:1077–1087. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and E. London. 1998. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 164:103–114. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and J.K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 68:533–544. [DOI] [PubMed] [Google Scholar]
- Camilli, A., H. Goldfine, and D.A. Portnoy. 1991. Listeria monocytogenes mutants lacking phosphatidylinositol-specific phospholipase C are avirulent. J. Exp. Med. 173:751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X., and M.D. Resh. 2002. Cholesterol depletion from the plasma membrane triggers ligand-independent activation of the epidermal growth factor receptor. J. Biol. Chem. 277:49631–49637. [DOI] [PubMed] [Google Scholar]
- Coconnier, M.H., M. Lorrot, A. Barbat, C. Laboisse, and A.L. Servin. 2000. Listeriolysin O-induced stimulation of mucin exocytosis in polarized intestinal mucin-secreting cells: evidence for toxin recognition of membrane-associated lipids and subsequent toxin internalization through caveolae. Cell. Microbiol. 2:487–504. [DOI] [PubMed] [Google Scholar]
- Cossart, P., and P.J. Sansonetti. 2004. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 304:242–248. [DOI] [PubMed] [Google Scholar]
- Cossart, P., M.F. Vicente, J. Mengaud, F. Baquero, J.C. Perez-Diaz, and P. Berche. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 57:3629–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart, P., J. Pizarro-Cerda, and M. Lecuit. 2003. Invasion of mammalian cells by Listeria monocytogenes: functional mimicry to subvert cellular functions. Trends Cell Biol. 13:23–31. [DOI] [PubMed] [Google Scholar]
- Dramsi, S., and P. Cossart. 1998. Intracellular pathogens and the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 14:137–166. [DOI] [PubMed] [Google Scholar]
- Dramsi, S., and P. Cossart. 2003. Listeriolysin O-mediated calcium influx potentiates entry of Listeria monocytogenes into the human Hep-2 epithelial cell line. Infect. Immun. 71:3614–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi, S., I. Biswas, E. Maguin, L. Braun, P. Mastroeni, and P. Cossart. 1995. Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol. Microbiol. 16:251–261. [DOI] [PubMed] [Google Scholar]
- Edidin, M. 2003. Lipids on the frontier: a century of cell-membrane bilayers. Nat. Rev. Mol. Cell Biol. 4:414–418. [DOI] [PubMed] [Google Scholar]
- Garner, M.J., R.D. Hayward, and V. Koronakis. 2002. The Salmonella pathogenicity island 1 secretion system directs cellular cholesterol redistribution during mammalian cell entry and intracellular trafficking. Cell. Microbiol. 4:153–165. [DOI] [PubMed] [Google Scholar]
- Gatfield, J., and J. Pieters. 2000. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 288:1647–1650. [DOI] [PubMed] [Google Scholar]
- Giurisato, E., D.P. McIntosh, M. Tassi, A. Gamberucci, and A. Benedetti. 2003. T cell receptor can be recruited to a subset of plasma membrane rafts, independently of cell signaling and attendantly to raft clustering. J. Biol. Chem. 278:6771–6778. [DOI] [PubMed] [Google Scholar]
- Harder, T., and K. Simons. 1997. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr. Opin. Cell Biol. 9:534–542. [DOI] [PubMed] [Google Scholar]
- Ilangumaran, S., A. Briol, and D.C. Hoessli. 1998. CD44 selectively associates with active Src family protein tyrosine kinases Lck and Fyn in glycosphingolipid-rich plasma membrane domains of human peripheral blood lymphocytes. Blood. 91:3901–3908. [PubMed] [Google Scholar]
- Ireton, K., B. Payrastre, and P. Cossart. 1999. The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositide 3-kinase. J. Biol. Chem. 274:17025–17032. [DOI] [PubMed] [Google Scholar]
- Jutras, I., L. Abrami, and A. Dautry-Varsat. 2003. Entry of the lymphogranuloma venereum strain of Chlamydia trachomatis into host cells involves cholesterol-rich membrane domains. Infect. Immun. 71:260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei, T., T. Matozaki, T. Sakisaka, A. Kodama, S. Yokoyama, Y.F. Peng, K. Nakano, K. Takaishi, and Y. Takai. 1999. Coendocytosis of cadherin and c-Met coupled to disruption of cell-cell adhesion in MDCK cells–regulation by Rho, Rac and Rab small G proteins. Oncogene. 18:6776–6784. [DOI] [PubMed] [Google Scholar]
- Kilsdonk, E.P., P.G. Yancey, G.W. Stoudt, F.W. Bangerter, W.J. Johnson, M.C. Phillips, and G.H. Rothblat. 1995. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 270:17250–17256. [DOI] [PubMed] [Google Scholar]
- Krasemann, S., M.H. Groschup, S. Harmeyer, G. Hunsmann, and W. Bodemer. 1996. Generation of monoclonal antibodies against human prion proteins in PrP0/0 mice. Mol. Med. 2:725–734. [PMC free article] [PubMed] [Google Scholar]
- Lafont, F., G. Tran Van Nhieu, K. Hanada, P. Sansonetti, and F.G. van der Goot. 2002. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44-IpaB interaction. EMBO J. 21:4449–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit, M., H. Ohayon, L. Braun, J. Mengaud, and P. Cossart. 1997. Internalin of Listeria monocytogenes with an intact leucine-rich repeat region is sufficient to promote internalization. Infect. Immun. 65:5309–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit, M., R. Hurme, J. Pizarro-Cerda, H. Ohayon, B. Geiger, and P. Cossart. 2000. A role for alpha- and beta-catenins in bacterial uptake. Proc. Natl. Acad. Sci. USA. 97:10008–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou, G.I., S. Matragoon, S. Samuel, M.A. Behzadian, N.T. Tsai, X. Gu, P. Roon, D.M. Hunt, R.C. Hunt, R.B. Caldwell, and D.M. Marcus. 2002. MAP kinase and beta-catenin signaling in HGF induced RPE migration. Mol. Vis. 8:483–493. [PubMed] [Google Scholar]
- London, E., and D.A. Brown. 2000. Insolubility of lipids in Triton X-100: physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim. Biophys. Acta. 1508:182–195. [DOI] [PubMed] [Google Scholar]
- Manes, S., R. Ana Lacalle, C. Gomez-Mouton, and A.C. Martinez. 2003. From rafts to crafts: membrane asymmetry in moving cells. Trends Immunol. 24:320–326. [DOI] [PubMed] [Google Scholar]
- Mengaud, J., M. Lecuit, M. Lebrun, F. Nato, J.C. Mazie, and P. Cossart. 1996. a. Antibodies to the leucine-rich repeat region of internalin block entry of Listeria monocytogenes into cells expressing E-cadherin. Infect. Immun. 64:5430–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud, J., H. Ohayon, P. Gounon, R.M. Mege, and P. Cossart. 1996. b. E-Cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell. 84:923–932. [DOI] [PubMed] [Google Scholar]
- Mora, R., V.L. Bonilha, A. Marmorstein, P.E. Scherer, D. Brown, M.P. Lisanti, and E. Rodriguez-Boulan. 1999. Caveolin-2 localizes to the golgi complex but redistributes to plasma membrane, caveolae, and rafts when co-expressed with caveolin-1. J. Biol. Chem. 274:25708–25717. [DOI] [PubMed] [Google Scholar]
- Naroeni, A., and F. Porte. 2002. Role of cholesterol and the ganglioside GM(1) in entry and short-term survival of Brucella suis in murine macrophages. Infect. Immun. 70:1640–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky, N., R. Stein, A. Yanai, G. Friedlander, and A. Taraboulos. 1997. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J. Biol. Chem. 272:6324–6331. [DOI] [PubMed] [Google Scholar]
- Nusrat, A., C.A. Parkos, P. Verkade, C.S. Foley, T.W. Liang, W. Innis-Whitehouse, K.K. Eastburn, and J.L. Madara. 2000. Tight junctions are membrane microdomains. J. Cell Sci. 113:1771–1781. [DOI] [PubMed] [Google Scholar]
- Orlandi, P.A., and P.H. Fishman. 1998. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J. Cell Biol. 141:905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron, P., C. Bordier, E.N. N'Diaye, and I. Maridonneau-Parini. 2000. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J. Immunol. 165:5186–5191. [DOI] [PubMed] [Google Scholar]
- Pizarro-Cerda, J., S. Sousa, and P. Cossart. 2004. Exploitation of host cell cytoskeleton and signalling during Listeria monocytogenes entry into mammalian cells. C. R. Biol. 327:115–123. [DOI] [PubMed] [Google Scholar]
- Rietveld, A., and K. Simons. 1998. The differential miscibility of lipids as the basis for the formation of functional membrane rafts. Biochim. Biophys. Acta. 1376:467–479. [DOI] [PubMed] [Google Scholar]
- Schuck, S., M. Honsho, K. Ekroos, A. Shevchenko, and K. Simons. 2003. Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. USA. 100:5795–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seveau, S., R.J. Eddy, F.R. Maxfield, and L.M. Pierini. 2001. Cytoskeleton-dependent membrane domain segregation during neutrophil polarization. Mol. Biol. Cell. 12:3550–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y., M. Naujokas, M. Park, and K. Ireton. 2000. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell. 103:501–510. [DOI] [PubMed] [Google Scholar]
- Shin, J.S., Z. Gao, and S.N. Abraham. 2000. Involvement of cellular caveolae in bacterial entry into mast cells. Science. 289:785–788. [DOI] [PubMed] [Google Scholar]
- Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- Sousa, S., D. Cabanes, A. El-Amraoui, C. Petit, M. Lecuit, and P. Cossart. 2004. Unconventional myosin VIIa and vezatin, two proteins crucial for Listeria entry into epithelial cells. J. Cell Sci. 117:2121–2130. [DOI] [PubMed] [Google Scholar]
- Tilney, L.G., and D.A. Portnoy. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109:1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Boland, J.A., M. Kuhn, P. Berche, T. Chakraborty, G. Dominguez-Bernal, W. Goebel, B. Gonzalez-Zorn, J. Wehland, and J. Kreft. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilette, D., O. Andreoletti, F. Archer, M.F. Madelaine, J.L. Vilotte, S. Lehmann, and H. Laude. 2001. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc. Natl. Acad. Sci. USA. 98:4055–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias, D.A., J.D. Violin, A.C. Newton, and R.Y. Tsien. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 296:913–916. [DOI] [PubMed] [Google Scholar]