

Abstract
To avoid immune recognition by cytotoxic T lymphocytes (CTLs), human immunodeficiency virus (HIV)-1 Nef disrupts the transport of major histocompatibility complex class I molecules (MHC-I) to the cell surface in HIV-infected T cells. However, the mechanism by which Nef does this is unknown. We report that Nef disrupts MHC-I trafficking by rerouting newly synthesized MHC-I from the trans-Golgi network (TGN) to lysosomal compartments for degradation. The ability of Nef to target MHC-I from the TGN to lysosomes is dependent on expression of the μ1 subunit of adaptor protein (AP) AP-1A, a cellular protein complex implicated in TGN to endolysosomal pathways. We demonstrate that in HIV-infected primary T cells, Nef promotes a physical interaction between endogenous AP-1 and MHC-I. Moreover, we present data that this interaction uses a novel AP-1 binding site that requires amino acids in the MHC-I cytoplasmic tail. In sum, our evidence suggests that binding of AP-1 to the Nef–MHC-I complex is an important step required for inhibition of antigen presentation by HIV.
Introduction
The human immunodeficiency virus (HIV) establishes a chronic infection in T lymphocytes by evading the host immune response. To prevent recognition and killing by host cytotoxic T lymphocytes (CTLs), HIV reduces the cell surface expression of major histocompatibility complex class I molecules (MHC-I) (Schwartz et al., 1996), which are needed to present antigenic peptides to CTLs. The HIV Nef protein is required for the reduced MHC-I cell surface expression (Schwartz et al., 1996) and to thereby protect HIV-infected primary T cells from anti-HIV CTLs (Collins et al., 1998).
HIV-1 Nef is a 27–34-kD multifunctional protein that has no apparent enzymatic activity and is thought to function by acting as an adaptor protein (AP). Consistent with this, Nef binds to cytoplasmic tail domains (Harris and Neil, 1994; Greenway et al., 1995; Grzesiek et al., 1996; Rossi et al., 1996; Schaefer et al., 2000; Williams et al., 2002), and contains a number of potential protein–protein interaction domains that are required either for MHC-I or CD4 downmodulation. An NH2-terminal α helical domain, an acidic domain and a polyproline repeat are all required for MHC-I downmodulation (Greenberg et al., 1998b; Mangasarian et al., 1999). In contrast, dileucine and diacidic motifs within Nef are necessary for removal of cell surface CD4 (Aiken et al., 1994; Greenberg et al., 1998b).
Thus far, Nef has been shown to downmodulate all MHC-I HLA-A and HLA-B allotypes. However, Nef does not downmodulate MHC-I HLA-C and HLA-E (Le Gall et al., 1998; Cohen et al., 1999). This selective downmodulation results from amino acid sequence variation in the Nef binding domain within the cytoplasmic tails of these molecules (Williams et al., 2002). Because HLA-C and HLA-E are known to inhibit natural killer cell lysis, it has been proposed that maintenance of their cell surface expression may allow survival of the infected cell (Le Gall et al., 1998; Cohen et al., 1999).
The current model for how Nef affects CD4 cell surface expression is that Nef binds to the CD4 cytoplasmic tail and links it to cellular trafficking proteins that accelerate its endocytosis. Indeed, Nef is known to interact with a number of proteins that regulate intracellular trafficking. For example, Nef interacts with the heterotetrameric clathrin APs (AP-1, AP-2, and/or AP-3; Bresnahan et al., 1998; Greenberg et al., 1998a; Le Gall et al., 1998; Piguet et al., 1998; Craig et al., 2000; Erdtmann et al., 2000; Janvier et al., 2003a,b), through the same dileucine motif that is needed for CD4 downmodulation (Bresnahan et al., 1998; Greenberg et al., 1998a; Craig et al., 2000; Janvier et al., 2003a,b).
The adaptin complexes (Robinson and Bonifacino, 2001; Traub, 2003; Robinson, 2004) are each composed of four subunits; two large subunits (β1, β2, β3 plus γ, α, or δ), medium subunit (μ1A, μ1B, μ2, μ3) and one small subunit (σ1, σ2, σ3) for AP-1 (A or B), AP-2 and AP-3, respectively. There is evidence that all of these complexes sort proteins by binding recognition sequences in their cytoplasmic tails and linking them to clathrin. AP-1 is localized to the TGN and endosomes. Thus, it is thought to be important for trafficking between these compartments (Doray et al., 2002; Waguri et al., 2003), and for eventual sorting of some proteins (e.g., lysosomal hydrolases) to the lysosomes. AP-3 is localized to endosomes (Peden et al., 2004) and is needed for proper targeting of other proteins (e.g., lysosome-associated membrane protein [LAMP-1]) to lysosomes. Finally, AP-2 is localized to the cell surface and plays a role in endocytosis (Traub, 2003). Thus, one model is that Nef-dependent CD4 endocytosis occurs via recruitment of AP-2 through Nef's dileucine motif. Nef-dependent CD4 endocytosis may additionally require interaction with a subunit of the vacuolar ATPase (Lu et al., 1998), which may indirectly promote an association of Nef with AP-2 (Geyer et al., 2002). However, the mechanistic details of how interactions of Nef with various adaptin molecules accelerate CD4 endocytosis and degradation are not well understood.
There is increasing evidence that Nef disrupts MHC-I cell surface expression by a different mechanism than it uses to downmodulate CD4. The diacidic motif needed for Nef to interact with the vacuolar ATPase to promote CD4 down-regulation is not required for Nef to disrupt MHC-I trafficking (Greenberg et al., 1998b; Lu et al., 1998). In addition, the dileucine motif within Nef that is required for Nef to directly bind adaptin complexes is also dispensable (Greenberg et al., 1998b; Mangasarian et al., 1999). Moreover, in HIV-infected T lymphocytes and astrocytic cells, the primary effect of Nef on MHC-I is to disrupt its transport to the cell surface rather than to promote its endocytosis (Swann et al., 2001; Kasper and Collins, 2003). The molecular mechanism underlying this effect of Nef is unknown.
To determine how Nef disrupts the transport of MHC-I to the cell surface, we used biochemical and cell biological approaches to examine MHC-I trafficking in Nef-expressing T cells. We found that Nef redirected MHC-I from the TGN to lysosomes. Moreover, we demonstrated that the AP, AP-1, which is required for proper sorting of lysosomal hydrolases from the TGN to lysosomes, was required for Nef to disrupt MHC-I trafficking. Finally, in HIV-infected primary T cells, we found that Nef stabilized an interaction between MHC-I and AP-1. This interaction used a novel domain that required sequences from the NH2-terminal α helix of Nef and from the MHC-I cytoplasmic tail.
Results
To perform large scale biochemical experiments to examine the effects of Nef on MHC-I trafficking in T cells, we developed a system that permitted transient, uniform, expression of Nef in T cells. This was accomplished by using a replication-defective adenoviral vector expressing HIV-1 Nef, which disrupts MHC-I transport in a manner that is indistinguishable from HIV (Kasper and Collins, 2003). In addition, to specifically detect an allotype of MHC-I that is responsive to Nef, we attached an HA tag to the NH2-terminal extracellular domain of HLA-A2. Based on a number of biochemical measurements, the tagged molecule was folded properly, matured through the secretory pathway normally, and was equally responsive to HIV-1 Nef (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200407031/DC1).
Trafficking of MHC-I in Nef-expressing T cells
It has been previously shown that Nef does not affect the transport of MHC-I into the medial Golgi apparatus (Schwartz et al., 1996), but does inhibit its transport to the cell surface (Kasper and Collins, 2003). To further define where in the secretory pathway Nef exerts its effects, we asked whether MHC-I was transported normally into the TGN. This was accomplished by monitoring the rate at which HLA-A2 acquired sialic acid residues in this compartment, which can be measured by testing sensitivity to neuraminidase digestion. We detected sialylation of HLA-A2 as a slight increase in HLA-A2 molecular weight (Fig. 1 A, lanes 4 and 7) that was eliminated by neuraminidase treatment (Fig. 1 A, lanes 5 and 8). Based on these data (quantified in Fig. 1 B), Nef does not delay the transport of HLA-A2 into the TGN. As a control, we also confirmed the previously published observation that Nef does not affect transit into the medial Golgi (Schwartz et al., 1996), as assessed by sensitivity to endoglycosidase H (endo H) digestion (Fig. 1, A and B).
Figure 1.
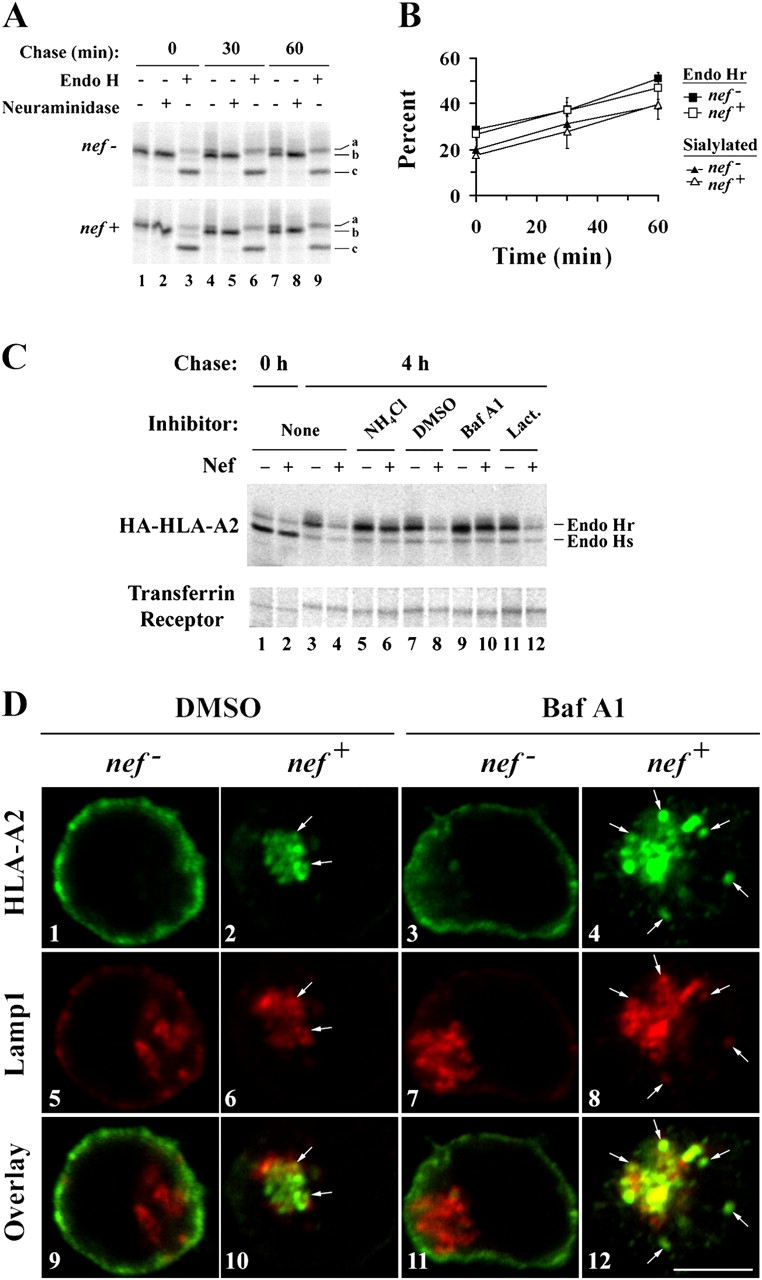
HIV-1 Nef redirects MHC-I from the TGN to lysosomes. (A and B) Nef does not affect the maturation of HLA-A2 through the TGN. CEM HA-HLA-A2 cells were transduced with a control adenovirus (nef −) or an adenovirus expressing HIV-1 Nef (nef +) and subjected to a pulse-chase metabolic labeling assay to follow proteins through the biosynthetic pathway. HA-HLA-A2 species with different gel mobilities were identified based on enzymatic digestion profiles: (a) N-glycosylated and sialylated HA-HLA-A2; (b) N-glycosylated HA-HLA-A2; and (c) core HA-HLA-A2 protein. (B) Quantitation of the relative percentage of endo H resistant and sialylated HA-HLA-A2. The mean percentage ± SD from two independent experiments is plotted over time. (C) Nef-induced degradation of mature MHC-I is blocked by inhibitors of acidic degradation. CEM HA-HLA-A2 cells were treated with adenovirus, pulsed with radioactive amino acids, and were either collected immediately (lanes 1 and 2) or chased for 4 h in media containing the indicated chemical inhibitor (lanes 3–12). Cellular lysates were divided equally, and HA-HLA-A2 or transferrin receptor was recovered by immunoprecipitation. All samples were digested with endo H before SDS-PAGE. The results are representative of three independent experiments. (D) Bafilomycin A1 treatment increases the degree of colocalization between HLA-A2 and LAMP-1. Cells transduced with the indicated adenovirus were treated with either solvent alone (DMSO) or bafilomycin A1 (Baf A1) for 4 h before staining. HLA-A2 and LAMP-1 were detected by indirect immunofluorescence using mAbs as described in Materials and methods. Arrows indicate colocalization between HLA-A2 and LAMP-1. Images were collected using a confocal microscope. Individual z-sections are shown. Bar, 5 μm.
HIV-1 Nef causes HLA-A2 to be degraded in lysosomal compartments
There is consensus that Nef causes the accumulation of MHC-I in the TGN of multiple non-T cell lines (Schwartz et al., 1996; Greenberg et al., 1998b; Le Gall et al., 1998; Piguet et al., 2000; Swann et al., 2001; Blagoveshchenskaya et al., 2002). However, it is controversial as to whether MHC-I is ultimately targeted to other organelles in which it is degraded at an accelerated rate (Schwartz et al., 1996; Blagoveshchenskaya et al., 2002; Williams et al., 2002). In agreement with other studies performed in T cells (Schwartz et al., 1996), we found that Nef expression accelerated the degradation of mature, endo H resistant HLA-A2, but did not affect the stability of immature, endo H–sensitive molecules (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200407031/DC1). In addition, we found that two inhibitors of lysosomal protein degradation, ammonium chloride and bafilomycin A1, blocked HLA-A2 degradation. Whereas, lactacystin, a proteasome inhibitor, had no effect on HLA-A2 degradation (Fig. 1 C). Thus, after reaching the TGN, HLA-A2 appears to be targeted for lysosomal degradation in Nef-expressing T cells.
To provide supporting evidence that MHC-I is ultimately targeted to lysosomes, we examined the colocalization of HLA-A2 with LAMP-1, a marker of lysosomal compartments (Fig. 1 D). In T cells expressing Nef, we observed a very small amount of colocalization of HLA-A2 with LAMP-1 (Fig. 1 D, 10). The majority colocalized with markers of the TGN (not depicted). However, when lysosomal degradation was inhibited by treatment of cells with bafilomycin A1, the amount of HLA-A2 colocalizing with LAMP-1 dramatically increased in Nef-expressing cells (Fig. 1 D, compare 10 with 12). These data support the model that MHC-I is ultimately directed to lysosomal compartments in Nef-expressing T cells, where it is rapidly degraded.
Nef targets HLA-A2 to lysosomes using an AP-1A–dependent pathway
Transport of lysosomal hydrolases and LAMP-1 to the lysosomes is known to require the APs AP-1 and AP-3, respectively. Interestingly, both of these adaptors are known to interact with HIV-1 Nef. Therefore, to explore a possible requirement for these complexes, we transfected cells with siRNAs directed at the μ subunit of AP-1A and AP-3. As shown in Fig. 2 (A and B), we were able to reduce expression of these molecules in both astrocytic cells and T cells. The effect was more dramatic in astrocytic cells, probably because we were able to transfect them more efficiently than T cells. In both cell types, we found that inhibiting the expression of AP-1A, but not that of AP-3, reduced the effect of Nef on MHC-I cell surface expression (Fig. 2, C and D). In addition, AP-1A expression was required for HLA-A2 degradation (Fig. 2 E). However, reducing the expression of μ1A and μ3 did not affect Nef's ability to downmodulate CD4 (Fig. 2, C and D), nor did it affect Nef expression (Fig. 2, A, B, and E). These results suggest that AP-1A expression is specifically required for Nef to disrupt MHC-I cell surface expression and to target it for degradation.
Figure 2.
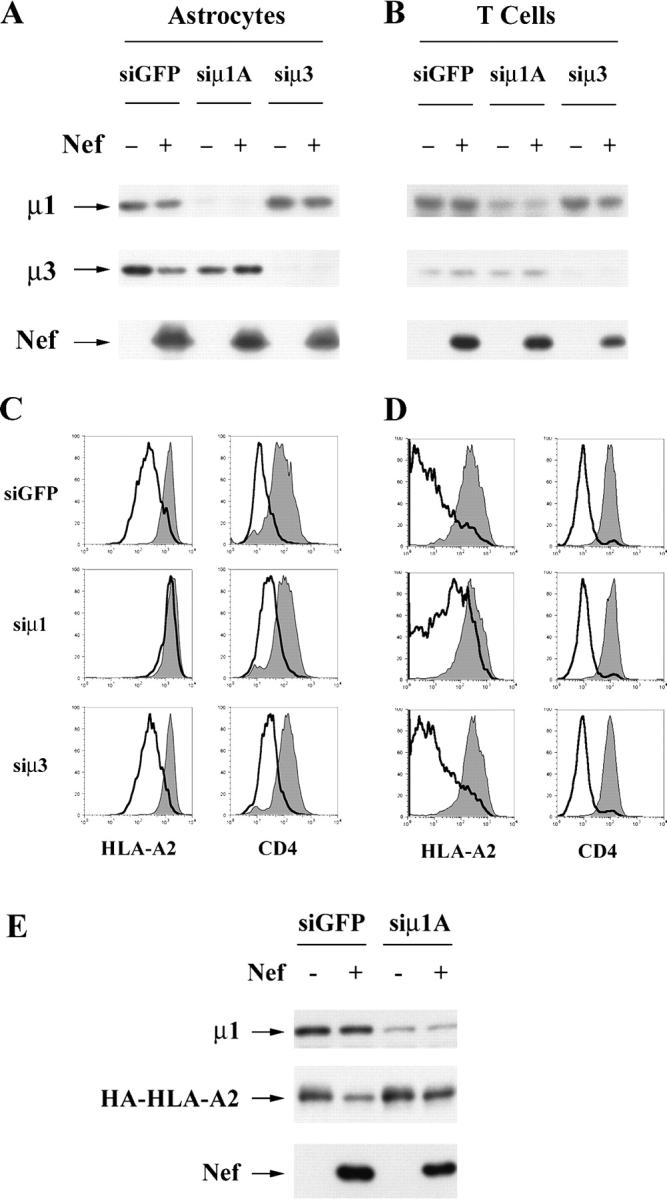
HIV-1 Nef targets MHC-I into an AP-1–dependent pathway. (A and B) Western blot analysis of adaptin and Nef expression in siRNA-treated cells. Astrocytic cells (A) or CEM T cells (B) were transfected with the indicated siRNA and transduced with control or Nef-expressing adenoviruses as described in Materials and methods. Lysates were harvested and immunoblotted for the indicated protein. (C and D) Flow cytometric analysis of siRNA-treated cells. Astrocytic cells (C) or CEM T cells (D) were stained for surface HLA-A2 expression (left column) or surface CD4 expression (right column); shaded curve, control adenovirus; black line, Nef-expressing adenovirus. (E) Depletion of AP-1 inhibits Nef-dependent HLA-A2 degradation. CEM T cells were transduced and treated with siRNAs as described above except that an additional siRNA transfection was included before transduction. Lysates were prepared and analyzed by immunoblot to examine the expression levels of the indicated proteins.
Nef promotes the formation of a complex containing MHC-I and AP-1
To determine whether Nef functions as an AP linking MHC-I to AP-1, we immunoprecipitated endogenous HLA-A2 from HIV-infected primary T cells and then immunoblotted for Nef and for endogenous subunits of AP-1. As expected, we detected Nef protein coprecipitating with HLA-A2 (Fig. 3 A). In addition, we also detected the μ and γ subunits of AP-1 coprecipitating with HLA-A2. This result is particularly striking because only ∼25% of the primary T cells were infected in this experiment. The interaction was highly specific based on the fact that the related AP, AP-3, did not coprecipitate with HLA-A2 (Fig. 3 B). Thus, these studies strongly indicate that Nef promotes the formation of a complex containing MHC-I and AP-1.
Figure 3.

Nef expression results in coprecipitation of AP-1 with HLA-A2. (A) HLA-A2+ primary human T cells or control CEM-SS cells that did not express HLA-A2 (Ctrl) were transduced with an HIV molecular clone expressing Nef (nef +), or a matched control HIV that did not express Nef (nef−). Cells were harvested, and HLA-A2 was immunoprecipitated as described in Materials and methods. Proteins that coprecipitated with HLA-A2 were detected by Western blotting as indicated. Western blots of protein inputs are also shown as a control for relative protein levels in each sample before immunoprecipitation (Input Controls). Results are typical of two independent experiments. (B) AP-3 does not coprecipitate with HLA-A2. CEM-SS cells (lanes 1 and 4) or CEM HA-HLA-A2 cells (lanes 2, 3, 5, and 6) were infected with HIV, and HLA-A2 was recovered by immunoprecipitation as described in Materials and methods. Coprecipitating proteins were detected by Western blotting (lanes 1–3). A fraction of the protein input before immunoprecipitation was also analyzed to ensure similar protein expression levels (lanes 4–6). Western blots of δ-adaptin (AP-3) and γ-adaptin (AP-1) were performed simultaneously using the same antibody concentrations. Apparent molecular masses of protein standards are denoted in kilodaltons on the left.
AP-1 binds to the Nef–MHC-I complex in a dileucine-independent manner
Previous studies that have examined the direct interaction of Nef with AP-1 have found that it depends on the Nef dileucine motif (Bresnahan et al., 1998; Craig et al., 2000; Janvier et al., 2003a,b). However, we found that the dileucine motif was not needed for coprecipitation of AP-1 with MHC-I in CEM T cells treated with adeno-Nef (Fig. 4 A) or HIV (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200407031/DC1). These data are consistent with the fact that the dileucine motif is not necessary for disruption of MHC-I trafficking (Greenberg et al., 1998a; Mangasarian et al., 1999), and suggests that there is an alternative AP-1 binding site in the Nef–MHC-I complex.
Figure 4.

Analysis of MHC-I and Nef domains that contribute to AP-1 recruitment. (A) Recruitment of AP-1 requires an intact Nef–MHC-I complex and is independent of the dileucine motif in Nef. CEM-SS cells (lane 1) or CEM HA-HLA-A2 cells (lanes 2–5) were transduced with the indicated adenoviral vector. The cells were harvested at 72 h and subjected to the coimmunoprecipitation assay. Cells were transduced with each adenovirus to achieve equivalent Nef expression levels (Input Controls): wild-type NL4-3 Nef (MOI = 25); NL4-3 Nef with a NH2-terminal deletion (V10EΔ17-26, MOI = 300); HXB Nef with a mutation in the dileucine motif (LL164,165AA, MOI = 25). (As shown, HXB Nef migrates more slowly on SDS PAGE than NL4-3 Nef. Wild-type HXB Nef and HXB NefLL164,165AA are directly compared in Fig. S3.) All results are typical of at least two independent experiments. (B) HLA-A2 tyrosine 320 (Y320) is required for efficient Nef binding, AP-1 recruitment, and targeting for degradation. CEM-SS (lanes 1 and 2), CEM HA-HLA-A2 (lanes 3 and 4), or CEM cells expressing the MHC-I mutant, HA-HLA-A2 Y320A (lanes 5 and 6), were transduced with a control (−) or Nef-expressing adenovirus (+). At 72 h after transduction, HLA-A2 immunoprecipitations were recovered and analyzed by Western blotting as described in Fig. 3.
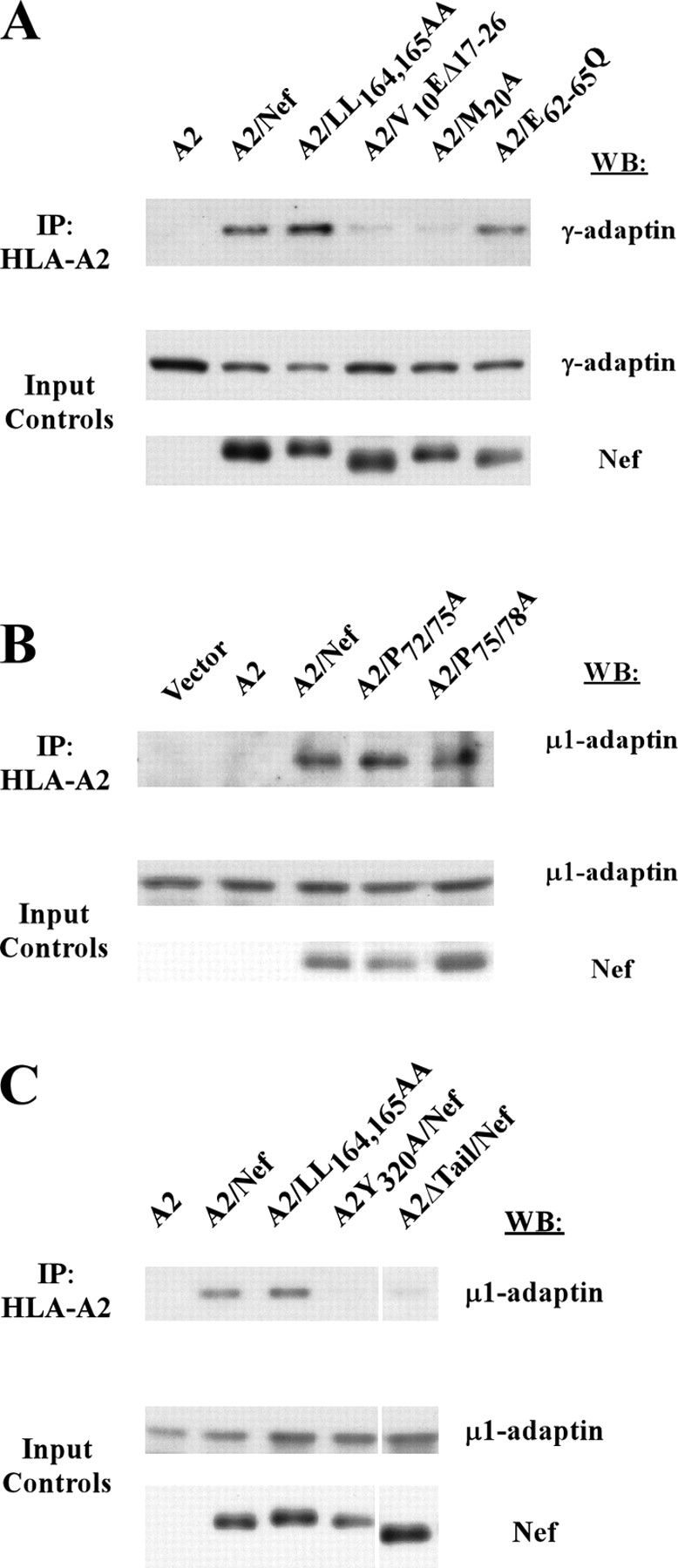
To rule out the possibility that the interaction we observed was nonspecific, and to verify that it was dependent on Nef domains important for MHC-I trafficking to lysosomes, we asked whether AP-1 would coprecipitate with MHC-I in cells expressing a Nef mutant (V10EΔ17-26) that was unable to affect MHC-I trafficking and to bind MHC-I (Mangasarian et al., 1999; Williams et al., 2004). As shown in Fig. 4 A, we found that AP-1 did not coprecipitate with HLA-A2 under these conditions.
In addition, we asked whether AP-1 would coprecipitate with an HLA-A2 mutant lacking a tyrosine residue at position 320 that is necessary for responsiveness to Nef (Greenberg et al., 1998b; Le Gall et al., 1998). As shown in Fig. 4 B, this mutant was expressed well (Fig. 4 B, input controls, bottom). However, it was resistant to Nef-dependent MHC-I degradation, coprecipitated Nef less efficiently, and was defective in AP-1 recruitment (Fig. 4 B). Thus, these studies indicate that coprecipitation of AP-1 with HLA-A2 in Nef-expressing cells was highly specific and depended on amino acid sequences in Nef and MHC-I that are functionally important for Nef's effects on MHC-I cell surface expression.
Recruitment of AP-1 requires both the cytoplasmic tail of HLA-A2 and the NH2-terminal α-helix in Nef
The domains of HIV Nef specifically involved in MHC-I trafficking have been well characterized. It is known that disruption of Nef's NH2-terminal α helix by deletion (V10EΔ17-26) or point mutation (M20A), mutation of an acidic cluster (E62-65A), or mutation of a polyproline repeat (P69/72/75/78A) specifically affects this activity (Greenberg et al., 1998b; Mangasarian et al., 1999). We have recently reported that each of these mutants fails to coprecipitate with MHC-I (Williams et al., 2004) and thus binding to MHC-I may be the primary role of these domains. However, it is also possible that one or more of these domains plays an additional role. To determine whether any of these domains might also be involved in AP-1 recruitment directly, we bypassed the requirement for Nef binding by directly fusing Nef to the COOH terminus of the HLA-A2 cytoplasmic tail (A2/Nef; Fig. 5 A). As expected, Nef was able to reduce the cell surface expression of HLA-A2 in cis (Fig. 5 B, left), and to promote its colocalization with markers of the Golgi apparatus (Fig. 5 C). In addition, the A2/Nef fusion protein was able to efficiently coprecipitate AP-1 from T cell lysates in a manner that was independent of the Nef dileucine motif (Fig. 6 A). However, the fusion protein was not capable of reducing MHC-I surface expression in trans (Fig. 5 B, right). Thus, A2/Nef was a useful reagent to explore which amino acids in Nef and MHC-I were important for AP-1 recruitment to the Nef–MHC-I complex.
Figure 5.
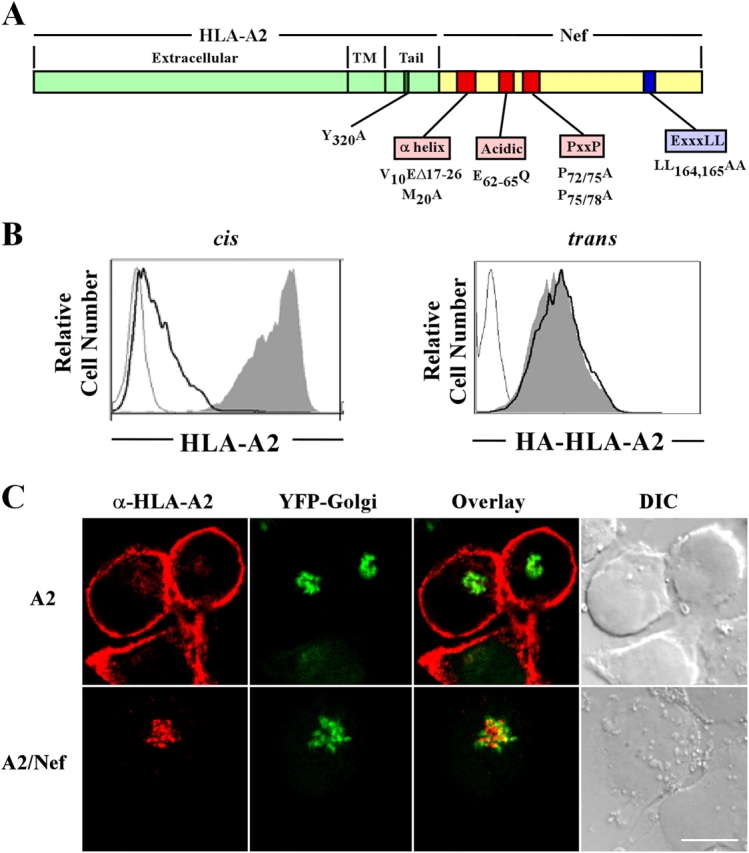
Characterization of the A2/Nef fusion protein. (A) Schematic diagram of the A2/Nef fusion protein. (B) The addition of Nef to the COOH terminus of HLA-A2 results in a reduction of cell surface expression in cis but not trans. In the left panel, the effect of Nef in cis was measured by transducing CEM-SS cells with a bi-cistronic murine retrovirus encoding GFP and HLA-A2 (shaded curve), GFP and A2/Nef (black line), or GFP alone (gray line). HLA-A2 was detected by staining with mAb BB7.2 and using flow cytometry to gate on the GFP-positive cells. In the right panel, the effect of A2/Nef in trans was measured by examining the effect of the fusion protein (black line) or wild-type HLA-A2 (shaded curve) on stably expressed HA tagged HLA-A2 (HA-HLA-A2). The negative control is CEM-SS cells transduced with virus expressing GFP only (gray line). HA-HLA-A2 was detected by staining with an mAb directed against HA and using flow cytometry to gate on the GFP-positive cells. Results are representative of three independent experiments. (C) The A2/Nef fusion protein accumulates in the TGN. CEM-SS cells that stably expressed a tagged late Golgi resident protein (EYFP-Golgi) were transduced with retroviral constructs (A2 or A2/Nef). Cells were stained for HLA-A2 and analyzed by confocal microscopy as described in Materials and methods. Bar, 5 μm.
Figure 6.
The cytoplasmic domain of HLA-A2 and the NH 2 -terminal α helix of Nef are both required for AP-1 binding. (A and B) The NH2-terminal α-helix in Nef is necessary for AP-1 recruitment. CEM-SS cells were transduced with the murine retrovirus encoding HLA-A2 or the indicated A2/Nef fusion constructs. Coprecipitating proteins were detected by Western blotting of anti-HLA-A2 immunoprecipitations. (C) AP-1 binding to the A2/Nef fusion protein requires tyrosine 320 (Y320) in the cytoplasmic tail of HLA-A2. The indicated mutations in the A2/Nef fusion protein were tested for involvement in AP-1 binding using the assay described above. A2ΔTail is an A2/Nef fusion protein that lacks the entire HLA-A2 cytoplasmic tail. All results are representative of at least three independent experiments. White lines indicate that intervening lanes have been spliced out.
As shown in Fig. 6 A, these studies revealed that mutation of amino acids in the NH2-terminal α-helix (A2/V10EΔ17-26 and A2/M20A) inhibited coprecipitation of AP-1 with the fusion protein. In contrast, disruption of the acidic (A2/E62-65Q) and polyproline (A2/P72/75A and A2/P75/78A) domains in Nef had no effect (Fig. 6, A and B). Surprisingly, deletion of the HLA-A2 cytoplasmic tail (A2ΔTail/Nef) or mutation of tyrosine 320 in the HLA-A2 cytoplasmic tail (A2Y320A/Nef) also dramatically inhibited coprecipitation of AP-1 by the fusion protein (Fig. 6 C). Thus, sequences in the Nef NH2-terminal α helix as well as in the HLA-A2 cytoplasmic tail are necessary to create a functional AP-1 binding site.
Discussion
In sum, we have found that in T cells, Nef disrupts MHC-I trafficking after it reaches the TGN to prevent MHC-I cell surface expression and to promote its degradation in lysosomes. RNAi treatment of Nef-expressing T cells and astrocytic cells revealed that this activity of Nef required the expression of AP-1A, but not AP-3. The requirement for AP-1A was further supported by the fact that we were able to isolate complexes containing MHC-I and AP-1 from Nef-expressing, HIV-infected primary T cells. The dileucine motif in Nef, which is necessary for Nef to directly bind AP-1 in other assay systems, was not needed for AP-1 to bind the Nef–MHC-I complex, suggesting an alternative means of association. Interestingly, when Nef was fused to HLA-A2, AP-1 was only recruited when the NH2-terminal α helical domain of Nef was intact and when a full-length, wild-type HLA-A2 cytoplasmic tail was present. Thus, amino acids within both Nef and MHC-I were necessary to create a binding site for AP-1 in the Nef–MHC-I complex.
The normal function of AP-1 is to sort proteins, such as lysosomal enzyme receptors at the TGN by linking clathrin to the cytoplasmic tails of cargo (for review see Dell'Angelica and Payne, 2001; Robinson and Bonifacino, 2001; Traub, 2003; Robinson, 2004). For example, the mannose 6-phosphate receptor (MPR), which binds soluble lysosomal enzymes, is sorted at the TGN by AP-1. It is then transported in vesicles to endosomes where the enzymes dissociate and are carried to the lysosomes. The MPRs are then recycled back to the TGN to transport more cargo. Thus, Nef-induced AP-1 binding of MHC-I would be expected to promote TGN accumulation and/or targeting of MHC-I into the endolysosomal pathway (Fig. 7).
Figure 7.

Model for MHC-I trafficking in Nef-expressing T cells. To disrupt MHC-I trafficking, Nef first binds to the MHC-I cytoplasmic tail in the secretory pathway. The formation of the Nef–MHC-I complex creates a binding site for AP-1 that is independent of the Nef dileucine motif. This tertiary complex leads to the recruitment of MHC-I into AP-1–positive clathrin-coated vesicles destined for degradation in the lysosomes.
Using the yeast two hybrid system (Le Gall et al., 1998; Piguet et al., 1998; Craig et al., 2000; Erdtmann et al., 2000; Janvier et al., 2003b), GST-Nef pull-downs (Bresnahan et al., 1998; Le Gall et al., 1998; Piguet et al., 1998; Janvier et al., 2003a) or overexpression of a Nef-CD8 chimera in 293 cells (Bresnahan et al., 1998), a number of investigators have found that the HIV-1 Nef protein interacts with adaptin subunits and whole adaptin complexes. A consensus binding domain for adaptin protein binding (D/EXXXLL) (Bonifacino and Traub, 2003) can be found in Nef and this motif is required for GST-Nef to pull down AP-1 complexes from mammalian cell lysates (Bresnahan et al., 1998; Janvier et al., 2003a). In addition, the leucine residues in this motif are required for Nef to downmodulate CD4, but not MHC-I (Greenberg et al., 1998a; Mangasarian et al., 1999). Thus, the dileucine-dependent binding to adaptins appears to be important for Nef's effects on CD4.
Our data showing that the dileucine motif is not required for Nef to promote an interaction between HLA-A2 and AP-1 indicates that there is an alternative binding site for AP-1 in the Nef–MHC-I complex. It should be noted that there is evidence for a second AP-1 binding site within Nef that is active in yeast two hybrid systems (Craig et al., 2000; Erdtmann et al., 2000). However, existing data suggest that this motif is not able to support coprecipitation of AP-1 with Nef in mammalian systems (Bresnahan et al., 1998; Janvier et al., 2003a). These data can be explained by our results showing that AP-1 recruitment to HLA-A2 in Nef-expressing cells appeared to result from the creation of a new binding site for AP-1 that was only active when Nef was bound to wild-type MHC-I.
The MHC-I cytoplasmic tail itself does not contain a consensus AP-1 binding site (D/EXXXLL or YXXΦ) (Bonifacino and Traub, 2003). However, the tyrosine residue at position 320 (Y320) that we show is necessary for Nef to recruit AP-1, is also required for the normal targeting of MHC-I to endolysosomal compartments in dendritic cells (Lizee et al., 2003). The mechanism by which MHC-I is normally targeted to these compartments is unknown, and it is interesting to speculate that in these cell types Y320 recruits an AP, such as AP-1, that targets MHC-I to the endolysosomal compartment. Thus, Nef may be taking advantage of an existing pathway for MHC-I trafficking that is normally only active in certain cell types.
Previous studies describing the mechanism of Nef's effects on MHC-I have focused on the ability of Nef to accelerate MHC-I endocytosis, rather than on the TGN to lysosomal pathway we describe (Greenberg et al., 1998b; Le Gall et al., 1998, 2000; Piguet et al., 2000; Blagoveshchenskaya et al., 2002; Kasper and Collins, 2003). This is most likely due to the fact that Nef is more active at disrupting transport from the TGN in T cells than it is in more commonly used cell lines, such as HeLa cells (Kasper and Collins, 2003). The effect of Nef on endocytosis has been attributed to the small GTPase ADP-ribosylation factor-6 (Blagoveshchenskaya et al., 2002), which is normally involved in MHC-I turnover from the cell surface (Caplan et al., 2002). However, there is new evidence that the effect of ADP-ribosylation factor-6 may be indirect (Larsen et al., 2004). In addition, the phosphofurin acidic cluster sorting protein (PACS-1), which binds Nef via the acidic domain, may target Nef and/or endocytosed Nef-MHC-I to the TGN in some cell types (Piguet et al., 2000; Blagoveshchenskaya et al., 2002). It is known that PACS-1 interacts with AP-1 to promote TGN recycling (Crump et al., 2001). However, it is unlikely that the AP-1 interaction we observed here is secondary to PACS-1 binding, because the PACS-1 binding region (i.e., the acidic domain) was not needed to recruit AP-1 in our system.
It should also be noted that Nef associates with lipid rafts and that this localization is important for its effects on MHC-I trafficking (Alexander et al., 2004). It has been proposed that Nef association with rafts in the Golgi could trap MHC-I there and perhaps promote its association with required cellular proteins, such as AP-1. Further studies are needed to determine whether lipid rafts are important for Nef–MHC-I–AP-1 complex formation.
In sum, our results indicate that in T cells, the primary effect of Nef on MHC-I is to promote TGN retention and to ultimately direct MHC-I to lysosomes. We have provided evidence that Nef accomplishes this by acting as an AP, which stabilizes a physical interaction between AP-1 and MHC-I in order to target MHC-I into the endolysosomal pathway. These results have important implications for understanding HIV disease pathogenesis and for possible pharmaceutical approaches in combating the development of AIDS.
Materials and methods
Cell culture
CEM T cell lines were maintained in R10 (RPMI supplemented with 10% FBS, 10 mM Hepes, 2 mM penicillin, streptomycin, and glutamine (P/S/G). The astrocytic cell lines stably expressing CD4 were maintained in DME supplemented with 10% FBS and P/S/G. Stable astrocytic and CEM T cell lines were generated by transduction with MSCV retroviral constructs pseudotyped with VSV-G as described previously (Kasper and Collins, 2003). Primary T lymphocytes were isolated from buffy coats as described previously (Collins et al., 1998).
DNA constructs
A nine–amino acid HA epitope tag (YPYDVPDYA) was added to the NH2 terminus of HLA-A2 and HLA-B71 (just after the leader sequence cleavage site and before the NaeI site; Swann et al., 2001) using the following primers and HLA-B3501 as the DNA template: forward primer, 5′-GGGAATTCCTCAGAATCTCCTCAGACGCCGAG-3′; reverse primer, 5′-TCCCGCCGGCATAGTCGGGTACGTCATACGGATAGGACCCGGCCCAG-GTCTCGGT-3′. The PCR product was then digested with EcoRI and NaeI and subcloned into a shuttle vector (New England Biolabs, Inc.). Next, the following primers were used to amplify the 3′ region of HLA-A2 and HLA-B71: HLA-A2 forward primer, 5′-GAATTCATGGTACCGTGCACG-3′; HLA-B71 forward primer, 5′-GGCTCGAGTCAAGCTGTGAGAGACACATC-3′; HLA-A2 reverse primer, 5′-CCGCTCGAGTCACACTTTACAAGCTGT-3′; HLA-B71 reverse primer, 5′-CTGGGCCGGCTCCCACTCCATGAGGTATTT-3′. The resulting PCR products were digested with NaeI and XhoI. Finally, the HLA-A2/B71 PCR products and the EcoRI–NaeI fragment purchased from New England Biolabs, Inc. were ligated into MSCV 2.1 (cut with EcoRI and XhoI) in a three-way ligation to generate MSCV HA-HLA-A2. MSCV HA-HLA-A2 Y320A was cloned by amplifying a portion of HLA-A2 Y320A (Swann et al., 2001) using the following primers: forward primer, 5′-GCAGCTCAGACCACCAAGCACAAG-3′; reverse primer 5′-CCGCTCGAGTCACACTTTACAAGCTGT-3′. The PCR product was then digested with PmlI and XhoI, and was ligated into the same sites in MSCV HA-HLA-A2 to replace the wild-type sequence.
An adenoviral vector expressing HXB Nef LL164,165AA was generated by PCR mutagenesis of the HIV molecular clone, HXB-PI (Chen et al., 1996), using the following primers: mutant forward primer, 5′-AATAAAGGAGAGAACACCAGCGCTGCTCACCCTGTGAGCCTGCATGG-3′; with reverse primer (3′hxb-xba), 5′-GCTCTAGATGCTAGAGATTTTCCACACTG-3′, and mutant reverse primer, 5′-TCCATGCAGGCTCACAGGGTGAGCAGCGCTGGTGTTCTCTCCTTTATT-3′; with forward primer (5′ nef) 5′-CGGGATCCATGGGTGGCAAGTGGTCAAA-3′. The resulting mutant PCR products were mixed and reamplified with 3′hxb-xba and 5′ nef primers, digested with XbaI and XhoI and cloned into the same sites of HXB-PI. The mutated Nef was then amplified and cloned into an adenoviral vector shuttle plasmid as described previously (Swann et al., 2001). The adenoviral vector expressing Nef V10EΔ17-26 has been described previously (Williams et al., 2004).
An HIV molecular clone containing the Nef LL164,165AA mutation was generated by PCR mutagenesis of the HIV molecular clone, HXB-EP (Chen et al., 1996), using the following primers: mutant forward primer, 5′-GAAGGAGAGAACACCCGCGCGGCACACCCTGTGAGCCTGCAT-3′; with reverse primer (3′hxb-xba), 5′-GCTCTAGATGCTAGAGATTTTCCACACTG-3′, and mutant reverse primer, 5′-ATGCAGGCTCACAGGGTGTGCCGCGCGGGTGTTCTCTCCTTC-3′; with forward primer (5′ nef) 5′-GACAGATCCATTCGATTAGTG-3′. The resulting mutant PCR products were mixed and reamplified with 3′hxb-xba and 5′ nef primers, digested with XbaI and BamHI and cloned into the same sites of HXB-EP.
The A2/Nef fusion constructs were generated as follows: a PCR product containing the HLA-A2 sequence was generated with the primers; HLA-A2 5′-CGGGATCCACCATGGTACCGTGCACG-3′ and 5′-GGACTAGTCACTTTACAAGCTGTGAGAGA-3′. (For HLA-A2ΔTail, a different 3′ oligonucleotide was used to generate the truncated molecule 5′-GGACTAGTCTTCCTCCTCCACATCACAGC-3′). A PCR product containing wild-type or mutant Nef sequence (Williams et al., 2004) was generated with the primers; Nef 5′-GGACTAGTATGGGTGGCAAGTGGTCAAAA-3′ and 5′-GCGAATTCTCAGCAGTTCTTGAAGTACTC-3′. The HLA-A2 PCR product was digested with BamHI and SpeI, the Nef PCR product was digested with SpeI and EcoRI. These fragments were then cloned into the BglII and EcoRI sites of a bi-cistronic retroviral vector expressing an IRES GFP cassette (pMIG) (Van Parijs et al., 1999) in a three-way ligation. The control HLA-A2 IRES GFP construct was generated using the following primers 5′-CGGGATCCACCATGGTACCGTGCACG-3′ and 5′-GGTCAACTAGTCACTTTACAAGCTGTGAGAGA-3′. This PCR product was digested with BamHI and ligated into the BglII and HpaI sites of pMIG. A2 Y320A/Nef was generated from the A2/Nef template using the following mutant primers: 5′ primer; 5′-AGAAAAGGAGGGAGCGCCTCTCAGGCTGCA-3′, 3′ primer; 5′-TGCAGCCTGAGAGGCGCTCCCTCCTTTTCT-3′. A2/LL164,165AA was generated from the A2/Nef template using the following mutant primers: 5′ primer; 5′-ATGCAGGCTCACAGGGTGTGCCGCGCTGGTGTTCTCTCCTTT-3′, 3′ primer; 5′-AAAGGAGAGAACACCAGCGCGGCACACCCTGTGAGCCTGCAT-3′.
MSCV-EYFP-Golgi was constructed as follows: EYFP-Golgi (CLONTECH Laboratories, Inc.) was digested with NheI and HpaI. This fragment was ligated into the XbaI and EcoRV sites of pShuttle (Stratagene). The resulting construct was then digested with XhoI and BglII and ligated into the same sites in MSCV 2.1.
Viral transduction of T cells
CD8-depleted PHA-activated primary T cells or CEM-SS cells were transduced with VSV-G-pseudotyped HIV molecular clones (HXB-EP nef − and nef +), VSVG-pseudotyped murine retroviruses or adenoviral vectors as described previously (Collins et al., 1998; Swann et al., 2001; Kasper and Collins, 2003). Recombinant replication defective E3-deleted adenovirus expressing HIV-1 NL4-3 Nef, Nef V10EΔ17-26, Nef LL164,165AA, or no insert was generated by the University of Michigan Vector Core. For adenoviral transductions, CEM cell lines were resuspended in R2 media (RPMI, 2% FBS, P/S/G) at a concentration of 106 cells/ml. Adenovirus stocks were added to a relative multiplicity of infection (MOI; based on 293 cell infectivity) of 50–200 virus particles/cell. Cells were transferred to 96-well plates [100 μl (105 cells) per well]. After 2 h at 37°C, 200 μl of R10 media was added. Cells were harvested at 24–72 h after transduction.
Metabolic labeling and immunoprecipitation
Adeno-transduced CEM T cells were collected and washed twice with D-PBS (Invitrogen). Cells were incubated in prelabel media [RPMI –Cys –Met (Specialty Media, Inc.) + 10% dialyzed FBS (Invitrogen)] for 15 min at 37°C. Cells were pulsed with labeling media [prelabel media + 150 μCi/ml Pro-mix-l [35S] (>1,000 Ci/mmol; Amersham Biosciences)] for 15 min at 37°C. Cells were washed twice with R10 and chased in R10 for the indicated period of time (5 × 106 cells were used per time point). For some pulse-chase experiments, bafilomycin A1 (100 nM; Sigma-Aldrich), NH4Cl (25 mM; Fisher Scientific), or lactacystin (20 μM; Sigma-Aldrich) was included in the chase media. Cells were collected and washed once with D-PBS and lysed in 1 ml of lysis buffer (50 mM Tris, pH 8, 1% NP-40 (vol/vol), 5 mM MgCl2, 1 mM PMSF) for 30 min on ice, followed by centrifugation to remove insoluble material. Cellular lysates were precleared overnight at 4°C with 1.5 μg of mouse IgG1 (BD Biosciences) and 30 μl of protein A/G agarose (50% slurry; Calbiochem). For immunoprecipitations, 5 μg of anti–HLA-A2 (BB7.2) or 3.5 μg of anti-transferrin receptor antibody (Oncogene), was added to the precleared lysate, followed by the addition to 30 μl of protein A/G agarose beads. After 2 h at 4°C, the beads were washed three times with 1 ml RIPA buffer (Kasper and Collins, 2003). Endo Hf (New England Biolabs, Inc.) or neuraminidase (New England Biolabs, Inc.) digestion of immunoprecipitated material was performed according to the manufacturer's protocols. Samples were separated by 10% SDS-PAGE and the gels were dried and exposed to BioMaxMS films at −80°C. Radio-labeled proteins were quantitated using a Phosphor storage screen and Typhoon Scanner followed by processing with Image Quant software.
Immunofluorescence microscopy
CEM cells were allowed to adhere to chambered glass slides coated with poly-l-lysine (Sigma-Aldrich) for 1 h at 37°C. Cells were then fixed at RT for 15 min in D-PBS + 2% PFA. Next, cell membranes were permeabilized by incubation in D-PBS + 0.2% Tween 20 for 15 min at 37°C. Cells were washed twice with wash buffer (D-PBS, 10 mM Hepes, 10% goat serum [Sigma-Aldrich], 0.025% sodium azide) followed by blocking in wash buffer (adjusted to 10% goat serum and 10% human serum; Sigma-Aldrich) for 30 min on ice. For Fig. 1, mouse mAbs were used to detect LAMP-1 (clone H4A3; BD Biosciences; 1:500) and HLA-A2 (BB7.2, 20 μg/ml). Isotype-specific secondary antibodies were used to distinguish between these antibodies (anti–mouse IgG1 Alexa-Fluor546 [1:250; Molecular Probes] and anti–mouse IgG2b Alexa-Fluor488 [1:250; Molecular Probes], respectively). All antibody incubations were performed in wash buffer on ice for 20 min. Slides were mounted with coverslips using Prolong antifade reagent (Molecular Probes). Images were collected using a confocal microscope (model LSM 510; Carl Zeiss MicroImaging, Inc.) and processed using Adobe Photoshop 6.0 software. For inhibitor treatment, adeno-transduced CEM HA-HLA-A2 cells were resuspended in fresh R10 containing 100 nM bafilomycin A1 (Sigma-Aldrich) or solvent alone (DMSO) and incubated at 37°C for 3 h before adherence.
RNAi treatment
The following duplex siRNAs (Ambion) were used in this study: siGFP, sense 5′-GCUGACCCUGAAGUUCAUCTT-3′, antisense 5′-GAUGAACUUCAGGGUCAGCTT-3′; siμ3, sense 5′-GGACUACUUUGGUGAGUGUTT-3′, antisense 5′-ACACUCACCAAAGUAGUCCTG-3′. The μ1A siRNA was described previously (Hirst et al., 2003). Astrocytoma cells stably expressing CD4 (373-CD4) were transfected with annealed duplex siRNAs using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. In brief, cells were plated onto 6-well plates (105 cells per well) and the next day each well was transfected using 0.16 nmol siRNA and 4 μl of Lipofectamine 2000 reagent. 24 h later, cells were replated onto 6-well plates (105 cells per well) and the cells were transfected with siRNA again 24 h after replating. 4–6 h after transfection, the media was removed and the cells were transduced with adenovirus (MOI = 50). Cells were harvested and analyzed 24 h after transduction.
For experiments using CEM T cells, the cells were first transduced with adenovirus (MOI = 50). 24 h later, 5 × 106 cells were electroporated with 1 nmole siRNA according to the manufacturer's protocol (Amaxa Biosystems). The cells were then incubated for 48 h before FACS and Western blot assays. For Fig. 2 E, the cells were subjected to an initial siRNA transfection 1 d before the adenoviral transduction, and the experiment was then performed as described above.
Antibodies and Western blot analyses
IP-Western experiments were performed as described previously (Williams et al., 2004). HA-HLA-A2 was detected using the anti-HA antibody, HA.11 (1:5,000; Covance). Coprecipitating Nef protein was detected by Western blotting using antibodies obtained via the NIH AIDS Research and Reference Reagent Repository, Division of AIDS, NIAID, NIH: polyclonal anti-Nef antibody (2949, 1:5,000, a gift from R. Swanstrom, The University of North Carolina at Chapel Hill, Chapel Hill, NC; Shugars et al., 1993) or monoclonal anti-Nef (AG11, 1:500 for IP-Westerns and 1:10,000 for crude lysate, a gift from J. Hoxie, University of Pennsylvania, Philadelphia, PA; Chang et al., 1998). The adaptin subunit, μ1, was detected using RY/1 (1:2,500, a gift from L. Traub, University of Pittsburgh, Pittsburgh, PA; Traub et al., 1995). Other antibodies to AP subunits were purchased from BD Biosciences: γ-adaptin (1:100 for IP-Westerns; 1:1,000 for crude lysate); δ-adaptin (1:100 for IP-Westerns; 1:1,000 for crude lysate); and μ3 (p47A, 1:500). The HLA-A2–specific mAb, BB7.2 (Parham and Brodsky, 1981), and anti-Nef antibody, AG11 (Chang et al., 1998), were purified from ascites fluid as described previously (Kasper and Collins, 2003).
Online supplemental material
Fig. S1 shows that the addition of an HA tag does not affect MHC-I folding, maturation through the secretory pathway and responsiveness to Nef. Fig. S2 demonstrates that Nef targets mature MHC-I for degradation in HIV-infected T cells. Fig. S3 is coimmunoprecipitation and Western blot analysis demonstrating that the dileucine motif is not required for the Nef-MHC-I to recruit AP-1 in HIV-infected T cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200407031/DC1.
Acknowledgments
We are grateful to Dr. Linton Traub for antibody to μ1, to the NIH AIDS Reagent Repository for antibodies, to the University of Michigan vector core for adenovirus preparation, to Dr. Randy Schekman for helpful suggestions and to Drs. John Moran and Mark Benson for critical reading of the manuscript.
This research was supported by National Institutes of Health grant RO1 AI46998. M.R. Kasper was supported by the University of Michigan Genetics Training Program. M. Williams and J. Roeth were supported by the University of Michigan Cellular and Molecular Biology Training Program. M. Williams was also supported by a Rackham predoctoral fellowship and the Microbial Pathogenesis Training Program.
Abbreviations used in this paper: AP, adaptor protein; CTLs, cytotoxic T lymphocytes; endo H, endoglycosidase H; HIV, human immunodeficiency virus; HLA, human leukocyte antigen; LAMP-1, lysosome-associated membrane protein; MHC-I, major histocompatibility complex class I molecules; MOI, multiplicity of infection; MPR, mannose 6-phosphate receptor; PACS-1, phosphofurin acidic cluster sorting protein; P/S/G, penicillin, streptomycin, and glutamine.
References
- Aiken, C., J. Konner, N.R. Landau, M.E. Lenburg, and D. Trono. 1994. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell. 76:853–864. [DOI] [PubMed] [Google Scholar]
- Alexander, M., Y.C. Bor, K.S. Ravichandran, M.L. Hammarskjold, and D. Rekosh. 2004. Human immunodeficiency virus type 1 Nef associates with lipid rafts to downmodulate cell surface CD4 and class I major histocompatibility complex expression and to increase viral infectivity. J. Virol. 78:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagoveshchenskaya, A.D., L. Thomas, S.F. Feliciangeli, C.H. Hung, and G. Thomas. 2002. HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell. 111:853–866. [DOI] [PubMed] [Google Scholar]
- Bonifacino, J.S., and L.M. Traub. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72:395–447. [DOI] [PubMed] [Google Scholar]
- Bresnahan, P.A., W. Yonemoto, S. Ferrell, D. Williams-Herman, R. Geleziunas, and W.C. Greene. 1998. A dileucine motif in HIV-1 Nef acts as an internalization signal for CD4 downregulation and binds the AP-1 clathrin adaptor. Curr. Biol. 8:1235–1238. [DOI] [PubMed] [Google Scholar]
- Caplan, S., N. Naslavsky, L.M. Hartnell, R. Lodge, R.S. Polishchuk, J.G. Donaldson, and J.S. Bonifacino. 2002. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J. 21:2557–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, A.H., J.A. Hoxie, S. Cassol, M. O'Shaughnessy, and F. Jirik. 1998. Construction of single-chain antibodies that bind an overlapping epitope of HIV-1 Nef. FEBS Lett. 441:307–312. [DOI] [PubMed] [Google Scholar]
- Chen, B., R. Gandhi, and D. Baltimore. 1996. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J. Virol. 70:6044–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, G.B., R.T. Gandhi, D.M. Davis, O. Mandelboim, B.K. Chen, J.L. Strominger, and D. Baltimore. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 10:661–671. [DOI] [PubMed] [Google Scholar]
- Collins, K., B. Chen, S. Kalams, B. Walker, and D. Baltimore. 1998. HIV-1 Nef protein protects infected primary human cells from killing by cytotoxic T lymphocytes. Nature. 391:397–401. [DOI] [PubMed] [Google Scholar]
- Craig, H.M., T.R. Reddy, N.L. Riggs, P.P. Dao, and J.C. Guatelli. 2000. Interactions of HIV-1 nef with the mu subunits of adaptor protein complexes 1, 2, and 3: role of the dileucine-based sorting motif. Virology. 271:9–17. [DOI] [PubMed] [Google Scholar]
- Crump, C.M., Y. Xiang, L. Thomas, F. Gu, C. Austin, S.A. Tooze, and G. Thomas. 2001. PACS-1 binding to adaptors is required for acidic cluster motif-mediated protein traffic. EMBO J. 20:2191–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica, E.C., and G.S. Payne. 2001. Intracellular cycling of lysosomal enzyme receptors: cytoplasmic tails' tales. Cell. 106:395–398. [DOI] [PubMed] [Google Scholar]
- Doray, B., P. Ghosh, J. Griffith, H.J. Geuze, and S. Kornfeld. 2002. Cooperation of GGAs and AP-1 in packaging MPRs at the trans-Golgi network. Science. 297:1700–1703. [DOI] [PubMed] [Google Scholar]
- Erdtmann, L., K. Janvier, G. Raposo, H.M. Craig, P. Benaroch, C. Berlioz-Torrent, J.C. Guatelli, R. Benarous, and S. Benichou. 2000. Two independent regions of HIV-1 Nef are required for connection with the endocytic pathway through binding to the mu 1 chain of AP1 complex. Traffic. 1:871–883. [DOI] [PubMed] [Google Scholar]
- Geyer, M., H. Yu, R. Mandic, T. Linnemann, Y.H. Zheng, O.T. Fackler, and B.M. Peterlin. 2002. Subunit H of the V-ATPase binds to the medium chain of adaptor protein complex 2 and connects Nef to the endocytic machinery. J. Biol. Chem. 277:28521–28529. [DOI] [PubMed] [Google Scholar]
- Greenberg, M., L. DeTulleo, I. Rapoport, J. Skowronski, and T. Kirchhausen. 1998. a. A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr. Biol. 8:1239–1242. [DOI] [PubMed] [Google Scholar]
- Greenberg, M., A. Iafrate, and J. Skowronski. 1998. b. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 17:2777–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenway, A., A. Azad, and D. McPhee. 1995. Human immunodeficiency virus type 1 Nef protein inhibits activation pathways in peripheral blood mononuclear cells and T-cell lines. J. Virol. 69:1842–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek, S., S.J. Stahl, P.T. Wingfield, and A. Bax. 1996. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry. 35:10256–10261. [DOI] [PubMed] [Google Scholar]
- Harris, M.P., and J.C. Neil. 1994. Myristoylation-dependent binding of HIV-1 Nef to CD4. J. Mol. Biol. 241:136–142. [DOI] [PubMed] [Google Scholar]
- Hirst, J., A. Motley, K. Harasaki, S.Y. Peak Chew, and M.S. Robinson. 2003. EpsinR: an ENTH domain-containing protein that interacts with AP-1. Mol. Biol. Cell. 14:625–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janvier, K., H. Craig, D. Hitchin, R. Madrid, N. Sol-Foulon, L. Renault, J. Cherfils, D. Cassel, S. Benichou, and J. Guatelli. 2003. a. HIV-1 Nef stabilizes the association of adaptor protein complexes with membranes. J. Biol. Chem. 278:8725–8732. [DOI] [PubMed] [Google Scholar]
- Janvier, K., Y. Kato, M. Boehm, J.R. Rose, J.A. Martina, B.Y. Kim, S. Venkatesan, and J.S. Bonifacino. 2003. b. Recognition of dileucine-based sorting signals from HIV-1 Nef and LIMP-II by the AP-1 γ-sigma1 and AP-3 Δ-sigma3 hemicomplexes. J. Cell Biol. 163:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper, M.R., and K.L. Collins. 2003. Nef-mediated disruption of HLA-A2 transport to the cell surface in T cells. J. Virol. 77:3041–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, J.E., R.H. Massol, T.J. Nieland, and T. Kirchhausen. 2004. HIV Nef-mediated major histocompatibility complex class I down-modulation is independent of Arf6 activity. Mol. Biol. Cell. 15:323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gall, S., L. Erdtmann, S. Benichou, C. Berlloz-Torrent, L. Liu, R. Benarous, J. Heard, and O. Schwartz. 1998. Nef interacts with mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity. 8:483–495. [DOI] [PubMed] [Google Scholar]
- Le Gall, S., F. Buseyne, A. Trocha, B.D. Walker, J.M. Heard, and O. Schwartz. 2000. Distinct trafficking pathways mediate Nef-induced and clathrin-dependent major histocompatibility complex class I down-regulation. J. Virol. 74:9256–9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizee, G., G. Basha, J. Tiong, J.P. Julien, M. Tian, K.E. Biron, and W.A. Jefferies. 2003. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat. Immunol. 4:1065–1073. [DOI] [PubMed] [Google Scholar]
- Lu, X., H. Yu, S. Liu, F. Brodsky, and B. Peterlin. 1998. Interactions between HIV1 Nef and vacuolar ATPase facilitate the internalization of CD4. Immunity. 8:647–656. [DOI] [PubMed] [Google Scholar]
- Mangasarian, A., V. Piguet, J.K. Wang, Y.L. Chen, and D. Trono. 1999. Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J. Virol. 73:1964–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham, P., and F.M. Brodsky. 1981. Partial purification and some properties of BB7.2. A cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum. Immunol. 3:277–299. [DOI] [PubMed] [Google Scholar]
- Peden, A.A., V. Oorschot, B.A. Hesser, C.D. Austin, R.H. Scheller, and J. Klumperman. 2004. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 164:1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piguet, V., Y.L. Chen, A. Mangasarian, M. Foti, J.L. Carpentier, and D. Trono. 1998. Mechanism of Nef-induced CD4 endocytosis: Nef connects CD4 with the mu chain of adaptor complexes. EMBO J. 17:2472–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piguet, V., L. Wan, C. Borel, A. Mangasarian, N. Demaurex, G. Thomas, and D. Trono. 2000. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2:163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M.S. 2004. Adaptable adaptors for coated vesicles. Trends Cell Biol. 14:167–174. [DOI] [PubMed] [Google Scholar]
- Robinson, M.S., and J.S. Bonifacino. 2001. Adaptor-related proteins. Curr. Opin. Cell Biol. 13:444–453. [DOI] [PubMed] [Google Scholar]
- Rossi, F., A. Gallina, and G. Milanesi. 1996. Nef-CD4 physical interaction sensed with the yeast two-hybrid system. Virology. 217:397–403. [DOI] [PubMed] [Google Scholar]
- Schaefer, T.M., I. Bell, B.A. Fallert, and T.A. Reinhart. 2000. The T-cell receptor zeta chain contains two homologous domains with which simian immunodeficiency virus Nef interacts and mediates down-modulation. J. Virol. 74:3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, O., V. Marechal, S. Le Gall, F. Lemonnier, and J. Heard. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2:338–342. [DOI] [PubMed] [Google Scholar]
- Shugars, D.C., M.S. Smith, D.H. Glueck, P.V. Nantermet, F. Seillier-Moiseiwitsch, and R. Swanstrom. 1993. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J. Virol. 67:4639-4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann, S.A., M. Williams, C.M. Story, K.R. Bobbitt, R. Fleis, and K.L. Collins. 2001. HIV-1 Nef blocks transport of MHC class I molecules to the cell surface via a PI 3-kinase-dependent pathway. Virology. 282:267–277. [DOI] [PubMed] [Google Scholar]
- Traub, L.M. 2003. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J. Cell Biol. 163:203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub, L.M., S. Kornfeld, and E. Ungewickell. 1995. Different domains of the AP-1 adaptor complex are required for Golgi membrane binding and clathrin recruitment. J. Biol. Chem. 270:4933–4942. [DOI] [PubMed] [Google Scholar]
- Van Parijs, L., Y. Refaeli, J.D. Lord, B.H. Nelson, A.K. Abbas, and D. Baltimore. 1999. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity. 11:281–288. [DOI] [PubMed] [Google Scholar]
- Waguri, S., F. Dewitte, R. Le Borgne, Y. Rouille, Y. Uchiyama, J.F. Dubremetz, and B. Hoflack. 2003. Visualization of TGN to endosome trafficking through fluorescently labeled MPR and AP-1 in living cells. Mol. Biol. Cell. 14:142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, M., J.F. Roeth, M.R. Kasper, R.I. Fleis, C.G. Przybycin, and K.L. Collins. 2002. Direct binding of human immunodeficiency virus type 1 Nef to the major histocompatibility complex class I (MHC-I) cytoplasmic tail disrupts MHC-I trafficking. J. Virol. 76:12173–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, M., J.F. Roeth, M.R. Kasper, T.M. Filzen, and K.L. Collins. 2004. HIV-1 Nef domains required for disruption of MHC-I trafficking are also necessary for coprecipitation of Nef with HLA-A2. J. Virol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]