

Abstract
Phosphorylation of connexin43 (Cx43) on serine368 (S368) has been shown to decrease gap junctional communication via a reduction in unitary channel conductance. Examination of phosphoserine368 (pS368) in normal human skin tissue using a phosphorylation site–specific antibody showed relatively even distribution throughout the epidermal layers. However, 24 h after wounding, but not at 6 or 72 h, pS368 levels were dramatically increased in basal keratinocytes and essentially lost from suprabasal layers adjacent to the wound (i.e., within 200 μm of it). Scratch wounding of primary human keratinocytes caused a protein kinase C (PKC)-dependent increase in pS368 in cells adjacent to the scratch, with a time course similar to that found in the wounds. Keratinocytes at the edge of the scratch also transferred dye much less efficiently at 24 h, in a manner dependent on PKC. However, keratinocyte migration to fill the scratch required early (within <6 h) gap junctional communication. Our evidence indicates that PKC-dependent phosphorylation of Cx43 at S368 creates dynamic communication compartments that can temporally and spatially regulate wound healing.
Introduction
Gap junctions are tightly packed clusters of intercellular channels that directly connect the cytoplasms of adjacent cells. They coordinate cell-to-cell communication within tissues and allow for the transport of molecules <1,000 D among cells such as ions, amino acids, nucleotides, and second messengers (e.g., Ca2+, cAMP, cGMP, and IP3) (Simon et al., 1998; Saez et al., 2003). Gap junctions play significant regulatory roles in embryonic development, electrical coupling, metabolic transport in nonvascularized tissue, apoptosis, differentiation, and tissue homeostasis (Willecke et al., 2002; Saez et al., 2003). Dynamic communication compartments created by regulation of gap junctional communication have been implicated in the control of wound healing and differentiation (Clark, 1985; Grinnell, 1992; Gailit and Clark, 1994; Goliger and Paul, 1995; Martin, 1997; Kretz et al., 2003).
Gap junctions are composed of integral membrane proteins called connexins. There are ∼20 connexin gene family members in humans, many of which have been cloned and characterized (Willecke et al., 2002; Saez et al., 2003). During intercellular channel formation, six connexin proteins oligomerize into a hexameric hemichannel, or connexon, that traffics to the plasma membrane. One hemichannel docks with a second, in an opposing cell, to form an intact channel. These channels can be gated in response to various stimuli, including changes in voltage, pH, and connexin phosphorylation (Saez et al., 2003).
Connexin43 (Cx43) is phosphorylated at multiple serine residues in a variety of cell types (Crow et al., 1990; Musil et al., 1990; Brissette et al., 1991; Laird et al., 1991; Berthoud et al., 1992). Phosphorylation of Cx43 can affect trafficking, assembly, degradation, and channel gating. After treatment with the PKC activator PMA, Cx43 phosphorylation is increased, and gap junctional communication is decreased, in several different cell types (Brissette et al., 1991; Berthoud et al., 1992, 1993; Reynhout et al., 1992; Lampe, 1994). Phosphorylation of Cx43 on serine368 (S368; phosphoserine368 [pS368]) increases upon PMA treatment and throughout the cell cycle, and results in a reduction in unitary channel conductance (50 picosiemens channels are favored over 100 picosiemens channels) (Lampe et al., 2000; Solan et al., 2003).
Expression of connexin genes is tissue specific and although several connexins can be found in skin, Cx43 is the predominant connexin in human epidermis and in cultures of human keratinocytes (Fitzgerald et al., 1994; Di et al., 2001). Keratinocyte proliferation occurs in the basal layer of the epidermis and keratinocytes undergo terminal differentiation as they migrate through the suprabasal and granular layers to the skin surface. Connexin proteins are differentially expressed in skin, with lower expression in the proliferative regions and higher expression upon differentiation (Risek et al., 1992; Goliger and Paul, 1994; Salomon et al., 1994; Lampe et al., 1998; Kretz et al., 2003).
Wounding of the epidermis activates cell migration across the wound bed, increases proliferation, and promotes changes in cell-to-cell communication (Clark, 1985; Grinnell, 1992; Gailit and Clark, 1994; Goliger and Paul, 1995; Martin, 1997; Kretz et al., 2003). It has been suggested that gap junctional intercellular communication may regulate certain aspects of the wound healing process, including initiation/synchronization of cellular migration (Goliger and Paul, 1995; Lampe et al., 1998; Kretz et al., 2003). After wounding, connexin expression is decreased at the wound edge but enhanced at unwounded adjacent areas and upon wound closure when the cells differentiate (Goliger and Paul, 1995; Saitoh et al., 1997; Lampe et al., 1998). Cx43 antisense application to wounds accelerated keratinocyte migration and the rate of wound repair, resulting in less scarring (Qiu et al., 2003). Closure of wounds was 1 d faster in Cx43-deficient mice (Kretz et al., 2003). These results indicate that Cx43 regulation plays an important role in wound repair.
We examined Cx43 phosphorylation on S368 in normal and wounded human skin tissue. In unwounded skin, basal keratinocytes, which show the lowest expression of total Cx43, showed similar levels of staining for pS368 to those shown by the suprabasal cells. Remarkably, 24 h after wounding, pS368 labeling was dramatically increased but strictly limited to the basal cells. The levels and distribution of pS368 returned to normal at 72 h. We “scratch wounded” primary human foreskin keratinocytes (HFKs) to test for changes in gap junctional communication and delineate the signaling pathways involved. Efficient migration to fill the scratch was dependent on gap junctional communication within the first 6 h. Scratch wounding increased phosphorylation on S368 at 24 h, and communication at the wound edge was reduced in a manner dependent on PKC. Thus, this report documents dynamic changes in Cx43 phosphorylation at a specific site that affects intercellular communication during the complex process of wound healing in HFKs and in human skin. The observation that basal cells near the wound edge show a temporally and spatially regulated change in their communication properties indicates that restriction of specific signals between epithelial cells occurs during wound healing.
Results
Phosphorylation of Cx43 at S368 is increased in basal keratinocytes
Expression of connexin genes is tissue and species specific. In mouse epidermis, at least nine different connexins can be expressed at different levels throughout the skin layers during development (for review see Richard, 2000). Although many of the same proteins are present, the connexin expression pattern in human epidermis is different. The most abundant connexin expressed in human epidermis and cultures of human keratinocytes is clearly Cx43 (Fitzgerald et al., 1994; Di et al., 2001). In human skin, Cx43 is expressed at lower levels in the proliferative basal cell region and at higher levels upon differentiation in the suprabasal layers (Salomon et al., 1994; Lampe et al., 1998). Immunohistochemistry and differential interference contrast microscopy (Fig. 1 a) confirmed extensive Cx43 levels in the suprabasal cells. Basal cells, attached directly to the basement membrane (Fig. 1 a, dashed line), show only low levels of staining for Cx43.
Figure 1.
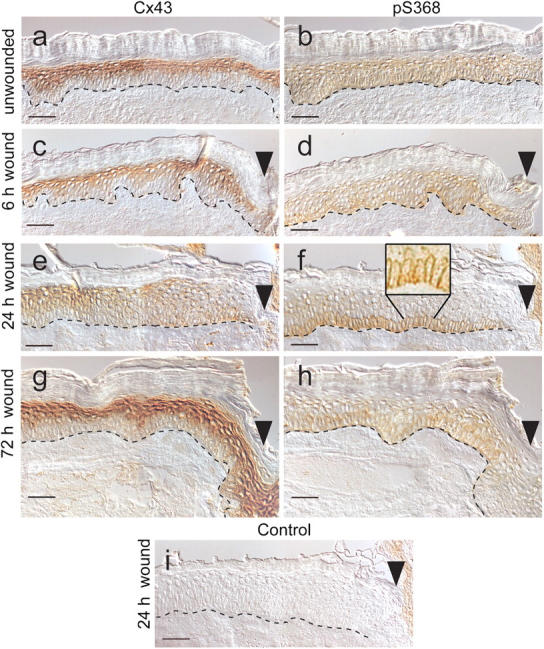
Immunohistochemistry staining of normal and wounded human skin collected 6, 24, and 72 h after wounding. Serial sections of skin tissue collected at 6, 24, and 72 h after wounding were reacted with antibodies to either Cx43 or pS368, and their localization is indicated by brown DAB staining. The rabbit primary antibody was omitted from the staining procedure for a 24-h wound section as a control (i). Dashed line indicates the basement membrane border between the dermis (lower) and epidermis (upper). Dead cornified cells (uppermost layers) lack Cx43 staining. (c–i) Wound edge is indicated by an arrowhead. Bars, 50 μm.
Several studies have shown that Cx43 is phosphorylated on multiple different serine residues throughout its life cycle in homeostatic cells (Crow et al., 1990; Musil et al., 1990; Brissette et al., 1991; Kadle et al., 1991; Laird et al., 1991; Berthoud et al., 1992). Of particular interest, phosphorylation of Cx43 on S368 increases in response to PMA treatment and throughout the cell cycle, and results in reduced unitary channel conductance (Lampe et al., 2000; Solan et al., 2003). Serial tissue sections were examined by immunohistochemistry using two antibodies; one that recognized total Cx43 and one that recognized Cx43 only when it was phosphorylated at S368 (Solan et al., 2003). Basal keratinocytes, which show the lowest expression of total Cx43, showed the same level of phosphorylation at S368 as did more differentiated cell layers (Fig. 1, compare a with b).
Phosphorylation of Cx43 at S368 is limited to basal keratinocytes 24 h after wounding
Total Cx43 levels are diminished in the suprabasal cells near the wound edge after wounding (Goliger and Paul, 1995; Saitoh et al., 1997; Lampe et al., 1998). To determine the temporal regulation of S368 phosphorylation, we performed immunostaining on a time course of human wounds. We investigated Cx43 and pS368 levels in the wound bed region of wounds that had healed for 6 (Fig. 1, c and d), 24 (Fig. 1, e and f), and 72 h (Fig. 1, g and h). 6 h after wounding, little change in the normal distribution of Cx43 or pS368 antibody staining was observed, except a reduction in Cx43 levels that occurred in suprabasal cells within 50 μm of the wound (Fig. 1, c and d). At 24 h, an extensive reduction in suprabasal Cx43 staining and slight increases in basal cell levels occurred, yielding a more even distribution up to 300 μm from the wound bed (Fig. 1 e). In contrast, the levels of phosphorylation at S368 24 h after wounding were dramatically increased in basal keratinocytes and essentially absent throughout the rest of the epidermis (Fig. 1 f). This unique staining pattern extended up to 400 μm from the wound site, beyond which point pS368 and Cx43 levels normalized to the unwounded levels. In the magnified inset in Fig.1 f, the staining is present at interfaces between basal cells and at basal cell–suprabasal cell interfaces (shown without differential interference contrast optics or the line indicating the basement membrane). As a control, 24-h wounds were processed without the mouse or rabbit antibody, and little staining was observed when either was omitted from the staining procedure (Fig. 1 i, rabbit control shown). By 72 h after wounding, S368 phosphorylation expression patterns and levels were similar to those in unwounded skin. Thus, 24-h wounds showed a unique basal cell staining pattern for Cx43 phosphorylation at S368.
Cx43 phosphorylation on S368 in primary HFKs
Primary HFKs were used to allow examination of gap junctional communication and signaling pathways associated with the wounding process. To determine whether wounding of HFKs would affect phosphorylation at S368 consistent with in vivo wounds, we scratch wounded HFK monolayers with a pipette tip in a manner consistent with previous studies (e.g., Yamada et al., 2000). We performed immunofluorescence to determine whether changes elicited by scratch wounding paralleled those found in the in vivo wounds; i.e., whether pS368 levels increased near the scratch edge. Fig. 2 shows immunofluorescence of total Cx43 (A) and pS368 (B) in keratinocytes 24 h after scratch wounding. Although a reduction in Cx43 in the differentiating suprabasal cells was observed in wounds (Fig. 1 f), little change in Cx43 levels was observed near the scratch (Fig. 2 A), possibly because the cultured cells, like basal cells, are not differentiated. The ratio of pS368 to Cx43 signal, most clearly demonstrated in the overlay panel (Fig. 2 C), shows that the relative pS368 levels are highest 1–4 cell diameters from the scratch. Note that the signal for pS368 appears to be plasma membrane–associated and continuous throughout the cell–cell interfaces, similar to the basal cell staining in the in situ wounds (Fig. 1 f, inset). The total Cx43 antibody used for this staining has a preference for the Cx43 present in gap junctions (Solan et al., 2003), so the continuous pS368 staining may represent protein that is not yet assembled into gap junctions. This possibility would be consistent with the observation that PKC activation inhibits the formation of new gap junctions (Lampe, 1994). Further away from the scratch, most of the pS368 signal overlays with the total Cx43 signal.
Figure 2.

Changes in Cx43 and pS368 levels 24 h after scratch wounding HFKs. Immunofluorescence for total Cx43 (A) and pS368 (B) staining and their colocalization (C). DAPI nuclear staining (blue) is also included in the overlay. Bar, 50 μm.
Whether the level of pS368 throughout the scratch-wounded culture was generally elevated is difficult to determine by immunofluorescence, so we performed a time course experiment and immunoblotted for pS368 and total Cx43 at 3, 6, 10, 24, 48, and 72 h after the scratch wound (Fig. 3 A). The phosphorylation level at S368 increased up to 24 h and leveled at 48 h, after which proliferation and migration completely closed the scratch wound. The extent of phosphorylation then decreased, at 72 h, to control levels. Given the immunofluorescence results, we would expect that the cells near the scratch contribute most of the increase in pS368 signal.
Figure 3.

pS368 levels rise relative to total Cx43 in response to scratch wounding in a manner dependent on PKC activity. (A) Time course after wounding of the ratio of pS368 to total Cx43, normalized to the zero time level. (B) pS368/Cx43 levels 24 h after wounding in the presence and absence of kinase inhibitors, normalized to the control level. Control was not scrape wounded but was harvested at the same time as the wounded samples. BIM, bisindolylmaleimide (200 nM); Ro-31, Ro-31-8220 (100 nM); and Gö6976 (100 nM). (A and B) Error bars represent standard error of the mean. (C) The COOH-terminal region of Cx43 (residues 246–382) was phosphorylated with PKCɛ and immunoblotted as described in Materials and methods. Phosphorylation and total Cx43 were detected with antibodies to pS368 and the COOH-terminal region of Cx43. (D) Representative immunoblot of the PKC inhibitor data summarized in B. Note that the migration of the pS368 signal overlays with the fastest migrating species of Cx43 and there is a higher level of total Cx43 in the control because cells were not lost during scratch wounding.
We wanted to determine which signaling pathways might be responsible for phosphorylation at S368, so we scratch wounded HFKs in the presence and absence of protein kinase inhibitors and assayed by immunoblot after 24 h. Inhibitors such as PD98059, Gö6983, chelerythrine, and K252a had little effect (unpublished data), but some PKC inhibitors were very effective at reducing the relative level of phosphorylation at S368. For example, relatively low concentrations of bisindolylmaleimide (BIM) and Ro-31-8220 reduced the extent of phosphorylation at S368 by ∼45% (Fig. 3, B and D), to a level >30% lower than that in the control (unscratched, also harvested at 24 h). However, other PKC inhibitors, such as Gö6976, had less effect. Comparison of the specificity of these PKC inhibitors can yield clues to the identity of the PKC isoforms that phosphorylated Cx43 at S368. BIM and Gö6976 have similar specificities, except Gö6976 has little effect on the novel PKCs. Of the novel PKCs, both BIM and, especially, Ro-31-8220 affect primarily PKCɛ. To determine whether PKCɛ was capable of phosphorylating Cx43 at S368, we tested whether the COOH-terminal region of Cx43 (residues 246–382) was a substrate for PKCɛ in vitro. Purified PKCɛ and fusion protein were reacted in the presence of lipid vesicles, ATP, PMA, and Mg2+. After electrophoresis and immunoblotting, detection of total Cx43 and pS368 (Fig. 3 C) showed significant pS368 signal in the lane where PKCɛ was used to phosphorylate Cx43. Therefore, the in vitro phosphorylation and inhibition studies in HFK cultures indicate that PKCɛ is active and capable of phosphorylating S368 in cells.
Gap junctional communication was assayed by microinjection of Alexa Fluor 488 (with a molecular weight of 570.5) in keratinocytes present in 24-h scratch-wounded cultures, to determine whether phosphorylation at S368 affected the ability of HFKs to communicate. Examples of injections distant from and near the scratch edge and of the resulting dye transfer are shown in Fig. 4 A. In untreated cells (Fig. 4 B, CON), communication was limited to ∼4 cells when dye was injected near the scratch, compared with ∼25 cells in unwounded areas. When BIM was added to the scratch-wounded cells, gap junctional communication adjacent to the scratch occurred to ∼10 cells (Fig. 4 B) and was 2.4-fold more extensive (Fig. 4 C) than for the untreated cells injected at the scratch edge. Because cells injected adjacent to the scratch can only transfer away from the wound, the dye transfer levels would be expected to be less than half the level they are in cells that are injected internal to the wound. Thus, the BIM-treated cells adjacent to the scratch transferred dye nearly as efficiently as cells distant from the wound, whereas BIM had little apparent effect on the extent of dye transfer in cells injected far from the wound. Although Fig. 2 shows low levels of phosphorylation of Cx43 at S368 in cells distant from the scratch, apparently these levels are below a threshold level of phosphorylation necessary for detection of a reduction in gap junctional communication by dye transfer. Given the high specificity of BIM, these results indicate that PKC is likely integral to the wound signaling pathway responsible for the temporal reduction of communication of keratinocytes adjacent to human wounds.
Figure 4.

Gap junctional communication near and distant from the scratch wound edge. (A) Phase (top) and Alexa Fluor 488 fluorescence (bottom) views of dye spread after microinjection (injected cell marked with arrows) of scratch edge (left and middle) or distant cells (right). Bar, 25 μm. (B) Average number of cells that received dye after microinjection into untreated (CON) or 200 nM BIM-treated cells at the scratch edge or distant from the scratch (Internal). Error bars represent standard error of the mean. (C) The ratio of the number of recipient cells for BIM-treated cells over control at the scratch edge or distant from the scratch (Internal).
Because gap junctional communication is known to regulate many different cellular processes, phosphorylation at S368 could regulate multiple wound healing processes, including proliferation, differentiation, and migration. Previous results by others (Noszczyk and Majewski, 2001; Onuma et al., 2001) and staining of our wounds (unpublished data) have shown that proliferation is most extensive 2–7 d after wounding and, thus, that any link with S368 phosphorylation would likely be indirect. We examined migration of keratinocytes up to 48 h after scratch wounding in the presence of BIM or a gap junctional communication inhibitor, carbenoxolone (CBX; Rozental et al., 2001). 6 h after scratch wounding, essentially no migration had occurred under all conditions (Fig. 5 A), consistent with little change in Cx43 expression in the in situ wounds (Fig. 1, c and d). Treatment of keratinocytes with BIM 2 h before scratch wounding (BIM-0h) significantly (P < 0.001) reduced migration at 24 and 48 h, to ∼70% of the control level at the same time points (Fig. 5 A). If BIM was added 6 h after scratch wounding (Fig. 5 A, BIM-6h), little difference was observed in total migration, compared with the control. However, after 48 h, the migration front in BIM-treated cells was noticeably more irregular than in untreated cells, indicating nonuniform migration/outgrowth (Fig. 5 B). We quantified this difference by measuring the length of a line drawn along the front edge of the leading cells after 48 h of migration, relative to the length of a line drawn along the front immediately after the scratch was performed (Fig. 5 B). Cells treated with BIM had average migration front lengths per millimeter of scratch length of 1.19 (BIM-0h) and 1.30 mm (BIM-6h), and both lengths were significantly (Fig. 5 B, P < 0.0001) longer than in the control (1.07) or after CBX treatment (1.06), implying a potential role for PKC regulation in the organization of migration.
Figure 5.
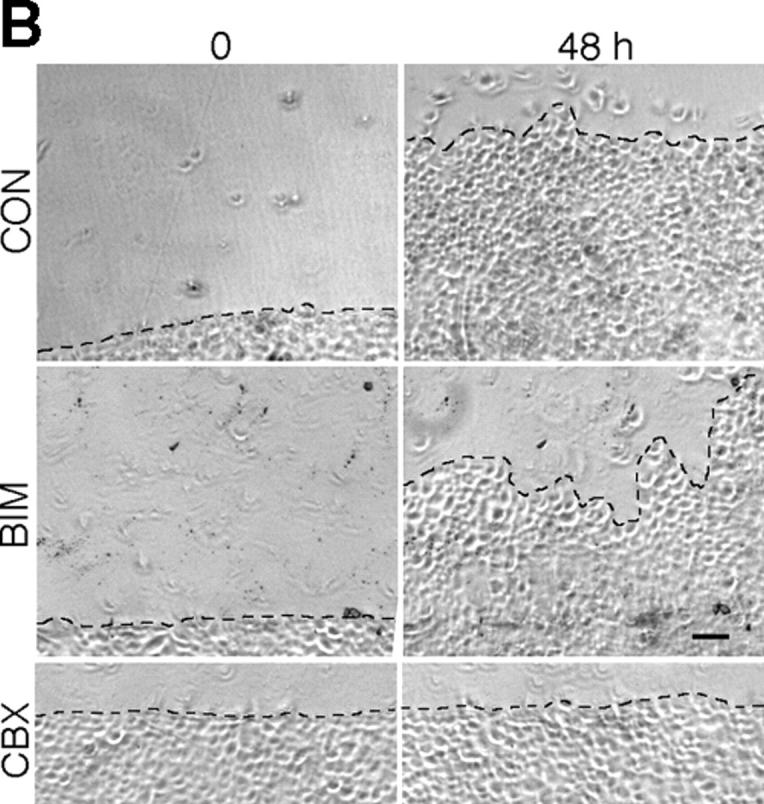

Migration of scratch-wounded keratinocytes treated with inhibitors of gap junctional communication or PKC. (A) HFKs were either pretreated for 2 h with 15 μM carbenoxolone (CBX-0h) or 200 nM bisindolylmaleimide (BIM-0h), or untreated (CON), and then were scratch wounded and incubated for 48 h. In some cases, 15 μM CBX or 200 nM BIM was added 6 h after scratch wounding to previously untreated cultures (CBX-6h and BIM-6h, respectively). Error bars represent standard error of the mean. (B) Relative position of untreated (CON), BIM-treated, or CBX-treated cells immediately after scratch wounding (0 time) compared with 48 h later. The lengths of the dashed lines indicating the migration fronts at 48 h were measured and compared with their respective zero time lengths immediately after scratch wounding. Bar, 100 μm.
Treatment with CBX led to very different results depending on whether the inhibitor was added 2 h before (CBX-0h) or 6 h after (CBX-6h) scratch wounding (Fig. 5 A). If the inhibitor was added before scratch wounding (Fig. 5 A, CBX-0h), very little migration (Fig. 5 B) was observed up to 48 h (significantly different from the results for CON, CBX-6h, BIM-0h, or BIM-6h; Fig. 5 A, P < 0.0001). However, if the inhibitor was added 6 h after the scratch, migration was similar to the control level (Fig. 5 A, compare CON with CBX-6h). These results indicate that migration to fill a scratch wound requires gap junctional communication early after wounding.
Discussion
We have shown previously that phosphorylation of Cx43 on S368 reduces unitary channel conductance of gap junction channels (Lampe et al., 2000; Solan et al., 2003). Here, we report that phosphorylation at S368 is highly regulated during wound repair. Cx43 phosphorylated on S368 was found to be evenly distributed throughout unwounded human epidermal layers. However, a striking change was observed 24 h after wounding, when pS368 levels were found to be dramatically increased in basal keratinocytes and essentially lost from suprabasal layers adjacent to the wound (i.e., within 300 μm). The changes in pS368 were temporally regulated, as they were observed only at 24 h after wounding and were not present in 6- or 72-h wounds. We could mimic activation of keratinocytes using a scratch wound technique, and we observed increased phosphorylation of S368 in cells adjacent to the scratch, with a time course that was similar to that of the in situ wounds, in a manner dependent on PKC, likely the ɛ isoform. These keratinocytes at the edge of the scratch transferred dye much less efficiently, in a manner that was dependent on PKC. Furthermore, gap junctional communication within the first 6 h after scratch wounding was necessary for efficient keratinocyte migration to close the scratch. Thus, changes in Cx43 phosphorylation at S368 were tightly linked, temporally, with changes in gap junctional communication and keratinocyte migration.
Connexin proteins have distinct expression patterns in skin, with more expression upon differentiation (Risek et al., 1992; Goliger and Paul, 1994; Salomon et al., 1994). Similarly, the high level of S368 phosphorylation in basal cells near the wound delineates a communication compartment distinct from suprabasal cells. For example, signals produced by suprabasal cells that inhibit the initiation of migration or promote differentiation might not be passed to basal cells that are highly phosphorylated at S368. The highly specific temporal regulation is particularly noteworthy, as the only times these signals would be restricted are at ∼24–48 h. Indeed, consistent with feedback control as observed in many signaling pathways, the levels of Cx43 and pS368 returned to the unwounded level after 72 h. Thus, in both the in situ wounds and scratch-wounded HFKs, phosphorylation at S368 appears to be a transient process that regulates communication in a time-dependent manner.
The purpose of Cx43 phosphorylation at S368 may also be to inhibit the ability of basal keratinocytes to communicate signals that initiate differentiation. Communication of different growth and differentiation signals by two related connexins has been shown in the lens, where connexin46 (Cx46) and connexin50 (Cx50) appear to transmit different signaling molecules (White, 2002). Knockout of either connexin results in cataracts, but lack of Cx50 also results in reduced ocular growth that is not relieved by knocking Cx46 into the Cx50 promoter. However, the knockin mouse no longer forms cataracts, indicating that Cx50 transmits different growth and differentiation signals from those transmitted by Cx46. Given the change in conductance properties associated with phosphorylation of Cx43 at S368, we believe that different differentiation/activation/migration signals may be transmitted by the basal keratinocytes as compared with the more differentiated layers, and that this regulation can be highly dynamic, as indicated by the wound studies.
During wound healing, epidermal cells must undergo changes in communication, proliferation, differentiation, migration, and activation (Clark, 1985; Grinnell, 1992; Gailit and Clark, 1994; Martin, 1997). Dynamic changes occur in the expression of specific proteins during wound repair. For example, wounding temporally alters expression of E-cadherin, laminin 5, p16, and integrin α6β4 in basal keratinocytes at the wound edge (Stepp et al., 1996; Lampe et al., 1998; Natarajan et al., 2003). However, the timing and the highly specific compartmentalized localization of pS368 in basal cells distinctly point to specific migration/differentiation roles. Clearly, gap junctional communication that occurs before and after the 24–48-h window would not be affected by S368 phosphorylation. Thus, early events such as increased production of laminin 5 and other activation events could still be signaled/regulated by gap junctional communication. Indeed, gap junctional communication before 6 h appears to be critical for migration, consistent with previous results indicating that cellular migration was inhibited in endothelial cells expressing dominant-negative Cx43 (Kwak et al., 2001) and that directionality of movement was reduced in Cx43-null neural crest cells (Xu et al., 2001). Furthermore, the reduction in gap junctional communication at 24–48 h is a change in conductance, not a complete closure (Lampe et al., 2000). Therefore, the transfer of smaller, more permeable molecules will not be eliminated by this phosphorylation event. BIM treatment could then lead to temporally inappropriate transfer of larger, gap junction–permeant molecules that might be important in regulating differentiation or coordination of migration. Alternatively, Cx43 phosphorylation might affect the interaction of Cx43 with proteins important in migration or differentiation. Consistent with these ideas, Xu et al. (2001) suggested that transfer of carboxyfluorescein, a relatively small dye with a molecular weight of 376.3, may not be a suitable gauge of the molecules important in neural crest cell motility, or Cx43-interacting proteins important in regulating motility might be affected by Cx43 conformational changes—possibly caused by connexin phosphorylation. In any case, these results must be viewed with caution because dye transfer can be a poor predictor of the transfer of biologically important molecules (Goldberg et al., 2002) and BIM would be expected to have multiple effects on PKC-activated pathways that could influence migration and differentiation. For example, BIM pretreatment reduced keratinocyte migration by ∼30%, even though dye transfer increased. Thus, PKC-dependent pathways may enhance migration independent of Cx43.
A reduction in gap junctional communication several hours after wounding appears to be important in healing, as is evidenced by changes in connexin expression near the wound bed (Clark, 1985; Grinnell, 1992; Gailit and Clark, 1994; Goliger and Paul, 1995; Martin, 1997). Cx43 expression patterns and response to wounding differ in rodents and humans (Fitzgerald et al., 1994; Salomon et al., 1994; Goliger and Paul, 1995; Saitoh et al., 1997; Lampe et al., 1998; Richard, 2000; Di et al., 2001). However, the importance of tight regulation of gap junctional communication during the wounding process is evident. Wound closure occurred 1 d earlier (2 vs. 3 d) in Cx43-deficient mice, compared with wild-type controls (Kretz et al., 2003). Furthermore, Cx43 antisense application to wounds accelerated the rate of keratinocyte migration, reduced inflammation, and increased the rate of wound repair, resulting in less scarring (Qiu et al., 2003). Multiple signaling pathways regulate wound repair. Calcium levels play an important role in the regulation of wounded skin (Lansdown, 2002). Mitogen-activated protein kinases have been shown to coordinate migration and proliferation (Sharma et al., 2003). Many reports show that PKC plays a central regulatory role in wound repair, and PKC is activated by integrin–extracellular matrix interactions after wounding (Clark and Brugge, 1995; Wallis et al., 2000). PKC can phosphorylate Cx43 at S368 (Fig. 3; Lampe et al., 2000), suggesting that PKC is a key component of the regulatory pathway that links the wounding event to changes in intercellular communication. The distinct distribution of pS368 in basal cells, the transient nature of the phosphorylation event, and the resulting change in gap junctional communication and migration all imply important roles for Cx43 in the regulation of epidermal cell activation after wounding and a role for gap junctions in forming a basal cell–specific “communication compartment” to spatially regulate these processes.
Materials and methods
Preparation of human skin tissues
Simplate II bleeding-time devices (Organon Teknika Corp.) were used to create uniform incisional wounds (5 mm long and 1 mm deep) on the lateral lower legs of 11 normal male and 3 normal female volunteers. This human wound model has been described previously in detail (Olerud et al., 1995). Wounds were harvested by 4-mm punch biopsies at 6, 24, and 72 h after wounding, using local 1% lidocaine for anesthesia; immediately frozen in O.C.T. (Sakura Finetek); and stored at −70°C. Volunteers were recruited using methods approved by the University of Washington Institutional Review Board.
Immunostaining of tissues
6-μm cryostat sections of incisional wounds were immunolabeled using indirect immunoperoxidase, as described previously (Lampe et al., 1998). All tissue sections were postfixed in cold acetone and then labeled with primary antibodies mouse anti-Cx43 (Cx43NTI; described in Goldberg et al., 2002; Solan et al., 2003) or anti-phospho–connexin 43 Ser368 (Cell Signaling Technology) at dilutions of 1:1,200 and 1:250, respectively. We detected signal using biotinylated goat anti–rabbit and biotinylated horse anti–mouse (Vector Laboratories) at dilutions of 1:400 and 1:200, respectively, with a streptavidin complex kit (SABC; Vector Laboratories), 0.12% 3, 3′ diaminobenzidine for the chromogen, and Glycergel (DakoCytomation) as mounting media. All tissue images were viewed with a microscope (Microphot-SA; Nikon), using brightfield and differential interference contrast optics.
Cells and cell culture
Normal primary HFKs were prepared as described previously (Boyce and Ham, 1983). Cells were maintained in serum-free keratinocyte growth medium (Clonetics Corp.) containing insulin, epidermal growth factor, hydrocortisone, and bovine pituitary extract in a humidified 5% CO2 incubator at 37°C. To mimic wounding, parallel 90–95% confluent HFK cultures grown on 60-mm culture dishes were left untreated or were scratch wounded with a plastic pipette tip. Immunofluorescence was performed as described previously (Brown et al., 2004), using formaldehyde fixation and Alexa Fluor 488–labeled goat anti–rabbit and Alexa Fluor 594 goat anti–mouse (both highly cross-adsorbed; Molecular Probes), on a scanning confocal microscope (Meta Multi-Photon, LSM 510; Carl Zeiss MicroImaging, Inc.) using Argon (488 nm) and HeNe (543 nm) lasers with a 40×/1.3 oil objective (Plan Neofluar). For immunoblot analysis, cultures were consistently “scratched” in a spiral pattern followed by a crosshatch pattern in the absence (control) or presence of the kinase inhibitors PD98059, Gö6983, chelerythrine, K252a, BIM, and Ro-31-8220 (all from Calbiochem). Cells were washed once with PBS and harvested on ice-cold 1% Triton X-100 in PBS or 2% SDS sample buffer, both containing 500 μM Na3VO4, 50 mM NaF, 2 mM PMSF, and 1× Complete protease inhibitors (Roche Diagnostics), as performed previously (Solan et al., 2003).
Immunoblot detection of Cx43 and pS368 levels
Cellular proteins were separated by SDS-PAGE (10% acrylamide). Proteins were transferred to nitrocellulose, the membrane was blocked in 1% milk in PBS, and Cx43 and pS368 were detected simultaneously on the same blot using mouse anti-Cx43 (Cx43NT1) at a dilution of 1:5,000 and rabbit anti-phosphoCx43 S368 at a dilution of 1:1,000, in blocking solution. Membranes were washed three times with PBS for 15 min per wash, and primary antibodies were detected using IRDye800–donkey anti–mouse (Rockland) and goat anti–rabbit Alexa Fluor 680 (Molecular Probes) secondary antibodies diluted at 1:2,500. The absolute levels of these antibodies were directly detected by fluorescence using the Odyssey image analysis system (LI-COR Biotechnology). To determine the ratio of Cx43 to pS368, fluorescence values were ratioed and then divided by the zero time for the time course experiment or the control value for the drug treatment experiment to yield the relative ratio for each time point. The relative ratios were averaged across experiments and the SEM was calculated for each time point.
In vitro phosphorylation and detection of pS368 were performed with the COOH-terminal portion (amino acids 246–382) of Cx43 containing six NH2-terminal histidine residues (Cooper and Lampe, 2002) and purified PKCɛ (Panvera), according to previously published methods (Lampe et al., 2000).
Microinjection and migration studies
HFKs were grown on 60-mm dishes to confluency and, where indicated (Fig. 4), were either scratch wounded as described above or left unwounded, in the presence or absence of 200 nM BIM. Injections were performed on an inverted microscope (Diaphot, model TE300; Nikon) with phase and fluorescence optics, using a micromanipulator (Narashigi). Micropipettes were drawn from borosilicate glass capillaries (Kwik-Fil; World Precision Instruments, Inc.; 1.0-mm OD × 0.75 mm). 10 mM Alexa Fluor 488 (570.5 Mr) in 0.2 M KCl was injected until the injected cell was brightly fluorescent, and then the pipette was removed. The number of cells receiving dye when coupled (i.e., the extent of dye coupling) was assessed 3 min after microinjection with a cooled digital camera (MicroMax; Princeton Instruments) driven by an attached PC and Metamorph imaging software. For migration studies, HFKs were grown on 100-mm dishes and scratch wounded with a cell lifter along marked lines. Specific areas for measurement were also marked, and digital images were taken with the inverted microscope (Nikon) immediately after scraping and at 6, 24, and 48 h. Where indicated (Fig. 5), 15 μM CBX (3-hydroxy-11-oxoolean-12-en-30-oicacid 3-hemisuccinate) or 200 nM BIM was added. After alignment, images were superimposed and migration distances were measured. Three random positions per image were measured and at least 20 images were collected per experiment. Results from three separate experiments were compiled by taking the ratio of the distance migrated at 6 and 24 h relative to the distance the untreated cells migrated at 48 h. Standard error was calculated and results were compared with a two-tailed, unequal variance t test. For measurement of migration front regularity, the length of a line drawn along the front edge of the leading cells after 48 h of migration was measured relative to the length of a line drawn immediately after the scratch was performed. These measurements were compared between BIM-treated and untreated cultures using a two-tailed, unequal variance t test.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants GM55632 (to P.D. Lampe), AR47963 (to W.G. Carter), and DK59221 (to J.E. Olerud). T.S. Richards was supported in part by NIH training grant T32-AI07509.
T.S. Richards and C.A. Dunn contributed equally to this paper.
Abbreviations used in this paper: BIM, bisindolylmaleimide; CBX, carbenoxolone; Cx43, connexin43; Cx46, connexin46; Cx50, connexin50; HFK, human foreskin keratinocyte; pS368, phosphoserine368; S368, serine368.
References
- Berthoud, V.M., M.L.S. Ledbetter, E.L. Hertzberg, and J.C. Saez. 1992. Connexin43 in MDCK cells: Regulation by a tumor-promoting phorbol ester and calcium. Eur. J. Cell Biol. 57:40–50. [PubMed] [Google Scholar]
- Berthoud, V.M., M. Rook, E.L. Hertzberg, and J.C. Saez. 1993. On the mechanism of cell uncoupling induced by a tumor promoter phorbol ester inclone 9 cells, a rat liver epithelial cell line. Eur. J. Cell Biol. 62:384–396. [PubMed] [Google Scholar]
- Boyce, S.T., and R.G. Ham. 1983. Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture. J. Invest. Dermatol. 81:335–405. [DOI] [PubMed] [Google Scholar]
- Brissette, J.L., N.M. Kumar, N.B. Gilula, and G.P. Dotto. 1991. The tumor promoter 12-O-tetradecanoylphorbol-13-acetate and the ras oncogene modulate expression and phosphorylation of gap junction proteins. Mol. Cell. Biol. 11:5364–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, T.A., T.M. Yang, T. Zaitsevskaia, Y. Xia, C.A. Dunn, R.O. Sigle, B. Knudsen, and W.G. Carter. 2004. Adhesion or plasmin regulates tyrosine phosphorylation of a novel membrane glycoprotein p80/gp140/CUB domain-containing protein 1 in epithelia. J. Biol. Chem. 279:14772–14783. [DOI] [PubMed] [Google Scholar]
- Clark, R.A. 1985. Cutaneous tissue repair: basic biological considerations. J. Am. Acad. Dermatol. 13:701–725. [DOI] [PubMed] [Google Scholar]
- Clark, E.A., and J.S. Brugge. 1995. Integrins and signal transduction pathways: the road taken. Science. 268:233–239. [DOI] [PubMed] [Google Scholar]
- Cooper, C.D., and P.D. Lampe. 2002. Casein kinase 1 regulates connexin43 gap junction assembly. J. Biol. Chem. 277:44962–44968. [DOI] [PubMed] [Google Scholar]
- Crow, D.S., E.C. Beyer, D.L. Paul, S.S. Kobe, and A.F. Lau. 1990. Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol. Cell. Biol. 10:1754–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di, W.L., E.L. Rugg, I.M. Leigh, and D.P. Kelsell. 2001. Multiple epidermal connexins are expressed in different keratinocyte subpopulations including connexin 31. J. Invest. Dermatol. 117:958–964. [DOI] [PubMed] [Google Scholar]
- Fitzgerald, D.J., N.E. Fusenig, P. Boukamp, C. Piccoli, M. Mesnil, and H. Yamasaki. 1994. Expression and function of connexin in normal and transformed human keratinocytes in culture. Carcinogenesis. 15:1859–1865. [DOI] [PubMed] [Google Scholar]
- Gailit, J., and R.A. Clark. 1994. Wound repair in the context of extracellular matrix. Curr. Opin. Cell Biol. 6:717–725. [DOI] [PubMed] [Google Scholar]
- Goldberg, G.S., A.P. Moreno, and P.D. Lampe. 2002. Heterotypic and homotypic gap junction channels mediate the selective transfer of endogenous molecules between cells. J. Biol. Chem. 277:36725–36730. [DOI] [PubMed] [Google Scholar]
- Goliger, J.A., and D.L. Paul. 1994. Expression of gap junction proteins Cx26, Cx31.1, Cx37, and Cx43 in developing and mature rat epidermis. Dev. Dyn. 200:1–13. [DOI] [PubMed] [Google Scholar]
- Goliger, J.A., and D.L. Paul. 1995. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol. Biol. Cell. 6:1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinnell, F. 1992. Wound repair, keratinocyte activation and integrin modulation. J. Cell Sci. 101:1–5. [DOI] [PubMed] [Google Scholar]
- Kadle, R., J.T. Zhang, and B.J. Nicholson. 1991. Tissue-specific distribution of differentially phosphorylated forms of Cx43. Mol. Cell. Biol. 11:363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretz, M., C. Euwens, S. Hombach, D. Eckardt, B. Teubner, O. Traub, K. Willecke, and T. Ott. 2003. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J. Cell Sci. 116:3443–3452. [DOI] [PubMed] [Google Scholar]
- Kwak, B.R., M.S. Pepper, D.B. Gros, and P. Meda. 2001. Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol. Biol. Cell. 12:831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird, D.W., K.L. Puranam, and J.P. Revel. 1991. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 273:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe, P.D. 1994. Analyzing phorbol ester effects on gap junction communication: a dramatic inhibition of assembly. J. Cell Biol. 127:1895–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe, P.D., B.P. Nguyen, S. Gil, M. Usui, J. Olerud, Y. Takada, and W.G. Carter. 1998. Cellular interaction of integrin α3β1 with laminin 5 promotes gap junctional communication. J. Cell Biol. 143:1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe, P.D., E.M. TenBroek, J.M. Burt, W.E. Kurata, R.G. Johnson, and A.F. Lau. 2000. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 126:1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdown, A.B. 2002. Calcium: a potential central regulator in wound healing in the skin. Wound Repair Regen. 10:271–285. [DOI] [PubMed] [Google Scholar]
- Martin, P. 1997. Wound healing–aiming for perfect skin regeneration. Science. 276:75–81. [DOI] [PubMed] [Google Scholar]
- Musil, L.S., B.A. Cunningham, G.M. Edelman, and D.A. Goodenough. 1990. Differential phosphorylation of gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J. Cell Biol. 111:2077–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan, E., M. Saeb, C.P. Crum, S.B. Woo, P.H. McKee, and J.G. Rheinwald. 2003. Co-expression of p16(INK4A) and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am. J. Pathol. 163:477–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noszczyk, B.H., and S.T. Majewski. 2001. p63 expression during normal cutaneous wound healing in humans. Plast. Reconstr. Surg. 108:1242–1247. [DOI] [PubMed] [Google Scholar]
- Olerud, J.E., G.F. Odland, E.M. Burgess, C.R. Wyss, L.D. Fisher, and F.A. Matsen III. 1995. A model for the study of wounds in normal elderly adults and patients with peripheral vascular disease or diabetes mellitus. J. Surg. Res. 59:349–360. [DOI] [PubMed] [Google Scholar]
- Onuma, H., C. Mastui, and M. Morohashi. 2001. Quantitative analysis of the proliferation of epidermal cells using a human skin organ culture system and the effect of DbcAMP using markers of proliferation (BrdU, Ki-67, PCNA). Arch. Dermatol. Res. 293:133–138. [DOI] [PubMed] [Google Scholar]
- Qiu, C., P. Coutinho, S. Frank, S. Franke, L.Y. Law, P. Martin, C.R. Green, and D.L. Becker. 2003. Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol. 13:1697–1703. [DOI] [PubMed] [Google Scholar]
- Reynhout, J.K., P.D. Lampe, and R.G. Johnson. 1992. An activator of protein kinase C inhibits gap junction communication between cultured bovine lens cells. Exp. Cell Res. 198:337–342. [DOI] [PubMed] [Google Scholar]
- Richard, G. 2000. Connexins: a connection with the skin. Exp. Dermatol. 9:77–96. [DOI] [PubMed] [Google Scholar]
- Risek, B., F.G. Klier, and N.B. Gilula. 1992. Multiple gap junction genes are utilized during rat skin and hair development. Development. 116:639–651. [DOI] [PubMed] [Google Scholar]
- Rozental, R., M. Srinivas, and D.C. Spray. 2001. How to close a gap junction channel. Efficacies and potencies of uncoupling agents. Methods Mol. Biol. 154:447–476. [DOI] [PubMed] [Google Scholar]
- Saez, J.C., V.M. Berthoud, M.C. Branes, A.D. Martinez, and E.C. Beyer. 2003. Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83:1359–1400. [DOI] [PubMed] [Google Scholar]
- Saitoh, M., M. Oyamada, Y. Oyamada, T. Kaku, and M. Mori. 1997. Changes in the expression of gap junction proteins (connexins) in hamster tongue epithelium during would healing and carcinogenesis. Carcinogenesis. 18:1319–1328. [DOI] [PubMed] [Google Scholar]
- Salomon, D., E. Masgrau, S. Vischer, S. Ullrich, E. Dupont, P. Sappino, J.-H. Saurat, and P. Meda. 1994. Topography of mammalian connexins in human skin. J. Invest. Dermatol. 103:240–247. [DOI] [PubMed] [Google Scholar]
- Sharma, G.D., J. He, and H.E. Bazan. 2003. p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between MAP kinase cascades. J. Biol. Chem. 278:21989–21997. [DOI] [PubMed] [Google Scholar]
- Simon, A.M., D.A. Goodenough, and D.L. Paul. 1998. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr. Biol. 8:295–298. [DOI] [PubMed] [Google Scholar]
- Solan, J.L., M.D. Fry, E.M. TenBroek, and P.D. Lampe. 2003. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J. Cell Sci. 116:2203–2211. [DOI] [PubMed] [Google Scholar]
- Stepp, M.A., L. Zhu, and R. Cranfill. 1996. Changes in beta 4 integrin expression and localization in vivo in response to corneal epithelial injury. Invest. Ophthalmol. Vis. Sci. 37:1593–1601. [PubMed] [Google Scholar]
- Wallis, S., S. Lloyd, I. Wise, G. Ireland, T.P. Fleming, and D. Garrod. 2000. The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol. Biol. Cell. 11:1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, T.W. 2002. Unique and redundant connexin contributions to lens development. Science. 295:319–320. [DOI] [PubMed] [Google Scholar]
- Willecke, K., J. Eiberger, J. Degen, D. Eckardt, A. Romualdi, M. Guldenagel, U. Deutsch, and G. Sohl. 2002. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 383:725–737. [DOI] [PubMed] [Google Scholar]
- Xu, X., W.E. Li, G.Y. Huang, R. Meyer, T. Chen, Y. Luo, M.P. Thomas, G.L. Radice, and C.W. Lo. 2001. N-cadherin and Cx43alpha1 gap junctions modulates mouse neural crest cell motility via distinct pathways. Cell Commun. Adhes. 8:321–324. [DOI] [PubMed] [Google Scholar]
- Yamada, T., Y. Aoyama, M.K. Owada, H. Kawakatsu, and Y. Kitajima. 2000. Scraped-wounding causes activation and association of C-Src tyrosine kinase with microtubules in cultured keratinocytes. Cell Struct. Funct. 25:351–359. [DOI] [PubMed] [Google Scholar]