

Abstract
Fas apoptosis inhibitory molecule (FAIM) is a protein identified as an antagonist of Fas-induced cell death. We show that FAIM overexpression fails to rescue neurons from trophic factor deprivation, but exerts a marked neurite growth–promoting action in different neuronal systems. Whereas FAIM overexpression greatly enhanced neurite outgrowth from PC12 cells and sympathetic neurons grown with nerve growth factor (NGF), reduction of endogenous FAIM levels by RNAi decreased neurite outgrowth in these cells. FAIM overexpression promoted NF-κB activation, and blocking this activation by using a super-repressor IκBα or by carrying out experiments using cortical neurons from mice that lack the p65 NF-κB subunit prevented FAIM-induced neurite outgrowth. The effect of FAIM on neurite outgrowth was also blocked by inhibition of the Ras–ERK pathway. Finally, we show that FAIM interacts with both Trk and p75 neurotrophin receptor NGF receptors in a ligand-dependent manner. These results reveal a new function of FAIM in promoting neurite outgrowth by a mechanism involving activation of the Ras–ERK pathway and NF-κB.
Introduction
The tyrosine kinase receptor TrkA binds the neurotrophin NGF and signals cell survival and differentiation (Chao, 2003). NGF and other neurotrophins interact with a second receptor, p75 neurotrophin receptor (p75NTR). p75NTR is a member of the tumor necrosis factor receptor superfamily that contains an intracellular death domain (Dechant and Barde, 2002; Chao, 2003). A variety of functions for p75NTR have been proposed, including induction of apoptosis, modulation of neurite growth, and regulation of synaptic transmission (Dechant and Barde, 2002). NGF induces activation of the transcription factor complex NF-κB in PC12 cells and neurons by a Trk and p75NTR-dependent mechanism. This pathway has been implicated in promoting both cell survival and neurite process formation in these cells (Wood, 1995; Yoon et al., 1998; Hamanoue et al., 1999; Foehr et al., 2000; Wooten et al., 2001).
Death receptors are critical regulators of cellular homeostasis and apoptosis (Peter and Krammer, 2003) and play a role in regulating cell death in the developing nervous system (Cheema et al., 1999; Martin-Villalba et al., 1999; Raoul et al., 1999, 2002). Recently, it has been reported that Fas can also induce neurite growth and nerve regeneration after injury in vivo via extracellular regulated kinase (ERK) activation (Desbarats et al., 2003).
Fas apoptosis inhibitory molecule (FAIM) is a Fas antagonist that was initially characterized by differential display as a gene that is up-regulated in B cells resistant to Fas-mediated cell death and functions as an inhibitor of Fas-induced cell death (Schneider et al., 1999). FAIM is highly conserved in evolution from Caenorhabditis elegans to humans and is broadly expressed in many tissues (Rothstein et al., 2000). Recently, an alternatively spliced isoform of FAIM, FAIM-L, has been identified, which is predominantly expressed in the brain (Zhong et al., 2001). However, FAIM function in the nervous system has not been elucidated.
Here, we demonstrate that overexpression of FAIM markedly increases NGF-induced neurite outgrowth in several neuronal models and that decrease in the level of endogenously expressed FAIM markedly reduces NGF-induced neurite outgrowth. FAIM activates the NF-κB signaling pathway, and this pathway together with the Ras–ERK pathway are necessary for the neurite growth–promoting effects of FAIM. These findings reveal a novel role for FAIM in the developing nervous system that contrasts with its established role in modulating Fas signaling and regulating cell survival in the immune system.
Results
Rat FAIM cloning and expression
FAIM is a newly identified antiapoptotic protein that is highly conserved among multiple animal phyla and is expressed in at least two isoforms, long and short (Schneider et al., 1999, Zhong et al., 2001). By searching publicly available EST databases (GenBank, NCBI, and NIH), we found clones encoding both the short and the long forms of rat FAIM. Fig. 1 A compares the FAIM sequences from Rattus norvegicus with Drosophila melanogaster, C. elegans, Gallus gallus, Danio rerio, Mus musculus, and Homo sapiens. The cloned rat sequences are available from GenBank/EMBL/DDBJ under accession no. NP_543171 (short form) and AAL77007 (long form). Alignment of FAIM vertebrate sequences shows more than 68% of homology. The percentage of homology is reduced to 30–50% when vertebrate sequences were aligned with D. melanogaster and C. elegans FAIM sequences, respectively (Table I).
Figure 1.
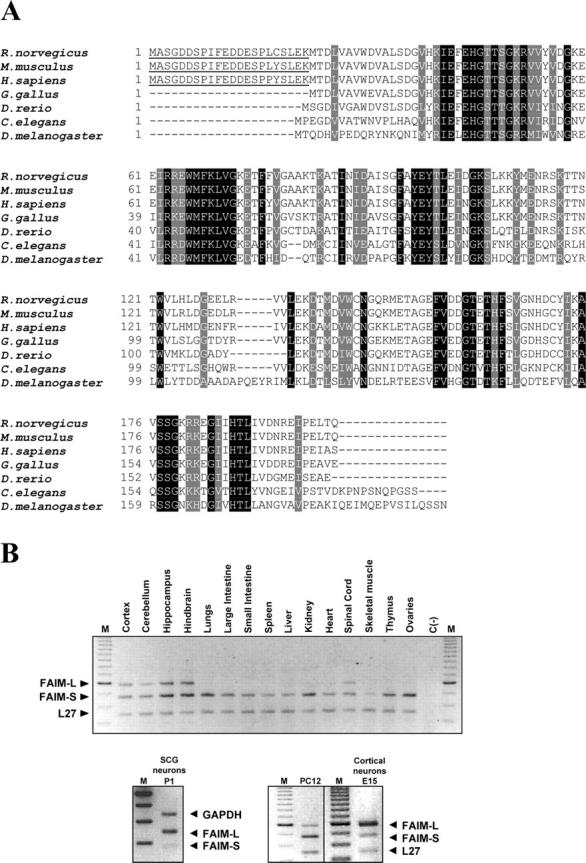
Protein alignments of FAIM. (A) Comparison of FAIM protein sequences from rat, mouse, human, C. elegans, D. melanogaster, G. Gallus, and D. rerio. Identical amino acids are shaded in black, and those with conservative changes in gray. The additional 22 amino acids of the long form are underlined. Some protein sequences are available from GenBank/EMBL/DDBJ under accession no. NP_543171(rat FAIM-S), AAL77007 (rat FAIM-L), NP_035940 (mouse FAIM-S), NP_060617 (human FAIM-S). The remaining sequence data were predicted from nucleotide sequences BE986872 (mouse FAIM-L), BQ638715 (5′ human FAIM-L), BU323531 (G. gallus), the combination of BG304437, AL923876 (D. rerio), AF003387 (C. elegans), and NM_137164 (D. melanogaster). (B) Semi-quantitative RT-PCR quantification of FAIM-S and FAIM-L expression in different rat tissues (top) and in P1 mouse SCG neurons, E15 mouse cortical neurons, and PC12 cells (bottom). As controls of the amplification GAPDH and L27 ribosomal proteins were used. C(−), control PCR reactions without reverse transcriptase; M, 50-bp DNA ladder marker.
Table I. Percentage of identity in a pairwise alignment between different species.
| R. norvegicus | M. musculus | H. sapiens | G. gallus | D. rerio | C. elegans | D. melanogaster | |
|---|---|---|---|---|---|---|---|
| R. norvegicus | — | 96 | 90 | 86 | 68 | 50 | 36 |
| M. musculus | 96 | — | 89 | 87 | 68 | 50 | 35 |
| H. sapiens | 90 | 89 | — | 85 | 65 | 49 | 35 |
| G. gallus | 86 | 87 | 85 | — | 68 | 50 | 36 |
| D. rerio | 68 | 68 | 65 | 68 | — | 50 | 38 |
| C. elegans | 50 | 50 | 49 | 50 | 50 | — | 32 |
| D. melanogaster | 36 | 35 | 35 | 36 | 38 | 32 | — |
Using RT-PCR, we found that FAIM mRNA is expressed in a wide variety of tissues including all the neural tissue samples examined (cortex, cerebellum, hippocampus, hindbrain, and spinal cord; Fig. 1 B). FAIM is also expressed in PC12 cells, mouse superior cervical ganglion (SCG) neurons, and cortical neurons (Fig. 1 B).
FAIM fails to protect neurons from neurotrophic factor deprivation but increases NGF-induced neurite outgrowth
NGF induces survival and neurite outgrowth from certain peripheral nervous system neurons and promotes the differentiation of PC12 cells to neuronal-like cells with neuritic processes. PC12 cells and SCG neurons require NGF for in vitro survival, and NGF withdrawal is rapidly followed by cell death. Because FAIM was initially cloned as an antagonist of Fas, and because Fas has been implicated in some types of neuronal cell death, we wanted to ascertain if FAIM was able to prevent cell death after NGF deprivation. To this end, we generated pools of PC12 cells stably transfected with either FAIM or the corresponding empty vector (Neo). These cells were grown in the presence of NGF for 72 h, and then were deprived of NGF for 24 h and cell viability was assessed. NGF-deprived, FAIM-transfected PC12 cells showed a reduction in survival (55.1 ± 8.0% survival) similar to that observed in NGF-deprived Neo cells (46.2 ± 6.4% survival). Similar results were obtained using mouse SCG neurons. For these experiments, SCG neurons were transiently transfected with FAIM or the empty vector and were grown with NGF for 24 h before NGF deprivation, and the number of surviving neurons was counted 24 h later. NGF-deprived, FAIM-transfected SCG showed a dramatic decrease in cell viability (8.1 ± 2.3% survival) similar to that found in NGF-deprived empty vector-transfected neurons (6.1 ± 1.4% survival). As a positive control, we transfected SCG cultures with Bcl-XL, a well characterized antiapoptotic molecule of the Bcl-2 family. Neurons overexpressing Bcl-XL survived NGF deprivation (81.6 ± 10.9% survival). These results demonstrate that FAIM is unable to prevent cell death after NGF deprivation in both PC12 and SCG neurons.
Despite the absence of an antiapoptotic function in PC12 cells and SCG neurons, we noticed that when these cells were transfected with FAIM and maintained in presence of NGF, there was a marked increase in neurite outgrowth compared with empty vector-transfected neurons. To ascertain if FAIM was able to modulate NGF-induced neurite outgrowth or influence neurite growth independently of NGF, pools of PC12 cells stably transfected with FAIM or the empty vector were grown with or without NGF for 1 d. Fig. 2 shows that in NGF-supplemented medium, PC12 cells overexpressing FAIM exhibited a fivefold increase in neurite length compared with control-transfected cells. However, overexpression of FAIM did not have any effect on neurite outgrowth from PC12 cells grown in complete medium without NGF. These results suggest that FAIM is able to regulate neurite outgrowth in NGF-differentiated PC12 cells. When the long form of FAIM was tested in the same assay, no enhanced neurite outgrowth was observed (Fig. 2) and, therefore, the studies using this splicing variant were no longer pursued.
Figure 2.

FAIM increases NGF-induced neurite outgrowth. (A) Phase-contrast micrographs of PC12 cells stably transfected with empty vector (top), FAIM-S (middle), or FAIM-L (bottom). PC12 cells treated with complete medium (left) or with NGF 100 ng/ml (right) for 1 d. Bar, 100 μm. (B) Histogram showing the neurite length measurements of cells shown in A. Significant differences in neurite length between Neo and FAIM-S are indicated (*, P < 0.001; t test).
To ascertain whether or not FAIM has a similar effect on NGF-induced neurite outgrowth in NGF-dependent primary neurons, we repeated these experiments using primary cultures of SCG neurons. Postnatal day (P1) mouse SCG neurons were plated and transiently cotransfected using gold carriers coated with both an eYFP expression plasmid (to visualize the transfected neurons) plus plasmids expressing FLAG-FAIM or the corresponding empty vector. The neurons were grown for 24 h in presence of NGF before the neuron arbors were captured in the confocal microscope and drawn using the confocal software. Fig. 3 (A and B) illustrates drawings and pictures of representative SCG neurons showing an increase in the neuritic arbor in FAIM-transfected neurons compared with the cultures transfected with the empty vector. Quantification of neurite length and number of branching points shows that FAIM induced a significant increase in these parameters (Fig. 3 C). These results indicate that FAIM is capable of regulating neurite length in sympathetic neurons.
Figure 3.

FAIM increases NGF-induced neurite outgrowth in mouse SCG neurons. Ballistic transfections of cultured neurons with an eYFP expression plasmid together with the Neo control vector or FAIM-S plasmids. After 24-h incubation with NGF (10 ng/ml), eYFP-labeled SCG were visualized and digitally acquired. (A) Representative drawings of the neurons corresponding to the 25, 50, 75, and 100 percentiles of the neurite length are shown. (B) Representative micrographs showing the FAIM-increased neuritic outgrowth. Bars, 50 μm. (C) Quantification of neurite length (top) and branching points (bottom). Significant differences in neurite length between Neo and FAIM are indicated (*, P < 0.05 and **, P < 0.001, respectively; t test).
Endogenous FAIM affects NGF-induced neurite outgrowth
To ascertain the role of endogenous FAIM in NGF-induced neurite outgrowth, we generated two RNAi that target different sites of FAIM sequence. To check the ability of the RNAi constructs to knockdown FAIM expression, double stably transfected PC12 cell lines with FAIM plus the RNAi1, RNAi2, or the pSUPER empty vector were generated. Because anti-FAIM antibodies capable of detecting endogenous FAIM are not currently commercially available, we generated an anti-FAIM antibody (see Materials and methods). This antibody was only capable of recognizing overexpressed FAIM but was not sensitive enough to detect endogenous FAIM. Both RNAi1 and RNAi2 dramatically decreased the level of overexpressed FAIM protein in PC12 cells, RNAi2 being more effective than RNAi1 (Fig. 4 A). We also checked the level FAIM mRNA under these experimental conditions. As shown in Fig. 4 B, transfection with the FAIM expression plasmid was associated with increased FAIM mRNA, whereas transfection with RNAi caused a decrease in endogenous FAIM mRNA.
Figure 4.
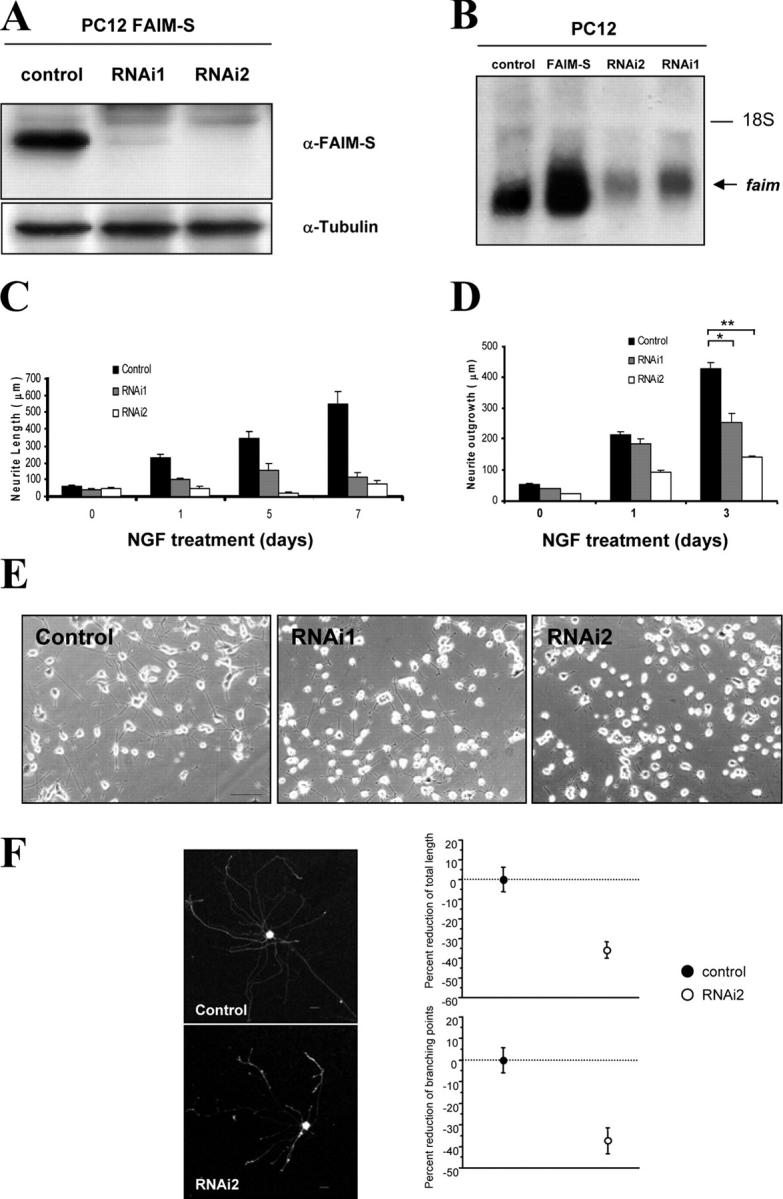
FAIM-S is necessary for NGF-induced PC12 cells differentiation. (A) PC12 cells that stably express FAIM-S were transfected with pSUPER containing sequences encoding RNAi against FAIM (RNAi1 or RNAi2) or the empty vector (control). Stable pools of transfected cells were obtained with puromycin. Extracts were probed with the anti-FAIM antibody by Western blot analysis (top). Membranes were stripped and reprobed with an antibody to α-tubulin used to control the loading of lanes. (B) PC12 cells were stably transfected with an empty vector (control), with the FAIM-S expression plasmid, or with the plasmid RNAi1 or RNAi2. Total RNA (15 μg) was extracted, electrophoresed, and hybridized with a [32P]dCTP-labeled FAIM probe. Images were obtained by autoradiography for 40 h at −80°C. (C) PC12 cells were transiently transfected with eYFP and either RNAi1 or RNAi2, or with the empty vector (control). After 4 d, cells were treated with 100 ng/ml NGF for 1 d. Histogram shows the neurite length measurements of the labeled and digitally acquired cultures transfected with the indicated cDNA. (D and E) PC12 cells were stably transfected with either an empty vector (control) or with either of the two FAIM RNAi (RNAi1 and RNAi2). (D) Cells were treated with NGF for 1 or 3 d or left untreated and total neurite length was measured. (E) Representative images of NGF-induced differentiation on stably transfected cells after 1 d of treatment. Bar, 100 μm. (F) Rat SCG neurons were cotransfected with eYFP and either the empty pSUPER (control) or the vector containing RNAi2. After 60 h of NGF treatment (10 ng/ml), eYFP-labeled SCG were visualized, digitally acquired (representative images are shown on the left), and neurite outgrowth was quantified. Neurite length (top) and the branching points (bottom) were quantified. Bars, 25 μm.
To analyze the effect of RNAi on neurite length, PC12 cells were transfected with eYFP and either the FAIM RNAi1 and RNAi2 or the empty vector. After 4 d, the cells were treated with 100 ng/ml NGF plus 0.5% heat-inactivated horse serum for the indicated times after which neurite length was measured. Fig. 4 C shows that both RNAi1 and RNAi2 decreased neurite outgrowth, the inhibition of outgrowth being significantly greater with RNAi2 than RNAi1. Similar results were obtained with PC12 cells stably transfected with the same constructs but in this case we observed a homogeneous effect on the entire population of transfected cells (Fig. 4, D and E). These decreases in neurite outgrowth with the RNAi constructs correlate with the level of FAIM protein knockdown and demonstrate that endogenous FAIM plays a role in regulating neurite outgrowth in NGF-stimulated PC12 cells.
To explore the relevance of endogenous FAIM in primary neurons, rat SCG neurons were transiently transfected with either RNAi2 or an empty vector (pSUPER). After 60 h of incubation with NGF, RNAi2 produced >30% significant decreases in total neurite length and branch point number compared with control-transfected neurons (Fig. 4 F). These results show that endogenous FAIM plays a significant role in NGF-induced neurite growth in sympathetic neurons.
ERK pathway is required for FAIM-induced neurite outgrowth
Because ERK signaling has been implicated in neurotrophin-induced neurite outgrowth, we investigated the role of this signaling pathway in mediating the neurite outgrowth–inducing effects of FAIM by using pharmacological agents to inhibit ERK signaling. Addition of PD98059, a MEK inhibitor, markedly reduced the extent of neurite outgrowth in FAIM-transfected cells (Fig. 5 A). These results suggest that activation of the ERK pathway is required for the neurite growth–inducing effect of FAIM overexpression. To confirm that the doses of PD98059 completely abolished ERK activation in FAIM-transfected cells as well as in the Neo cells, Western blot analysis was performed using a specific anti–phospho-ERK1/2 antibody. 30-min pretreatment of the stably transfected PC12 cells with 50 μM PD98059 completely abolished ERK1/2 phosphorylation (Fig. 5 B).
Figure 5.

Effects of FAIM on ERK pathway stimulation. (A) PC12 cells stably transfected with empty vector or with FAIM-S were treated with NGF (100 ng/ml) or with NGF plus 50 μM of PD98059 for 1 d. Histogram shows the neurite length measurements of the cultures transfected with the indicated cDNA. Significant differences are indicated (*, P < 0.05; **, P < 0.001; t test). (B) PC12 cells stably transfected with empty vector (N) or FAIM-S (S) were serum starved for 12 h, pretreated (+) or not (−) with 50 μM PD98059 for 30 min, and stimulated (+) or not (−) with 100 ng/ml NGF for 5 min. Protein extracts were analyzed by Western blotting with an anti–phospho-ERK antibody (top) and stripped and reprobed with anti–pan-ERK and anti-tubulin antibodies (middle) to control the protein loading. Anti-FLAG immunoblotting (bottom) confirmed FAIM-S overexpression in transfected PC12 cells. White lines indicate that intervening lanes have been spliced. (C) PC12 cells stably transfected with empty vector (N), FAIM-S (S), or RNAi2 (R) were serum starved for 12 h and stimulated with 100 ng/ml NGF for the indicated times. Protein extracts were processed as in B.
Although the ERK pathway is required for the effects of FAIM overexpression on neurite outgrowth, to determine if FAIM stimulates ERK signaling, we used Western blotting to assess the level of ERK phosphorylation in NGF-stimulated PC12 cells stably transfected with FAIM, with the RNAi2 construct, or with an empty vector. NGF induced a peak of ERK phosphorylation after 5 min of stimulation. This increase in ERK1/2 phosphorylation was detectable after 15 min and 1 h and undetectable after 12 h. Our results show that the level of phosphorylation of ERK was not different between Neo, FAIM, or RNAi2 transfected cells at any of the time points studied (Fig. 5 C).
The absence of clear differences in the level and the duration of ERK activation between wild-type and FAIM-overexpressing or FAIM knockdown neurons raises the possibility that other signaling pathways play a role in mediating FAIM-induced neurite outgrowth.
FAIM enhances NGF-mediated activation of NF-κB
NGF signaling through p75NTR and TrkA has been shown to be critical in the regulation of neurite outgrowth (Davies, 2000), and one of the pathways activated by NGF through these receptors is the NF-κB pathway (Wood, 1995; Carter et al., 1996; Yoon et al., 1998; Hamanoue et al., 1999; Foehr et al., 2000). To investigate whether or not FAIM-induced neurite outgrowth was mediated by NF-κB, FAIM stably transfected PC12 cells were transiently transfected with an NF-κB–dependent luciferase reporter construct or a control construct lacking the NF-κB site (Rodriguez et al., 1996) and assayed for luciferase activity after NGF treatment. FAIM-transfected cells showed a marked increase in luciferase activity after 6–9 h of stimulation with NGF compared with the Neo-transfected cells, indicating that NF-κB activation is potentiated by FAIM expression (Fig. 6 A). In cells that were stably transfected with RNAi2, the levels of luciferase activity were found to be significantly decreased at all the time points analyzed (Fig. 6 A).
Figure 6.
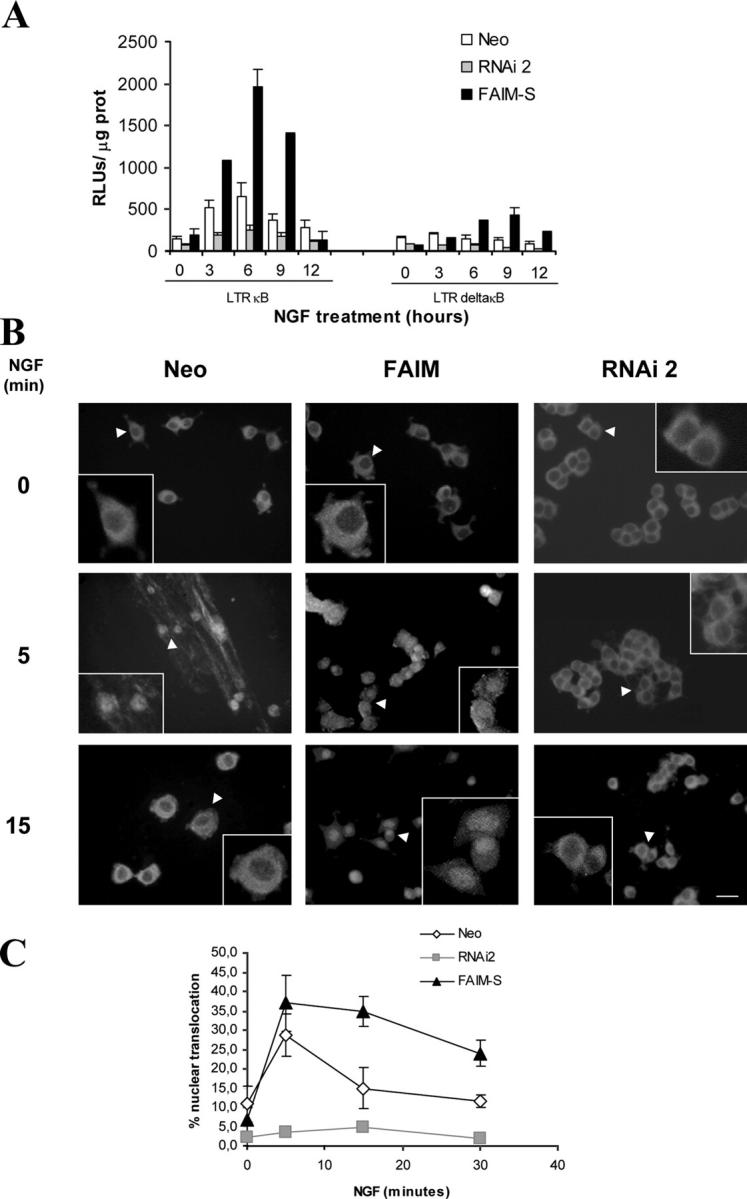
NF-κB activation is modulated by FAIM. (A) PC12 cells were stably transfected with an empty vector (Neo), with the FAIM-S expression plasmid, or with the FAIM RNAi plasmid. Cells were then transiently transfected with an NF-κB–dependent reporter vector (LTR KB) or with the equivalent control vector lacking NF-κB binding sites (LTR deltaKB). After 24 h, the cells were treated with NGF (100 ng/ml) for the indicated times and luciferase activity was measured in the cell lysates using a Luciferase Assay System. Luciferase values were normalized to protein concentration (RLU/μg of protein). (B) Immunocytochemistry was performed to detect the p65 subunit of NF-κB in similar cells as described in A. Representative low and high magnification photomicrographs are shown (arrowheads indicate the selected cells illustrated in the insets). Bar, 25 μm. (C) Graph showing the percentage of nuclear p65 staining in the different experimental conditions. The mean ± SEM of three independent experiments are shown.
To determine an additional parameter of NF-κB activation, we checked p65 translocation to the nucleus after NGF stimulation. p65 is one of the two subunits most ubiquitous forms of NF-κB dimer. To this end, we stained cultures of FAIM-, RNAi2-, or Neo-transfected cells with an antibody against p65. Before NGF stimulation very few Neo-, FAIM-, or RNAi2-transfected cells showed nuclear staining for p65. After 5 min of NGF stimulation, cells stably transfected with FAIM or with the empty vector (Neo) have a similar activation of the NF-κB pathway as inferred from p65 nuclear translocation. Differences between Neo and FAIM are evident at longer stimulation. For example, at 15 min the percentage of Neo cells that show p65 translocation has dropped to ∼15%, but remains high (∼35%) in FAIM-transfected cells (Fig. 6, B and C). However, no increase was observed in RNAi2-transfected cells at any of the time points analyzed (Fig. 6, B and C). These results show that nuclear translocation of NF-κB following NGF stimulation is enhanced in cells overexpressing FAIM and completely prevented in cells in which FAIM expression was reduced by RNAi2. Together, these findings raised the possibility that NF-κB signaling plays a regulatory role in FAIM-induced neurite outgrowth and also suggest that endogenous FAIM is necessary for the NGF-induced NF-κB activation.
NF-κB signaling is required for FAIM-induced neurite outgrowth
Having ascertained that NF-κB activation is increased in FAIM-expressing PC12 cells stimulated with NGF, we investigated whether or not preventing NF-κB activation would interfere with FAIM neurite outgrowth responses. We stably (Fig. 7, A–D) or transiently (Fig. 7 E) transfected PC12 cells with a construct encoding a form of IκBα carrying serine-to-alanine mutations at residues 32 and 36, named SR-IκBα. These mutations prevent IκBα phosphorylation and subsequent proteasome-mediated degradation, thereby preventing release and nuclear translocation of NF-κB (Rodriguez et al., 1996). Expression of SR-IκBα effectively prevented nuclear translocation of the p65 subunit of NF-κB even when the cells were treated with TNFα, one of the most potent activators of this pathway (Fig. 7, B and C). In these cells, NGF-induced neurite outgrowth was significantly reduced (Neo, 198 ± 18 μm, vs. SR-IκBα, 108 ± 5 μm; Fig. 7 D) to a similar extent to that previously reported for NF-κB inhibition in PC12 cells (Foehr et al., 2000), implying that NF-κB is involved in NGF-induced neurite outgrowth in PC12 cells.
Figure 7.

SR-IκBα transfection attenuates the NGF-induced differentiation and abolishes the effects of FAIM overexpression on neurite outgrowth. (A) PC12 cells stably transfected with empty vector (Neo) or with SR-IκBα. Total cell lysates were analyzed by Western blot using an antibody against IκBα (top). Membranes were stripped and reprobed with an antibody to α-tubulin to normalize the loading content per lane (bottom). (B–D) PC12 cells stably transfected with the empty vector (Neo) or with the vector expressing SR-IκBα and were treated with NGF for 5 min, TNFα for 15 min, or left untreated. Quantification of immunocytochemical detection of nuclear p65 is shown in B. Representative pictures of low and high magnification (arrows point to selected cells shown in insets) are shown in C. Bar, 25 μm. Quantification of neurite outgrowth was performed 1 d after NGF treatment (D). Asterisks indicate a significant (P < 0.001; t test) reduction of neurite outgrowth in PC12 cells expressing the SR-IκBα. (E) Neo or FAIM-S stably transfected cells were additionally transfected transiently with eYFP alone or with eYFP plus SR-IκBα. After 24 h, they were treated with NGF (100 ng/ml) for an additional 24-h period and neurite length was quantified. Significant differences are indicated (*, P < 0.001; t test).
We also analyzed whether or not this NF-κB is involved in the increased neurite outgrowth observed in FAIM-overexpressing PC12 cells after NGF stimulation. PC12 cells stably overexpressing FAIM-S were transiently transfected with the SR-IκBα together with eYFP. After 24 h, the cells were treated with NGF and neurite outgrowth was quantified 24 h later. SR-IκBα completely abolished FAIM-S–induced neurite outgrowth (Fig. 7 E). These results demonstrate that NF-κB signaling mediates the enhanced neurite outgrowth induced by FAIM overexpression.
p65-deficient cortical neurons show impaired FAIM-induced neurite outgrowth
To further confirm the role of NF-κB in mediating FAIM-induced neurite outgrowth, we compared the neurite arbors in cultures established from wild-type embryos and embryos with a null mutation of the p65 gene (Beg et al., 1995). We used primary cultures of cortical neurons instead of SCG neurons for these studies because the latest stage at which these experiments could be performed was at day 15 of gestation (E15), since p65 −/− embryos die in utero shortly after this stage. After 3-h culture, E15 cortical neurons were ballistically transfected with gold particles coated with FAIM plus eYFP or pcDNA3 plus eYFP, and neurite arbors were analyzed after a further 24-h incubation. Fig. 8 shows that p65 +/+ cortical neurons overexpressing FAIM had more extensive (50–60% longer) and more branched neurite arbors (80–120% more branch points) than control transfected neurons. This increase in neurite growth and complexity between FAIM-transfected and control-transfected neurons was not observed in cultures established from p65 −/− littermates. These results additionally indicate that a functional NF-κB is required for FAIM-induced neurite outgrowth in cortical neurons.
Figure 8.

FAIM-induced increase in neurite outgrowth is eliminated in cortical neurons from p65 − / − mouse embryos. Cultured cortical neurons from p65+/+ (WT) or p65−/− (KO) mice were transfected with eYFP alone or with eYFP plus FAIM-S. After 24 h, labeled cortical neurons were visualized, digitally acquired, and measured. (A) Drawings of neurons that represent the 25, 50, 75, and 100 percentiles of neurite length measurements. (B) Micrograph showing representative cells of p65+/+ (WT) or p65−/− genotypes (KO) transfected with the indicated plasmids. (C) Quantification of the neurite outgrowth performed by tracing the neuritic arbors and measuring the neuritic length (top) and branching points (bottom). Bars, 50 μm. *, P < 0.001; t test.
FAIM interacts with TrkA and p75NTR receptors in an NGF-dependent manner
NF-κB activation after NGF stimulation has been described to be mediated by TrkA and p75NTR (Carter et al., 1996; Hamanoue et al., 1999; Foehr et al., 2000; Wooten et al., 2001). To examine the possibility that FAIM interacts with the NGF receptors TrkA or p75, we performed immunoprecipitation experiments. Fig. 9 shows that FLAG-tagged FAIM is able to coimmunoprecipitate with both HA-tagged TrkA (Fig. 9 A) and HA-tagged p75 (Fig. 9 B) receptors upon NGF stimulation when these molecules are transfected in PC12 cells. Lysates from cells that were not stimulated with NGF failed to show any interaction, indicating that this interaction requires NGF receptor activation. We also used immunoprecipitation to investigate whether FAIM is able to interact with endogenous TrkA and whether this interaction is dependent on ligand binding. PC12 cells were transfected with FLAG-tagged FAIM and stimulated with and without NGF. Fig. 9 C shows that FLAG-tagged FAIM was able to coimmunoprecipitate with endogenous Trk but only when the cells had been stimulated with NGF. These data raise the possibility that FAIM influences NGF-induced neurite outgrowth by modulating activation of intracellular signaling pathways downstream of TrkA and p75.
Figure 9.

FAIM interacts with TrkA and p75NTR receptors in an NGF-dependent manner. Wild-type PC12 cells were electroporated with FLAG-tagged FAIM-S and either TrkA-HA (A) or p75NTR-HA (B). After 48 h, the cells were stimulated (+) or not (−) with 100 ng/ml NGF for 15 min. TrkA-HA (A) or FAIM-FLAG (B) were immunoprecipitated from 1 mg of cell lysate followed by Western blotting with either FLAG or HA antibody. An IP with myc antibody was used as a negative IP control in B. A fraction of the lysate was blotted with FLAG, HA, or phospho-ERKs antibody (top). (C) Wild-type PC12 cells were electroporated with FLAG-FAIM-S and were stimulated (+) or not (−) with 100 ng/ml NGF for 15 min. FLAG-FAIM-S was immunoprecipitated from 1 mg of cell lysate with the FLAG antibody, and Western blotting was used to detect endogenous Trk with anti-Trk antibody 203 in the precipitates. A fraction of the lysate was blotted with FLAG or phospho-ERKs antibody (top).
Discussion
Our work provides evidence for the involvement of FAIM, a previously identified Fas apoptosis inhibitory molecule, in regulating neurite growth. Whereas overexpression of FAIM markedly enhances neurite growth from peripheral and central neurons and from the PC12 neuronal cell line, reduction of endogenous FAIM by RNAi significantly decreases NGF-stimulated neurite growth from PC12 cells and SCG neurons. Furthermore, in contrast to work showing the antiapoptotic function of FAIM in B-lymphocytes (Schneider et al., 1999; Rothstein et al., 2000), we find that modulating FAIM expression has no effect on neuronal survival. Fas and Fas ligand (FasL) are widely expressed in the nervous system in both neurons and glial cells (Park et al., 1998; Spanaus et al., 1998; Raoul et al., 1999; Casha et al., 2001, Choi et al., 1999; Wohlleben et al., 2000; Desbarats et al., 2003). Fas-induced apoptosis plays a role in promoting the death of certain neurons during development and after ischemia or axotomy in the adult. Blocking interaction between Fas and FasL reduces the death of embryonic cerebellar granule cells and spinal motoneurons following neurotrophic factor deprivation in vitro. Fas is constitutively expressed in these neurons and FasL is induced after neurotrophic factor deprivation (Brunet et al., 1999; Raoul et al., 1999). FasL is rapidly induced following ischemia in the adult rat central nervous system, and there is evidence that this plays a role in bringing about neuronal apoptosis (Martin-Villalba et al., 1999; Felderhoff-Mueser et al., 2000; Matsushita et al., 2000). Fas also plays a role in axotomy-induced death of motoneurons (Ugolini et al., 2003). However, Fas does not play a ubiquitous role in bringing about neuronal death in the developing nervous system. For example, FasL/Fas interactions are apparently not required for naturally occurring neuronal death in the SCG (Putcha et al., 2002). This finding is relevant to the present work because we showed no effect of FAIM overexpression in regulating the survival of SCG neurons and PC12 cells, which are thought to be a model of developing sympathetic neurons. Thus, in future work it will be important to explore more widely in the nervous system the potential involvement of FAIM in regulating neuronal survival.
In addition to its well established role in regulating cell survival, Fas has been implicated in neural regeneration and regulation of neurite growth. Whereas FasL and agonist Fas antibodies induce neurite outgrowth from neonatal mouse dorsal root ganglion (DRG) explants in vitro and accelerate regeneration following sciatic nerve crush in vivo, recovery from sciatic nerve crush is delayed in Fas-deficient lpr mice (Desbarats et al., 2003). Our findings seem at odds with these results if FAIM were acting predominantly as an inhibitor of Fas signaling. However, neurite outgrowth in our experimental models was induced without Fas engagement, so it is unlikely that FAIM affected neurite by modulating Fas signaling. Rather, our demonstration that FAIM interacts with both Trk and p75NTR NGF receptors in a ligand-dependent manner and that FAIM enhances neurite outgrowth only when NGF is present in the culture medium raises the possibility that FAIM influences neurite growth by regulating NGF receptor signaling. Both TrkA and p75NTR receptor components are capable of activating NF-κB signaling in response to NGF (Carter et al., 1996; Hamanoue et al., 1999, Foehr et al., 2000). In PC12 cells, NF-κB activation by NGF involves the formation of a TRAF6-p62 complex, which acts as a bridge linking both TrkA and p75NTR signaling, and activation of IKK by p62 via participation of atypical protein kinase C (Sanz et al., 2000; Wooten et al., 2001). Although blocking TrkA-mediated NF-κB activation reduces neurite process formation in NGF-stimulated PC12 cells, NF-κB activation alone is not sufficient to induce neurite outgrowth (Foehr et al., 2000). In our present work we have demonstrated that FAIM overexpression promotes nuclear translocation of NF-κB and enhanced transcriptional activity. Moreover, blocking NF-κB activation in PC12 cells and SCG neurons with SR-IκBα or carrying out experiments on cortical neurons from mice that lack the p65 NF-κB subunit has demonstrated that FAIM-induced neurite outgrowth is dependent on NF-κB activation. More importantly, when we used an RNAi strategy to down-regulate endogenous FAIM expression, activation of NF-κB by NGF was completely abolished. Moreover, NGF-induced neurite outgrowth was substantially reduced by RNAi in both PC12 cells and SCG neurons. Although we have not ascertained the molecular mechanism by which FAIM enhances NF-κB activation in NGF-stimulated neurons, the fact that FAIM binds both TrkA and p75 suggests that FAIM influences NF-κB activation at the level of receptor signal transduction.
We have also shown that the effect of FAIM overexpression on neurite outgrowth is blocked by pharmacological inhibition of MEK1/MEK2 of the ERK signaling pathway. This pathway has been widely implicated in promoting neurite outgrowth in NGF-stimulated PC12 cells (Traverse et al., 1992; Cowley et al., 1994; Marshall, 1995; Vaillancourt et al., 1995). Furthermore, cross-linking Fas on SY-SH5Y neuroblastoma cells or DRG neurons promotes sustained activation of ERK signaling and up-regulation of p35, which in turn plays a key role in mediating the neurite growth–promoting effects of Fas activation in DRG neurons (Desbarats et al., 2003). However, in our experimental paradigm FAIM transfection does not promote a sustained increased in ERK activation. Thus, although ERK signaling is required for FAIM-induced neurite outgrowth in NGF-stimulated PC12 cells, it seems unlikely that FAIM enhances neurite outgrowth in these cells by modulating ERK signaling directly.
In summary, we provide the first evidence for the involvement of a previously identified inhibitor of Fas-induced apoptosis in the regulation of neurite growth. We show that FAIM overexpression enhances neurite outgrowth from NGF-stimulated cells and that endogenous FAIM is required for NGF-induced neuritogenesis. Activation of both ERK and NF-κB signaling pathways are required for the neurite growth–promoting effects of FAIM, but only NF-κB activation is modulated by FAIM. The effect of FAIM on neurite growth is not mediated by its effect on Fas signaling. Our demonstration that FAIM interacts with p75 and TrkA in an NGF-dependent manner suggests that FAIM influences neurite growth by affecting NGF signal transduction at the cell surface receptor complex.
Materials and methods
Materials
All biochemicals were obtained from Sigma-Aldrich unless otherwise indicated. Purified recombinant human NGF was obtained from Genentech, Inc., and mouse 7S NGF was prepared from male submandibular salivary glands (Mobley et al., 1976).
Cell culture
PC12 cells were grown on 100-mm tissue culture dishes (Falcon Discovery Labware; BD Biosciences) in DME supplemented with 6% heat-inactivated FCS, 6% heat-inactivated horse serum (GIBCO BRL), 10 mM Hepes, 20 units/ml penicillin, and 20 μg/ml streptomycin.
SCG neurons were dissected from P1 CD1 mice or rat as indicated, trypsinized (0.25% trypsin for 25 min at 37°C), and dissociated by trituration. The neurons were plated in defined, serum-free medium on a poly-ornithine/laminin substratum in 35-mm tissue culture dishes (Davies et al., 1993). Ballistic transfection of cultured neurons was performed 3 h after plating.
For the studies of the role of the p65 NF-κB subunit in regulating neurite growth, cortical neuron cultures were established from mice that possess a null mutation in the p65 gene (Beg et al., 1995). Heterozygous mice were crossed to obtain p65 −/−, p65 −/+, and p65 +/+ embryos. At E15, pregnant females were killed and separate dissociated cultures of cortical neurons were established from each embryo (the embryo genotypes were subsequently ascertained by a PCR-based approach). The dissected cerebral cortices from each embryo were collected in ice-cold HBSS (Invitrogen) and triturated with a fire-polished Pasteur pipette. The resulting cells were plated in poly-l-ornithine/laminin-coated 35-mm culture dishes and cultured in Ham's F14 (Sigma-Aldrich) supplemented with 2 mM glutamine and 2% B27 (Invitrogen). After 24 h in culture, the cells from the p65 −/− and p65 +/+ embryos were transfected with expression plasmids.
Plasmids
Rat FAIM, human TrkA, human p75, and super-repressor IκBα (SR-IκBα) cDNAs were expressed under the control of a CMV constitutive promoter in the pcDNA3 expression vector (Invitrogen). eYFP was obtained from BD Biosciences. The NF-κB–dependent reporter vector (HIV-LTR-Luciferase), the equivalent control vector lacking κB binding sites (ΔκB HIV-LTR-Luciferase), and SR-IκBα cDNA (Rodriguez et al., 1996) were obtained from R. Hay (University of St. Andrews, Fife, Scotland). TrkA and p75 were obtained from J. Moscat (Centro de Biología Molecular Severo Ochoa, Madrid, Spain). FAIM-S cDNA was obtained from the EST UI-R-BJ1-awa-h-09-0-UI (GenBank/EMBL/DDBJ accession no. BE112969) and the long form was cloned from clones UI-R-BJ1-awg-h-12-0-UI (5′ end) and UI-R-BJ1-awa-h-09-0-UI (3′ end) found in dbEST as GenBank/EMBL/DDBJ accession no. BE113668 and BE112969, respectively (Invitrogen). FAIM was tagged with 3× FLAG at the amino terminus and was subcloned into the pcDNA3 mammalian-expressing vector.
For RNAi experiments, constructs were obtained into the pSUPER. retro.puro plasmid (OligoEngine) using specific oligonucleotides of the FAIM sequence, indicated by capital letter, as follows: RNAi1, (forward) gatccccGATGTTCAAATTGGTGGGCttcaagagaGCCCACCAATTTGAACATCttttt and (reverse) agctaaaaaGATGTTCAAATTGGTGGGCtctcttgaaGCCCACCAATTTGAACATCggg; or RNAi2, (forward) gatc-cccGGCAAACGAGTTGTGTACGttcaagagaCGTACACAACTCGTTTGCCttttt and (reverse) agctaaaaaGGCAAACGAGTTGTGTACGtctcttgaaCGTACACAACTCGTTTGCCggg. Oligonucleotides were obtained from Sigma-Aldrich.
Cell transfection
Unless otherwise indicated, PC12 cells were transfected with the desired expression plasmid using Lipofectamine 2000 (GIBCO BRL). Pools of cells transfected with FAIM-S, FAIM-L, SR-IκBα, or the empty pcDNA3 were performed by adding G-418 (Geneticin) to the medium at a final concentration of 500 μg/ml (GIBCO BRL). Pools of cells transfected with RNAi1, RNAi2, or the empty pSUPER vector were performed by adding puromycin at 1 μg/ml.
Ballistic transfection of neuronal cultures was performed at indicated times using the hand-held gene-gun (Helios Gene-gun; Bio-Rad Laboratories). Gold particle cartridges were prepared as follows: 20 mg of gold were resuspended in 100 μl of 50 mM spermidine and 20 μg of each plasmid cDNA(s). 100 μl of 2 M CaCl2 was added dropwise to the suspension to precipitate the particles, which were washed three times with 100% ethanol. The coated particles were resuspended in 1.2 ml of 100% ethanol with 0.01 mg/ml polivinylpirrolidone and loaded into the Teflon cartridge tubing that was cut into multiple cartridges for the Gene-gun. The coated gold particles were shot into the cultured neurons with the gun pressurized at 200 psi. A 70-μm nylon mesh screen was placed between the gun and the culture to protect the tissue from the shock wave.
RT-PCR analysis
cDNA was reverse transcribed from RNA extracted from multiple rat tissues, PC12 cells, P1 SCGs neurons, and E15 cortical neurons. Multiplex PCR was performed by coamplification of FAIM and the house keeping L27 ribosomal protein or GAPDH as indicated. Primers used to amplify 189- and 246-bp specific fragments corresponding to FAIM-S and FAIM-L were GACAGCTGCTGACTACGTCG (forward) and TCCTTCCCATCCACGTACAC (reverse). The L27 ribosomal protein primers were AGCTGTCATCGTGAAGAA (forward) and CTTGGCGATCTTCTTCTTGCC (reverse). GAPDH primers were CAGTGGCAAAGTGGAGATTG (forward) and CGTGGTTCACACCCATCAC (reverse).
Northern blotting
The efficacy of FAIM RNAi constructs in reducing FAIM expression was checked by Northern blotting. In brief, 15 μg of total RNA was electrophoresed in a 1% agarose gel containing 1.1% formaldehyde, transferred into Hybond-N+ nylon membranes (Amersham Biosciences), cross-linked by UV exposure, and hybridized overnight at 65°C with a [32P]dCTP-labeled FAIM PCR product in a 0.5 M phosphate-buffered solution containing 7% SDS, 1 mM EDTA, and 1% BSA, pH 6.8. The membrane was washed twice with 1× SSC, 0.1% SDS, for 15 min at RT, and once with 0.1× SSC, 0.1% SDS, for 30 min at 65°C. Images were obtained by exposing the radiolabeled membrane against SuperRX film (Fuji) for 40 h at −80°C.
Neurite measurement
For PC12 cells, photographs of random fields were taken of three different dishes using an inverted microscope (Olympus) equipped with epifluorescence optics and a camera (model OM-4 Ti; Olympus). Neurite outgrowth was assessed using the Adobe Photoshop 6.0 software. In every experiment, no less than 100 cells were measured per condition.
Labeled SCG or cortical neurons were visualized and digitally acquired in a laser scanning confocal microscope (model Axioplan; Carl Zeiss MicroImaging, Inc.) after an incubation period of 24 or 60 h. For every condition studied, between 40 and 50 cells were captured. Neuritic arbors were traced using the LSM510 software. The obtained traces were analyzed by direct counting of the branching points, metric neuritic length, and other topological parameters.
Western blot
Cells were rinsed with ice-cold PBS at pH 7.2 after stimulation. Cell lysate was harvested with prewarmed (95°C) 2% SDS-125 mM Tris, pH 6.8. Protein concentration was quantified by a modified Lowry assay (Bio-Rad Dc protein assay; Bio-Rad Laboratories).
The different cell lysates (50 μg of protein) were resolved in SDS-polyacrylamide gels and were transferred onto Polyvinylidene difluoride Immobilon-P transfer membrane (Millipore). After blocking with TBS with Tween-20 containing 5% nonfat dry milk for 1 h at RT, the membranes were probed with the appropriated primary antibodies against the phosphorylated forms of ERK1 and ERK2 (New England Biolabs, Inc.), pan-ERK (BD Biosciences), anti-FLAG M2 mAb (Sigma-Aldrich), anti–α-tubulin (clone B-5-1-2; Sigma-Aldrich), or a polyclonal anti-IκBα antibody (Cell Signaling Technology).
After incubation with specific peroxidase-conjugated secondary antibodies, the membranes were developed with the ECL SuperSignal West Dura Extended Duration Substrate (Pierce Chemical Co.).
A rabbit polyclonal antibody against rat FAIM (full-length protein) was generated by injecting a GST-FAIM recombinant protein produced by E. coli cells into white New Zealand rabbits (Harlow and Lane, 1988). The quality and specificity of the antibody was checked using lysates of PC12 cells transfected with a pcDNA3 plasmid expressing rat FAIM or using different tissue lysates from adult rats.
Coimmunoprecipitation experiments
For coimmunoprecipitation experiments, a total of 20 × 106 PC12 cells were electroporated with 30 μg of plasmids encoding TrkA or p75 tagged with HA at their NH2-terminal region plus 10 μg of NH2-terminally FLAG-tagged FAIM using a Gene Pulser II (Bio-Rad Laboratories). After 48 h, cells were harvested in lysis buffer PD (40 mM Tris, pH 78.0, 500 mM sodium chloride, 6 mM EDTA, 6 mM EGTA, 0.1% NP-40, 10 mM β-glycerophophate, 10 mM NaF, 300 μM Na3VO4, 1 mM DTT, 2 μM PMSF, 1 mM benzamidine, 10 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin), and lysates were clarified by centrifugation and quantified by Bradford assay (Bio-Rad Laboratories). As a control, 25 μg of the lysate was blotted with anti-FLAG M2 (Sigma-Aldrich), an in-house monoclonal anti-HA from J. Moscat, or phospho-ERKs (New England Biolabs, Inc.) antibody to check the expression levels of the exogenous proteins and the correct stimulation with NGF. Alternatively, 1 mg of cleared supernatants was subjected to immunoprecipitation with 1 μg of the anti-FLAG, anti-myc monoclonal (9E10) (SC-40; Santa Cruz Biotechnology, Inc.) or anti-HA (Y-11) (SC-805; Santa Cruz Biotechnology, Inc.) antibodies overnight at 4°C. Antibodies were recovered with protein G or A according to the immunoglobulin isotype and were resolved in SDS-PAGE. For the semiendogenous approach (Fig. 9 C), the Western blotting was probed with an anti-pan Trk antibody 203 (provided by D. Martin-Zanca, Centro Superior de Investigaciones Científicas, Salamanca, Spain; Martin-Zanca et al., 1989).
NF-κB activity
PC12 cells were transfected by electroporation using 12 μg of the NF-κB–dependent reporter vector (HIV-LTR-Luciferase) or the equivalent control vector lacking κB binding sites (ΔκB HIV-LTR-Luciferase). Cells were stimulated with 100 ng/ml NGF for the indicated time. Cell lysates were obtained with 50 μl of Cell Culture Lysis Reagent (Luciferase Assay System Kit; Promega). Aliquots of supernatant were transferred to a standard 96-well plate for protein concentration determination using Protein Dye reagent (Bio-Rad Laboratories) following the manufacturer's instructions, and luciferase values were normalized to protein concentration (RLU/μg of protein).
Immunocytochemistry
Stably transfected PC12 cells were treated with 100 ng/ml NGF at the indicated times. The cells were fixed in methanol 100% for 5 min at RT, incubated with polyclonal anti-p65 (C-20) (sc-372; Santa Cruz Biotechnology, Inc.) for 1 h at RT and further incubated with anti-rabbit secondary antibody conjugated with Alexa Fluor 488 (Molecular Probes) for 1 h at RT protected from light. Micrographs were obtained using an inverted microscope (Olympus) equipped with epifluorescence optics and a camera (model OM-4 Ti; Olympus).
Statistical analysis
All the experiments were performed at least three times. Values were expressed as mean ± SEM. t tests were used to determine the statistical significance of differences in neurite length, branching points numbers, and luciferase activity (P < 0.001 was considered significant).
Acknowledgments
We thank Dr. Ron Hay for the super-repressor IκBα plasmid and Dr. Dionisio Martin-Zanca for providing 203 antibody. We thank our colleagues of the Group of Cell Signaling and Apoptosis for their unconditional support.
C. Sole and M.F. Segura hold postgraduate fellowships and X. Dolcet holds a postdoctoral fellowship from the Spanish Government, Misterio de Educación, Cultura y Deporte de España. This work has been supported by grants FIS (PI020051) from Ministerio de Sanidad y Consumo, Fundació La Caixa (Convocatòria de Malalties Neurodegeneratives), Suport als Grups de Recerca, and Distinció Joves Investigadors from Generalitat de Catalunya to J.X. Comella, the Wellcome Trust to A.M. Davies, and the European Commission (QLG3-CT-1999-00602) to J.X. Comella and A.M. Davies.
X. Dolcet, M.F. Segura, and H. Gutierrez contributed equally to this paper.
A.M. Davies and J.X. Comella contributed equally to this paper.
Abbreviations used in this paper: DRG, dorsal root ganglion; ERK, extracellular regulated kinase; FAIM, Fas apoptosis inhibitory molecule; FasL, Fas ligand; P1, postnatal day; p75NTR, p75 neurotrophin receptor; SCG, superior cervical ganglion.
References
- Beg, A.A., W.C. Sha, R.T. Bronson, S. Ghosh, and D. Baltimore. 1995. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 376:167–170. [DOI] [PubMed] [Google Scholar]
- Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C. Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. [DOI] [PubMed] [Google Scholar]
- Carter, B.D., C. Kaltschmidt, B. Kaltschmidt, N. Offenhauser, R. Bohm-Matthaei, P.A. Baeuerle, and Y.A. Barde. 1996. Selective activation of NF-kappa B by nerve growth factor through the neurotrophin receptor p75. Science. 272:542–545. [DOI] [PubMed] [Google Scholar]
- Casha, S., W.R. Yu, and M.G. Fehlings. 2001. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience. 103:203–218. [DOI] [PubMed] [Google Scholar]
- Chao, M.W. 2003. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4:299–309. [DOI] [PubMed] [Google Scholar]
- Cheema, Z.F., S.B. Wade, M. Sata, K. Walsh, F. Sohrabji, and R.C. Miranda. 1999. Fas/Apo [apoptosis]-1 and associated proteins in the differentiating cerebral cortex: induction of caspase-dependent cell death and activation of NF-kappaB. J. Neurosci. 19:1754–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, C., J.Y. Park, J. Lee, J.H. Lim, E.C. Shin, Y.S. Ahn, C.H. Kim, S.J. Kim, J.D. Kim, I.S. Choi, and I.H. Choi. 1999. Fas ligand and Fas are expressed constitutively in human astrocytes and the expression increases with IL-1, IL-6, TNF-alpha, or IFN-gamma. J. Immunol. 162:1889–1895. [PubMed] [Google Scholar]
- Cowley, S., H. Paterson, P. Kemp, and C.J. Marshall. 1994. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 77:841–852. [DOI] [PubMed] [Google Scholar]
- Davies, A.M. 2000. Neurotrophins: neurotrophic modulation of neurite growth. Curr. Biol. 10:R198–R200. [DOI] [PubMed] [Google Scholar]
- Davies, A.M., K.F. Lee, and R. Jaenisch. 1993. p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron. 11:565–574. [DOI] [PubMed] [Google Scholar]
- Dechant, G., and Y.A. Barde. 2002. The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat. Neurosci. 5:1131–1136. [DOI] [PubMed] [Google Scholar]
- Desbarats, J., R.B. Birge, M. Mimouni-Rongy, D.E. Weinstein, J.S. Palerme, and M.K. Newell. 2003. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat. Cell Biol. 5:118–125. [DOI] [PubMed] [Google Scholar]
- Felderhoff-Mueser, U., D.L. Taylor, K. Greenwood, M. Kozma, D. Stibenz, U.C. Joashi, A.D. Edwards, and H. Mehmet. 2000. Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic-ischemic injury to the developing rat brain. Brain Pathol. 10:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehr, E.D., X. Lin, A. O'Mahony, R. Geleziunas, R.A. Bradshaw, and W.C. Greene. 2000. NF-kappa B signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J. Neurosci. 20:7556–7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanoue, M., G. Middleton, S. Wyatt, E. Jaffray, R.T. Hay, and A.M. Davies. 1999. p75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol. Cell. Neurosci. 14:28–40. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane, editors. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 726 pp.
- Marshall, C.J. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 80:179–185. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba, A., I. Herr, I. Jeremias, M. Hahne, R. Brandt, J. Vogel, J. Schenkel, T. Herdegen, and K.M. Debatin. 1999. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J. Neurosci. 19:3809–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Zanca, D., R. Oskam, G. Mitra, T. Copeland, and M. Barbacid. 1989. Molecular and biochemical characterization of the human trk proto-oncogene. Mol. Cell. Biol. 9:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita, K., Y. Wu, J. Qiu, L. Lang-Lazdunski, L. Hirt, C. Waeber, B.T. Hyman, J. Yuan, and M.A. Moskowitz. 2000. Fas receptor and neuronal cell death after spinal cord ischemia. J. Neurosci. 20:6879–6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley, W.C., A. Schenker, and E.M. Shooter. 1976. Characterization and isolation of proteolitically modified nerve growth factor. Biochemistry. 15:5543–5552. [DOI] [PubMed] [Google Scholar]
- Park, C., K. Sakamaki, O. Tachibana, T. Yamashima, J. Yamashita, and S. Yonehara. 1998. Expression of fas antigen in the normal mouse brain. Biochem. Biophys. Res. Commun. 252:623–628. [DOI] [PubMed] [Google Scholar]
- Peter, M.E., and P.H. Krammer. 2003. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10:26–35. [DOI] [PubMed] [Google Scholar]
- Putcha, G.V., C.A. Harris, K.L. Moulder, R.M. Easton, C.B. Thompson, and E.M. Johnson Jr. 2002. Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J. Cell Biol. 157:441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul, C., C.E. Henderson, and B. Pettmann. 1999. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J. Cell Biol. 147:1049–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul, C., A.G. Estevez, H. Nishimune, D.W. Cleveland, O. deLapeyriere, C.E. Henderson, G. Haase, and B. Pettmann. 2002. Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron. 35:1067–1083. [DOI] [PubMed] [Google Scholar]
- Rodriguez, M.S., J. Wright, J. Thompson, D. Thomas, F. Baleux, J.L. Virelizier, R.T. Hay, and F. Arenzana-Seisdedos. 1996. Identification of lysine residues required for signal-induced ubiquitination and degradation of I kappa B-alpha in vivo. Oncogene. 12:2425–2435. [PubMed] [Google Scholar]
- Rothstein, T.L., X. Zhong, B.R. Schram, R.S. Negm, T.J. Donohoe, D.S. Cabral, L.C. Foote, and T.J. Schneider. 2000. Receptor-specific regulation of B-cell susceptibility to Fas-mediated apoptosis and a novel Fas apoptosis inhibitory molecule. Immunol. Rev. 176:116–133. [DOI] [PubMed] [Google Scholar]
- Sanz, L., M.T. Diaz-Meco, H. Nakano, and J. Moscat. 2000. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 19:1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T.J., G.M. Fischer, T.J. Donohoe, T.P. Colarusso, and T.L. Rothstein. 1999. A novel gene coding for a Fas apoptosis inhibitory molecule (FAIM) isolated from inducibly Fas-resistant B lymphocytes. J. Exp. Med. 189:949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanaus, K.S., R. Schlapbach, and A. Fontana. 1998. TNF-alpha and IFN-gamma render microglia sensitive to Fas ligand-induced apoptosis by induction of Fas expression and down-regulation of Bcl-2 and Bcl-xL. Eur. J. Immunol. 28:4398–4408. [DOI] [PubMed] [Google Scholar]
- Traverse, S., N. Gomez, H. Paterson, C. Marshall, and P. Cohen. 1992. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem. J. 288:351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolini, G., C. Raoul, A. Ferri, C. Haenggeli, Y. Yamamoto, D. Salaün, C.E. Henderson, A.C. Kato, B. Pettmann, and A.O. Hueber. 2003. Fas/tumor necrosis factor receptor death signaling is required for axotomy-induced death of motoneurons in vivo. J. Neurosci. 23:8526–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillancourt, R.R., L.E. Heasley, J. Zamarripa, B. Storey, M. Valius, A. Kazlauskas, and G.L. Johnson. 1995. Mitogen-activated protein kinase activation is insufficient for growth factor receptor-mediated PC12 cell differentiation. Mol. Cell. Biol. 15:3644–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlleben, G., S.M. Ibrahim, J. Schmidt, K.V. Toyka, H.P. Hartung, and R. Gold. 2000. Regulation of Fas and FasL expression on rat Schwann cells. Glia. 30:373–381. [PubMed] [Google Scholar]
- Wood, J.N. 1995. Regulation of NF-kappa B activity in rat dorsal root ganglia and PC12 cells by tumour necrosis factor and nerve growth factor. Neurosci. Lett. 192:41–44. [DOI] [PubMed] [Google Scholar]
- Wooten, M.W., M.L. Seibenhener, V. Mamidipudi, M.T. Diaz-Meco, P.A. Barker, and J. Moscat. 2001. The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J. Biol. Chem. 276:7709–7712. [DOI] [PubMed] [Google Scholar]
- Yoon, S.O., P. Casaccia-Bonnefil, B. Carter, and M.V. Chao. 1998. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J. Neurosci. 18:3273–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, X., T.J. Schneider, D.S. Cabral, T.J. Donohoe, and T.L. Rothstein. 2001. An alternatively spliced long form of Fas apoptosis inhibitory molecule (FAIM) with tissue-specific expression in the brain. Mol. Immunol. 38:65–72. [DOI] [PubMed] [Google Scholar]