

Abstract
Although compelling evidence supports the central role of caspase-activated DNase (CAD) in oligonucleosomal DNA fragmentation in apoptotic nuclei, the regulation of CAD activity remains elusive in vivo. We used fluorescence photobleaching and biochemical techniques to investigate the molecular dynamics of CAD. The CAD-GFP fusion protein complexed with its inhibitor (ICAD) was as mobile as nuclear GFP in the nucleosol of dividing cells. Upon induction of caspase-3–dependent apoptosis, activated CAD underwent progressive immobilization, paralleled by its attenuated extractability from the nucleus. CAD immobilization was mediated by its NH2 terminus independently of its DNA-binding activity and correlated with its association to the interchromosomal space. Preventing the nuclear attachment of CAD provoked its extracellular release from apoptotic cells. We propose a novel paradigm for the regulation of CAD in the nucleus, involving unrestricted accessibility of chromosomal DNA at the initial phase of apoptosis, followed by its nuclear immobilization that may prevent the release of the active nuclease into the extracellular environment.
Introduction
Apoptosis, or programmed cell death, allows the elimination of unwanted cells during normal development and tissue homeostasis without affecting the viability of adjacent cells. At the molecular level, apoptosis comprises a cascade of proteolytic events mediated by a family of cysteine-aspartate proteases, the caspases, involved in both the signaling and the execution phase of cell death (Nicholson, 1999). Apoptotic cells undergo preprogrammed morphological changes, reflected by cell and nuclear shrinkage, chromatin condensation, and apoptotic body formation, ultimately engulfed by scavenger cells (Hanayama et al., 2002). One of the biochemical hallmarks of apoptosis is the cleavage of nuclear DNA into oligonucleosome-sized fragments, or DNA fragmentation, a mechanism proposed to be essential for preventing autoimmune diseases and horizontal gene transfer (Herrmann et al., 1998; Zhang and Xu, 2002).
Caspase-activated DNase (CAD) is activated by the caspase-3–dependent cleavage of the DNA fragmentation factor (DFF), which contains two subunits, the CAD and the inhibitor of CAD (ICAD). CAD and ICAD have an NH2-terminal homologous domain, the cell-inducing DFF45-like effector (CIDE) domain, responsible for the heterodimerization of ICAD and CAD (CAD/ICAD) (Aravind et al., 1999). Besides inhibiting CAD activity, ICAD is also essential for the folding of CAD by functioning as a chaperone (Enari et al., 1998; Sakahira et al., 1999, 2000). CAD appears to be the most important apoptotic nuclease because chromatin condensation and DNA fragmentation were substantially delayed in ICAD−/− mouse embryonic fibroblasts (MEFs) lacking functional CAD, in cells expressing caspase-resistant ICAD, as well as in CAD−/− thymocytes (Zhang et al., 1998; Hanayama et al., 2002; Nagata et al., 2003).
We and others have demonstrated that two NLSs, identified at the COOH terminus of both CAD and ICAD, are necessary and sufficient for the nuclear targeting of CAD/ICAD (Samejima and Earnshaw, 1998; Lechardeur et al., 2000; Scholz et al., 2002). After caspase-3–mediated cleavage of ICAD, activated CAD is released and generates double-stranded breaks in the chromatin (Halenbeck et al., 1998; Sakahira et al., 1998; Liu et al., 1999). In addition to CAD, endonuclease G and the apoptotic inducing factor are implicated in the cell-autonomous chromosomal DNA degradation (Penninger and Kroemer, 2003). However, complete cleavage of chromosomal DNA requires the activity of the lysosomal DNase II, after the phagocytic engulfment of apoptotic bodies (Nagata et al., 2003).
Both homo- and heteromeric protein–protein interactions have been invoked in the regulation of CAD activity. In vitro biochemical studies suggested that histone H1, as well as high mobility group (HMG) 1/2 proteins and topoisomerase II interact with CAD and may tether the CAD/ICAD complex to the chromatin (Zhang and Xu, 2002). These interactions were proposed to regulate the nuclease activity of CAD by influencing the DNA conformation and/or acting as allosteric activators (Liu et al., 1998, 1999; Toh et al., 1998; Zhang and Xu, 2002). Because histone H1 and HMG 1/2 bind to the linker region of the relatively immobile chromatin (Misteli et al., 2000; Kimura and Cook, 2001), it is anticipated that complex formation of CAD with these nuclear proteins would limit the substrate accessibility of the nuclease both in space and time. Other studies suggested that activated CAD forms large homo-oligomeric complexes (>1 MD), which could restrict its diffusion (Liu et al., 1999; Meiss et al., 2001; Widlak and Garrard, 2001; Zhou et al., 2001).
To resolve these contrasting observations and establish the molecular dynamics of CAD in living cells, we used a combination of morphological, biochemical, and biophysical techniques. Our results indicate that the mobility of the CAD/ICAD complex is similar to freely moving nuclear proteins in dividing cells, implying that there are no or only transient interactions with immobile nuclear constituents. Although activated CAD displays virtually free diffusion at the initial stage of apoptosis, movements of CAD become gradually restricted during the progression of apoptosis. This appears to be a consequence of CAD association with the nuclear matrix. We propose that immobilization of CAD may represent a protective mechanism against the release of the activated DNase into the extracellular space,
Results
CAD becomes resistant to detergent extraction in apoptotic nuclei
We used epitope-tagged CAD/ICAD to investigate whether the CAD/ICAD heterodimer binds to nuclear constituents by immunostaining. Both HA-tagged CAD and ICAD-myc transiently expressed in HeLa cells. Both proteins were confined to the nuclei (Fig. 1 A, left, −perm; Lechardeur et al., 2000). Permeabilization of the cells by the nonionic detergent Triton X-100 before fixation provoked the complete loss of HA-CAD/ICAD-myc from the nuclei of normal cells (Fig. 1 A, left, +perm). EGFP harboring three tandem repeats of the NLS of the SV40-antigen (GFP-NLS) was also depleted, validating the permeabilization procedure (Fig. 1 B, left; +perm). In contrast, association of core and linker histones with the chromosomal DNA was retained in permeabilized cells (Fig. 1 C, left; +perm). Collectively, these observations suggest the CAD/ICAD complex can be released from the nuclei similarly to soluble proteins such as GFP-NLS.
Figure 1.

Extractability of CAD from normal and apoptotic HeLa cells. (A–C) Indirect immunofluorescence of (A) HA-CAD/ICAD-myc, (B) GFP-NLS, and (C) histones in HeLa cells. The indicated proteins were immunostained in cells fixed with PFA before (−perm) or after (+perm) permeabilization in cytoskeletal buffer containing 0.2% Triton X-100. DNA was stained with DAPI. Apoptosis was induced with STS for 2 h. Single optical sections were acquired with a fluorescence microscope (DMI RE2; Leica) and OpenLab software. Bars, 10 μm. (D) Nuclear retention of endogenous CAD. Lysates were prepared from normal and apoptotic HeLa cells before (lane 1) or 5 min after (lane 3) permeabilization as in A–C. Extracellular release of CAD from the cells is shown in lanes 2 and 4. Apoptosis was induced with STS for 2 h. Cell lysates and medium corresponding to the same number of cells was processed for Western blot using an anti-CAD antibody.
Staurosporine (STS), a nonspecific inhibitor of PKC and an effective apoptosis-inducing agent, activates caspase-3 and triggers the proteolysis of ICAD (Samuelsson et al., 2002). A significant fraction of both endogenous and heterologous ICAD was degraded after 2 h of STS treatment (Fig. 2 A; Lechardeur et al., 2000). As a corollary, substantial DNA fragmentation occurred, as indicated by the appearance of the oligonucleosomal DNA ladder in apoptotic cells (Fig. 2 B). STS-induced apoptosis delayed the release of HA-CAD from permeabilized cells as compared with control (Fig. 1 A). Meanwhile, the extractability of neither GFP-NLS (Fig. 1 B, right panels) nor the cleaved endogenous nor the exogenous ICAD was altered (Fig. 1 A, right, +perm and unpublished data).
Figure 2.


Biogenesis and stability of CAD and ICAD in dividing and apoptotic cells. (A) The stability of endogenous ICAD and CAD in HeLa cells during STS-induced apoptosis. Cellular proteins (40 μg) were separated by SDS-PAGE and probed by immunoblotting with anti-CAD and anti-ICAD antibodies. (B) Internucleosomal DNA fragmentation after 2.5 h apoptosis (lane 3). The accumulation of 200-bp nucleosomal DNA fragments is characteristic of late stage of apoptosis. Soluble DNA fragments were absent in control HeLa cells (lane 2). 1-kb DNA ladder; lane 1. (C and D) HeLa cells were transiently transfected with the HA-CAD-GFP and ICAD-myc vectors or carrier DNA. (C) Cell lysates from normal (lane 2) and STS treated HeLa cells (lane 3) were immunoprecipitated with an anti-GFP antibody. Precipitates (top panel) and 10% of the lysates (bottom panels) were immunoblotted. (D) The expression and stability of ICAD-myc and CAD-GFP were determined as described in A. (E) Indirect immunofluorescence of HeLa cells expressing CAD-GFP/ICAD-myc fixed before (−perm) or after (+perm) permeabilization as in Fig. 1 A. (F) Number of cells retaining nuclear CAD-GFP and GFP-NLS under permeabilization. After in vivo permeabilization as in A, cells were fixed and chromatin was counter-stained with DAPI. Approximately 500 nuclei were scored for their fluorescence for each condition. The number of cells expressing CAD-GFP was expressed as the percentage of nuclei remaining fluorescent upon permeabilization of cells. (G) DNase activity of CAD-GFP. Transiently expressed HA-CAD-GFP/ICAD myc was immunoisolated from HeLa cell lysate with an anti-HA antibody on protein G–Sepharose. Purified HA-CAD-GFP/ICAD-myc complex was incubated with plasmid DNA alone (lane 3, −casp) or in the presence of recombinant caspase-3 (lane 4, + casp). Nontransfected cells were used as control (lanes 5 and 6).
Because anti-CAD antibodies have insufficient specificity for immunostaining, extractability of endogenous CAD was tested by Western blotting. Although CAD could not be detected in the extracellular medium of nonpermeabilized cells (Fig. 1 D, lanes 1 and 2), 90% of the cellular CAD content was released after permeabilization of nonapoptotic cells (Fig. 1 D, lanes 3 and 4). In contrast, apoptosis induction substantially decreased the amount of CAD released into the extracellular medium: more than 70% of the cellular CAD content remained associated with the apoptotic cells after Triton X-100 treatment (Fig. 1 D, bottom, lanes 3 and 4). The nuclear retention of CAD suggests that activated CAD has increased affinity to some of the immobile component(s) of the apoptotic nucleus. To test this hypothesis and compare the molecular dynamics of CAD in proliferating and apoptotic cells, we used fluorescence photobleaching techniques.
Characterization of CAD-GFP fusion protein
To characterize the diffusional mobility of CAD, an HA-CAD-EGFP (CAD-GFP) fusion protein was constructed. Compelling evidence indicates that CAD-GFP mimics the cellular behavior of endogenous CAD. First, CAD-GFP is targeted into the nucleus and was coprecipitated with ICAD-myc in normal cells (Fig. 2 C; Fig. 2 E, left, −perm). Second, the steady-state protein level of CAD-GFP was substantially increased (5–8-fold) upon its coexpression with ICAD-myc (Fig. 2 D; lanes 1 and 2), suggesting that the biosynthesis of CAD-GFP, similarly to HA-CAD and CAD, requires the chaperone function of ICAD. Because most of the endogenous ICAD is associated with endogenous CAD, the folding of CAD-GFP is severely impaired in the absence of ICAD. Third, rapid degradation of both endogenous and exogenous ICAD was observed in STS-treated HeLa cells, regardless of whether the CAD/ICAD heterodimer contained CAD-GFP or endogenous CAD (Fig. 2, A and D). STS induced ∼50% degradation of the endogenous ICAD and >50% of ICAD-myc after 2 and 1.5 h incubation, respectively (Fig. 2, A and D), demonstrating that CAD-GFP chimera does not interfere with the caspase-3–dependent proteolysis of ICAD. Fourth, permeabilization of the cells by Triton X-100 triggered the complete release of CAD-GFP from dividing, but not from apoptotic cells, similarly to that observed for HA-CAD (Fig. 2, E and F). Fluorescent nuclei of CAD-GFP/ICAD-myc– or GFP-NLS–expressing cells permeabilized as above were scored. Although CAD-GFP was released from nearly all of the dividing cells, 90% of the apoptotic nuclei retained CAD-GFP but not GFP-NLS (Fig. 2 F). Finally, purified HA-CAD-GFP/ICAD-myc heterodimer from HeLa cells exhibited nuclease activity after the proteolytic cleavage of ICAD by caspase-3, suggesting that the fusion protein retained nucleolytic activity (Fig. 2 G). Together, these data suggest that CAD-GFP is an authentic surrogate of the native CAD.
The CAD/ICAD heterodimer is mobile in the nucleus of proliferating cells
To compare the dynamics of CAD-GFP/ICAD with freely moving GFP and GFP-NLS in live cells, we measured their one-dimensional diffusion coefficient and mobile fraction by FRAP technique in HeLa cells. More than 80% of the initial fluorescence of CAD-GFP as well as GFP-NLS was recovered in 2–3 s, whereas complete recovery of GFP was attained in <1 s after the bleach (Fig. 3, A and B). CAD-GFP diffusional coefficient (DCAD-GFP = 1.34 ± 0.20 μm2/s; n = 32, mean ± SEM) was comparable to that of GFP-NLS (DGFP-NLS = 0.94 ± 0.10 μm2/s; n = 47), but was 6.6-fold slower than GFP (DGFP = 8.95 ± 0.95 μm2/s; n = 36) (Fig. 3 C). In comparison, the diffusional coefficient of histone H1 (H1.1-GFP,DH1.1GFP = 0.019 ± 0.003 μm2/s; n = 20) was ∼70-fold slower (Fig. 3, A and C). The slow mobility of H1.1-GFP confirms previously published results (Misteli et al., 2000; Kimura and Cook, 2001).
Figure 3.
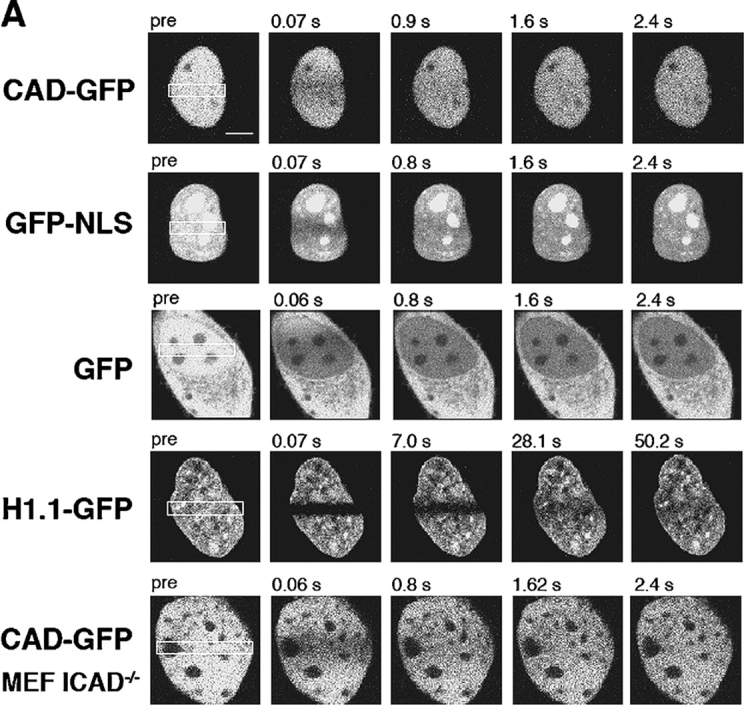


Nuclear dynamics of CAD in dividing HeLa cells. (A) Photobleaching of nuclear CAD-GFP, GFP-NLS, GFP, and H1.1-GFP in transiently transfected HeLa and MEF ICAD−/− cells. FRAP was performed as described in Materials and methods. Images show single x-y sections of the nucleus and were obtained before (pre) and after the bleach at indicated times. Although the laser intensity was sufficient to cause complete loss of fluorescence in PFA fixed cells, partial recovery occurred during the first scan due to the rapid lateral diffusion of the GFP chimeras. (B) Quantitative FRAP. Normalized fluorescence recovery was plotted as a function of time. Negligible recovery occurs on PFA-fixed HeLa cells (+PFA). (C) Diffusional coefficients of GFP and GFP chimeras in the nucleus of HeLa cells. The recovery curves were fitted according to a one-dimensional diffusion formula described in Materials and methods. Data are means ± SEM; n is indicated in brackets. (D) FLIP of CAD-GFP and GFP in HeLa cells. A 2-μm circle in the nucleus of HeLa cells expressing CAD-GFP or GFP was bleached in every 10 s. The entire nucleus was imaged after each bleach. Scans of the remaining fluorescence in the whole nucleus are shown for CAD-GFP– and GFP-expressing cells for the indicated times after the first bleach. Bar, 5 μm. (E) Quantitative FLIP. The ratio of the average fluorescence intensity of the half nucleus opposite to the bleach spot relative to the prebleach image was plotted at each time point. (F) Diffusional coefficients of CAD-GFP/ICAD in the nucleus of HeLa, BHK, MCF7, and MEF ICAD−/− cells. Diffusional coefficients were calculated as described in C. Data are means ± SEM.
To elucidate whether the entire nuclear pool of CAD-GFP/ICAD is mobile, fluorescence loss induced by photobleaching (FLIP) was performed. Repeated spot photobleaching completely depleted the fluorescence of CAD-GFP and GFP-NLS at comparable rates in the entire nucleus (Fig. 3, D and E; Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200404105/DC1).
The possibility that high-affinity binding sites are saturated by the heterologously expressed CAD/ICAD complex was tested in MEFs, derived from ICAD−/− mouse, lacking endogenous CAD/ICAD (Zhang et al., 1998). Quantitative FRAP analysis showed that the CAD-GFP/ICAD has a comparable diffusion constant (DCAD-GFP = 2.6 ± 0.50 μm2/s) in MEF ICAD−/− to that in BHK, HeLa, and MCF7 cells together with an absence of detectable size of immobile pool (Fig. 3 F and unpublished data). Finally, short-term ATP depletion of HeLa cells has only a marginal effect on the diffusional coefficient of CAD-GFP (DCAD-GFP = 0.60 ± 0.03 μm2/s; n = 10) as well as GFP-NLS (DGFP-NLS = 0.62 ± 0.04 μm2/s; n = 10), suggesting that the rapid movement of the CAD/ICAD complex is independent of metabolic energy. Although the possibility that CAD/ICAD undergoes a transient association with nuclear substrates cannot be precluded, the mobility measurements and the permeabilization studies suggested that CAD/ICAD, similarly to GFP-NLS, diffuses relatively freely in contrast to the slowly moving histone H1 and chromatin in proliferating cells.
Immobilization of CAD in apoptotic nuclei
The nuclear retention of both HA-CAD and CAD-GFP in permeabilized apoptotic cells is consistent with the notion that the diffusion of CAD became restricted during apoptosis (Fig. 1 A and Fig. 2 E). To further characterize this process, FRAP measurements were performed by spot photobleaching on HeLa nuclei. After an initial lag period (1–1.5 h), the immobile fraction of CAD-GFP progressively increased in the nucleus of a single cell (Fig. 4, A and B). To verify that CAD immobilization requires the activation of the apoptotic enzyme cascade, the translocation of phosphatidylserine into the outer leaflet of the plasma membrane was measured simultaneously with CAD mobility on STS-treated cells. Remarkably, the immobilization of CAD was observed only in those cells that displayed annexin V staining and plasma membrane blebbing (Fig. 4, C and D). To confirm this notion, the mobility of CAD was determined on a large number cells after induction of apoptosis, as well as in cells deficient of functional caspase-3.
Figure 4.
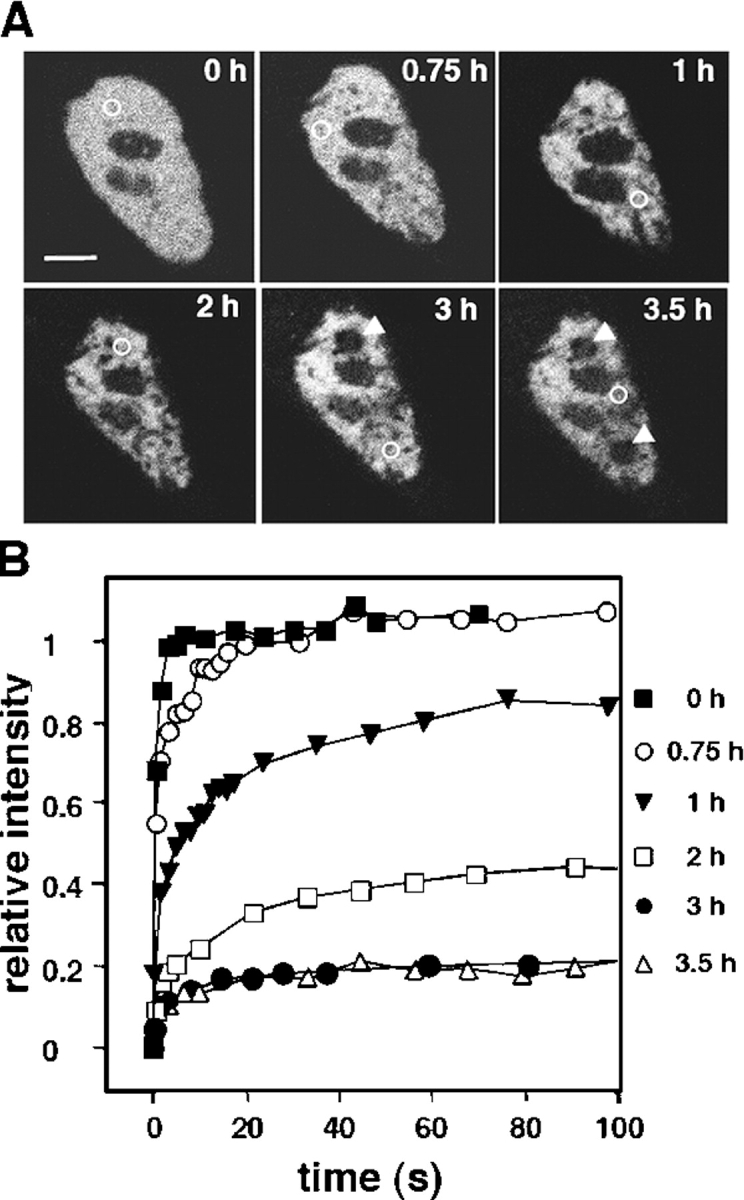
Immobilization of CAD in apoptotic nucleus. The mobility of CAD-GFP was monitored by spot photobleaching in the nucleus of transiently transfected HeLa cells exposed to STS for the indicated time. (A and B) FRAP of a single nucleus at different times of apoptosis. (A) Single x-y optical sections of nucleus. The circles indicate the bleached area. Bar (A), 5 μm. The corresponding recovery curves are plotted in B. Note that after 2 h of apoptosis, recovery in the bleached areas is negligible (indicated by arrowheads). (C and D) Phosphatidylserine translocation and CAD-GFP mobility. FRAP was performed as in A on cells incubated with Alexa 594–labeled annexin V. Cells were visualized by differential interference contrast microscopy. The corresponding recovery curves are shown in D. Bar (C), 10 μm.
As illustrated by the time-lapse images of CAD-GFP recoveries (Fig. 5 A) and the statistical analysis, STS provoked a modest immobilization of CAD during the first 2 h of apoptosis (Fig. 5, B and C). Extending the incubation time to 3 h increased the t1/2 by >20 fold (t1/2 = 23 ± 4.6 s; n = 18) relative to the control (t1/2 = 1.0 ± 0.05 s; n = 39) (Fig. 5 B). A fraction of CAD (40 ± 4.2%; n = 20) became immobile between 2–3 h of apoptosis induction (Fig. 5 C). Extending the incubation up to 3 h immobilized >80% of CAD-GFP (Fig. 5 C). Similar nuclear immobilization was observed in BHK and MEF ICAD−/− cells, as well as using the mouse CAD-GFP fusion protein, suggesting that CAD immobilization is cell- and species-independent (unpublished data). Inhibition of CAD diffusion was not restricted to specific regions, but affected the entire nucleus, as demonstrated by FLIP analysis (Video 1 and Fig. S2, A and B; available at http://www.jcb.org/cgi/content/full/jcb.200404105/DC1). In contrast, the diffusional mobility of GFP-NLS was not altered in apoptotic cells, as shown by the rapid fluorescence recovery rate and insignificant immobile fraction of GFP-NLS after 3 h of STS treatment (Fig. 5, B and C; Fig. S2). The lack of GFP-NLS immobilization cannot be attributed to the slower onset of nuclear apoptosis because a negligible effect on the mobility of GFP-NLS was observed in cells coexpressing HA-CAD/ICAD or using the DNase-deficient CADH242N-GFP fusion protein (unpublished data and see following paragraph).
Figure 5.
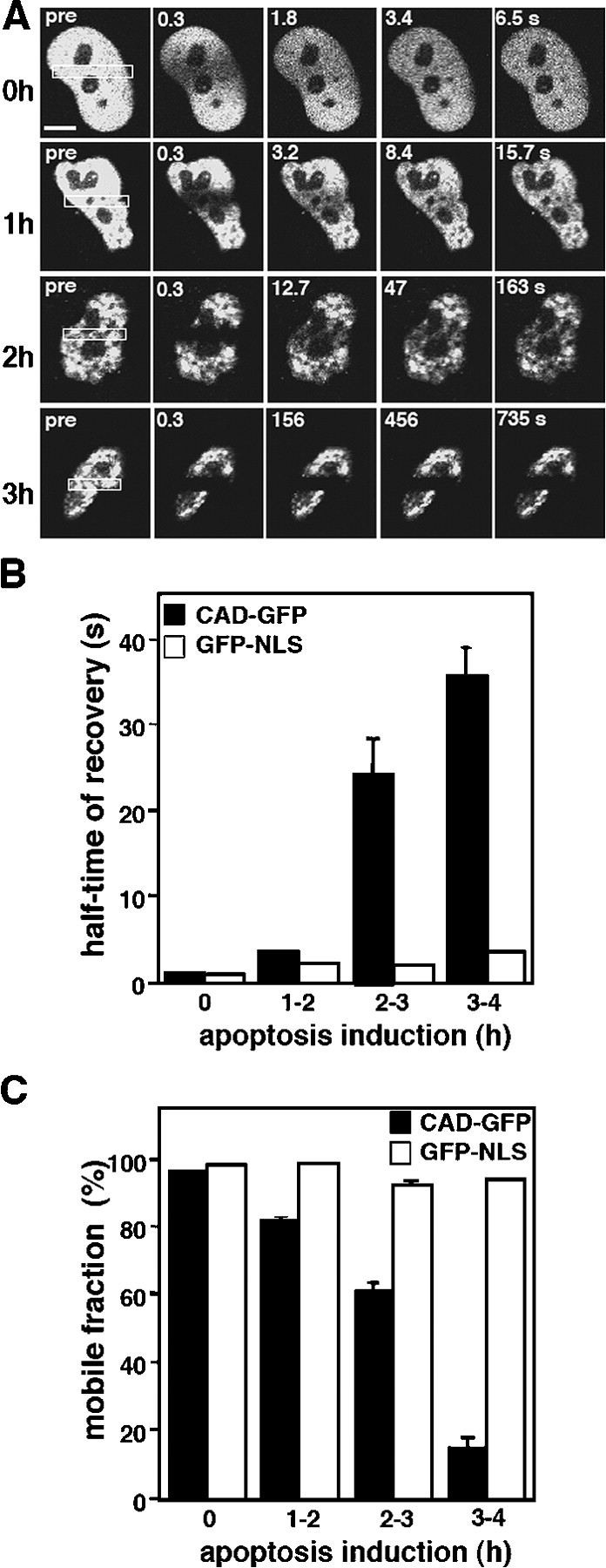
The diffusional mobility of CAD-GFP and GFP-NLS in HeLa cells undergoing apoptosis. FRAP experiments and confocal imaging were conducted as described in Fig. 3. (A) Qualitative FRAP during apoptosis. Representative nuclei of different cells, expressing CAD-GFP, were imaged before and during the recovery after photobleaching the boxed area. Near-complete immobilization of CAD-GFP was observed after 3 h of STS treatment. Bar, 5 μm. (B) Half-time of fluorescence recoveries and (C) immobile fractions of CAD-GFP and GFP-NLS after STS exposure for the indicated duration. Half-time of recoveries were calculated from normalized recovery curves of 30–40 individual cells for each time interval. The mobile fraction was calculated as the percentage of corrected total fluorescence intensity reached during the recovery phase (see Materials and methods).
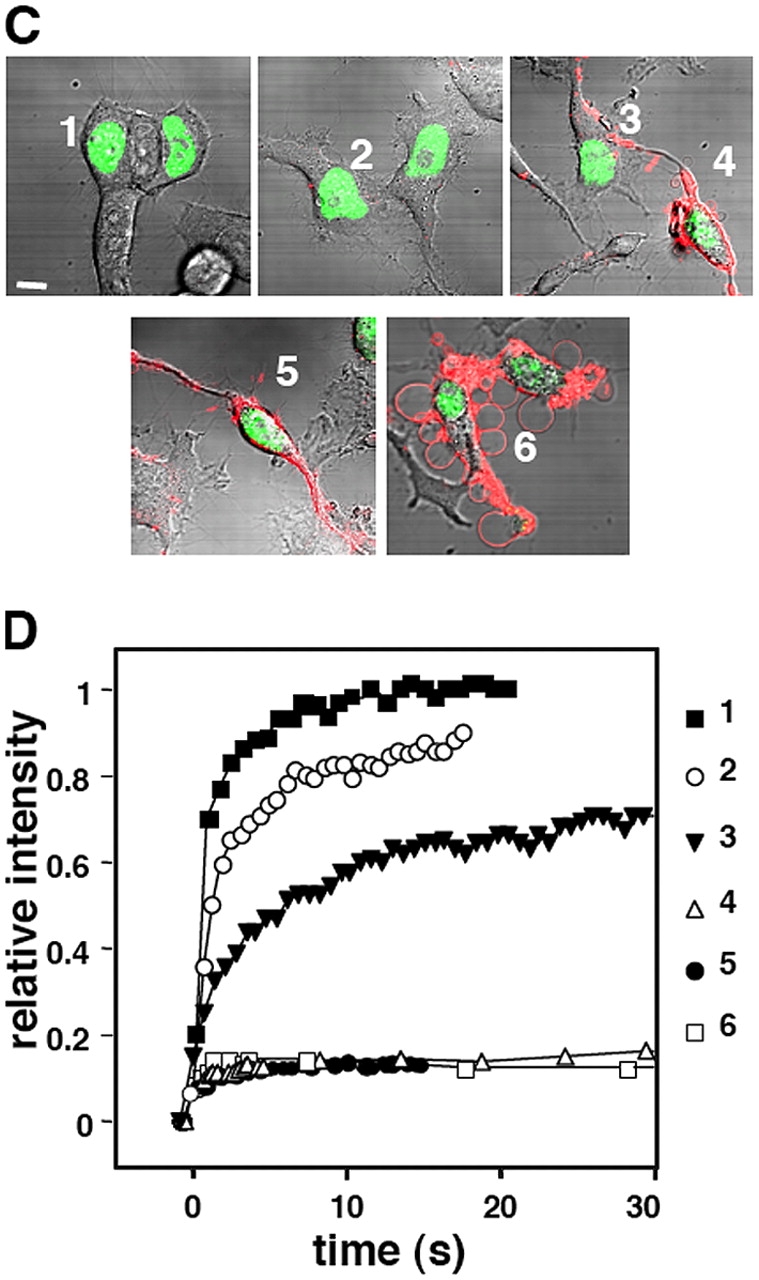
Several lines of evidence suggested that the immobilization of CAD required caspase-3 activation: (1) treatment of the cells with the pan-caspase inhibitor Z-VAD-fmk, or with the caspase-3–specific inhibitor DEVD-CHO abolished the immobilization of CAD upon STS treatment (Fig. 6 A); (2) the diffusional mobility of CAD was preserved in STS-treated MCF7 cells that lack functional caspase-3 (Fig. 6 B, top; Fig. 6 C and Video 1), but CAD-GFP was immobilized in MCF7 cells expressing functional caspase-3 (Fig. 6 B, bottom; Fig. 6 C); (3) immobilization of activated CAD is not restricted to cells undergoing STS-induced apoptosis: CAD was also immobilized to comparable extent in HeLa cells subjected to UV irradiation or CHX and TNFα treatment (Fig. S3, B, C, F, and G; available at http://www.jcb.org/cgi/content/full/jcb.200404105/DC1). Both inducers provoked the degradation of ICAD (Fig. S3, A and E) and their effects on CAD immobilization were prevented in the presence of the caspase-3 inhibitor DEVD-CHO (Fig. S3, D and H), implying that CAD activation occurred via the caspase-3–dependent pathway. Collectively, these results suggest that immobilization of CAD-GFP is a common feature of nuclei undergoing caspase-3–dependent apoptosis regardless of the upstream signaling mechanism.
Figure 6.


The immobilization of CAD in the apoptotic nucleus is caspase-3 dependent. (A) Effect of caspase inhibitors on CAD-GFP diffusional mobility. HeLa cells were exposed to STS for 3 h in the presence of DEVD-CHO (20 μM) or ZVAD-fmk (50 μM). Immobile fraction was determined by FRAP as described in Fig. 5. (B and C) CAD-GFP is immobilized only in exogenous caspase-3–expressing MCF7 cells. (B) x-y optical sections of MCF7 cells, cotransfected with CAD-GFP, ICAD-myc, and procaspase-3. Cells were incubated for 3.5 h with STS before the caspase-3 substrate Red-DEVD-fmk was added to the medium to identify cells containing only CAD-GFP (−casp3) or both CAD-GFP and active caspase-3 (+casp3). CAD-GFP mobility was measured by FLIP as described in Fig. 3 D. Bar, 10 μM. (C) Results from B were plotted as in Fig. 3 D. Control represents a nontreated MCF7 cell.
The mechanism of CAD immobilization in apoptotic nuclei
Four possible mechanisms were considered to account for CAD immobilization in apoptotic nuclei. The first possibility that increased viscosity of the nucleosol impedes the diffusion of CAD-GFP could be ruled out based on the observation that neither the immobile fraction nor the recovery kinetics of GFP-NLS and GFP were significantly altered in apoptotic HeLa cells (Fig. 5, B and C; Fig. S2). The second scenario that apoptosis facilitates the complex formation of CAD-GFP with chromatin could be also discounted because neither CAD-GFP nor CAD-HA colocalized with the condensed chromatin of apoptotic cells (Fig. 1, A and C; Fig. 2 E). The subnuclear distribution of CAD was distinct from endogenous histones (Fig. 1, compare A, E, and C). In addition, complete digestion of the chromosomal DNA by DNase did not cause the liberation of CAD from apoptotic nuclei (see following paragraph). Finally, replacement of His242 by Ala, an amino acid residue critical for DNA binding of CAD (Meiss et al., 2001), failed to prevent the immobilization of CADH242A-GFP (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200404105/DC1), confirming that direct DNA binding of the nuclease is not required.
As a third hypothesis, pronounced homo-oligomerization of CAD as reported with the recombinant DNase previously (Liu et al., 1999; Meiss et al., 2001; Widlak and Garrard, 2001; Zhou et al., 2001) into a high molecular weight complex was also ruled out because CAD oligomers were not detected by size-exclusion chromatography of apoptotic cell extracts (unpublished data).
Finally, the hypothesis that CAD binds to the chromatin-depleted subnuclear compartment, also called interchromatin space or nuclear matrix, was tested. Nuclear matrix preparation was obtained by the combination of successive detergent extraction, DNase digestion, and high salt washes of HeLa cells as described previously (Nickerson et al., 1997). Although CAD was largely depleted from the nuclear matrix preparation of nonapoptotic cells, a significant fraction of CAD-GFP remained associated with nonchromatin proteins of apoptotic cells according to immunoblot analysis (Fig. 7 A). The association of HA-CAD as well as CAD-GFP with nuclear interchromatin structures could be confirmed at the morphological level by fluorescence microscopy in apoptotic cells (Fig. 7, B and C), but not in control cells. CAD remained nuclear upon the complete digestion of the chromosomal DNA by DNase, verified by immunostaining of DAPI-negative nuclei (Fig. 7, B and C). In contrast, nuclear matrix preparations failed to retain GFP-NLS, visualized either by fluorescence microscopy or by immunoblotting (unpublished data). These observations are consistent with the notion that CAD, but not the heterodimeric CAD/ICAD, preferentially binds to the nuclear matrix of apoptotic cells.
Figure 7.
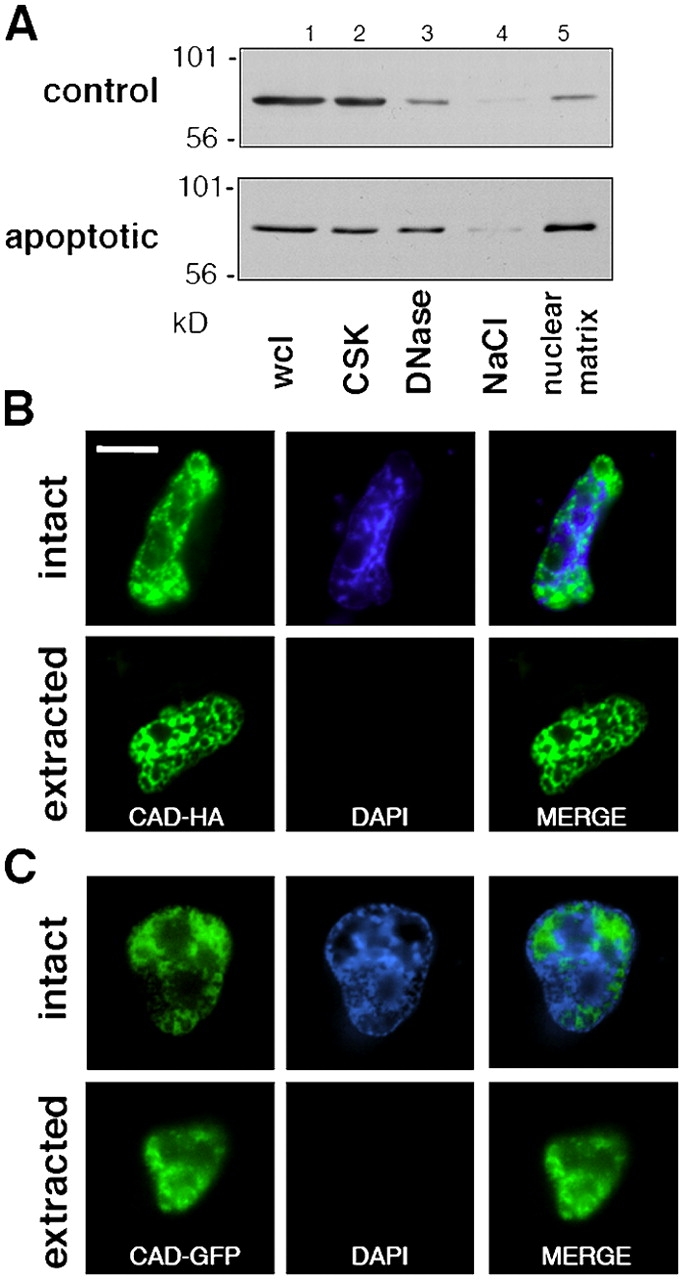

Association of CAD with the nuclear matrix in apoptotic HeLa cells. (A) Biochemical isolation of nuclear matrix from cells expressing the CAD-GFP/ICAD-myc complex. Apoptosis was induced with STS for 2 h. HeLa cells were biochemically fractionated and the CAD-GFP content of each fraction was visualized by anti-GFP immunoblotting. Wcl, whole-cell lysate (lane 1); CSK, extraction with the cytoskeletal buffer (lane 2); DNaseI digestion (lane 3); NaCl, high salt extraction (lane 4); and nuclear matrix proteins (lane 5). (B and C) In situ isolation of the nuclear matrix was performed as in A on HeLa cells grown on glass coverslips. CAD-HA and chromatin was visualized by indirect immunostaining and with DAPI, respectively. Single x-y optical sections are shown. (D) Colocalization of CAD-GFP and CAD-HA with NuMA. Apoptosis was induced with STS for 2 h. HeLa cells were immunostained with anti-HA and anti-NuMa antibodies. (E) Colocalization of CAD-HA with NuMA. Immunostaining was performed as in D. Inset shows a magnification of the images captured with a fluorescence microscope (DMI RE2; Leica), deconvolved, and analyzed using OpenLab software (Improvision). Single images were selected for presentation. Bars, 10 μm.
To confirm our hypothesis that CAD is tethered to the nuclear matrix, coimmunostaining of CAD and nuclear mitotic apparatus protein (NuMA), a component of the nuclear matrix (Fey et al., 1991), was performed. A significant overlap in the nuclear localization of NuMA and CAD-GFP, as well as CAD-HA, could be demonstrated by deconvolution fluorescence microscopy (Fig. 7, D and E; inset). Although these experiments are unable to distinguish between direct and indirect binding of CAD to the nuclear matrix, the results are consistent with the in vivo FRAP measurements, indicating that activated CAD associates with virtually immobile component(s) of apoptotic nuclei.
The NH2 terminus is required for nuclear immobilization of CAD
To elucidate the signal responsible for CAD association with the nuclear matrix, progressive COOH- and NH2-terminal truncations of CAD were generated and fused in frame to EGFP (Fig. 8 A), and the mobility of the chimeras was measured. Truncated variants of human CAD were designed based on sequence alignments of the human, mouse, chicken, and Drosophila CAD, respecting putative domain boundaries (Mukae et al., 2000) (Fig. 8 A). Western blot analysis verified the expected molecular mass of the truncated CAD-GFP fusion proteins (Fig. 8 B). The nuclear mobility of truncated CAD was assayed in proliferating and apoptotic HeLa cells. Four of the nine fusion proteins that were immobilized in STS-treated HeLa cells contained amino acid residues between 84 and 152 of CAD (CAD[1–152], [1–278], [84–152], and [84–203]) (Fig. 8, A and C). In contrast, fusion proteins incorporating peptide fragments of the COOH terminus of CAD ([152–203], [203–278], [278–338], and [203–338]) or the CIDE-N domain remained as mobile as GFP-NLS in apoptotic cells (Fig. 8 A and unpublished data). The partial immobilization of CAD[84–152] and CAD[84–203] was potentiated in the presence of the CIDE-N domain because both CAD[1–152] and CAD[1–278] were immobile in apoptotic nuclei (Fig. 8 C). These results suggest that the NH2-terminal 152 amino acid residues are necessary for the immobilization of CAD.
Figure 8.

Localization of the nuclear matrix targeting motif in CAD. (A) Schematic pictures of deletion mutants of CAD fused to the NH2 terminus of EGFP. All constructs were freely mobile in nonapoptotic cells. The relative mobilities of the truncated CAD in STS-treated cells are indicated by the size of the immobile pool on the right, representing at least 10 independent measurements for each construct. No or negligible effect, −; partial immobilization, +; and complete immobilization, ++. (B) Mutant CAD-GFP fusion proteins were transiently expressed in HeLa cells. The apparent molecular mass of fusion proteins was assessed by immunoblotting using an anti-GFP antibody. (C) FRAP studies of selected CAD-GFP fusion proteins. The mobility of the indicated fusion proteins was determined with FRAP as described in Fig. 3, before and after STS (2 h) treatment. Representative categories of the CAD chimera in apoptotic cells are demonstrated. CIDE-GFP, marginal inhibition of mobility (left); CAD[84–152]-GFP, partial immobilization (middle); and CAD[1–152], complete immobilization (right).
The possible role of CAD immobilization in apoptotic cells
It is well documented that the integrities of the plasma and nuclear membranes are compromised during the late stages of apoptosis (Faleiro and Lazebnik, 2000; Tzung et al., 2001). We hypothesized that nuclear immobilization of the DNase prevents its extracellular release. To test this assumption, we measured the accumulation of CAD in the extracellular compartment under conditions that interfered with its nuclear immobilization.
To prevent the immobilization of CAD, we took advantage of our previous observation demonstrating that deletion of the NLS in both CAD and ICAD caused the relocation of the CAD-ΔNLS/ICAD-ΔNLS complex into the cytoplasm and prevented nuclear uptake of CAD-ΔNLS (Lechardeur et al., 2000). Importantly, neither the dimerization nor the activation kinetics of CAD was altered by deleting the two NLSs (unpublished data). The extracellular liberation of HA-CAD from apoptotic HeLa cells expressing either CAD/ICAD or CAD-ΔNLS/ICAD-ΔNLS was determined by immunoblotting after the immunoprecipitation of the extracellular CAD with anti-HA antibody (Fig. 9). A significant fraction of CAD-ΔNLS, but not CAD, leaked into the medium from HeLa cells undergoing STS-induced apoptosis (Fig. 9, lanes 3 and 6). The absence of myc-tagged ICAD in the immunoprecipitates confirmed that the proteolysis of ICAD-myc preceded the extracellular egress of CAD (Fig. 9, bottom). Thus, nuclear entrapment of CAD may have a critical role in preventing the extracellular release of the active DNase from apoptotic cells.
Figure 9.

Interfering with nuclear entrapment of CAD facilitates its extracellular release from apoptotic cells. (A) HeLa cells, transiently expressing the mouse HA-CAD/ICAD-myc or the HA-CADΔNLS/ICADΔNLS-myc complex were exposed to STS for 2.5 h in serum-free medium. CAD was immunoprecipitated from both the medium (med) and the cell lysate (cell) with anti-HA antibody. The precipitates and the cell lysates (lys) were probed with anti-CAD and anti-myc antibodies to visualize exogenous CAD and ICAD. (B) As a control, HeLa cells were transfected with the HA-CADΔNLS/ICADΔNLS-myc and were incubated overnight in serum-free medium. CAD was then immunoprecipitated from the medium and probed with the indicated antibodies.
Discussion
The experiments presented in this paper were designed to determine the molecular dynamics of CAD in living cells, critical for our understanding of the regulation of the most important apoptotic nuclease. In the nucleus of nonapoptotic cells, the CAD/ICAD heterodimer was found to be as mobile as the nuclear targeted EGFP, an archetype of freely diffusing proteins (Houtsmuller et al., 1999; Kruhlak et al., 2000). The fast diffusional movements of CAD/ICAD were preserved after the acute ATP depletion of HeLa cells, as well as in MEF ICAD−/− cells, deficient of the endogenous CAD/ICAD complex (Zhang et al., 1999). These observations, together with the lack of discernible immobile pool of CAD/ICAD, imply that the cellular dynamics of the heterodimer could be ascribed as passive diffusion. Free diffusion could optimize the probability of activation of CAD/ICAD by providing an efficient scanning mechanism for caspase-3 in the nuclear space, as reported for several other nuclear proteins (Pederson, 2001; Carmo-Fonseca et al., 2002). Free diffusion would also ensure efficient catalytic binding of CAD to the immobile chromatin during the initial phase of apoptosis. The high mobility of the heterodimer is at variance with previous suggestions, invoking high-affinity interactions of CAD/ICAD with slowly moving or relatively immobile (e.g., histone-H1 and chromatin) nuclear constituents (Liu et al., 1998, 1999; Toh et al., 1998; Durrieu et al., 2000; Zhang and Xu, 2002). More surprisingly, the molecular dynamics of CAD undergo dramatic changes upon apoptosis. Data obtained by both biochemical and biophysical techniques indicated that the diffusional mobility of CAD is impeded in apoptotic nuclei. In contrast, the diffusion of GFP-NLS and other nuclear proteins, such as HMG N1, N2, and the transcription factor NF1, were preserved (Stenoien et al., 2000; Scaffidi et al., 2002). The activation of caspase-3 seems to be necessary for the immobilization of CAD. Whether ICAD cleavage by caspase-3 is sufficient or the modifications of other nuclear constituents are also required for CAD immobilization remains to be determined.
Biochemical fractionation and immunocolocalization studies with NuMA suggested that CAD associates with the nonchromatin subnuclear compartment, defined as the nuclear matrix in apoptotic cells (Fey et al., 1991; Nickerson et al., 1997). This conclusion is consistent with the observation that the nuclear retention of CAD was compromised in the absence of oxovanadium-modified ribonucleosides (unpublished data), a competitive ribonucleosidase inhibitor that is an efficient stabilizer of the nuclear matrix (Stenoien et al., 2001).
The nuclear matrix is proposed to consist of the nuclear envelope and a meshwork of nonchromatin nuclear proteins (Nickerson, 2001). The subnuclear compartment is involved in multiple nuclear functions such as chromatin dynamic organization and metabolism, RNA processing and regulation, or steroid hormone action (Nickerson, 2001; Heng et al., 2004). Modulation of protein partitioning between the nuclear matrix, in an immobilized nonfunctional state, and the nucleoplasm, in a mobile functional state, may constitute a fundamental regulatory mechanism. Short stretches of amino acids as well as large segments of polypeptides can serve as nuclear matrix targeting signals (NMTS) (Nickerson, 2001). Because no consensus NMTS could be identified in CAD, deletional mutagenesis was used to show that the NMTS encompasses aa [84–152] of CAD, connecting the regulatory CIDE domain and the COOH-terminal catalytic domain.
Dissociation of the heterodimer, by itself, seems likely to be insufficient for targeting CAD to the nuclear matrix considering the following observations. Immunoblot analysis showed that at least 50% of ICAD was degraded during the first 1.5 h of apoptosis, expressing the CAD-HA/ICAD-myc or the CAD-GFP/ICAD-myc complex (Fig. 2, B and E). In contrast, 50% of the CAD-GFP became immobile only after 3 h of STS exposure (Fig. 5 C). The degradation of ICAD presumably is completed shortly after its initiation at the single-cell level, similar to that described for various caspase-3 substrates using the FRET technique (Takemoto et al., 2003). The extensive DNA ladder formation in STS-treated HeLa cells also supports the notion that DNA fragmentation appears to take place before the immobilization of CAD (Fig. 2 C). Considering the degradation kinetics of ICAD are faster than the immobilization of CAD, it is conceivable that besides the dissociation of the CAD/ICAD heterodimer, conformational and/or compositional changes of binding partners are also required for nuclear entrapment of CAD.
If CAD diffusion is a prerequisite for its catalytic activity on internucleosomal chromosomal regions of the virtually immobile chromatin, our results suggest that CAD activity is limited to a relatively short period of time. Thus, CAD would primarily be responsible for the cleavage of the most susceptible A/T reach, matrix attachment regions of chromatin and the induction of genomic DNA condensation by destabilizing the chromatin structure, rather than for complete nucleosomal fragmentation.
Recent evidence suggests that nucleosomal fragmentation by CAD is indeed not an absolute requirement for the elimination of apoptotic nuclei. Kawane et al. (2003) demonstrated that phagocytosis plays an important role in the degradation process of apoptotic bodies derived from CAD−/− thymocytes (see also Nagata et al., 2003). Although most of the apoptotic cells are cleared by phagocytosis before losing membrane integrity, apoptotic bodies may escape their degradation and thus provoke an immune response. Supporting this notion, impeding nuclear apoptosis, as well as the degradation of apoptotic nuclei, provokes the activation of innate immunity in DNase II−/−CAD−/− mice (Nagata et al., 2003).
The accumulation of nuclear proteins (e.g., core histones) has been demonstrated not only in the cytosol, but also in the extracellular compartment as well when the phagocytic capacity of the organism is saturated by the number of apoptotic lymphocytes (Gabler et al., 2003). This process leads to the production of auto-antibodies against dsDNA and/or histones in diseases such as the autoimmune lymphoproliferative syndrome (Chun et al., 2002) and lupus (Dieker et al., 2002). These observations suggest that the nuclear–cytoplasmic barrier can be compromised in apoptotic cells (Faleiro and Lazebnik, 2000) and that specific mechanisms are in place to retain potentially harmful proteins in those cells that undergo programmed cell death. Recent observations showed that the pro-inflammatory transcription factor HMG B1 follows this paradigm. Although HMG B1 is bound loosely to chromatin in intact cells, it becomes tightly tethered to the condensed chromatin (Scaffidi et al., 2002). As a consequence, the extracellular release of HMG B1 is limited from apoptotic bodies, minimizing a systemic immune response (Bianchi and Manfredi, 2004). Finally, Brinkmann et al. (2004) have recently shown that activated neutrophils release proteins and chromatin fragments, forming an extracellular mesh called the neutrophil extracellular trap (NET), which has a bactericidal effect. This structure seems to be essential in the inflammatory response. The NET has been shown to be disintegrated by exogenous DNaseI. Release of the active DNase by apoptotic cells in the extracellular compartment could consequently compromise the integrity and bactericidal activity of the NET. In conclusion, we propose that retention of activated CAD in apoptotic nuclei may represent a safeguard against the accumulation of active DNase in the extracellular compartment.
Materials and methods
Plasmid construction
The cDNA, encoding the human and mouse CAD, were provided by Dr. R. Halenbeck (Chiron Corp., Emeryville, CA) and Dr. S. Nagata (Osaka University Medical School, Osaka, Japan), respectively. The cDNA of human ICAD was cloned as described previously (Lechardeur et al., 2000). The expression plasmid encoding the HA-CAD-EGFP fusion protein (CAD-GFP) was constructed by inserting the coding region of the HA-CAD into pEGFP-N1 plasmid (BD Biosciences). Other epitope-tagged CAD and ICAD were obtained as described previously (Lechardeur et al., 2000). Nuclear-targeted EGFP (GFP-NLS) was derived from pECFP-NUC (BD Biosciences). CADH242N-GFP and CADH242A-GFP, defective of nuclease activity and DNA-binding and nuclease activity, respectively, were constructed by overlapping PCR mutagenesis (Meiss et al., 2001). COOH- and NH2-terminal truncations of CAD were obtained by PCR and then were subcloned into pEGFP-N1 plasmid. The histone H1.1-GFP and the procaspase-3 vectors were gifts from Dr. J. Th'ng (Northwestern Ontario Regional Cancer Center, Thunder Bay, Ontario, Canada) and Dr. D. Andrews (McMaster University, Hamilton, Ontario, Canada), respectively. All constructs were verified by dideoxy chain termination DNA sequencing.
Cell culture, transfection, and induction of apoptosis
HeLa, COS-1, MCF7, and BHK-21 cells were grown in DME supplemented with 10% FBS at 37°C under an atmosphere of 5% CO2. MEFs were isolated from ICAD knockout (ICAD−/−) (Zhang et al., 1999). Transfections were performed with FuGENE (Roche) and harvested after 24–48 h. To limit the expression of CAD and to ensure that the exogenous ICAD is complexed with CAD, the ratio of CAD/ICAD plasmid DNA was 2:1 in transient transfections. Immunostaining of cells expressing HA-CAD/ICAD-myc by the polyclonal anti-ICAD antibody indicated that ICAD-myc was approximately fivefold overexpressed relative to the endogenous ICAD level. In light of the obligatory heterodimerization of CAD, this implies that CAD was not more than 4–5-fold overexpressed relative to its endogenous level. ATP depletion was achieved by incubating the cells in glucose-free medium with 4 μg/ml anti-mycin, 10 mM deoxy-d-glucose, and 10 μM carbonyl cyanide p-chlorophenyl hydrazone at 37°C.
Immunocytochemistry
Cells were fixed in 4% PFA and HA, Myc, and ICAD were visualized by indirect immunostaining as described previously (Lechardeur et al., 2000). NuMA was detected with anti-NuMA mAb (Oncogene Research Products) and histones with anti-histone mAb (Roche).
Apoptosis assays
Soluble DNA fragments were isolated according to the method described by Cao et al. (2001) and visualized on agarose gel by ethidium bromide staining. Phosphatidylserine translocation was monitored by incubating the cells with AlexaFluor 568–conjugated annexin V (Molecular Probes, Inc.), and activated caspase-3 was detected with the fluorescent red-DEVD-fmk caspase-3 inhibitor (Oncogene Research Products) incubated for 30 min preceding time-lapse imaging and FRAP analysis.
Immunoblotting and coimmunoprecipitation
Immunoblottings were performed on cells transiently transfected with the indicated CAD and ICAD expression vectors (Lechardeur et al., 2000). The CAD-GFP/ICAD was coimmunoprecipitated from the cell lysates using a polyclonal anti-GFP antibody (Molecular Probes, Inc.) or monoclonal anti-HA (Covance) and protein G–Sepharose. The Myc and HA epitopes were detected with monoclonal anti-epitope (Covance) and HRP-conjugated secondary antibodies (Amersham Biosciences). CAD and ICAD were immunoblotted with a rabbit anti-DFF40 (StressGen Biotechnologies) and anti-ICAD (Lechardeur et al., 2000) antibodies, respectively.
Nuclear matrix isolation
Nuclear matrix from plated cells was prepared in cytoskeletal buffer (10 mM Pipes, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, and 0.5% Triton X-100, pH 6.8) according to published protocol (Stenoien et al., 2001). For immunostaining, the nuclear matrix preparation was fixed in 4% PFA. Immunoblotting was performed on isolated nuclear matrix solubilized with RIPA buffer.
Confocal microscopy
Photobleaching experiments were performed on cells expressing the minimum amount fluorescence permitting the measurements. For FRAP and FLIP analysis, transfected cells were grown on 35-mm glass coverslips and kept in a thermostated chamber (Molecular Probes, Inc.) in bicarbonate-free α-MEM supplemented with 10 mM Hepes, pH 7.2, at 37°C during the measurements. Microscopy was performed with an inverted laser confocal fluorescence microscope (LSM 510; Carl Zeiss MicroImaging, Inc.) equipped with a Plan-Apochromat 63×/NA 1.4 objective and the FITC filter setting (488 nm/515 nm). A single x-y optical section was acquired with a 6× zoom setting, and photobleaching was performed using the 488-nm line of an argon laser at 100% power. Images were acquired at 0.1% attenuated laser power. For quantitative FRAP experiments, the bleach was performed using a 2-μm width stripe across the nucleus with 1–2 iterations, enough to decrease the fluorescence of PFA-fixed cells to the background fluorescence. Typically, the bleach time duration was <200 ms. The 0 time point was defined as the mid-point of the bleach period. For quantitative FRAP analysis, the average fluorescence intensity in the bleached area was measured using the LSM 510 software. The total loss of fluorescence during the FRAP measurements was corrected by monitoring the fluorescence outside the bleach area by setting the prebleach intensity to 1 in the bleaching area, according to the equation: Ibl,corr(t) = Ibl(t)/Icont(t)) × (Icont(<0)/Ibl(<0)), where Ibl,corr(t) is the corrected intensity value in the bleached area, Ibl(t) and Icont(t) are the raw intensities in the bleached and background area, respectively, and Ibl(<0) and Icon(<0) are the intensities in the same regions of the prebleach scan. The one-dimensional diffusion coefficients were determined as the D value after fitting the corrected recovery curves according to the mathematical equation Ibl,corr(t) = 1 − (w2x(w2 + 4πDt)−1)1/2, where w is the width (2 μm) of the bleached stripe and t is the time. The time required for the recovery of half the fluorescence was determined from the noncorrected intensity values as the time corresponding to the intensity Ibl(t1/2) = Ibl(inf)/2 where Ibl(inf) is the value of the intensity when the recovery is completed and assuming that Ibl (0) = 0.
For FLIP experiments, a 2-μm-diam spot was bleached in every 10 s for 20–30 iterations using similar settings as for FRAP, and the whole nucleus was scanned after each pulse. Bleaching and image acquisition was repeated until the fluorescence signal of the nucleus was decreased to the background level. The ratio of the average fluorescence intensity of the half nucleus opposite to the bleaching spot at each time point divided by its average intensity in the prebleach image was plotted versus time, and represents the rate of fluorescence decay during the FLIP experiment. Data were analyzed with SigmaPlot and images were exported into Adobe Photoshop.
Online supplemental material
Fig. S1 shows FLIP in normal and apoptotic HeLa nuclei expressing CAD-GFP. GFP-NLS remains mobile in apoptotic nuclei as demonstrated by FRAP in Fig. S2. CAD-GFP immobilization is induced by UV or TNFα+CHX treatment of HeLa cells (Fig. S3). Point mutation of an amino acid residue that is essential for DNA binding does not prevent CAD immobilization (Fig. S4). Video 1 illustrates CAD-GFP diffusional mobility by FLIP in normal and apoptotic HeLa and MCF7 cells. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200404105/DC1.
Acknowledgments
The authors are indebted to D. Andrews, R. Halenbeck, S. Nagata, and J. Th'ng for providing expression plasmids. We thank David Bazett-Jones for helpful comments. This work was supported by a grant from the Canadian Institute of Health Research to G.L. Lukacs.
Abbreviations used in this paper: CAD, caspase-activated DNase; CIDE, cell-inducing DFF45-like effector; DFF, DNA fragmentation factor; FLIP, fluorescence loss induced by photobleaching; HMG, high mobility group; ICAD, inhibitor of caspase-activated DNase; MEF, mouse embryonic fibroblast; NuMA, nuclear mitotic apparatus protein; STS, staurosporine.
References
- Aravind, L., V. Dixit, and E. Koonin. 1999. The domains of death: evolution of the apoptosis machinery. Trends Biochem. Sci. 24:47–53. [DOI] [PubMed] [Google Scholar]
- Bianchi, M., and A. Manfredi. 2004. Chromatin and cell death. Biochim. Biophys. Acta. 1677:181–186. [DOI] [PubMed] [Google Scholar]
- Brinkmann, V., U. Reichard, C. Goosmann, B. Fauler, Y. Uhlemann, D. Weiss, Y. Weinrauch, and A. Zychlinsky. 2004. Neutrophil extracellular traps kill bacteria. Science. 303:1532–1535. [DOI] [PubMed] [Google Scholar]
- Cao, G., W. Pei, J. Lan, R.A. Stetler, Y. Lua, T. Nagayama, S.H. Graham, X.-M. Yin, R.P. Simon, and J. Chen. 2001. Caspase-activated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in cerebral ischemia and in neuronal cultures. J. Neurosci. 21:4678–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca, M., M. Platani, and J.R. Swedlow. 2002. Macromolecular mobility inside the cell nucleus. Trends Cell Biol. 12:491–495. [DOI] [PubMed] [Google Scholar]
- Chun, H.J., L. Zheng, M. Ahmad, J. Wang, C.K. Speirs, R.M. Siegel, J.K. Dale, J. Puck, J. Davis, C.G. Hall, et al. 2002. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 419:395–399. [DOI] [PubMed] [Google Scholar]
- Dieker, J.W., J. Van Der Vlag, and J.H. Berden. 2002. Triggers for anti-chromatin auto-antibody production in SLE. Lupus. 11:856–864. [DOI] [PubMed] [Google Scholar]
- Durrieu, F., K. Samejima, J.M. Fortune, S. Kandels-Lewis, N. Osheroff, and W.C. Earnshaw. 2000. DNA topoisomerase IIa interacts with CAD nuclease and is involved in chromatin condensation during apoptotic execution. Curr. Biol. 10:923–926. [DOI] [PubMed] [Google Scholar]
- Enari, M., H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu, and S. Nagata. 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitors ICAD. Nature. 391:43–50. [DOI] [PubMed] [Google Scholar]
- Faleiro, L., and Y. Lazebnik. 2000. Caspases disrupts the nuclear-cytoplasmic barrier. J. Cell Biol. 151:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey, E.G., P. Bangs, C. Sparks, and P. Odgren. 1991. The nuclear matrix: defining structural and functional roles. Crit. Rev. Eukaryot. Gene Expr. 1:127–143. [PubMed] [Google Scholar]
- Gabler, C., J.R. Kalden, and H.-M. Lorenz. 2003. The putative role of apoptosis-modified histones for the induction of autoimmunity in Systemic Lupus Erythematosus. Biochem. Pharmacol. 66:1441–1446. [DOI] [PubMed] [Google Scholar]
- Halenbeck, R., H. MacDonald, A. Roulston, T.T. Chen, L. Conroy, and L.T. Williams. 1998. CPAN, a human nuclease regulated by the caspase-sensitive inhibitor DFF45. Curr. Biol. 8:537–540. [DOI] [PubMed] [Google Scholar]
- Hanayama, R., M. Tanaka, K. Miwa, A. Shinohara, A. Iwamatsu, and S. Nagata. 2002. Identification of a factor that links apoptotic cells to phagocytes. Nature. 417:182–187. [DOI] [PubMed] [Google Scholar]
- Heng, H.H.Q., S. Goetze, C.J. Ye, G. Liu, J.B. Stevens, S.W. Bremer, S.M. Wykes, J. Bode, and S.A. Krawetz. 2004. Chromatin loops are selectively anchored using scaffold/matrix-attachment regions. J. Cell Sci. 117:999–1008. [DOI] [PubMed] [Google Scholar]
- Herrmann, M., R.E. Voll, O.M. Zoller, M. Hagenhofer, B.B. Ponner, and J.R. Kalden. 1998. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis. Rheum. 41:1241–1250. [DOI] [PubMed] [Google Scholar]
- Houtsmuller, A.B., S. Rademakers, A.L. Nigg, D. Hoogstraten, J.H.J. Hoeijmakers, and W. Vermeulen. 1999. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science. 284:958–961. [DOI] [PubMed] [Google Scholar]
- Kawane, K., H. Fukuyama, H. Yoshida, H. Nagase, Y. Ohsawa, Y. Uchiyama, K. Okada, T. Iida, and S. Nagata. 2003. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat. Immunol. 4:138–144. [DOI] [PubMed] [Google Scholar]
- Kimura, H., and P.R. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak, M.J., M.A. Lever, W. Fischle, E. Verdin, D.P. Bazett-Jones, and M.J. Hendzel. 2000. Reduced mobility of the ASF splicing factor through the nucleoplasm and steady state speckle compartment. J. Cell Biol. 150:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechardeur, D., L. Drzymala, M. Sharma, J. Pacia, C. Hicks, N. Usmani, D. Zylka, J. Rommens, and G. Lukacs. 2000. Determinants of the nuclear localization of the heterodimeric DNA fragmentation factor (DFF). J. Cell Biol. 150:321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., P. Li, P. Widlak, H. Zou, X. Luo, W.T. Garrard, and X. Wang. 1998. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc. Natl. Acad. Sci. USA. 95:8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., H. Zou, P. Widlack, W. Garrard, and X. Wang. 1999. Activation of the apoptotic endonuclease DFF40 (caspase-activated DNase or nuclease). Oligomerization and direct interaction with histone H1. J. Biol. Chem. 274:13836–13840. [DOI] [PubMed] [Google Scholar]
- Meiss, G., S.R. Scholz, C. Korn, O. Gimadutdinow, and A. Pingoud. 2001. Identification of functionally relevant histidine residues in the apoptotic nuclease CAD. Nucleic Acids Res. 29:3901–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T., A. Gunjan, R. Hock, M. Bustin, and D.T. Brown. 2000. Dynamic binding of histone H1 to chromatin in living cells. Nature. 408:877–881. [DOI] [PubMed] [Google Scholar]
- Mukae, N., H. Yokoyama, T. Yokokura, Y. Sakoyama, H. Sakahira, and S. Nagata. 2000. Identification and development expression of inhibitor of caspase-activated DNase (ICAD) in Drosophila melanogaster. J. Biol. Chem. 275:21402–21408. [DOI] [PubMed] [Google Scholar]
- Nagata, S., H. Nagase, K. Kawane, N. Mukae, and H. Fukuyama. 2003. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 10:108–116. [DOI] [PubMed] [Google Scholar]
- Nicholson, D.W. 1999. Caspases structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 6:1028–1042. [DOI] [PubMed] [Google Scholar]
- Nickerson, J. 2001. Experimental observations of a nuclear matrix. J. Cell Sci. 114:463–474. [DOI] [PubMed] [Google Scholar]
- Nickerson, J.A., G. Krockmalnic, K.M. Wan, and S. Penman. 1997. The nuclear matrix revealed by eluting chromatin from a cross-linked nucleus. Proc. Natl. Acad. Sci. USA. 94:4446–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson, T. 2001. Protein mobility within the nucleus—what are the right moves? Cell. 104:635–638. [DOI] [PubMed] [Google Scholar]
- Penninger, J.M., and G. Kroemer. 2003. Mitochondria, AIF and caspases—rivaling for cell death execution. Nat. Cell Biol. 5:97–99. [DOI] [PubMed] [Google Scholar]
- Sakahira, H., M. Enari, and S. Nagata. 1998. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 391:96–99. [DOI] [PubMed] [Google Scholar]
- Sakahira, H., M. Enari, and S. Nagata. 1999. Functional differences of two forms of the inhibitor of caspase-activated DNase, ICAD-L, and ICAD-S. J. Biol Chem. 274:15740–15744. [DOI] [PubMed] [Google Scholar]
- Sakahira, H., A. Iwamatsu, and S. Nagata. 2000. Specific chaperone-like activity of inhibitor of caspase-activated DNase for caspase-activated DNase. J. Biol. Chem. 275:8091–8096. [DOI] [PubMed] [Google Scholar]
- Samejima, K., and W.C. Earnshaw. 1998. ICAD/DFF regulator of apoptotic nuclease is nuclear. Exp. Cell Res. 243:453–459. [DOI] [PubMed] [Google Scholar]
- Samuelsson, M.K., A. Pazirandeh, and S. Okret. 2002. A pro-apoptotic effect of the CDK inhibitor p57(Kip2) on staurosporine-induced apoptosis in HeLa cells. Bichem. Biophys. Res. Commun. 296:702–709. [DOI] [PubMed] [Google Scholar]
- Scaffidi, P., T. Misteli, and M.E. Bianchi. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 418:191–195. [DOI] [PubMed] [Google Scholar]
- Scholz, S.R., C. Korn, O. Gimadutdinow, M. Knoblauch, A. Pingoud, and G. Meiss. 2002. The effect of ICAD-S on the formation and intracellular distribution of a nucleolytically active caspase-activated DNase. Nucleic Acids Res. 30:3045–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien, D.L., S. Simeoni, Z.D. Sharp, and M.A. Mancini. 2000. Subnuclear dynamics and transcription factor function. J. Cell. Biochem. 35:99–106. [DOI] [PubMed] [Google Scholar]
- Stenoien, D.L., K. Patel, M.G. Mancini, M. Dutertre, C.L. Smith, B.W. O'Malley, and M.A. Mancini. 2001. FRAP reveals that mobility of oestrogen receptor-α is ligand- and proteasome-dependent. Nat. Cell Biol. 3:15–23. [DOI] [PubMed] [Google Scholar]
- Takemoto, K., T. Nagai, A. Miyawaki, and M. Miura. 2003. Spatio-temporal activation of caspase revealed by indicator that is insensitive to environmental effect. J. Cell Biol. 160:235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh, Y.T., X. Wang, and P. Li. 1998. Identification of the nuclear factor HMG2 as an activator for DFF nuclease activity. Biochem. Biophys. Res. Commun. 250:598–601. [DOI] [PubMed] [Google Scholar]
- Tzung, S.-P., K.M. Kim, G. Basanez, C.D. Giedt, J. Simon, J. Zimmerberg, K. Zhang, and D.M. Hockenbery. 2001. Antimycin A mimics a cell death-inducing Bcl-2 homology domain 3. Nat. Cell Biol. 3:183–191. [DOI] [PubMed] [Google Scholar]
- Widlak, P., and W.T. Garrard. 2001. Ionic and cofactor requirements for the activity of the apoptotic nuclease DFF40/CAD. Mol. Cell. Biochem. 218:125–130. [DOI] [PubMed] [Google Scholar]
- Zhang, J., and M. Xu. 2002. Apoptotic DNA fragmentation and tissue homeostasis. Trends Cell Biol. 12:84–89. [DOI] [PubMed] [Google Scholar]
- Zhang, J., X. Liu, D.C. Scherer, L. Kaer, X. Wang, and M. Xu. 1998. Resistance to DNA fragmentation and chromatin condensation in mice lacking the DNA fragmentation factor 45. Proc. Natl. Acad. Sci. USA. 95:12480–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J., X. Wang, K.E. Bove, and M. Xu. 1999. DNA fragmentation factor 45-deficient cells are more resistant to apoptosis and exhibit different dying morphology than wild-type control cells. J. Biol. Chem. 274:37450–37454. [DOI] [PubMed] [Google Scholar]
- Zhou, P., A.A. Lugovskoy, J.S. McCarty, P. Li, and G. Wagner. 2001. Solution structure of DFF40 and DFF45N-terminal domain complex and mutual chaperone activity of DFF40 and DFF45. Proc. Natl. Acad. Sci. USA. 98:6051–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]