

Abstract
Purified cytolytic T lymphocyte (CTL) proteases granzyme (gzm)A and gzmB with sublytic dose of perforin (perf) initiate distinct proapoptotic pathways. Their physiological relevance in CTL-mediated target cell apoptosis is elusive. Using ex vivo virus-immune CD8+ T cells from mice deficient in perf, gzmA and/or gzmB, and the Fas-resistant EL4.F15 tumor target cell, we show that (a) CTL from gzmA−/− or gzmB−/− mice similarly induced early proapoptotic features, such as phosphatidyl serine (PS) exposure on plasma membrane, ΔΨm loss, and reactive oxygen radical generation, though with distinct kinetics; (b) CTL from gzmA−/− but not from gzmB−/− mice activate caspase 3 and 9; (c) PS exposure induced by CTL from gzmA−/− or gzmB−/− mice is prevented, respectively, by caspase inhibitors or by reactive oxygen scavengers without interfering with target cell death; and (d) all gzm-induced apoptotic features analyzed depend critically on perf. Thus, perf is the principal regulator in CTL-mediated and gzm-facilitated intracellular processes. The ability of gzmA and gzmB to induce multiple independent cell death pathways may be the hosts response to circumvent evasion strategies of pathogens and tumors.
Introduction
Cytolytic leukocytes, Natural Killer (NK) and CD8+ T (cytotoxic T lymphocyte [CTL]) cells, are key components of the hosts immune system against intracellular parasites and tumors. NK and CTL exert their biological activity via two distinct mechanisms. One is the synthesis and release of soluble mediators such as IFN-γ (Boehm et al., 1997), TNF-α (Vassalli, 1992), and interleukins (Biron, 1994), which may act proximal and distal to the effector cell. The other is direct cytolysis and induction of programmed cell death, apoptosis, in target cells by either or both of two cytolytic pathways (Kagi et al., 1994; Lowin et al., 1994). One is the Fas pathway involving the Fas ligand (FasL) on the effector cell engaging the Fas receptor (Fas or CD95) on the target cell (Rouvier et al., 1993; Nagata and Golstein, 1995; Krammer, 1999). The other, executed via the granula exocytosis pathway, is mediated by perforin (perf; Podack, 1986; Henkart, 1994) and the two principal granzymes (gzm), gzmA and gzmB (Simon and Kramer, 1994; Tschopp, 1994). These components are stored in cytoplasmic granules and are released into the immunological synapse formed between effector cells and their targets (Podack, 1986; Henkart, 1994; Stinchcombe et al., 2001). Although gzmA and gzmB can get access to target cells independent of perf via the mannose 6-phosphate receptor pathway or by other means (Motyka et al., 2000; Trapani et al., 2003; Dressel et al., 2004), their delivery to the cytosol and/or nucleus, where they initiate alternative proteolytic processes to trigger apoptosis, is critically dependent on perf (Froelich et al., 1996; Trapani et al., 1998).
Significant advances in our understanding of the molecular basis for gzmA- and gzmB-induced cell death are based on experiments using the mast-cell exocytosis model (Nakajima et al., 1995) and from in vitro studies using purified effector molecules. It was concluded that gzmB initiates cell death through various pathways, either involving activation of caspases, directly or indirectly, and resulting in disruption of mitochondrial integrity (Barry et al., 2000; Heibein et al., 2000; Sutton et al., 2000, 2003; Alimonti et al., 2001; Goping et al., 2003; Metkar et al., 2003), by derepressing the endonuclease CAD (Thomas et al., 2000; Sharif-Askari et al., 2001) or by cleaving key structural proteins in the nuclear membrane or cytoskeleton (Browne et al., 2000; Zhang et al., 2001a,b). In contrast, gzmA seems to induce apoptosis by caspase-independent pathways (Beresford et al., 1999; Shresta et al., 1999). Besides other substrates of gzmA, like lamins (Zhang et al., 2001a) and histones (Zhang et al., 2001b), a special target seem to be the endoplasmic reticulum–associated SET complex (Fan et al., 2002). This complex contains three gzmA substrates, the nucleosome assembly protein SET, the DNA bending protein HMG-2, and the base excision repair endonuclease Ape1. The SET complex also contains the tumor suppressor protein pp32 and the DNase NM23-H1, the latter of which is released from inhibition by gzmA cleavage of SET and translocates to the nucleus (Fan et al., 2003a). Furthermore gzmA-mediated proteolysis of Ape1 interferes with its known oxidative repair function for DNA (Fan et al., 2003b).
These findings, principally obtained with purified effector molecules, emphasize the complexity of intracellular events triggered by gzmA and gzmB. However, it is unclear to what extent these in vitro observations reflect the biologically relevant interaction of NK and CTL with target cells in vivo. In particular, factors such as the quality of the proteins involved, the way and context of their delivery, and/or their accessibility to cellular compartments may play a crucial role in the outcome of cytolytic function. The present work uses ex vivo–derived virus-immune CD8+ T cells from mice with targeted gene defects in perf or gzmA and/or gzmB to examine for the first time the apoptotic pathways activated by CTL via the two gzms in an in vitro cytotoxic assay using Fas-resistant tumor target cells.
Results
Perf-mediated cytotoxicity of EL4.F15 by CTL: loss of plasma membrane integrity and mitochondrial membrane potential is similarly and independently induced by gzmA and gzmB
To study a physiologically relevant model for granule exocytosis–executed apoptotic cell death, in particular mediated by gzms and independent of Fas, we used an in vitro CTL-target cytotoxic assay. Ex vivo–derived virus-immune CD8+ T cells (day eight after infection) from B6 mice or those deficient in gzm(s) (gzmA−/−, gzmB−/−, and gzmA×B−/−) or perf (perf−/−), infected with LCMV 8 d before, were used as effector cells. As target cell, the tumor cell line EL4.F15 was chosen because it is Fas resistant and at the same time sensitive to both gzmA- and gzmB-induced apoptosis (Pardo et al., 2002). Confirming previous studies, splenocytes from 8-d LCMV-immune B6, gzmA−/−, and gzmB−/− mice contained similar numbers (11, 6, and 9%, respectively) of gp33/D btetramer+/CD8+ T cells (Fig. 1 Aa). As shown previously (Simon et al., 1997; Pardo et al., 2002), enriched CD8+ T cells from all three mouse strains readily induced specific nucleolysis in gp33-pulsed EL4.F15 target cells (4-h assay), which was similar in magnitude for B6 and gzmA−/− and somewhat lower for gzmB−/− T cells (Fig. 1 Ab).
Figure 1.


PS exposure, mitochondrial depolarization, and ROS generation is induced by gzmA − / − and gzmB − / − but not by perf − / − and gzmA×B − / − CTL. EL4.F15 cells were incubated with ex vivo virus-immune CD8+ cells (MACS selected, ≥95% CD8+ cells) from either B6, gzmA−/−, or gzmB−/− (A) or from B6, perf −/−, or gzmA×B−/− (B) mice, in the presence or absence of the LCMV peptide gp33. (Aa) percentage of virus-specific CD8+ cells, determined by staining with gp33-labeled tetramers (H-2Db; tet-PE). (Ab) EL4.F15 cells were incubated with effector cells for 4 h (open symbols, −gp33; closed symbols, +gp33) at different effector/target ratios, and DNA fragmentation was analyzed by 3H-thymidine release. (Ac and Ad) EL4.F15 cells were incubated with effector cells for 2 h at a 10:1 effector/target ratio. Subsequently, PS exposure on plasma membrane (annexin-V-FITC) and PI uptake (c) and, in parallel, ΔΨm loss (3,3-dihexyloxacarbocyanin [DiOC6(3)] staining) and ROS generation (2-hydroxiethidine [2-HE]; d) were analyzed by three-color flow cytometry in the cell population negative for CD8 expression (target cells). EL4.F15 cells were also incubated with 200 μM glyotoxin (c) or 200 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP; d) as positive controls for PS exposure and ΔΨm loss, respectively. Numbers indicate percentage of cells in each quadrant. (Ae) EL4.F15 cells were incubated with effector cells for 30, 60, or 120 min at a 10:1 effector/target ratio, and PS exposure and PI uptake or ΔΨm loss and ROS generation were analyzed as described in Ac and Ad. Data presented are the mean of at least three independent experiments and are given as the difference of percentages in the presence versus the absence of gp33 ± SD. (B) a, same as Aa; b, same as Ab; c, target cell death (open symbols, −gp33; closed symbols, +gp33) was monitored after incubation of CML for 2 h at an effector/target cell ratio of 10:1 by the cell survival assay. Bd, same as Ac; Be, same as Ad.
Changes of cellular parameters occurring in EL4.F15 cells were analyzed after incubation with LCMV-immune CD8+ T cells (2 h), by gating effector/target mixtures for CD8 negative cells. CD8+ T cell populations from B6, gzmA−/−, and gzmB−/− mice similarly induced loss of plasma membrane integrity of gp33-pulsed EL4.F15 cells, as monitored by exposure of phosphatidyl serine (PS) at the plasma membrane and incorporation of propidium iodide (PI). This finding is revealed by the fact that numbers of annexinV/PI-positive cells was increased in gp33-pulsed as compared with mock-treated targets (51, 40, and 40% vs. 9, 14, and 13% for B6, gzmA−/−, and gzmB−/− T cells, respectively; Fig. 1 Ac). Gliotoxin, a hydrophobic fungal metabolite with apoptotic potential for mammalian cells, served as positive control for induction of the aforementioned parameters (Waring et al., 1999; Fig. 1 Ac). Furthermore, upon specific CTL–target cell interaction (2 h), all three CD8+ T cell populations induced severe mitochondrial perturbation in EL4.F15 targets. This result can be deduced by the comparable reduction of the mitochondrial transmembrane potential (ΔΨm; 88 to 33%, 83 to 42%, and 81 to 45% for B6, gzmA−/−, and gzmB−/− T cells, respectively) and an increase in the generation of reactive oxygen species (ROS), when mock-treated were compared with gp33-pulsed EL4.F15 cells (3 to 38%, 4 to 35%, and 3 to 38% for B6, gzmA−/−, and gzmB−/− T cells, respectively; Fig. 1 Ad). The protoionophore, carbonyl cyanide m-chlorophenylhydrazone served as positive control for ΔΨm loss in EL4.F15 cells (Fig. 1 Ad). The data suggest that CTL-induced loss of plasma membrane and mitochondrial integrity can be elicited to the same extent by gzmA or gzmB and independently of each other. However, when tested at earlier time points of CD8+ T cell–target cell interaction (i.e., at 30 or 60 min) loss of plasma membrane and mitochondrial integrity was more pronounced with B6 and gzmA−/− as compared with gzmB−/− CD8+ T cells (Fig. 1 Ae). Moreover, at these two time points but not later (2 h), annexinV reactivity preceded membrane permeability with B6 and gzmA−/− effectors.
Induction of loss of plasma membrane integrity and mitochondrial membrane potential elicited by CTL via gzmA or gzmB is dependent on perf
To determine if loss of plasma membrane and mitochondrial integrity in EL4.F15 target cells induced by gzmA- or gzmB-deficient CTL is dependent on perf, ex vivo–derived (day eight after infection) and CD8-enriched LCMV-immune splenic T cells from B6, perf−/−, and gzmA×B−/− mice were tested under the same conditions (2 h) for their potential to induce the same apoptotic parameters described in the previous paragraph. Effector cell populations from all three mouse strains contained similar numbers of gp33/D btetramer+/CD8+ T cells (10, 11, and 9% for B6, perf−/−, and gzmA×B−/− T cells, respectively; Fig. 1 Ba). As previously found (Simon et al., 1997; Pardo et al., 2002), only CD8+ T cells from B6, but not from perf−/− or gzmA×B−/−, mice were able to induce specific nucleolysis in EL4.F15 target cells (Fig. 1 Bb, 4-h assay). Therefore, target cell survival upon interaction with either B6-, perf−/−-, or gzmA×B−/−-derived CTL was inversely proportional to nucleolysis (Fig. 1 Bc). From the three effector cell populations, only CD8+ T cells from B6 mice readily induced loss of plasma membrane integrity in EL4.F15 target cells, whereas CD8+ T cells from perf−/− or gzmA×B−/− mice were, if at all, only marginally effective (Fig. 1 Bd). Similarly, only CD8+ T cells from B6 mice were able to optimally induce ΔΨm loss (from 82 to 59%) and ROS generation (from 8 to 25%, in the absence vs. presence of gp33 peptide). In contrast, ΔΨm loss and ROS generation was much lower when CD8+ T cells from gzmA×B−/− mice were used (ΔΨm: 83 to 77%, ROS: 7 to 13%; in the absence vs. presence of gp33 peptide) and not detectable with CTL from perf−/− mice (Fig. 1 Be). These data demonstrate that for gzm-facilitated loss of plasma membrane and mitochondrial integrity in EL4.F15 target cells during specific CTL attack, the presence of perf is critical. In addition, in the absence of gzmA and gzmB, perf is able on its own or together with as yet unknown factors to induce, though only slightly, ΔΨm loss and ROS generation, however without affecting the survival of target cells (Fig. 1 B, e vs. c).
CTL-mediated activation of caspase 3 and 9 is dependent on gzmB but not gzmA
LCMV-immune CD8+ T cells from B6, gzmA−/−, and gzmB−/− mice were incubated with gp33-pulsed or mock-treated EL4.F15 target cells (2 h), and activation of intracellular caspase 3 was assessed using the mAb C92-605. Fig. 2 A shows that effector cells from B6 and gzmA−/− mice specifically and comparably induced activation of caspase 3 in ∼30% of target cells, whereas those from gzmB−/− mice did not (Fig. 2 Aa, top). The presence of blocking anti-FasL mAb did not alter activation of caspase 3 by the two CD8+ T cell populations (Fig. 2 Aa, bottom). This excludes a contribution of the Fas pathway and suggests that the CTL-mediated effect is solely elicited via the exocytosis pathway (Pardo et al., 2002). When caspase activation in EL4.F15 cells was tested under similar conditions by applying intracellular staining with the caspase 3–specific agent SRH-DEVD-fmk, comparable results were obtained (Fig. 2 Ab, top). Gp33-pulsed but not mock-treated target cells incubated with either B6- or gzmA−/−-derived LCMV-immune CD8+ T cells stained positive for active caspase 3 (44 vs. 6% and 60 vs. 15%, respectively). Again, the presence of blocking anti-FasL mAb did not alter activation of caspase 3 by B6 and gzmA−/− CTL (Fig. 2 Ab, middle, + gp33 peptide). In both cases, pretreatment of targets with the specific inhibitor Z-DEVD-fmk (Fig. 2 Ab, orange) leads to a drastic reduction in positively staining cells (Fig. 2 Ab, bottom). Surprisingly and in contrast to results obtained using the specific mAb for active caspase 3, a small fraction of gp33-pulsed EL4.F15 cells (30 vs. 17% for mock-treated cells) also stained positive with SRH-DEVD-fmk after incubation with gzmB−/− LCMV-immune CD8+ T cells (Fig. 2 Ab, top). However, the number of EL4.F15 cells staining for active caspase 3 were only marginally reduced by the specific inhibitor Z-DEVD-fmk (Fig. 2 Ab, bottom). This finding indicates that the apparent caspase 3 activation observed by using the fluorescent substrate (but not the mAb) is due to unspecific binding of the substrate to a molecule distinct from caspase 3 (Amstad et al., 2001).
Figure 2.
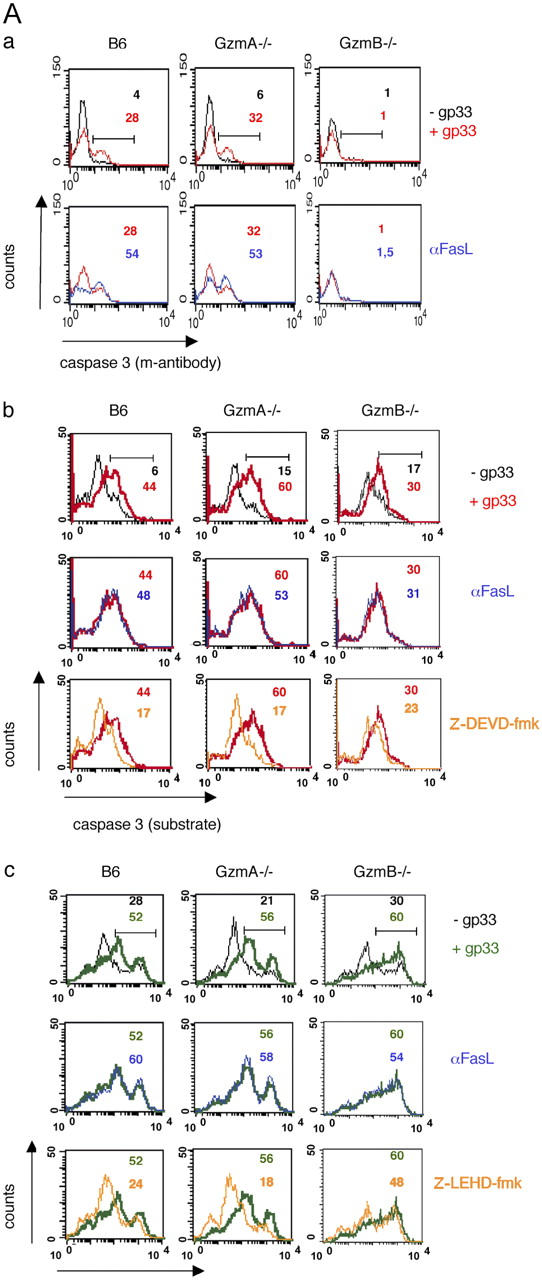
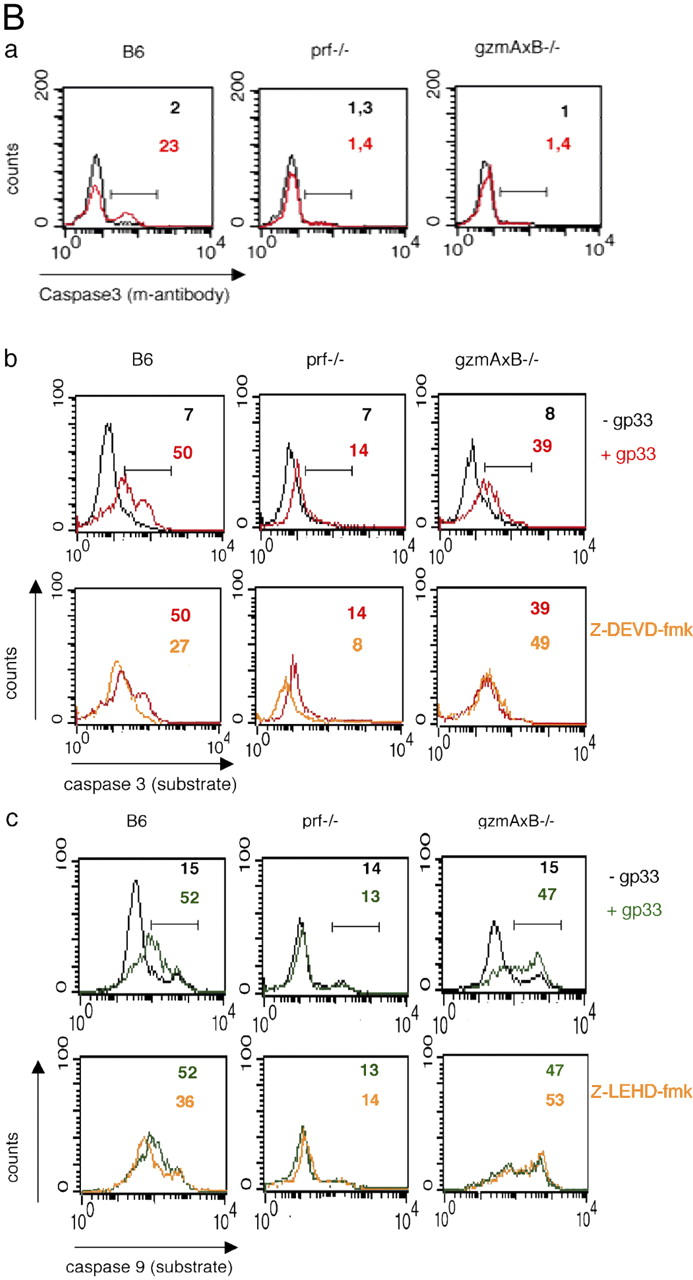
B6 and gzmA − / − but neither gzmB − / − , perf − / − , nor gzmA×B − / − CTL are able to induce caspase 3 and 9 activity. EL4.F15 cells were incubated with ex vivo virus-specific CD8+ cells (MACS selected, ≥95% CD8+ cells) from either B6, gzmA−/−, or gzmB−/− (A) or from B6, perf−/−, or gzmA×B−/− (B) mice (2 h, 10:1 effector/target ratio), in the presence (red and green) or absence (black) of the LCMV peptide gp33, as indicated. Activation of caspase 3 was monitored with either an FITC-labeled mAb against the active form of the enzyme (Aa and Ba) or the specific fluorescent substrates SRH-DEVD-fmk (Ab and Bb), by two-color flow cytometry in the cell population negative for CD8 expression (target cells) as described in Materials and methods. Similarly, activation of caspase 9 was monitored with the specific fluorescent substrates FAM-LEHD-fmk (Ac and Bc). CMLs were also developed in the presence of a blocking mAb anti-FasL, where indicated (Aa, bottom, dark blue; and Ab and Ac, middle, dark blue), and in the presence of the caspase 3 (100 μM Z-DEVD-fmk; b) or caspase 9 (100 μM Z-LEHD-fmk; c) inhibitors (Ab, Ac, Bb, and Bc, bottom, orange). Numbers correspond to the percentage of cells positive for the labeling in each case, indicated by the horizontal bars shown in the top panels.
The CTL-induced intracellular activation of caspase 9 was monitored by using the specific agent FAM-LEHD-fmk. As shown in Fig. 2 Ac (top) LCMV-immune CD8+ T cells from B6 and gzmA−/− mice specifically activated caspase 9 in a fraction of EL4.F15 cells (gp33 vs. mock peptide: 52% vs. 28% and 56% vs. 21%, respectively). In both cases, activation was totally inhibited upon pretreatment of EL4.F15 cells with the specific inhibitor of caspase 9, Z-LEHD-fmk (Fig. 2 Ac, bottom, orange). Again, incubation of EL4.F15 cells with gzmB−/−-derived LCMV-immune CD8+ T cells resulted in an increase of target cells staining with FAM-LEHD-fmk (Fig. 2 Ac, top). However, this effect was not or only marginally altered upon pretreatment with the specific inhibitor Z-LEHD-fmk (Fig. 2 Ac, bottom, orange), indicating that FAM-LEHD-fmk binds to a protein(s) distinct from caspase 9 (Amstad et al., 2001). Specific activation of caspase 9 by B6 and gzmA−/− CTL was not inhibited in the presence of anti-FasL mAb (Fig. 2 Ac, middle).
CTL-mediated activation of caspase 3 and 9 by gzmB is strictly dependent on perf
To determine whether or not CTL-induced and gzmB-facilitated activation of caspase 3 is also dependent on perf, EL4.F15 cells were cocultured with LCMV-immune CD8+ T cells from B6, perf−/−, and gzmA×B−/− mice. Fig. 2 Ba shows that only CTL from B6 mice were able to specifically induce activation of caspase 3, as monitored with the mAb C92-605 in a fraction of EL4.F15 cells (23 vs. 2% for mock-treated cells). Similar results were obtained when SRH-DEVD-fmk was used to detect active caspase 3 (Fig. 2 Bb, top). As expected, the number of cells staining positive with SRH-DEVD-fmk, upon incubation with B6 CTL, was drastically reduced after preincubation of EL4.F15 cells with Z-DEVD-fmk (Fig. 2 Bb, bottom, orange). A considerable increase in positive staining cells for SRH-DEVD-fmk was also seen after incubation with LCMV-immune gzmA×B−/−-derived CD8+ T cells (Fig. 2 Bb, top). However, fluorescence was not altered after preincubation of cells with Z-DEVD-fmk (Fig. 2 Bb, bottom), indicating that SRH-DEVD-fmk binds to a protein(s) distinct from caspase 3.
Similarly, only effector cells from B6 mice were able to specifically induce activation of caspase 9, as observed with FAM-LEHD-fmk (52 vs. 15% for mock-treated cells; Fig. 2 Bc, top), which was inhibited in the presence of Z-LEHD-fmk (52 to 36%; Fig. 2 Bc, bottom, orange). In contrast, LCMV-immune CD8+ T cells from perf−/− mice did not induce activation of caspase 9. Furthermore, the increase in intracellular staining of EL4.F15 cells for FAM-LEHD-fmk after incubation with gzmA×B−/−-derived LCMV-immune CD8+ T cells (47 vs. 15% for mock-treated cells) was not inhibited by Z-LEHD-fmk (47 vs. 53%; Fig. 2 Bc, bottom).
GzmB-activated caspases contribute to loss of plasma membrane integrity and mitochondrial membrane potential
We tested if in the course of CTL-mediated cytolysis, activation of caspases is essential for gzm-induced PS exposure on plasma membrane, cell death (PI incorporation), ΔΨm loss, and ROS generation. For this purpose, LCMV-immune CD8+ T cells from B6, gzmA−/−, or gzmB−/− mice were incubated with EL4.F15 cells in the presence of the universal caspase inhibitor Z-VAD-fmk or with the caspase 3 inhibitor Z-DEVD-fmk. As shown in Fig. 3 (A and B; mean of three independent experiments), the number of annexinV+ and PI+ cells was drastically reduced by either Z-DEVD-fmk or Z-VAD-fmk when gzmA−/−-derived CTL were used, only partially in the presence of B6 CTL, and not at all with gzmB−/− CTL. Furthermore, ΔΨm loss and ROS generation induced by B6- and gzmA−/−- but not gzmB−/−-derived CTL were also reduced, though only partially, in the presence of Z-DEVD-fmk or Z-VAD-fmk (Fig. 3, C and D). These data suggest that CTL-mediated induction of plasma membrane perturbation is critically dependent on functional active caspases when elicited by gzmB but not by gzmA and reveals caspase-dependent and -independent mechanisms of gzmB-induced mitochondrial depolarization. In both cases, a DEVD-sensitive caspase, in particular caspase 3, may be the main executor of these caspase-dependent processes.
Figure 3.

Caspase inhibitors completely block PS exposure induced by gzmA-deficient CTL. (A–F) EL4.F15 cells were incubated with ex vivo virus-specific CD8+ cells (MACS selected, ≥95% CD8+ cells) from B6, gzmA−/−, or gzmB−/− mice for 2 h at a 10:1 effector/target ratio in the presence or absence of the LCMV peptide gp33. (A–D) CML cultures were incubated in the presence or absence of 100 μM of the pan caspase inhibitor Z-VAD-fmk or the caspase 3 inhibitor Z-DEVD-fmk. Subsequently, PS exposure on plasma membrane (annexin-V-FITC; A) and PI uptake (B) and, in parallel, ΔΨm loss (DiOC6(3) staining; C) and ROS generation (2-HE; D) were analyzed by three-color flow cytometry in the cell population negative for CD8 expression (target cells). Results are the mean of three independent experiments and are given as the difference of percentages in the presence or absence of gp33 ± SD. (E and F) Inhibition of caspase 3 or 9 activity reduces, respectively, caspase 9 or 3 activation induced by B6 and gzmA−/− CTL. Caspase 3 activity was analyzed using an FITC-labeled mAb against the active form of the enzyme (E), whereas caspase 9 activity was analyzed by using the specific fluorescent substrate FAM-LEHD-fmk (F) by two-color flow cytometry in the cell population negative for CD8 expression (target cells). CMLs were also developed in the presence of the caspase 3 (100 μM Z-DEVD-fmk; F, bottom, gray) or caspase 9 (100 μM Z-LEHD-fmk; E, bottom, orange; and F, middle, orange) inhibitors. Numbers correspond to the percentage of cells positive for the labeling in each case, indicated by the horizontal bars shown in the top panels.
Synergy between caspase 3 and 9 in CTL-mediated target cell lysis
To clarify if the level of active caspase 3 generated during CTL-mediated cytolysis is regulated by caspase 9, which is only produced in its active form after mitochondrial perturbation and cytochrome c release (Li et al., 1997; Boatright et al., 2003), LCMV-immune CD8+ T cells from B6 and gzmA−/− mice were incubated with EL4.F15 cells in the presence of the caspase 9 inhibitor Z-LEHD-fmk. As shown in Fig. 3 E, Z-LEHD-fmk (orange) considerably reduced specific activation of caspase 3, as monitored with mAb C92-605. This result was independent of the effector cell population used (from 58 to 25% for B6 CTL and from 55 to 24% for gzmA−/− CTL; Fig. 3 E, bottom).
To see whether or not the generation of functional active caspase 9 as monitored with the specific substrate FAM-LEHD-fmk was dependent on previous activation of caspase 3, aliquots of the cytotoxicity assay (CML) cultures derived from B6 and gzmA−/− mice were incubated in the presence of inhibitors for either caspase 3 (Z-DEVD-fmk, gray) or caspase 9 (Z-LEHD-fmk, orange). Fig. 3 F shows that specific induction of caspase 9 activity by both CTL populations (top) is completely inhibited by Z-LEHD-fmk (from 36 to 13% for B6 CTL and from 36 to 14% for gzmA−/− CTL; Fig. 3 F, middle) and also by Z-DEVD-fmk (from 36 to 16% for B6 CTL and from 36 to 17% for gzmA−/− CTL; Fig. 3 F, bottom). Together, these results suggest that in CTL-mediated cytolysis activation of caspase 3 is a prerequisite for caspase 9 activation and that the generation of the former caspase is amplified by the latter.
Effect of oxidative stress on gzmA- and gzmB-induced loss of plasma membrane integrity and mitochondrial membrane potential
As shown in the first paragraph of Results, incubation of EL4.F15 cells with either B6, gzmA−/−, or gzmB−/− LCMV-immune CD8+ T cells leads to ROS generation. To evaluate the requirement for ROS generation in gzmA- and gzmB-mediated apoptotic processes, the respective CML cultures were incubated with targets (2 h) previously treated with the antioxidant N-acetylcysteine (NAC) and tested for the annexinV±/PI± phenotype, ΔΨm loss, and presence of active caspases 3 and 9. As expected, ROS formation is similarly suppressed after pretreatment of EL4.F15 cells with NAC, irrespective of the source of CTL (Fig. 4 D). The number of cells with the annexinV+/PI+ phenotype was significantly reduced in NAC-treated as compared with mock-treated EL4.F15 cells after culture with B6- and gzmA−/−-derived CTL and reduced to near background level when cultured with gzmB−/−-derived CTL (Fig. 4, A and B). NAC protected EL4.F15 cells only partially from ΔΨm loss, independent of the effector cell population (Fig. 4 C). Pretreatment of EL4.F15 cells with NAC had no effect on the activation of caspase 3, as monitored by mAb C92-605 fluorescence (Fig. 4 E, left), but resulted in considerably reduced activation of caspase 9 after incubation with either B6- or gzmA−/−-derived CTL, as determined by staining with FAM-LEHD-fmk (Fig. 4 E, right). Upon addition of both NAC and LEHD-fmk to cultures of B6 CTL and EL4.F15 target cells, inhibition of caspase 3 activation did not exceed that obtained with LEHD-fmk alone (Fig. 3 E and not depicted). The fact that suppression of plasma membrane perturbation (annexin+/PI± phenotype) by NAC is more pronounced when gzmB−/−- compared with gzmA−/−-derived CTL are used suggests that the generation of ROS is critical in gzmA-induced cell surface membrane perturbation, but less so in gzmB-mediated but caspase-dependent processes.
Figure 4.
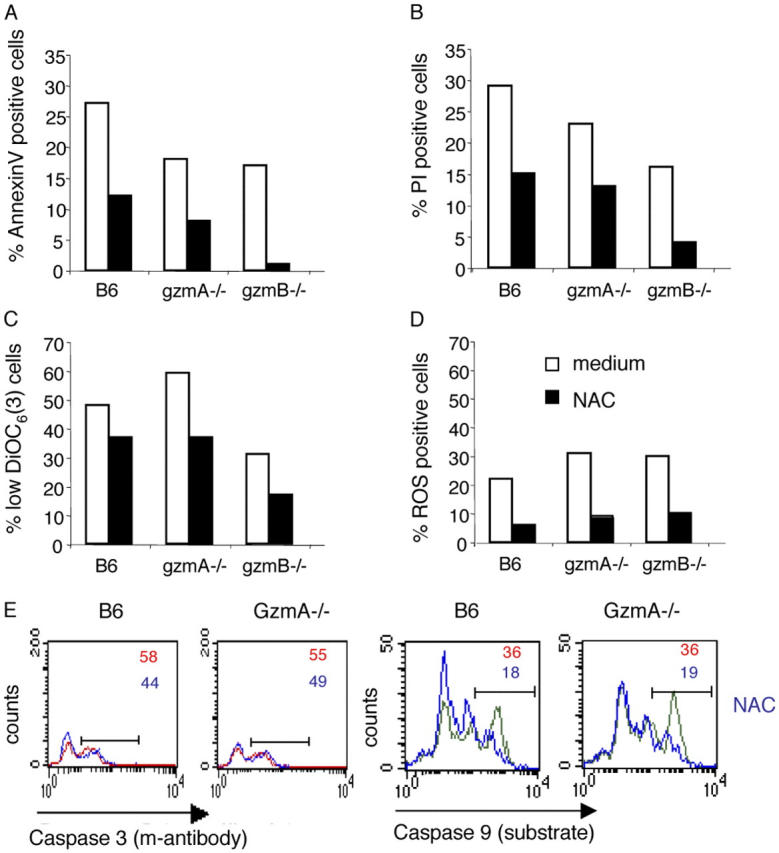
PS exposure induced by gzmB −/− CTL is blocked in the absence of ROS, but mitochondrial depolarization is still observed. Gp33-pulsed EL4.F15 cells were incubated with ex vivo virus-specific CD8+ cells (MACS selected, ≥95% CD8+ cells) from wild-type B6, gzmA−/−, and/or gzmB−/− mice for 2 h at a 10:1 effector/target ratio, in the presence or absence of the ROS scavenger NAC at 15 mM. PS exposure on plasma membrane (annexin-V-FITC; A) and PI uptake (B) and, in parallel, ΔΨm loss (DiOC6(3) staining; C) and ROS generation (2-HE; D) were analyzed by three-color flow cytometry in the cell population negative for CD8 expression. Results are the mean of two independent experiments and are given as the difference of percentages in the presence or absence of gp33 (SD was always lower that 5% of the mean). (E) Alternatively, CML cultures were incubated in the absence (red or green) or presence (blue) of NAC (15 mM). Caspase 3 activation was determined using an FITC-labeled mAb against the active form of the enzyme (left), whereas caspase 9 activity was determined by using the specific fluorescent substrate FAM-LEHD-fmk (right) by two-color flow cytometry in the cell population negative for CD8 expression (target cells). Numbers correspond to the percentage of cells positive for the labeling in each case, indicated by the horizontal bars.
CTL-mediated cleavage of Ape-1 is strictly dependent on perf but only partially on gzmA and/or gzmB
To test if the apurinic endonuclease-1, Ape-1, a known substrate of gzmA (Fan et al., 2003b), is cleaved during CTL-mediated lysis of targets, LCMV-immune CD8+ T cells from either B6, gzmA−/−, or gzmB−/− mice were incubated with gp33-pulsed EL4.F15 cells (4 h), and changes in Ape-1 expression were tested by monitoring intracellular staining of CD8-negative target cells with FITC-labeled Ape-1–reactive polyclonal antibody H-300. As shown in Fig. 5 A, 95% of mock-treated EL4.F15 cells stained with the antibody preparation. When gp33-pulsed target cells were incubated with either B6-, gzmA−/−-, or gzmB−/−-derived CTL, the numbers of Ape-1–positive cells were significantly reduced, compared with control mock-treated targets (from 85 to 65%, 74 to 48%, and 76 to 54%, respectively; Fig. 5 A). When, in a further experiment (Fig. 5 B), B6 CTL were compared with perf−/− and gzmA×B−/− CTL using similar conditions, the number of target cells staining for Ape-1 were also significantly reduced by gzmA×B−/− CTL (from 77 to 66%), though to a lesser extend compared with B6 CTL (from 79 to 54%). No reduction occurred with perf−/−-derived CTL. These results indicate that cleavage of Ape-1 in targets, following CTL attack, is strictly dependent on perf and also occurs, at least partially, in the absence of both gzms.
Figure 5.

Ape-1 degradation is induced by B6, gzmA − / − , gzmB − / − , and gzmA×B − / − but not perf − / − CTL. EL4.F15 cells were incubated with ex vivo virus-specific CD8+ cells (MACS selected, ≥95% CD8+ cells) from wild-type B6, gzmA−/−, or gzmB−/− mice (A) or from perf−/− or gzmA×B−/− mice (B) for 4 h at a 10:1 effector/target ratio in the presence (green) or absence (blue) of gp33 peptide. Subsequently, Ape-1 expression was analyzed in the cell population negative for CD8 expression by two-color flow cytometry. Red histograms indicate the cells labeled with the control isotype antibody. Numbers correspond to the percentage of cells positive for the labeling in each case, indicated by the horizontal bars.
Caspase activation or oxidative stress is not essential for CTL-mediated cell death
To further evaluate the requirement of caspase activation or ROS in gzm-mediated target cell death, LCMV-immune CD8+ T cells from either B6 gzmA−/− or gzmB−/− mice were incubated with gp33 peptide or mock-treated EL4.F15 cells (Fig. 6 A, top) or in the presence of either Z-VAD-fmk or Z-DEVD-fmk (Fig. 6 A, middle) or after pretreatment of target cells with NAC (Fig. 6 A, bottom). Target cell survival was estimated by extended culture under growth conditions unsuitable for CTL and by adding fresh inhibitors every 24 h. Neither treatment of target cells with Z-VAD-fmk nor Z-DEVD-fmk was able to rescue target cells from death, irrespective of the CTL population used. Furthermore, pretreatment of EL4.F15 cells with NAC did not prevent their death by either B6, gzmA−/−, or gzmB−/− CD8+ T cells. The lack of inhibition of target cell death was most likely not due to loss of inhibitor activity of any of the three agents. This is indicated by the finding that supernatant of cell cultures still retained most of the inhibitor activity after 24 h of culture, as tested by its effect on either anti-Fas mAb-mediated killing of L1210.Fas cells (Fig. 6 Ba, for Z-VAD-fmk and Z-DEVD-fmk activities) or on generation of gliotoxin-induced ROS in EL4.F15 cells (Fig. 6 Bb, for NAC activity). However, even with this control, the possibility that during CTL-mediated apoptosis caspases were not completely inhibited and contributed to cell death cannot be formally excluded.
Figure 6.

Cell death induced by B6, gzmA −/− , and gzmB − / − CTL is not affected by caspase inhibitors or ROS scavengers. (A) EL4.F15 cells were incubated with ex vivo LCMV-immune CD8+ T cells (MACS selected, ≥95% CD8+ cells) from B6, gzmA−/−, or gzmB−/− mice for 4 h at a 10:1 effector/target ratio, in the presence (opened symbols) or absence (closed symbols) of the viral protein gp33 (top panels). Alternatively, gp33-pulsed EL4.F15 cells were incubated with the respective effector cell populations in the presence of either 100 μM Z-VAD-fmk (square), 100 μM Z-DEVD-fmk (middle, triangle), or 15 mM NAC (bottom, circle). Subsequently, target cell death was monitored by propagating CML cultures at conditions unsuitable for CTL. (Ba) L1210.Fas cells were cultured in the presence or absence of αFas antibody (Jo-2; 1 μg/ml) for 20 h; cell cultures supplemented with Jo-2 were in addition treated with either Z-VAD-fmk or Z-DEVD-fmk (100 μM) or with 24-h SN of effector cell-target cell cultures (A; B6 CD8+ T cells +EL4.F15 + gp33). Cell death was analyzed by Trypan blue exclusion and results are expressed as the mean ± SD of two independent experiments. (Bb) EL4.F15 cells were cultured in the presence or absence of gliotoxin (100 or 200 μM) for 3 h; cell cultures supplemented with gliotoxin were in addition treated with either 15 mM NAC or with 24 h SN of effector cell-target cell cultures (A; B6 CD8+ T cells +EL4.F15 + gp33). Generation of ROS was analyzed by 2-HE staining. Data shown are representative of three independent experiments.
Discussion
The evolution of multiple strategies by intracellular pathogens and transformed cells to evade the immune system has brought about a multitude of effector mechanisms for CTL, the biologically most relevant being the ability to induce apoptosis. By using ex vivo–derived CTL from mice deficient in either gzmA and/or gzmB or perf, we show here that induction of cell death, as monitored by a cell survival assay, only occurs when at least one of the two gzms, gzmA and gzmB, is present and that perf is the ultimate regulator of the underlying processes. The data reveal that gzmA and gzmB independently induce several similar or distinct morphological and biochemical events now recognized as characteristic features of the apoptotic program, including PS exposure at the plasma membrane, mitochondrial depolarization, caspase activation, and Ape-1 cleavage. Moreover, results obtained with inhibitors of caspases or ROS generation suggest that individual gzm-facilitated proapoptotic pathways are effective on their own in inducing cell death but that the latter process is amplified by their concerted action. The finding that gzmA×B−/− but not perf−/−-derived CTL express residual potential to induce certain apoptotic features indicates the involvement of additional effector molecules, besides gzmA and gzmB, in perf-mediated cell death. However, these factors do not contribute to cell death of the target used in the present study.
The findings that during CTL-target cell killing, perf-induced plasma membrane disturbance in EL4.F15 target cells, as revealed by PS exposure, occurs faster with gzmB as compared with gzmA and that in the case of gzmB annexinV reactivity precedes membrane permeabilization are new. Together with the fact that this proapoptotic program was greatly repressed by universal and specific caspase inhibitors only in case of gzmA−/−, but not gzmB−/−, CTL supports recent findings with purified perf and gzms (Sarin et al., 1998; Heibein et al., 1999) and suggests that activation of caspase(s) contributes, together with other pathways (i.e., mitochondria; see below in Discussion), to the accelerated process by gzmB. This finding also emphasizes the notion that CTL-mediated apoptosis elicited in the presence of both gzms is greatly influenced by the quality of the individual target cell (Pardo et al., 2002).
The result that PS exposure on EL4.F15 cells was only marginally increased upon incubation with gzmA×B−/− CTL makes it unlikely that additional molecules, besides gzmA and gzmB, contribute to plasma membrane perturbation. However, because the targeted null mutation of gzmB also affects other downstream genes in the domain, including gzmC, -D, -E, -F, and -G (Pham et al., 1996), their involvement in this CTL-mediated pathway cannot be excluded. Especially as it was recently shown that purified gzmC together with perf induces PS exposure on plasma membrane in a process distinct from gzmB (Johnson et al., 2003). In addition, other gzms, termed orphan gzms (Grossman et al., 2003), such as gzmK and gzmM, with cellular expression patterns distinct from gzmA and gzmB may also facilitate this proapoptotic process (Shresta et al., 1997; MacDonald et al., 1999; Kelly et al., 2004).
The finding that the ROS scavenger NAC completely blocks the induction of PS exposure on plasma membrane of EL4.F15 cells by gzmB−/− but not gzmA−/− CTL is novel and indicates that ROS synergize with gzmA but not gzmB in CTL-induced plasma membrane perturbation. This finding is remarkable in the context of a recent paper demonstrating the coupling of actin cytoskeleton dynamics and the production of ROS in cell death of yeast and most probably also mammalian cells (Gourlay et al., 2004). Thus, we speculate that gzmA is destabilizing the actin cytoskeleton in target cells by cleaving a yet unknown substrate, leading to depolarization of mitochondrial membrane and ROS generation, and that the latter amplifies PS exposure at the plasma membrane, thereby accelerating cell death and/or uptake of targets by phagocytes via the PS receptor (Savill and Fadok, 2000; Danial and Korsmeyer, 2004). Alternatively, as the SET complex, a major substrate of gzmA (Fan et al., 2002, 2003a,b), is associated with the ER, gzmA may initiate apoptotic processes by perturbing ER membrane and inducing Ca2+ release from ER stores. This cell death pathway is elicited by interorganelle flow of Ca2+ and facilitated by direct physical connections between ER and mitochondria (Newmeyer and Ferguson-Miller, 2003), leading to caspase-independent ΔΨm loss and ROS generation (Demaurex and Distelhorst, 2003; Orrenius et al., 2003).
Mitochondria are implicated in CTL-mediated pathways of apoptosis, but the contribution of ΔΨm loss and ROS generation to this process is still controversial (MacDonald et al., 1999; Barry et al., 2000; Heibein et al., 2000; Danial and Korsmeyer, 2004). The data that gzmA−/− and gzmB−/−, but not perf−/−, CTL induce ΔΨm reduction and ROS generation in EL4.F15 target cells to a similar extent indicates that both processes are dependent on perf, but are similarly and independently elicited by gzmA or gzmB. In addition, the finding that activation of the mitochondrial pathway is inhibited, at least partially, by Z-VAD-fmk and DEVD-fmk in the case of gzmA−/− but not gzmB−/− CTL argues that gzmA and gzmB induce ΔΨm loss via distinct molecular pathways. Furthermore, it indicates that gzmA induces mitochondrial depolarization in a caspase-independent manner, whereas gzmB may use both caspase-dependent and -independent pathways and supports previous findings (Heibein et al., 1999; Barry et al., 2000; Alimonti et al., 2001; Pinkoski et al., 2001; Goping et al., 2003; Sutton et al., 2003). Understanding how these complex processes induced by gzmA and gzmB interrelate and facilitate target cell lysis by CTLs requires further analysis.
The present demonstration that CTL from B6 and gzmA−/−, but not from gzmB−/−, perf−/−, or gzmA×B−/−, mice induced activation of caspase 3 and 9 in EL4.F15 cells corroborates and extends previous work with purified proteins and CTL on the differential effect of gzmA and gzmB in the perf-facilitated caspase pathway (Heibein et al., 1999; Pardo et al., 2002; Goping et al., 2003; Metkar et al., 2003; Sutton et al., 2003). However, because the targeted null mutation of gzmB also affects other downstream gzm genes in the domain (gzmC–G; Pham et al., 1996), their putative effect on these processes cannot be assessed. Activation of caspase 3 and 9 by wild-type B6 and gzmA−/− CTL was completely blocked by pretreatment of EL4.F15 cells with the caspase 3–specific inhibitor Z-DEVD-fmk. In contrast, using similar conditions, the caspase 9–specific inhibitor Z-LEHD-fmk completely inhibited activation of caspase 9, but only partially that of caspase 3. Thus we conclude that in gzmB-mediated cytolysis, activation of caspase 3 is an early event in target cell apoptosis and is a prerequisite for the generation of the caspase 9 apoptosome—most likely via mitochondrial permeabilization—which in turn cleaves additional procaspase 3. Such an amplification loop between caspase 3 and 9 has been proposed before with purified gzmB and perf (Metkar et al., 2003).
Ape-1, a multifunctional component of the SET complex (Fan et al., 2003b) expressing both endonuclease activity and the potential to repair oxidative damage to DNA and oxidative changes to transcription factors (Bennett et al., 1997; Hirota et al., 1997; Evans et al., 2000), is similarly cleaved in EL4.F15 cells, irrespective of whether CTL from either B6, gzmA−/−, or gzmB−/− mice are used as effectors. Cleavage of Ape-1 was also observed, though at reduced levels, with gzmA×B−/− CTL, but was absent in perf−/− CTL. Fan et al. (2003b) have shown that gzmA specifically binds Ape-1 and destroys its known oxidative repair function by proteolytic cleavage. Our data indicate that CTL contain additional enzymatic activity(ies) distinct from gzmA and gzmB–G, such as gzmK (Shresta et al., 1997), which is able to block cellular repair in a perf-dependent manner. However, this gzmA/gzmB-independent process does not seem to be relevant for the induction of cell death, as demonstrated by survival of EL4.F15 cells after their incubation with gzmA×B−/− CTL (Fig. 2 C).
The lack of inhibition of CTL-mediated cell death of EL4.F15 cells, as monitored by target cell survival, by either the universal caspase inhibitor Z-VAD-fmk, the caspase 3 inhibitor DEVD-fmk, or the antioxidant agent NAC when effector cells from B6, gzmA−/−, or gzmB−/− mice were used emphasizes the multitude of gzmA- and gzmB-facilitated effector pathways elicited by CTL. Moreover, the data reiterate the notion that gzmB-induced cell death similarly occurs via caspase-dependent and -independent intracellular processes, whereas all gzmA-induced cell death events are caspase independent and also occur in the absence of ROS generation. This enormous versatility of CTL probably evolved in response to the multiple strategies of pathogens and tumors to evade immune-mediated apoptosis (Benedict et al., 2002; Igney and Krammer, 2002; Trapani and Smyth, 2002).
In conclusion, the present study is the first account on gzmA- and/or gzmB-initiated intracellular processes during CTL-target cell interaction, a model of which is schematically represented in Fig. 7. Although the biological significance of the multiple overlapping and nonoverlapping proapoptotic pathways observed with ex vivo–derived CTLs reported herein for their in vivo performance is still elusive, this type of in vitro cytotoxic assay should help to provide a physiologically relevant perspective for gzm-mediated control of infections and cancer.
Figure 7.

Model for apoptotic pathways activated by gzmB or gzmA during CTL-induced target cell death. Perf is central to all CTL-mediated and gzmA- and gzmB-dependent apoptotic processes. GzmA and gzmB are similarly able to induce PS exposure on plasma membrane (Membrane damage) and the mitochondrial pathway(s), including ΔΨm loss and ROS generation. Only gzmB but not gzmA activates both caspase 3 and 9. GzmB-dependent activation of caspase 9 requires previous activation of caspase 3 and induction of ROS, whereas activation of caspase 3 is amplified by caspase 9 and the mitochondrial pathway. Gzm-induced PS exposure is critically dependent on caspase activation when elicited by gzmB, but not gzmA, and dependent on ROS generation when elicited by gzmA, but not gzmB. GzmB induces the mitochondrial pathway (ΔΨm loss) both via caspase-dependent and -independent pathways, and this process seems to be amplified by ROS, whereas gzmA acts via a caspase-independent pathway also amplified by ROS. Ape-1, an oxidative protein relevant for DNA repair (Fan et al., 2003b), is readily cleaved in the presence of either gzmA or gzmB, but also occurs in the absence of both gzms. However, the latter perf-facilitated and gzm-independent process(es) does not lead to target cell death. Target cell death can also occur even when the caspase pathways or the generation of ROS is inhibited, suggesting alternative, undefined gzm-elicited death pathways.
Materials and methods
Mouse strains
Inbred C57BL/6 (B6) and mouse strains deficient for perf (perf−/−), gzmA (gzmA−/−), gzmB (gzmB−/−), and gzmA×B (gzmA×B−/−) bred on the B6 background were maintained at the Max-Planck-Institut für Immunbiologie and analyzed for their genotypes as described previously (Simon et al., 1997; Balkow et al., 2001). Male mice of 8 to 10 wk of age were used in all experiments and were conducted in accordance with the ethical guidelines of the Federation of European Laboratory Animal Science Association.
Generation of ex-vivo CD8+ cells
Mice were infected with 105 pfu LCMV-WE i.p. according to established protocols (Balkow et al., 2001). On day eight after infection, CD8+ cells were positively selected from spleen using α-CD8-MicroBeads (Miltenyi Biotec) with an autoMACS (Miltenyi Biotec) and resuspended in MEM/2 mg/ml BSA before use in cytotoxic assays. Purity of selected CD8+ cells was assessed by FACS staining and found to be between 95–98%.
Target cells
The mouse tumor lines EL4.F15 and L1210.Fas were used as target cells (Pardo et al., 2002). Cell lines were cultured in MEM supplemented with 10% heat-inactivated FCS and 2-mercaptoethanol (10−5 M) at 7% CO2, as described previously (Pardo et al., 2002).
CMLs
The DNA release assay and the target cell survival assay was performed as described previously (Müllbacher et al., 1999). For LCMV-immune CTL-mediated lysis, target cells were pretreated with the synthetic peptide KAVYNTATC (gp33) as described previously (Balkow et al., 2001).
Online supplemental material
For analysis of intracellular apoptotic processes in target cells, effector and target cells were incubated at a ratio of 10:1 (effector/target) and aliquots of CML cultures (105 targets/experimental point) were stained with anti-CD8 mAb. CD8-negative targets were subsequently analyzed for proapoptotic processes and cell death, including plasma membrane disintegration, mitochondrial perturbation, caspase activation, Ape-1 cleavage, and thymidine release/cell survival. Online supplemental material consists of the following methods: 3[H] DNA release and cell survival assays; analysis of inhibitor stability; flow cytometry, including cell surface phenotype analysis, exposure/incorporation of annexin-V/PI, and reduction of mitochondrial membrane potential/generation of ROS (i.e., ↓ΔΨm/ROS); activation of caspases; and proteolytic degradation of Ape-1. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200406115/DC1.
Acknowledgments
This work was supported in part by a grant of the Deutscher Akademischer Austauschdienst (Acciones Integradas Hispano-Alemanas; to M.M. Simon and A. Anel) and by Project SAF2001-1774 from Ministerio de Ciencia y Tecnologia (Spain). J. Pardo has a Fellowship ascribed to the SAF2001-1774 Project.
Abbreviations used in this paper: CML, cytotoxicity assay; CTL, cytotoxic T lymphocyte; DiOC6(3), 3,3-dihexyloxacarbocyanin; FasL, fas ligand; gzm, granzyme; HE, hydroxiethidine; NAC, N-acetylcysteine; NK, Natural Killer; perf, perforin; PI, propidium iodide; PS, phosphatidyl serine; ROS, reactive oxygen species.
References
- Alimonti, J.B., L. Shi, P.K. Baijal, and A.H. Greenberg. 2001. Granzyme B induces BID-mediated cytochrome c release and mitochondrial permeability transition. J. Biol. Chem. 276:6974–6982. [DOI] [PubMed] [Google Scholar]
- Amstad, P.A., G. Yu, G.L. Johnson, B.W. Lee, S. Dhawan, and D.J. Phelps. 2001. Detection of caspase activation in situ by fluorochrome-labeled caspase inhibitors. Biotechniques. 31:608–610, 612, 614, passim. [DOI] [PubMed] [Google Scholar]
- Balkow, S., A. Kersten, T.T. Tran, T. Stehle, P. Grosse, C. Museteanu, O. Utermohlen, H. Pircher, F. von Weizsacker, R. Wallich, et al. 2001. Concerted action of the FasL/Fas and perforin/granzyme A and B pathways is mandatory for the development of early viral hepatitis but not for recovery from viral infection. J. Virol. 75:8781–8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry, M., J.A. Heibein, M.J. Pinkoski, S.F. Lee, R.W. Moyer, D.R. Green, and R.C. Bleackley. 2000. Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol. Cell. Biol. 20:3781–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict, C.A., P.S. Norris, and C.F. Ware. 2002. To kill or be killed: viral evasion of apoptosis. Nat. Immunol. 3:1013–1018. [DOI] [PubMed] [Google Scholar]
- Bennett, R.A., D.M. Wilson III, D. Wong, and B. Demple. 1997. Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc. Natl. Acad. Sci. USA. 94:7166–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beresford, P.J., Z. Xia, A.H. Greenberg, and J. Lieberman. 1999. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity. 10:585–594. [DOI] [PubMed] [Google Scholar]
- Biron, C.A. 1994. Cytokines in the generation of immune responses to, and resolution of, virus infection. Curr. Opin. Immunol. 6:530–538. [DOI] [PubMed] [Google Scholar]
- Boatright, K.M., M. Renatus, F.L. Scott, S. Sperandio, H. Shin, I.M. Pedersen, J.E. Ricci, W.A. Edris, D.P. Sutherlin, D.R. Green, and G.S. Salvesen. 2003. A unified model for apical caspase activation. Mol. Cell. 11:529–541. [DOI] [PubMed] [Google Scholar]
- Boehm, U., T. Klamp, M. Groot, and J.C. Howard. 1997. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 15:749–795. [DOI] [PubMed] [Google Scholar]
- Browne, K.A., R.W. Johnstone, D.A. Jans, and J.A. Trapani. 2000. Filamin (280-kDa actin-binding protein) is a caspase substrate and is also cleaved directly by the cytotoxic T lymphocyte protease granzyme B during apoptosis. J. Biol. Chem. 275:39262–39266. [DOI] [PubMed] [Google Scholar]
- Danial, N.N., and S.J. Korsmeyer. 2004. Cell death: critical control points. Cell. 116:205–219. [DOI] [PubMed] [Google Scholar]
- Demaurex, N., and C. Distelhorst. 2003. Apoptosis: The calcium connection. Science. 300:65–67. [DOI] [PubMed] [Google Scholar]
- Dressel, R., S.M. Raja, S. Honing, T. Seidler, C.J. Froelich, K. von Figura, and E. Gunther. 2004. Granzyme-mediated cytotoxicity does not involve the mannose 6-phosphate receptors on target cells. J. Biol. Chem. 279:20200–20210. [DOI] [PubMed] [Google Scholar]
- Evans, A.R., M. Limp-Foster, and M.R. Kelley. 2000. Going APE over ref-1. Mutat. Res. 461:83–108. [DOI] [PubMed] [Google Scholar]
- Fan, Z., P.J. Beresford, D. Zhang, and J. Lieberman. 2002. HMG2 interacts with the nucleosome assembly protein SET and is a target of the cytotoxic T-lymphocyte protease granzyme A. Mol. Cell. Biol. 22:2810–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z., P.J. Beresford, D.Y. Oh, D. Zhang, and J. Lieberman. 2003. a. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 112:659–672. [DOI] [PubMed] [Google Scholar]
- Fan, Z., P.J. Beresford, D. Zhang, Z. Xu, C.D. Novina, A. Yoshida, Y. Pommier, and J. Lieberman. 2003. b. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat. Immunol. 4:145–153. [DOI] [PubMed] [Google Scholar]
- Froelich, C.J., K. Orth, J. Turbov, P. Seth, R. Gottlieb, B. Babior, G.M. Shah, R.C. Bleackley, V.M. Dixit, and W. Hanna. 1996. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J. Biol. Chem. 271:29073–29079. [DOI] [PubMed] [Google Scholar]
- Goping, I.S., M. Barry, P. Liston, T. Sawchuk, G. Constantinescu, K.M. Michalak, I. Shostak, D.L. Roberts, A.M. Hunter, R. Korneluk, and R.C. Bleackley. 2003. Granzyme B-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity. 18:355–365. [DOI] [PubMed] [Google Scholar]
- Gourlay, C.W., L.N. Carpp, P. Timpson, S.J. Winder, and K.R. Ayscough. 2004. A role for the actin cytoskeleton in cell death and aging in yeast. J. Cell Biol. 164:803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman, W.J., P.A. Revell, Z.H. Lu, H. Johnson, A.J. Bredemeyer, and T.J. Ley. 2003. The orphan granzymes of humans and mice. Curr. Opin. Immunol. 15:544–552. [DOI] [PubMed] [Google Scholar]
- Heibein, J.A., M. Barry, B. Motyka, and R.C. Bleackley. 1999. Granzyme B-induced loss of mitochondrial inner membrane potential (Delta Psi m) and cytochrome c release are caspase independent. J. Immunol. 163:4683–4693. [PubMed] [Google Scholar]
- Heibein, J.A., I.S. Goping, M. Barry, M.J. Pinkoski, G.C. Shore, D.R. Green, and R.C. Bleackley. 2000. Granzyme B–mediated cytochrome c release is regulated by the Bcl-2 family members bid and Bax. J. Exp. Med. 192:1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkart, P.A. 1994. Lymphocyte-mediated cytotoxicity: two pathways and multiple effector molecules. Immunity. 1:343–346. [DOI] [PubMed] [Google Scholar]
- Hirota, K., M. Matsui, S. Iwata, A. Nishiyama, K. Mori, and J. Yo. 1997. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. USA. 94:3633–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igney, F.H., and P.H. Krammer. 2002. Immune escape of tumors: apoptosis resistance and tumor counterattack. J. Leukoc. Biol. 71:907–920. [PubMed] [Google Scholar]
- Johnson, H., L. Scorrano, S.J. Korsmeyer, and T.J. Ley. 2003. Cell death induced by granzyme C. Blood. 101:3093–3101. [DOI] [PubMed] [Google Scholar]
- Kagi, D., F. Vignaux, B. Ledermann, K. Burki, V. Depraetere, S. Nagata, H. Hengartner, and P. Golstein. 1994. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 265:528–530. [DOI] [PubMed] [Google Scholar]
- Kelly, J.M., N.J. Waterhouse, E. Cretney, K.A. Browne, S. Ellis, J.A. Trapani, and M.J. Smyth. 2004. Granzyme M mediates a novel form of perforin-dependent cell death. J. Biol. Chem. 279:22236–22242. [DOI] [PubMed] [Google Scholar]
- Krammer, P.H. 1999. CD95(APO-1/Fas)-mediated apoptosis: live and let die. Adv. Immunol. 71:163–210. [DOI] [PubMed] [Google Scholar]
- Li, P., D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, and X. Wang. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91:479–489. [DOI] [PubMed] [Google Scholar]
- Lowin, B., M. Hahne, C. Mattmann, and J. Tschopp. 1994. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 370:650–652. [DOI] [PubMed] [Google Scholar]
- MacDonald, G., L. Shi, C. Vande Velde, J. Lieberman, and A.H. Greenberg. 1999. Mitochondria-dependent and -independent regulation of Granzyme B-induced apoptosis. J. Exp. Med. 189:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metkar, S.S., B. Wang, M.L. Ebbs, J.H. Kim, Y.J. Lee, S.M. Raja, and C.J. Froelich. 2003. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J. Cell Biol. 160:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyka, B., G. Korbutt, M.J. Pinkoski, J.A. Heibein, A. Caputo, M. Hobman, M. Barry, I. Shostak, T. Sawchuk, C.F. Holmes, et al. 2000. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell. 103:491–500. [DOI] [PubMed] [Google Scholar]
- Müllbacher, A., P. Waring, R.T. Hia, T. Tran, S. Chin, T. Stehle, C. Museteanu, and M.M. Simon. 1999. Granzymes are the essential downstream effector molecules for the control of primary virus infections by cytolytic leukocytes. Proc. Natl. Acad. Sci. USA. 96:13950–13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata, S., and P. Golstein. 1995. The Fas death factor. Science. 267:1449–1456. [DOI] [PubMed] [Google Scholar]
- Nakajima, H., H.L. Park, and P.A. Henkart. 1995. Synergistic roles of granzymes A and B in mediating target cell death by rat basophilic leukemia mast cell tumors also expressing cytolysin/perforin. J. Exp. Med. 181:1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer, D.D., and S. Ferguson-Miller. 2003. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 112:481–490. [DOI] [PubMed] [Google Scholar]
- Orrenius, S., B. Zhivotovsky, and P. Nicotera. 2003. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4:552–565. [DOI] [PubMed] [Google Scholar]
- Pardo, J., S. Balkow, A. Anel, and M.M. Simon. 2002. The differential contribution of granzyme A and granzyme B in cytotoxic T lymphocyte-mediated apoptosis is determined by the quality of target cells. Eur. J. Immunol. 32:1980–1985. [DOI] [PubMed] [Google Scholar]
- Pham, C.T., D.M. MacIvor, B.A. Hug, J.W. Heusel, and T.J. Ley. 1996. Long-range disruption of gene expression by a selectable marker cassette. Proc. Natl. Acad. Sci. USA. 93:13090–13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkoski, M.J., N.J. Waterhouse, J.A. Heibein, B.B. Wolf, T. Kuwana, J.C. Goldstein, D.D. Newmeyer, R.C. Bleackley, and D.R. Green. 2001. Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J. Biol. Chem. 276:12060–12067. [DOI] [PubMed] [Google Scholar]
- Podack, E.R. 1986. Molecular mechanisms of cytolysis by complement and by cytolytic lymphocytes. J. Cell. Biochem. 30:133–170. [DOI] [PubMed] [Google Scholar]
- Rouvier, E., M.F. Luciani, and P. Golstein. 1993. Fas involvement in Ca2+-independent T cell–mediated cytotoxicity. J. Exp. Med. 177:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin, A., E.K. Haddad, and P.A. Henkart. 1998. Caspase dependence of target cell damage induced by cytotoxic lymphocytes. J. Immunol. 161:2810–2816. [PubMed] [Google Scholar]
- Savill, J., and V. Fadok. 2000. Corpse clearance defines the meaning of cell death. Nature. 407:784–788. [DOI] [PubMed] [Google Scholar]
- Sharif-Askari, E., A. Alam, E. Rhéaume, P.J. Beresford, C. Scotto, K. Sharma, D. Lee, W.E. DeWolf, M.E. Nuttall, J. Lieberman, and R.P. Sékaly. 2001. Direct cleavage of the human DNA fragmentation factor-45 by granzyme B induces caspase-activated DNase release and DNA fragmentation. EMBO J. 20:3101–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shresta, S., P. Goda, R. Wesselschmidt, and T.J. Ley. 1997. Residual cytotoxicity and granzyme K expression in granzyme A-deficient cytotoxic lymphocytes. J. Biol. Chem. 272:20236–20244. [DOI] [PubMed] [Google Scholar]
- Shresta, S., T.A. Graubert, D.A. Thomas, S.Z. Raptis, and T.J. Ley. 1999. Granzyme A initiates an alternative pathway for granule-mediated apoptosis. Immunity. 10:595–605. [DOI] [PubMed] [Google Scholar]
- Simon, M.M., and M.D. Kramer. 1994. Granzyme A. Methods Enzymol. 244:68–79. [DOI] [PubMed] [Google Scholar]
- Simon, M.M., M. Hausmann, T. Tran, K. Ebnet, J. Tschopp, R. ThaHla, and A. Mullbacher. 1997. In vitro- and ex vivo-derived cytolytic leukocytes from granzyme A × B double knockout mice are defective in granule-mediated apoptosis but not lysis of target cells. J. Exp. Med. 186:1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe, J.C., G. Bossi, S. Booth, and G.M. Griffiths. 2001. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 15:751–761. [DOI] [PubMed] [Google Scholar]
- Sutton, V.R., J.E. Davis, M. Cancilla, R.W. Johnstone, A.A. Ruefli, K. Sedelies, K.A. Browne, and J.A. Trapani. 2000. Initiation of apoptosis by granzyme B requires direct cleavage of bid, but not direct granzyme B–mediated caspase activation. J. Exp. Med. 192:1403–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, V.R., M.E. Wowk, M. Cancilla, and J.A. Trapani. 2003. Caspase activation by granzyme B is indirect, and caspase autoprocessing requires the release of proapoptotic mitochondrial factors. Immunity. 18:319–329. [DOI] [PubMed] [Google Scholar]
- Thomas, D.A., C. Du, M. Xu, X. Wang, and T.J. Ley. 2000. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity. 12:621–632. [DOI] [PubMed] [Google Scholar]
- Trapani, J.A., and M.J. Smyth. 2002. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2:735–747. [DOI] [PubMed] [Google Scholar]
- Trapani, J.A., D.A. Jans, P.J. Jans, M.J. Smyth, K.A. Browne, and V.R. Sutton. 1998. Efficient nuclear targeting of granzyme B and the nuclear consequences of apoptosis induced by granzyme B and perforin are caspase-dependent, but cell death is caspase-independent. J. Biol. Chem. 273:27934–27938. [DOI] [PubMed] [Google Scholar]
- Trapani, J.A., V.R. Sutton, K.Y. Thia, Y.Q. Li, C.J. Froelich, D.A. Jans, M.S. Sandrin, and K.A. Browne. 2003. A clathrin/dynamin- and mannose-6-phosphate receptor–independent pathway for granzyme B–induced cell death. J. Cell Biol. 160:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp, J. 1994. Granzyme B. Methods Enzymol. 244:80–87. [DOI] [PubMed] [Google Scholar]
- Vassalli, P. 1992. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 10:411–452. [DOI] [PubMed] [Google Scholar]
- Waring, P., D. Lambert, A. Sjaarda, A. Hurne, and J. Beaver. 1999. Increased cell surface exposure of phosphatidylserine on propidium iodide negative thymocytes undergoing death by necrosis. Cell Death Differ. 6:624–637. [DOI] [PubMed] [Google Scholar]
- Zhang, D., P.J. Beresford, A.H. Greenberg, and J. Lieberman. 2001. a. Granzymes A and B directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc. Natl. Acad. Sci. USA. 98:5746–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D., M.S. Pasternack, P.J. Beresford, L. Wagner, A.H. Greenberg, and J. Lieberman. 2001. b. Induction of rapid histone degradation by the cytotoxic T lymphocyte protease Granzyme A. J. Biol. Chem. 276:3683–3690. [DOI] [PubMed] [Google Scholar]