

Abstract
The protective antigen (PA) of anthrax toxin binds to a cell surface receptor, undergoes heptamerization, and binds the enzymatic subunits, the lethal factor (LF) and the edema factor (EF). The resulting complex is then endocytosed. Via mechanisms that depend on the vacuolar ATPase and require membrane insertion of PA, LF and EF are ultimately delivered to the cytoplasm where their targets reside. Here, we show that membrane insertion of PA already occurs in early endosomes, possibly only in the multivesicular regions, but that subsequent delivery of LF to the cytoplasm occurs preferentially later in the endocytic pathway and relies on the dynamics of internal vesicles of multivesicular late endosomes.
Keywords: diphtheria toxin; LBPA; ALIX; MAPK; multivesicular; COP
Introduction
Anthrax toxin, one of the two main virulence factors produced by Bacillus anthracis, is an A-B type toxin, where the B subunit, called the protective antigen (PA), is involved in cell binding and the A subunits, of which there are two, lethal factor (LF) and edema factor (EF), bare the enzymatic toxic activities (Collier and Young, 2003). LF, a metalloprotease that targets MAPK kinases (MAPKKs), is responsible for lethality of the toxin (Collier and Young, 2003). EF, a CaM-dependent adenylate cyclase that elevates intracellular levels of cAMP (Collier and Young, 2003), is responsible for edema observed in anthrax patients.
PA (83 kD) binds to one of the two identified anthrax toxin receptors, ANTXR1 and ANTXR2 (Collier and Young, 2003), and is then processed at the NH2 terminus by the endoprotease furin, leaving a 63-kD form bound to the receptor. PA63 subsequently heptamerizes giving rise to a complex (PAheptamer) that is able to bind up to three molecules of LF and/or EF (Collier and Young, 2003). Heptamerization is accompanied by a spatial redistribution of the receptor from the glycerophospholipid region of the plasma membrane to specialized microdomains, so-called lipid rafts (Abrami et al., 2003). This redistribution triggers endocytosis of the PAheptamer–EF/LF complex (Abrami et al., 2003). Upon encounter of a sufficiently acidic milieu, PAheptamer undergoes a conformational change that leads to membrane insertion, which allows translocation of LF/EF across the endosomal membrane and delivery to the cytoplasm (Collier and Young, 2003). It is not clear at which stations of the endocytic pathway membrane insertion of PAheptamer, translocation of the enzymatic units, and their release into the cytoplasm occur. Here, we show that membrane insertion of PAheptamer can be uncoupled from cytoplasmic delivery of LF, each occurring at different stages of the endocytic pathway.
Results and discussion
We have previously shown that upon heptamerization, PAheptamer is internalized, transported to early endosomes, and then rapidly degraded (Abrami et al., 2003) indicating efficient transport to lysosomes and exclusion from the recycling pathway. Here, we investigated whether PAheptamer undergoes pH-induced membrane insertion in early or in late endosomes. Early and late endosomes were isolated from toxin-treated BHK cells using a well-established subcellular fractionation protocol (Aniento et al., 1993; Gruenberg, 2001). The SDS-resistant PAheptamer, which only forms after the pH-dependent conformational change, was highly enriched in early endosomes (Fig. 1 A), co-fractionating with the small GTPase rab5 (Gruenberg, 2001), indicating that membrane insertion already occurred in early endosomes. In contrast, little SDS-resistant PAheptamer was detected in late endosomes containing rab7, presumably because degradation is extremely rapid (Abrami et al., 2003). Interestingly, LF was abundant in early endosomes and clearly detectable in late endosomes (Fig. 1 A).
Figure 1.
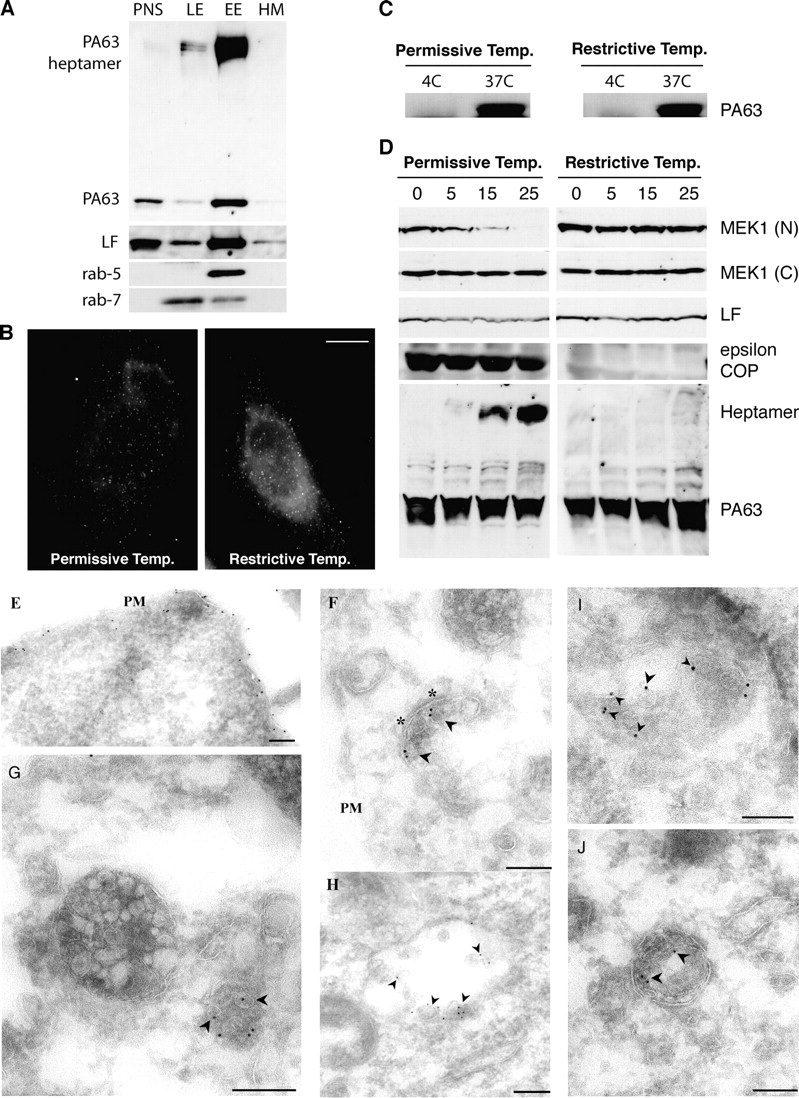
Conversion of PAheptamer to an SDS-resistant form occurs in early endosomes and is COPI dependent. (A) BHK cells were incubated at 4°C, 1 h with 500 ng/ml trypsin-nicked PA (PAn; Abrami et al., 2003) and 20 ng/ml LF, transferred to a toxin-free medium (37°C,1 h) and treated with 5 U/ml trypsin,10 min 37°C, to remove surface-bound toxin. Subcellular fractionation of postnuclear supernatants (PNS) was performed to separate early (EE) from late endosomes (LE) and heavy membranes (HM). 15 μg of protein from each fraction were analyzed by Western blotting for the presence of PA and PAheptamer, LF, rab5, and rab7. (B–D) LdlF cells were grown at the permissive (34°C) or restrictive temperature (40°C) for 18 h. (B) Cells were incubated for 20 min at 4°C with 500 ng/ml PASNKE, a PA variant with a mutated furin cleavage site, submitted to antibody cross-linking at 4°C and further incubated at 37°C for 30 min. Surface-bound toxin was removed by an acid wash (which lead to the observed background staining). Bar, 10 μm. (C) Cells were incubated with 500 ng/ml PAn, 1 h 4°C, washed and incubated at 4°C or 37°C with a toxin-free medium for 15 min. Surface toxin was removed as in A. 40 μg of PNS was analyzed by a SDS-PAGE gel followed by Western blotting against PA. (D) ldlF CHO cells were incubated at 4°C for 1 h with 500 ng/ml PAn and 500 ng/ml LF, transferred to 37°C for different periods of time (in min) in a toxin-free medium. 40 μg of total cell extracts were analyzed by Western blotting to detect LF processed MEK1 (anti–NH2-terminal antibody), total MEK1 (anti–COOH-terminal antibody), LF, ε-COP, and PA. (E–J) BHK cells were incubated with 10 μg/ml PAn for 1 h at 4°C, warmed to 37°C for either 10 min (E) or 40 min (F–J) before fixation and preparation of ultrathin sections. Sections were labeled with rabbit anti-PA followed by 10 nm protein A gold. Labeling is evident on the plasma membrane (E, PM) and in putative early endosomal structures (F), recognized by the presence of a characteristic patch of coat material (asterisks). At later times (G–J) labeling is evident within a subset of multivesicular endosomal structures with enrichment on internal vesicles (arrowheads). Bars, 200 nm.
The presence of LF in late endosomes and the fact that inhibitors of the vacuolar ATPase can delay the death of macrophages even when added long after LF (Menard et al., 1996) prompted us to investigate whether transport to late endocytic compartments was required for intoxication. Transport from early to late endosomes occurs via multivesicular intermediates, here after designated as endosomal carrier vesicles/multivesicular bodies (ECV/MVB), which detach from early endosomes and move toward late endosomes on microtubules (Gruenberg, 2001; see Fig. 5 for a schematic view of the endocytic pathway). To this end, we made use of the ldlF cell line, which contains a temperature-sensitive defect in the ε COPI coatomer subunit (ε-COP), so that ε-COP is rapidly degraded at the restrictive temperature (40°C). Then the generation of ECV/MVBs and transport to late endosomes is inhibited (Whitney et al., 1995) but bulk internalization and recycling remain normal (Daro et al., 1997; Gu et al., 1997). As expected, internalization of PA still occurred in ldlF cells (40°C). MEK1 was protected from LF, not for the anticipated reasons, but due to the lack of conversion to SDS-resistant form of PAheptamer (Fig. 1 D), which did occur in wild-type cells grown at 40°C (not depicted). The lack of SDS-resistant form is not due to an acidification defect, because early endosomes acidify normally in ldlF cells at 40°C (Daro et al., 1997; Gu et al., 1997; Piguet et al., 1999). There is however a plausible explanation. When lacking ε-COP (40°C), ldlF early endosomes are dramatically altered, appearing like clusters of thin tubules devoid of vesicular/multivesicular regions (Gu et al., 1997). Hence, it is attractive to speculate that such regions are required for PAheptamer membrane insertion.
Therefore, we analyzed PA-treated cells by electron microscopy. Specific labeling was initially observed on the PM (Fig. 1 E) but, after warming the cells to 37°C, labeling was increasingly found on the intraluminal vesicles of a subset of multivesicular endosomes (Fig. 1, G–J, arrowheads). Interestingly, at intermediate times, a consistent observation was labeling in the region of the coat patch (Fig. 1 F; not depicted) implicated in sorting of receptors into multivesicular endosomes (Sachse et al., 2002).
To further address whether cytoplasmic release of LF required delivery to late endosomes, we inhibited microtubule-dependent transport using the depolymerizing agent nocodazole. LF-dependent MEK1 cleavage was delayed, without affecting the formation of SDS-resistant PAheptamer (Fig. 2 A, note that degradation was inhibited as expected because access to late endosomes and lysosomes is impaired). To rule out the possibility that this delay was somehow linked to the presence of some MEK1 on late endosomes (Wunderlich et al., 2001), we also followed LF-induced cleavage of another MAPKK, MKK3, which is involved in the p38 MAPK signaling cascade, different from the MEK1-dependent ERK pathway. As for MEK1, MKK3 cleavage was delayed in nocodazole-treated cells (Fig. 2 B). Interestingly, the kinetics of cleavage of MKK3 were slower than those of MEK1 (Fig. 2 C).
Figure 2.

Efficient encounter between LF and its target occurs from late endosomes. (A) CHO cells were incubated or not with 10 μM nocodazole for 2 h at 37°C (nocodazole was then present throughout the entire experiment), followed by 1 h at 4°C with 500 ng/ml PAn and 20 ng/ml LF, transferred to 37°C for different periods of time (in min) in a toxin-free medium. 40 μg of PNS was analyzed by Western blotting against PA, the NH2 terminus of MEK1 (N) and LF. The amount of intact MEK1 (A) and MKK3 (B) was quantified by densitometry and normalized to the amount at time t = 0 (n = 4, errors bars represent SDs). (C) The kinetics of decrease in intact MEK1 were compared with that of decrease in MKK3. Western blots (inset) were quantified by densitometry as in A and B. (D) RAW 264 macrophages were incubated or not with 10 μM nocodazole (2 h at 37°C), followed by 1 h at 4°C with 500 ng/ml PAn and 100 ng/ml LF. 40 μg of PNS was analyzed by Western blotting against the NH2 termini of MEK1 and MKK3. (E) HeLa cells were transfected or not with dominant-negative rab7N125I cDNA, incubated at 4°C for 1 h with 500 ng/ml PAn and 500 ng/ml LF and transferred to 37°C for different periods of time (in min) in a toxin-free medium. 40 μg of PNS was analyzed as in A (n = 4).
Next, we investigated the effects of nocodazole on MAPKK cleavage kinetics in macrophages, considered as one of the important targets of the anthrax lethal toxin (Collier and Young, 2003). LF-induced MEK1 cleavage, and to a lesser but reproducible extent MKK3 cleavage, were delayed when RAW 264 macrophages were treated with nocodazole (Fig. 2 D) indicating that the requirement for transport of LF to late endosomes is a general feature of anthrax intoxication. Interestingly, as in CHO cells, kinetics of MKK3 cleavage were slower than those of MEK1 cleavage.
Because the above observations indicated that LF delivery preferentially occurs from late endosomes, we decided to affect this organelle by overexpressing a dominant-negative mutant of rab7 (N125I), a small GTPase known to be involved in late endosome function and dynamics (Gruenberg, 2001). Although formation (Fig. 2 E) and degradation (not depicted) of SDS-resistant PAheptamer occurred normally, cleavage of MEK1 was again delayed (Fig. 2 E) confirming the involvement of late endosomes in cytoplasmic delivery of LF.
Altogether, the above experiments support the following sequence of events: the LF–PAheptamer complex is internalized; in early endosomes, PAheptamer undergoes membrane insertion and mediates translocation of LF, in vitro studies indeed indicate that channel formation by PA is sufficient to allow translocation of LF. At that stage however, LF remains associated with early endosomes and microtubule-dependent transport to late endosomes is required for efficient delivery to the cytoplasm where LF can reach MAPKKs. The question arises why translocated LF can reach the cytoplasm from late endosomes but not from early endosomes. One possibility is that PAheptamer preferentially inserts into the membrane of intraluminal vesicles as suggested by the electron microscopy images (Fig. 1, G–J), which would lead to translocation of LF into the lumen of these vesicles.
Sorting into and formation of intraluminal vesicles occurs in early endosomes and seems to be, at that stage, a one-way street (Katzmann et al., 2002; Gruenberg and Stenmark, 2004). Once these intraluminal vesicles have reached late endosomes, some apparently acquire the ability to undergo regulated back fusion with the limiting membrane. The membrane of intraluminal vesicles indeed not only contains proteins destined to be degraded but also proteins in transit to other destinations in the cell (Kobayashi et al., 2000; Chow et al., 2002), which must get back to the limiting membrane from which budding of outgoing vesicles occurs (Gruenberg, 2001; Murk et al., 2003). To test whether this localized ability of back fusion of intraluminal vesicles could be used by LF to reach the cytoplasm, we affected one of the abundant and important components of intraluminal vesicles, the unconventional lipid lysobisphosphatidic acid (LBPA; Gruenberg, 2001). This lipid is unique to late endosomes and it was shown that feeding cells with a monoclonal antibody against LBPA, 6c4, impairs sorting of proteins and lipids leading to a traffic jam in the compartment (Kobayashi et al., 1999). We found that incubating cells with the 6c4 antibody did not affect the kinetics of formation of SDS-resistant PAheptamer as expected, but significantly delayed cleavage of MEK1 by LF (Fig. 3 A).
Figure 3.

Alteration of the dynamics of late endosomal intraluminal vesicles leads to a delay in MEK1 cleavage by LF. (A) CHO cells were incubated or not for 18 h with the anti-LBPA antibody 6c4 (50 μg/ml), washed, further incubated at 4°C for 1 h with 500 ng/ml PAn and 250 ng/ml LF, and transferred to 37°C for different periods of time (in min) in a toxin-free medium. 20 μg of PNS was analyzed by Western blotting to detect PAheptamer, the NH2 terminus of MEK1 and LF. The amount of intact MEK1 was quantified as in Fig. 2 A (n = 4). (B) HeLa cells were transfected with siRNA against ALIX, the efficiency of which was examined 78 h later by Western blotting using antibodies against ALIX. Cells were then incubated at 4°C for 1 h with 500 ng/ml PAn and 100 ng/ml LF, transferred to 37°C for different periods of time (in min) in a toxin-free medium, homogenized, and 20 μg of PNS was analyzed to detect the presence of PAheptamer and the NH2 terminus of MEK1. Quantifications were performed as in A. (C) HeLa cells were treated as in B. 80 μg of PNS were centrifuged at 100,000 g for 1 h and the pellet analyzed by Western blotting for LF. (D) CHO cells were treated as in A and incubated with 500 ng/ml PAn and 500 ng/ml FP59. 20 μg of cell lysates were loaded on native or SDS-PAGE before Western blotting against EF-2. (E) ALIX was knocked down in HeLa cells as in B, incubated with 500 ng/ml PAn with 500 ng/ml FP59, and the modification of EF2 analyzed as in D.
To further address the importance of intraluminal vesicles in LF delivery, we knocked down by RNAi the expression of ALIX, the mammalian homologue of the yeast class E vacuolar protein sorting vps31, involved in multivesicular body sorting and biogenesis (Odorizzi et al., 2003). Interestingly, ALIX has recently been proposed to directly regulate the dynamics of late endosome intraluminal vesicles (Matsuo et al., 2004). Formation of SDS-resistant PAheptamer, as well as its degradation, occurred normally in ALIX siRNA-treated cells (Fig. 3 B). Remarkably however, MEK1 cleavage was strongly delayed (Fig. 3 B), as was that of MKK3 (not depicted). To test whether LF remained trapped in late endosomes in ALIX RNAi-treated cells, we separated membrane-bound organelles from cytosol by high speed centrifugation. Membrane associated LF decreased with time in control cells as expected due to its release into the cytoplasm (Fig. 3 C). In contrast, LF remained largely associated with the organellar fraction in ALIX RNAi-treated cells. Note that whereas PAheptamer was normally degraded between 20 and 30 min in ALIX siRNA-treated cells (Fig. 3 B), LF remained intact during that time, in agreement with its proposed localization to the lumen of intraluminal vesicles—topologically equivalent to the cytoplasmic space—where it would be protected from lysosomal enzymes.
To rule out that the effects of 6c4 and ALIX siRNA were somehow related to the metalloprotease activity of LF rather than the trafficking of the toxin, we used a hybrid (called fusion protein 59; FP59) in which the metalloprotease domain of LF had been replaced by the ADP-ribosyltransferase domain of Pseudomonas exotoxin A, which modifies elongation factor 2 (EF-2). Because this modification leads to a change in charge of EF-2 it can be monitored on native gels (Liu and Leppla, 2003b). Incubation of cells with nocodazole (not depicted), anti-LBPA antibody 6c4, as well as the knockdown of ALIX, delayed the kinetics of ADP-ribosylation of EF-2 by FP59 (Fig. 3, D and E).
To exclude that the various treatment had a gross effect on the endocytic pathway, we repeated the studies using a different toxin, namely diphtheria toxin (DT), known to translocate into the cytoplasm at the level of early endosomes (Papini et al., 1993; Lemichez et al., 1997). DT is also an A-B toxin, where the A subunit is, as Pseudomonas exotoxin A, an ADP-ribosyltransferase that modifies EF-2. We first tested ldlF cells at 40°C and found that they were insensitive to DT (Fig. 4 A) because ADP-ribosylation of EF-2 did not occur (Fig. 4 B). However, in contrast to what was observed for anthrax lethal toxin and PA+FP59, treatment with nocodazole had no effect on the kinetics of substrate modification by DT (Fig. 4 C), nor did incubation with the anti-LBPA antibody 6c4 (Fig. 4 D) or RNAi against ALIX (Fig. 4 E).
Figure 4.
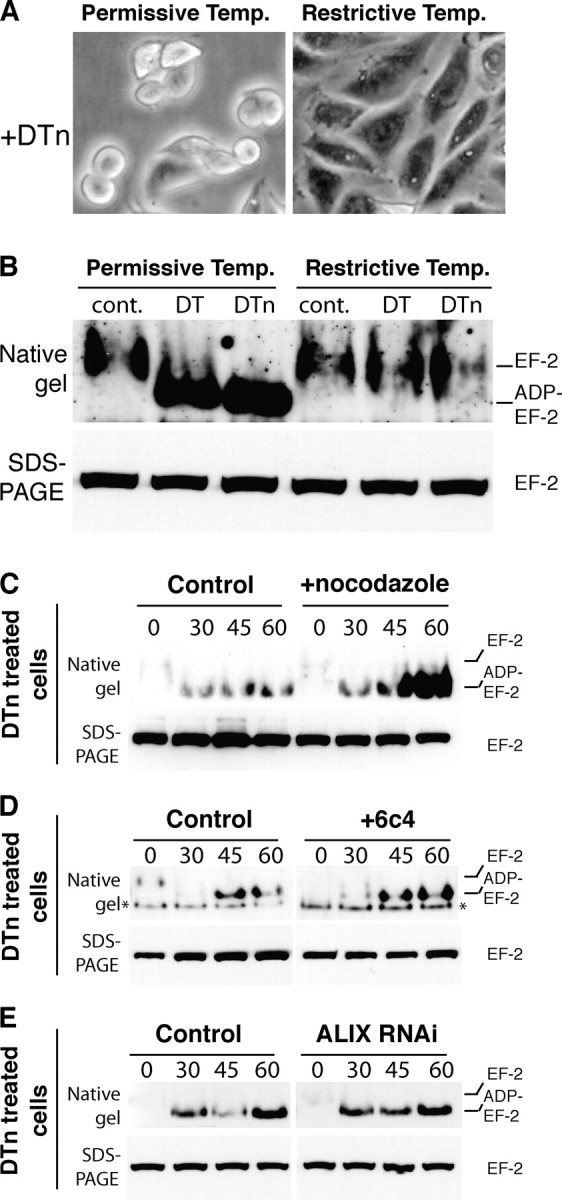
Cytosolic delivery of DT is COPI dependent but does not require transport to late endosomes. (A and B) ldlF cells were grown at the permissive or restrictive temperature for 16 h, incubated for 7 h (A) or 2.5 h (B) at 37°C in serum-free medium with 500 ng/ml of DT or trypsin-nicked DT (DTn) or left untreated (cont.). (B) Cell lysates were prepared and the modification of EF2 analyzed as in Fig. 3 D. (C) Wild-type CHO cells were incubated or not with 10 μM nocodazole for 2 h at 37°C and 15 min at 4°C, followed by 1 h at 4°C with 500 ng/ml DTn, washed and further incubated for different times at 37°C. The modification of EF2 was analyzed as above. (D) CHO cells were treated with 6c4 antibody as in Fig. 3 A and incubated with 500 ng/ml DTn and modification of EF2 analyzed. The band labeled with an asterisk is a cross-reacting protein, apparent in certain experiments. (E) ALIX was knocked down in HeLa cells as in Fig. 3 B, incubated with 500 ng/ml DTn, and modification of EF2 analyzed.
Based on the present data, we would like to propose the following models for the modes of delivery of anthrax lethal and DTs. Both toxins are internalized via clathrin-coated pits (Moya et al., 1985; Abrami et al., 2003) and transported to early endosomes where they are preferentially sorted to the vesicular regions (Fig. 5, A and B) as suggested by the fact that neither toxin affects lflF cells (40°C), which lack vesicular regions in early endosomes due to degradation of ε-COP. From that point, the pathways of the two toxins diverge. DT remains in the limiting membrane of early endosomes. Thus, upon membrane insertion, the ADP-ribosyltransferase is directly translocated from the endosomal lumen to the cytoplasm where it can reach EF-2 (Fig. 5 B). In contrast, the LF–PAheptamer complex is sorted into nascent intraluminal vesicles. Thus, upon PAheptamer-mediated membrane translocation, LF does not reach the cytoplasm but the lumen of the intraluminal vesicles (Fig. 5 A). Upon subsequent budding of ECV/MVB from early endosomes, PAheptamer and LF containing intraluminal vesicles are transported to late endosomes. There, PAheptamer is rapidly degraded but this has no consequences because PA has already fulfilled its translocation function. LF, being in the lumen of the intraluminal vesicles is protected from degradative enzymes and awaits back fusion events with the limiting membrane to be freed into the cytoplasm.
Figure 5.

Endocytic routes of anthrax and diphtheria toxins. Based on the present work and the literature, we propose the following models for the delivery of anthrax lethal toxin (A) and DT (B) to the cytoplasm. Both toxins are internalized, transported to early endosomes, and sorted into the vesicular regions. (A) Anthrax toxin would be sorted into intraluminal vesicles whereby membrane insertion of PA and translocation of LF would result in trapping of the metalloprotease in the lumen of intraluminal vesicles. Once transported to late endosomes, back fusion of intraluminal vesicles with the limiting membrane would deliver LF to the cytoplasm. (B) In contrast, in early endosomes, DT would preferentially insert into the limiting membrane allowing direct delivery to the cytoplasm.
There might be a biological rational, aimed at increasing toxin efficiency, behind the preferential delivery of LF at the level of late endosomes in the perinuclear region rather than at the periphery of the cells from early endosomes, and/or it might have consequences as to the physiological outcome of the toxins action. As mentioned above, a MEK1 containing catalytic scaffolding complex, important for MAPK signaling, was found associated to late endosomes (Wunderlich et al., 2001). Similarly other scaffolding domains are likely to localize specifically to the perinuclear region of cells. Interestingly, LF-mediated cleavage of MEK1 was more rapid than that of MKK3 (Fig. 2 C). Along the same line, LF-induced inhibition of ERK1 activation, involving MEK1, occurs at fivefold lower LF concentrations than p38 inactivation, involving MKK3 (Park et al., 2002). These examples support the notion that delivery of LF from late endosomes in the close proximity of a target, in this case MEK1, is more efficient than relying on stochastic encounter with distant targets, in this case MKK3, by diffusion through the cytoplasm. Target encounter by the diffusion is generally assumed for delivered bacterial toxins, a notion that might have to be revised. Delivery at a specific site in the cell might be particularly important at the low toxin concentrations encountered during the early stages of infection. It is not clear at present whether one specific MAPKK is a dominant LF target in a given cell type, nor whether unidentified, non-MAPKK, LF targets exist. However, it is tempting to speculate that late endosomes act as encounter platforms between LF and early targets in the perinuclear region of the cell.
Materials and methods
Cells and reagents
Wild-type PA, PASNKE, LF, FP59, a recombinant fusion toxin consisting of LF amino acids 1–254, fused to the ADP-ribosylation domain of Pseudomonas exotoxin A, and DT were produced as described previously (Liu and Leppla, 2003a, b) and the corresponding polyclonal antibodies used (Abrami et al., 2003). Antibodies against the COOH terminus of MEK1 were purchased from Santa Cruz Biotechnology, Inc.; the NH2 terminus of MEK1 was purchased from Upstate Biotechnology; the NH2 terminus of MKK3, EF-2, and rab5 was purchased from Santa Cruz Biotechnology Inc.; and rab7, ɛ-COP, and LBPA were gifts from J. Gruenberg (University of Geneva). Antibodies against ALIX were a gift from R. Sadoul (University of Grenoble, Grenoble, France). Nocodazole was purchased from Sigma-Aldrich. HeLa, CHO, and mutant ldlF cells (provided by M. Krieger; Massachusetts Institute of Technology, Cambridge, MA), and RAW 264 cells were maintained as described previously (Abrami et al., 1998, 2003). The dominant-negative Rab7 N125I mutant and siRNA against ALIX (Matsuo et al., 2004) were gifts from P. Boquet (Institut National de la Santé et de la Recherche Médicale and University of Nice, Nice, France) and J. Gruenberg, respectively.
Biochemical methods
Early and late endosomes were isolated using sucrose density gradients (Kobayashi et al., 1999). For SDS-PAGE analysis, samples were boiled for 5 min. The various subunits and forms of the anthrax toxin and ADP-ribosylation of EF-2 were detected by Western blotting of SDS-PAGE and native on 4–20% acrylamide gradient gels (Abrami et al., 2003; Liu and Leppla, 2003b). Transient transfection experiments in HeLa cells were performed 48 h (1 μg cDNA/9.6 cm2 plate) or 72 h (200 pmoles siRNA/9.6 cm2 plate) using Fugene (Roche Diagnostics Corporation) and oligofectamine (Invitrogen) transfection reagents, respectively.
Immunofluorescence
CHO cells were incubated with 500 ng/ml PASNKE, submitted to an antibody sandwich and incubated at 37°C, submitted to an acid wash to remove remaining surface-bound PASNKE and fixed with 3% PFA (Abrami et al., 2003). Images were acquired using a 100× lens on an Axiophot (Carl Zeiss MicroImaging, Inc.), equipped with a cooled camera (Hamamatsu) using the Openlab acquisition software.
Acknowledgments
We thank M. Krieger, P. Boquet, J. Gruenberg, and R. Sadoul for reagents; D. Hsu for making toxins; and J. Gruenberg, I. Le Blanc, and M. Moayeri for critical reading of the manuscript.
This work was supported the Swiss National Science Foundation, the EMBO Young Investigator Program, and the National Institutes of Health (AI053270-01).
Abbreviations used in this paper: DT, diphtheria toxin; DTn, trypsin-nicked DT; ε-COP, ε COPI coatomer subunit; ECV/MVB, endosomal carrier vesicles/multivesicular bodies; EF, edema factor; EF-2, elongation factor 2; FP59, fusion protein 59; LBPA, lysobisphosphatidic acid; LF, lethal factor; MAPKK, MAPK kinase; PA, protective antigen; PAn, trypsin-nicked PA; PNS, postnuclear supernatants.
References
- Abrami, L., M. Fivaz, E. Decroly, N.G. Seidah, J. François, G. Thomas, S. Leppla, J.T. Buckley, and F.G. van der Goot. 1998. The pore-forming toxin proaerolysin is processed by furin. J. Biol. Chem. 273:32656–32661. [DOI] [PubMed] [Google Scholar]
- Abrami, L., S. Liu, P. Cosson, S.H. Leppla, and F.G. van der Goot. 2003. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 160:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento, F., N. Emans, G. Griffiths, and J. Gruenberg. 1993. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol. 123:1373–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow, A., D. Toomre, W. Garrett, and I. Mellman. 2002. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature. 418:988–994. [DOI] [PubMed] [Google Scholar]
- Collier, R.J., and J.A. Young. 2003. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 19:45–70. [DOI] [PubMed] [Google Scholar]
- Daro, E., D. Sheff, M. Gomez, T. Kreis, and I. Mellman. 1997. Inhibition of endosome function in CHO cells bearing a temperature-sensitive defect in the coatomer (COPI) component epsilon-COP. J. Cell Biol. 139:1747–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg, J. 2001. The endocytic pathway: a mosaic of domains. Nat. Rev. Mol. Cell Biol. 2:721–730. [DOI] [PubMed] [Google Scholar]
- Gruenberg, J., and H. Stenmark. 2004. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 5:317–323. [DOI] [PubMed] [Google Scholar]
- Gu, F., F. Aniento, R.G. Parton, and J. Gruenberg. 1997. Functional dissection of COP-I subunits in the biogenesis of multivesicular endosomes. J. Cell Biol. 139:1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann, D.J., G. Odorizzi, and S.D. Emr. 2002. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3:893–905. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., M.H. Beuchat, M. Lindsay, S. Frias, R.D. Palmiter, H. Sakuraba, R.G. Parton, and J. Gruenberg. 1999. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1:113–118. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., U.M. Vischer, C. Rosnoblet, C. Lebrand, M. Lindsay, R.G. Parton, E.K. Kruithof, and J. Gruenberg. 2000. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol. Biol. Cell. 11:1829–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemichez, E., M. Bomsel, G. Devilliers, J. vanderSpek, J.R. Murphy, E.V. Lukianov, S. Olsnes, and P. Boquet. 1997. Membrane translocation of diphtheria toxin fragment A exploits early to late endosome trafficking machinery. Mol. Microbiol. 23:445–457. [DOI] [PubMed] [Google Scholar]
- Liu, S., and S.H. Leppla. 2003. a. Cell surface tumor endothelium marker 8 cytoplasmic tail-independent anthrax toxin binding, proteolytic processing, oligomer formation and internalization. J. Biol. Chem. 278:5227–5234. [DOI] [PubMed] [Google Scholar]
- Liu, S., and S.H. Leppla. 2003. b. Retroviral insertional mutagenesis identifies a small protein required for synthesis of diphthamide, the target of bacterial ADP-ribosylating toxins. Mol. Cell. 12:603–613. [DOI] [PubMed] [Google Scholar]
- Matsuo, H., J. Chevallier, N. Mayran, I. Le Blanc, C. Ferguson, J. Faure, N.S. Blanc, S. Matile, J. Dubochet, R. Sadoul, et al. 2004. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science. 303:531–534. [DOI] [PubMed] [Google Scholar]
- Menard, A., K. Altendorf, D. Breves, M. Mock, and C. Montecucco. 1996. The vacuolar ATPase proton pump is required for the cytotoxicity of Bacillus anthracis lethal toxin. FEBS Lett. 386:161–164. [DOI] [PubMed] [Google Scholar]
- Moya, M., A. Dautry-Varsat, B. Goud, D. Louvard, and P. Boquet. 1985. Inhibition of coated pit formation in Hep2 cells blocks the cytotoxicity of diphtheria toxin but not that of ricin toxin. J. Cell Biol. 101:548–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murk, J.L., B.M. Humbel, U. Ziese, J.M. Griffith, G. Posthuma, J.W. Slot, A.J. Koster, A.J. Verkleij, H.J. Geuze, and M.J. Kleijmeer. 2003. Endosomal compartmentalization in three dimensions: implications for membrane fusion. Proc. Natl. Acad. Sci. USA. 100:13332–13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi, G., D.J. Katzmann, M. Babst, A. Audhya, and S.D. Emr. 2003. Bro1 is an endosome-associated protein that functions in the MVB pathway in Saccharomyces cerevisiae. J. Cell Sci. 116:1893–1903. [DOI] [PubMed] [Google Scholar]
- Papini, E., R. Rappuoli, M. Murgia, and C. Montecucco. 1993. Cell penetration of diphtheria toxin. Reduction of the interchain disulfide bridge is the rate-limiting step of translocation in the cytosol. J. Biol. Chem. 268:1567–1574. [PubMed] [Google Scholar]
- Park, J.M., F.R. Greten, Z.W. Li, and M. Karin. 2002. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 297:2048–2051. [DOI] [PubMed] [Google Scholar]
- Piguet, V., F. Gu, M. Foti, N. Demaurex, J. Gruenberg, J.L. Carpentier, and D. Trono. 1999. Nef-induced CD4 degradation: a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell. 97:63–73. [DOI] [PubMed] [Google Scholar]
- Sachse, M., S. Urbe, V. Oorschot, G.J. Strous, and J. Klumperman. 2002. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol. Biol. Cell. 13:1313–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney, J.A., M. Gomez, D. Sheff, T.E. Kreis, and I. Mellman. 1995. Cytoplasmic coat proteins involved in endosome function. Cell. 83:703–713. [DOI] [PubMed] [Google Scholar]
- Wunderlich, W., I. Fialka, D. Teis, A. Alpi, A. Pfeifer, R.G. Parton, F. Lottspeich, and L.A. Huber. 2001. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J. Cell Biol. 152:765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]