

Abstract
Millions of patients suffer from Alzheimer's disease, and intensive efforts to find a cure for this devastating disorder center on the proteases, which release the deadly amyloid β-peptide from its precursor. The cutting procedure is thought to be cholesterol dependent and strategies to lower cholesterol as therapeutic treatment are under intensive investigation. Recent findings suggest that the complete proteolytic machinery required for amyloid β-peptide generation is located within lipid rafts. Data by Dotti and colleagues (Abad-Rodriguez et al., 2004), in this issue, suggest that rafts isolate the cutting machinery away from its deadly substrate. These findings describe a novel mechanism for controlling proteolytic activity by building a lipid boundary between proteases and their substrates.
In all developed countries, humans live longer and longer. Although we all wish to enjoy increased longevity, a longer life is unfortunately associated with a dramatic increase for the risk of Alzheimer's disease (AD). This has been widely recognized for years and numerous scientists have studied the cellular mechanisms causing AD with the goal of finally identifying targets for treatment. Indeed, it is likely that all genes directly involved in the generation of the deadly amyloid β-peptide (Aβ), which forms the disease-defining amyloid plaques, have now been identified (Haass, 2004). Currently, it is clear that Aβ is generated by proteolytic processing of β-amyloid precursor protein (APP), and two amyloidogenic secretases, β- and γ-secretase, are involved (Fig. 1). The β-secretase, β-site APP cleaving enzyme (BACE), is a typical aspartyl protease. γ-Secretase, however, is an unusual aspartyl protease complex composed of four individual proteins (presenilin, nicastrin, APH-1, and PEN-2), with presenilin carrying the protease active site (Haass, 2004). γ-Secretase is uncommon not only in its molecular composition but also in its proteolytic activity, since it is able to cleave its substrate within the membrane (Fig. 1). A prerequisite of this intramembrane cut is the release of the ectodomain of APP via cleavage by BACE or α-secretase. Removal of the ectodomain by BACE results in the production of Aβ following γ-secretase cleavage, whereas ectodomain cleavage by α-secretase is nonamyloidogenic since it cuts within the Aβ domain (Fig. 1).
Figure 1.

Processing of APP by secretases. α-Secretase cleavage occurs within the Aβ domain and prevents amyloidogenesis. However, a small peptide (p3) is generated by the subsequent cleavage of the C83 fragment by γ-secretase. Besides p3, the large ectodomain (APPs-α) is secreted. A shorter APPs species is secreted upon cleavage by BACE (APPs-β). The resulting C99 fragment is cleaved by γ-secretase to produce Aβ. The γ-secretase cut releases the APP intracellular domain (AICD), which may be involved in nuclear signaling.
Amyloidogenesis is known to be dependent on cholesterol levels (Simons et al., 2001), and moreover, this seems to have a direct relevance for AD, since high cholesterol is correlated with an increased risk for the disease (Kuo et al., 1998). In addition, the apolipoprotein 4 allele is known as a major genetic risk factor for AD (Corder et al., 1993). At the cellular level, Aβ generation was found to occur in special cholesterol-rich membrane subdomains called DRMs (detergent-resistant membranes), and lowering cholesterol levels by treatment with the cholesterol synthesis inhibitors, statins, resulted in a strong decrease in Aβ production (Simons et al., 1998; Fassbender et al., 2001; Ehehalt et al., 2003). This is likely due in part to an increase in nonamyloidogenic cleavage by α-secretase (Kojro et al., 2001). In animal models, statin treatment was also associated with a significant drop in Aβ load (Fassbender et al., 2001). Furthermore, a clear correlation between cholesteryl-ester levels and Aβ generation was reported in cultured cells (Puglielli et al., 2001). More recently, it was demonstrated that inhibition of the enzyme that regulates the conversion of free cholesterol into cholesteryl-esters not only leads to a dramatic lowering of AD pathology in animal models but is also associated with a reversion of cognitive deficits (Hutter-Paier et al., 2004). Not surprisingly, efforts are under way to use cholesterol-lowering drugs in patients to decrease Aβ production. Indeed, preliminary epidemiological studies and small clinical trials suggest that lowering cholesterol levels may reduce the risk for AD (Wolozin et al., 2000; Simons et al., 2002).
The strong cholesterol dependency of Aβ generation suggested that secretases might be located within DRMs and this may regulate the processing of APP. Indeed, Ehehalt et al. (2003) demonstrated that antibody cross-linking induced BACE and APP to copatch within cholesterol-rich microdomains, and this resulted in increased Aβ production. This proposed localization of secretases is supported by the recent findings of two different laboratories. Here, Abad-Rodriguez et al. (2004) report that they were able to identify endogenous BACE within DRMs of primary hippocampal neurons. Elsewhere, Vetrivel et al. (2004) recently demonstrated that the completely assembled, biologically active γ-secretase complex—consisting of the presenilin fragments APH-1, mature Nicastrin, and PEN-2—resides within DRMs. Moreover, γ-secretase was found in syntaxin 6–, syntaxin 13–, and VAMP4-positive vesicles, demonstrating that γ-secretase accumulates in the DRMs of late Golgi and endosomes (Vetrivel et al., 2004), exactly where BACE is thought to be biologically active (Haass et al., 1995; Vassar et al., 1999; Walter et al., 2001). Similarly, monomeric and oligomeric Aβ were concentrated in DRMs in the brains of a mouse model for AD (Lee et al., 1998; Kawarabayashi et al., 2004). Thus, it appears that the complete Aβ generating proteolytic machinery coexists within DRMs of the same vesicles.
In this issue, the researchers provide a novel and unexpected explanation for why secretases are localized to cholesterol rich membrane domains. They present data suggesting that the DRM association of BACE restricts its access to APP, which they demonstrate accumulates in detergent sensitive membrane domains outside of DRMs (see Fig. 2). This may indicate aberrant access of APP to DRMs, and hence aberrant Aβ production, during AD or it may indicate that even under physiological conditions some APP molecules come into close contact with DRMs. The latter seems more likely, since Aβ is a physiologically normal product and not produced just in the brains of AD patients (Haass, 2004). These findings also demonstrate a completely novel cellular mechanism for controlling protease activity. Cells undergo major efforts to prevent proteases from contacting proteins not destined to be digested. This is accomplished by numerous mechanisms including synthesis of inactive proforms to be activated at appropriate sites, tagging protease substrates with ubiquitin, sequestering proteases in membrane surrounded environments (endosomes/lysosomes), or hiding the active sites of proteases within narrow tunnels (proteasomes). Abad-Rodriguez et al. (2004) and Vetrivel et al. (2004) add yet another control mechanism. They show that lipids can build an invisible boundary, corralling the γ-secretase complex and BACE away from their substrate, APP. Certainly, this mechanism did not evolve to protect us from AD. So why do the secretases concentrate within DRMs? A probable explanation is that γ-secretase is involved in several signaling pathways including Notch signaling (Selkoe and Kopan, 2003; Haass, 2004) and concentrating the proteolytic machinery in small membrane domains facilitates these processes. This would, however, imply that physiological substrates such as Notch and others must gain access to DRMs, an observation yet to be made.
Figure 2.
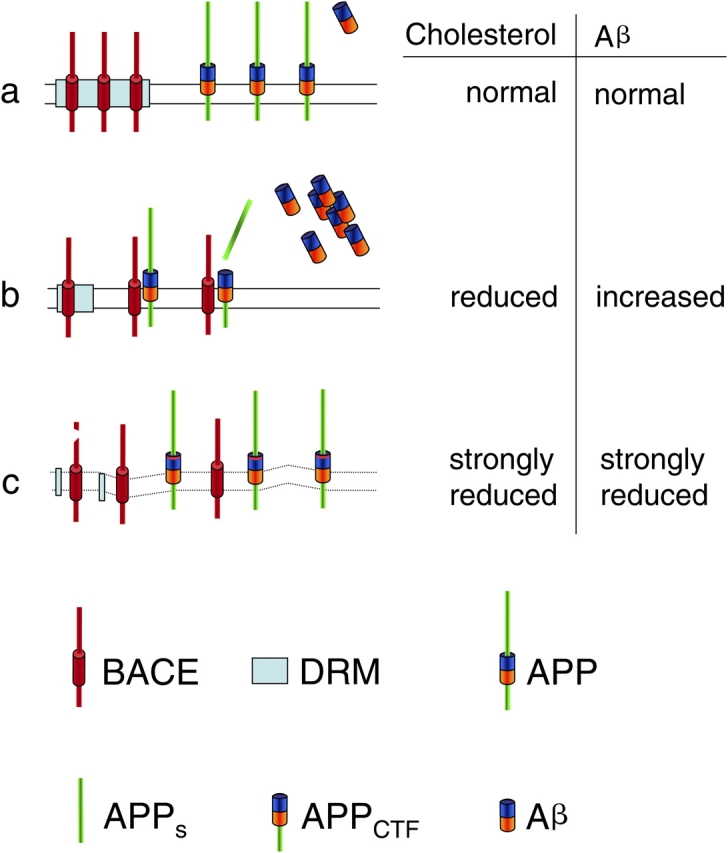
A model describing the effects of cholesterol reduction on Aβ generation. For details, see text.
There is, however, one obvious discrepancy between the results of the current study and earlier ones that must be addressed. Abad-Rodriguez et al. (2004) demonstrate that, upon moderate lowering of cholesterol, Aβ generation surprisingly increased, whereas a large body of previously published data (see above) clearly demonstrated reduced Aβ generation upon cholesterol reduction. How could that be the case, and does that mean that all previous approaches were simply wrong? The truth may be found on both sides. The findings Dotti and colleagues (Abad-Rodriguez et al., 2004) are indeed in apparent disagreement with studies showing that lowering cholesterol inhibits amyloidogenesis, both in cultured cells and in animal models (Simons et al., 1998; Fassbender et al., 2001; Ehehalt et al., 2003). However, in these studies, cholesterol was always strongly depleted and the cells that were used overexpressed APP. In contrast, the current study investigated neurons, the cells most affected by the deadly activity of Aβ. Moderate reduction of cholesterol led to increased Aβ levels, whereas a strong cholesterol reduction resulted in a significant drop in Aβ generation (Fig. 2), similar to that observed before (Simons et al., 1998; Fassbender et al., 2001; Ehehalt et al., 2003). Thus, the effects of cholesterol reduction are dose dependent, and two apparently independent cellular mechanisms are affected. A moderate reduction in cholesterol causes a disorganization of DRMs, allowing more BACE to contact APP and resulting in enhanced Aβ generation (Fig. 2). A strong reduction in cholesterol inhibits BACE/γ-secretase activity and results in a dramatic drop in Aβ generation, even though BACE and γ-secretase can now contact APP directly (Fig. 2). In clear contrast to this model, Ehehalt et al. reported that BACE cleavage of APP occurs within lipid DRMs (Ehehalt et al., 2003), This discrepancy might be due to the fact that when overexpressed a fraction of APP may be mislocalized to DRMs, and then cleaved by BACE, an observation also made by Abad-Rodriguez et al. (2004). More work is needed to settle this controversy since the effects of lowering brain cholesterol on the production of endogenous Aβ in mouse neurons or in human brains have not been directly demonstrated. Although the current findings raise some interesting questions, they do not necessarily challenge cholesterol-lowering tactics as AD therapy/prevention since animal studies to date are in clear favor of this strategy (Fassbender et al., 2001; Hutter-Paier et al., 2004). Moreover, cholesterol lowering drugs are taken by numerous patients with enormous beneficial effects on cardiovascular disorders and no side effects concerning Aβ generation or dementia have been reported. Cholesterol-dependent processing of APP provides a striking example how disease-based research can dramatically advance basic knowledge in cell biology, and vice versa. Who would have thought, only 10 years ago, that DRMs might contain and regulate the deadly machinery responsible for the most abundant human neurodegenerative disorder and that this machinery might function via a novel and totally unexpected mechanism of intramembrane proteolysis?
Abbreviations used in this paper: Aβ, amyloid β-peptide; AD, Alzheimer's disease; APP, β-amyloid precursor protein.
References
- Abad-Rodriguez, J., M.D. Ledesma, K. Craessaerts, S. Perga, M. Medina, A. Delacourte, C. Dingwall, B. De Strooper, and C.G. Dotti. 2004. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 167:953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder, E.H., A.M. Saunders, W.J. Strittmatter, D.E. Schmechel, P.C. Gaskell, G.W. Small, A.D. Roses, J.L. Haines, and M.A. Pericak-Vance. 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 261:921–923. [DOI] [PubMed] [Google Scholar]
- Ehehalt, R., P. Keller, C. Haass, C. Thiele, and K. Simons. 2003. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender, K., M. Simons, C. Bergmann, M. Stroick, D. Lutjohann, P. Keller, H. Runz, S. Kuhl, T. Bertsch, K. von Bergmann, et al. 2001. Simvastatin strongly reduces levels of Alzheimer's disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 98:5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass, C. 2004. Take five-BACE and the γ-secretase quartet conduct Alzheimer's amyloid β-peptide generation. EMBO J. 23:483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass, C., C.A. Lemere, A. Capell, M. Citron, P. Seubert, D. Schenk, L. Lannfelt, and D.J. Selkoe. 1995. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1:1291–1296. [DOI] [PubMed] [Google Scholar]
- Hutter-Paier, B., H.J. Huttunen, L. Puglielli, C.B. Eckman, D.Y. Kim, A. Hofmeister, R.D. Moir, S.B. Domnitz, M.P. Frosch, M. Windisch, and D.M. Kovacs. 2004. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer's disease. Neuron. 44:227–238. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi, T., M. Shoji, L.H. Younkin, L. Wen-Lang, D.W. Dickson, T. Murakami, E. Matsubara, K. Abe, K.H. Ashe, and S.G. Younkin. 2004. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 24:3801–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojro, E., G. Gimpl, S. Lammich, W. Marz, and F. Fahrenholz. 2001. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc. Natl. Acad. Sci. USA. 98:5815–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, Y.M., M.R. Emmerling, C.L. Bisgaier, A.D. Essenburg, H.C. Lampert, D. Drumm, and A.E. Roher. 1998. Elevated low-density lipoprotein in Alzheimer's disease correlates with brain abeta 1-42 levels. Biochem. Biophys. Res. Commun. 252:711–715. [DOI] [PubMed] [Google Scholar]
- Lee, S.J., U. Liyanage, P.E. Bickel, W. Xia, P.T. Lansbury Jr., and K.S. Kosik. 1998. A detergent-insoluble membrane compartment contains A beta in vivo. Nat. Med. 4:730–734. [DOI] [PubMed] [Google Scholar]
- Puglielli, L., G. Konopka, E. Pack-Chung, L.A. Ingano, O. Berezovska, B.T. Hyman, T.Y. Chang, R.E. Tanzi, and D.M. Kovacs. 2001. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 3:905–912. [DOI] [PubMed] [Google Scholar]
- Selkoe, D., and R. Kopan. 2003. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu. Rev. Neurosci. 26:565–597. [DOI] [PubMed] [Google Scholar]
- Simons, M., P. Keller, B. De Strooper, K. Beyreuther, C.G. Dotti, and K. Simons. 1998. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA. 95:6460–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, M., P. Keller, J. Dichgans, and J.B. Schulz. 2001. Cholesterol and Alzheimer's disease: is there a link? Neurology. 57:1089–1093. [DOI] [PubMed] [Google Scholar]
- Simons, M., F. Schwarzler, D. Lutjohann, K. von Bergmann, K. Beyreuther, J. Dichgans, H. Wormstall, T. Hartmann, and J.B. Schulz. 2002. Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann. Neurol. 52:346–350. [DOI] [PubMed] [Google Scholar]
- Vassar, R., B.D. Bennett, S. Babu-Khan, S. Kahn, E.A. Mendiaz, P. Denis, D.B. Teplow, S. Ross, P. Amarante, R. Loeloff, et al. 1999. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 286:735–741. [DOI] [PubMed] [Google Scholar]
- Vetrivel, K.S., H. Cheng, W. Lin, T. Sakurai, T. Li, N. Nukina, P.C. Wong, H. Xu, and G. Thinakaran. 2004. Association of {gamma}-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279:44945–44954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, J., R. Fluhrer, B. Hartung, M. Willem, C. Kaether, A. Capell, S. Lammich, G. Multhaup, and C. Haass. 2001. Phosphorylation regulates intracellular trafficking of beta-secretase. J. Biol. Chem. 276:14634–14641. [DOI] [PubMed] [Google Scholar]
- Wolozin, B., W. Kellman, P. Ruosseau, G.G. Celesia, and G. Siegel. 2000. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 57:1439–1443. [DOI] [PubMed] [Google Scholar]