

Abstract
During chemotaxis, receptors and heterotrimeric G-protein subunits are distributed and activated almost uniformly along the cell membrane, whereas PI(3,4,5)P3, the product of phosphatidylinositol 3-kinase (PI3K), accumulates locally at the leading edge. The key intermediate event that creates this strong PI(3,4,5)P3 asymmetry remains unclear. Here, we show that Ras is rapidly and transiently activated in response to chemoattractant stimulation and regulates PI3K activity. Ras activation occurs at the leading edge of chemotaxing cells, and this local activation is independent of the F-actin cytoskeleton, whereas PI3K localization is dependent on F-actin polymerization. Inhibition of Ras results in severe defects in directional movement, indicating that Ras is an upstream component of the cell's compass. These results support a mechanism by which localized Ras activation mediates leading edge formation through activation of basal PI3K present on the plasma membrane and other Ras effectors required for chemotaxis. A feedback loop, mediated through localized F-actin polymerization, recruits cytosolic PI3K to the leading edge to amplify the signal.
Introduction
Eukaryotic cells of diverse origin exhibit common mechanisms for sensing and migrating to a chemoattractant source. Cells can respond to chemoattractant gradients as shallow as a 2–5% difference between the anterior and posterior of the cell by converting this shallow external gradient into a steep intracellular gradient of signaling components (Parent and Devreotes, 1999; Chung et al., 2001a). Of major importance is understanding the mechanisms by which cells amplify this shallow extracellular gradient into a steep intracellular gradient that then mediates directional movement and the formation of a stable cellular polarity.
Part of the regulatory pathway that controls directional sensing is the establishment of a steep intracellular gradient at the leading edge of PI(3,4)P2 and PI(3,4,5)P3, the products of Class I phosphatidylinositol 3-kinases (PI3Ks), important mediators of chemotaxis (Iijima et al., 2002; Merlot and Firtel, 2003). Chemoattractant stimulation elicits a rapid, transient translocation to the activated plasma membrane of a subfamily of PH domain-containing proteins that preferentially bind PI(3,4)P2/PI(3,4,5)P3 (Parent et al., 1998; Meili et al., 1999; Servant et al., 2000). These proteins preferentially localize to the leading edge of chemotaxing cells; their spatially restricted cortical localization can be induced by placing a micropipette emitting a chemoattractant near a responsive cell.
These observations lead to a model in which a shallow chemoattractant gradient becomes highly amplified at the leading edge, in part, through a preferential activation of PI3K and localization of these PH domain-containing proteins. Studies in Dictyostelium discoideum cells and in mammalian leukocytes indicate that chemoattractant receptors and heterotrimeric G-protein subunits remain distributed fairly uniformly along the cell membrane (Jin et al., 2000; Iijima et al., 2002; Merlot and Firtel, 2003). Whereas Gβγ plays an essential role in mediating the biochemical responses resulting in leading edge formation, the absence of a highly polarized gradient of free Gβγ implies it is not the signal responsible for intracellular amplification of the chemoattractant gradient and localized activation of PI3K. Furthermore, activation of the G-protein is persistent, whereas PI3K activation is transient (Janetopoulos et al., 2001; Funamoto et al., 2002; Huang et al., 2003), indicating a different component regulates the localized turning on and off of PI3K. Thus, this initial amplification step lies downstream of G-protein activation and upstream of the generation of the 3-phosphoinositides (Merlot and Firtel, 2003).
The production and regulation of PI(3,4,5)P3 requires PI3Ks and the 3-phosphoinositide phosphatase PTEN. The first insight into the mechanisms of locally accumulating PI(3,4,5)P3 came from studies of the subcellular distribution of PI3Ks and PTEN in Dictyostelium (Funamoto et al., 2002; Iijima and Devreotes, 2002). Dictyostelium PI3K transiently translocates to the plasma membrane in response to chemoattractant stimulation and to the leading edge in chemotaxing cells, whereas PTEN exhibits a reciprocal pattern of localization. Loss of PTEN causes extended and nonspatially restricted PI3K activity, indicating that PTEN is required for temporally and spatially restricting PI(3,4,5)P3 at the leading edge. Cells expressing a membrane-tagged form of PI3K, which is uniformly distributed along the plasma membrane, form pseudopodia randomly along the plasma membrane when placed in a chemoattractant gradient, demonstrating that PI3K activation can directly lead to pseudopod formation. Subsequent studies in neutrophils have identified a feedback loop between PI(3,4,5)P3 and F-actin accumulation and have confirmed that the posterior localization of PTEN is conserved in some amoeboid chemotaxing mammalian cell types (Wang et al., 2002; Weiner et al., 2002; Li et al., 2003; Xu et al., 2003). These regulatory events are at least partially responsible for eliciting the asymmetry in signaling that results in directional sensing.
One of the important challenges in studying chemotaxis is to identify the upstream components that recruit and activate PI3K at the leading edge. Intensive study provides compelling evidence for a critical role of Ras in cell growth, differentiation, and survival (Hancock, 2003), yet the roles of Ras in chemotaxis remain to be defined. Ras has been proposed to be an activator of Class I mammalian and Dictyostelium PI3Ks (Pacold et al., 2000; Funamoto et al., 2002; Suire et al., 2002). Ras also interacts with other families of proteins required for chemotaxis and cell polarity (Lee et al., 1999), suggesting a broader regulatory role for Ras beyond PI3K. Furthermore, null mutations of specific Ras proteins in Dictyostelium have been implicated in cell movement and chemotaxis (Tuxworth et al., 1997; Lim et al., 2001).
Here, we examine the regulatory mechanisms by which the amplified gradient of PI(3,4,5P)3/PI(3,4)P2 is established at the leading edge. We demonstrate that Ras is an upstream component of the cell's chemotaxis compass; abolition of Ras function causes a loss of directional cell movement. We further demonstrate that the translocation of PI3K to the plasma membrane requires the F-actin cytoskeleton and is not part of the “initial” regulatory response. Our data suggest a model in which leading edge formation is controlled through an initial local response that includes localized Ras activation, which stimulates a small amount of preexisting, membrane-associated PI3K, causing F-actin polymerization. Positive feedback loops, one of which involves further recruitment of PI3K from the cytosol to the leading edge, reinforces this pathway to stabilize the nascent leading edge.
Results
RasG regulates PI3K activity and is uniformly distributed along the plasma membrane
Of the five Dictyostelium Ras proteins, RasB, RasD, and RasG are most closely related to mammalian H-Ras and K-Ras and have a conserved effector domain (Lim et al., 2002). RasG and RasD have been suggested to activate PI3K, as RasG- and RasD-GTP interact with the RBDs of PI3K1 and PI3K2 (Funamoto et al., 2002), and cell movement defects of rasG null cells partially overlap with those of cells lacking PI3K1 and PI3K2 (Tuxworth et al., 1997; Funamoto et al., 2002; Lim et al., 2002). Other studies (Lim et al., 2001) show that rasC null cells exhibit chemotaxis defects and reduced Akt/PKB activation, but our studies have been unable to demonstrate an interaction of RasC-GTP with the RBDs of PI3K1 and PI3K2, suggesting that PI3K may not be a direct RasC effector (Lee et al., 1999).
To test whether or not RasG is involved in PI3K activation, we measured the activation of the PI3K effector Akt/PKB in rasG null cells in response to chemoattractant stimulation. Fig. 1 A shows that chemoattractant-induced Akt/PKB activation was decreased in rasG null cells, and this effect was complemented by expressing FLAG- or GFP-tagged wild-type RasG cDNA from a constitutive actin promoter in rasG null cells (Fig. 1 B and not depicted). FLAG- and GFP-RasG also complemented the rasG null cell cytokinesis defect (unpublished data). GFP-RasG displayed uniform localization along the plasma membrane and cytosol in chemotaxing cells, unstimulated cells, and cells that were stimulated globally with a uniform, rapidly applied, saturating level of chemoattractant (Fig. 1 C). As rasG null cells only show a partial reduction in Akt/PKB activation, we expect other Ras proteins are involved in PI3K activation.
Figure 1.
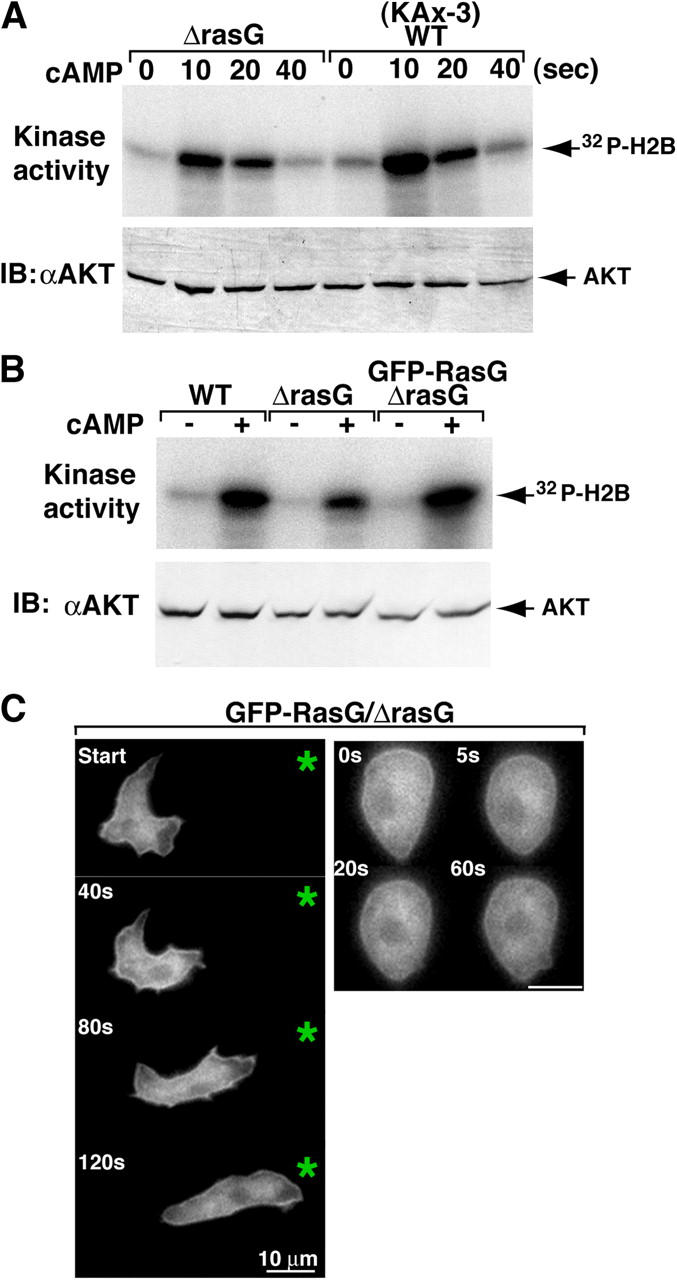
Ras regulates PI3K signaling and distribution along the membrane. (A and B) Activation of Akt/PKB is shown. Aggregation-stage cells (see Materials and methods) were treated with 10 μM cAMP for the indicated time (A) or at 10 s (B) and then lysed, Akt was immunoprecipitated with anti-Akt antibody, and Akt activity was assayed (Meili et al., 1999). Akt protein levels were determined in each sample by Western blot analysis (bottom panels). (C) Fluorescent images of GFP-RasG/rasG null cells exposed to a chemoattractant gradient (left) or to uniform chemoattractant stimulation (right). An asterisk indicates the position of the micropipette. The numbers in the top left corners represent the time after initiation when the image was captured. The data are representative of eight separate experiments.
Rapid and transient activation of Ras in response to cAMP
The partial requirement of RasG for PI3K activation suggests that RasG and possibly other Dictyostelium Ras proteins are activated in response to chemoattractant stimulation. To examine this suggestion, we used a GST-RBD of Raf1 in a pull-down assay that has been used to determine Ras activation in mammalian cells (Taylor et al., 2001). The rationale for using the RBD of Raf1 was that Dictyostelium RasG (and RasB and RasD) and mammalian H-Ras and K-Ras have identical effector domains (Lim et al., 2002). The RBD of Raf1 bound strongly to Dictyostelium constitutively activated (Q61L) RasG, RasB, and RasD but not to their dominant negative forms (S17N) in yeast two-hybrid and in vitro binding assays (unpublished data). Interestingly, RasGQ61L bound more strongly to the Raf1 RBD than to either the PI3K1 or PI3K2 RBD (unpublished data). Fig. 2 A illustrates that the RasG activation level is rapidly stimulated in response to cAMP, peaking at 5 s, after which the level of RasG-GTP rapidly decreases. The same results were obtained by using rasG null cells expressing GFP-tagged RasG (unpublished data). The kinetics of activation and subsequent adaptation are consistent with those of cAMP-stimulated PI3K activity (Huang et al., 2003). While we were preparing our manuscript for submission, an independent report also demonstrated chemoattractant-mediated RasG activation using the RBD of Byr2 (Kae et al., 2004; see Discussion).
Figure 2.
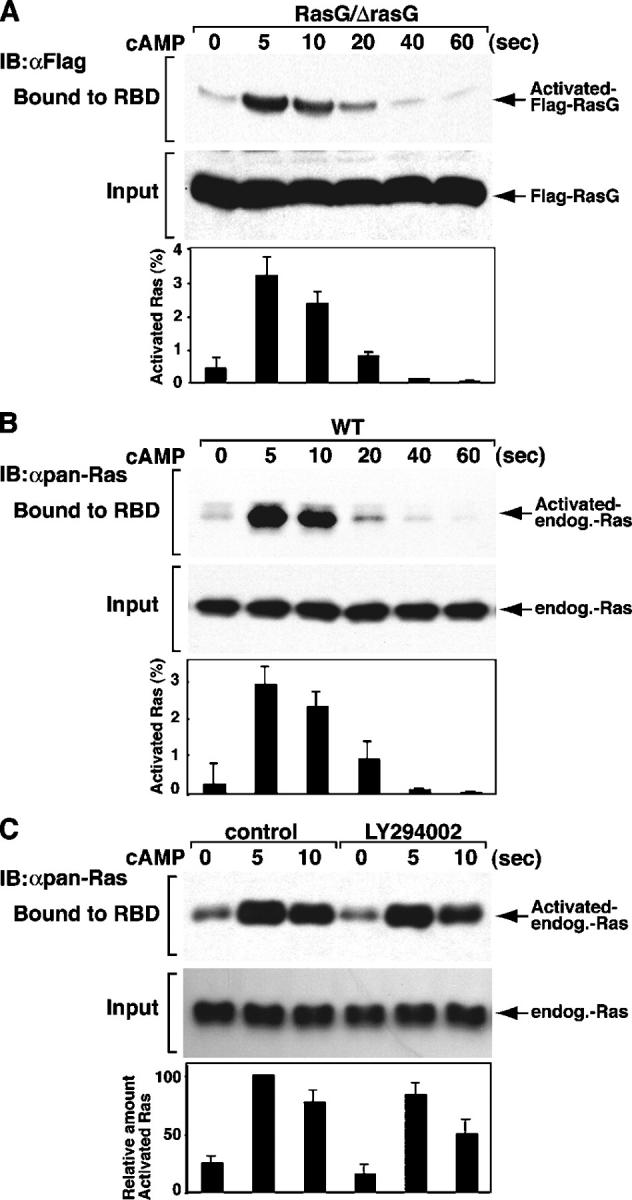
Activation of Dictyostelium Ras. The FLAG-RasG or endogenous Ras activation level in response to cAMP was assayed by a GST-RBD pull-down assay (see Materials and methods). Cells were stimulated with cAMP for the indicated duration, and the amount of Ras protein bound to GST-RBD was determined by Western blotting with the indicated antibody. The activated Ras was quantified by densitometry and normalized with total Ras. Cells were treated with 50 μM LY294002 or DMSO as a control for 20 min before the addition of cAMP (C). Similar results were observed over at least three independent experiments.
We also measured the activation level of endogenous Ras proteins (those whose GTP-bound state binds to the Raf1 RBD) by using an anti–pan-Ras antibody that should recognize some or all Dictyostelium Ras proteins. Fig. 2 B indicates that endogenous Ras proteins are activated by cAMP stimulation with timing kinetics similar to those of RasG. We expect our assay does not recognize RasC, as it binds poorly to the Raf1 RBD (Kae et al., 2004). The activation level of endogenous Ras proteins is only partially reduced in rasG null cells (unpublished data), indicating that Ras proteins in addition to RasG are activated. These data demonstrate that multiple endogenous Ras proteins are rapidly and transiently activated in response to chemoattractant stimulation and potentially provide an explanation of why rasG null cells may still exhibit a moderate level of chemoattractant-mediated Akt/PKB activation.
Several papers concluded that Ras is activated downstream of PI3K signaling (Chan et al., 2002; Yart et al., 2002). We used LY294002, a pharmacological PI3K inhibitor, to examine this possibility. Fig. 2 C illustrates that Ras proteins can be activated in the presence of LY294002. In addition, we observed Ras activation in pi3k1/2 null cells (unpublished data). These results indicate that Ras can be initially activated without PI3K signaling. However, we cannot eliminate the existence of a feedback loop between PI3K downstream signaling and Ras activation, as the Ras activation level is decreased by LY294002 treatment.
Ras activation in living cells
To determine the spatial-temporal kinetics of Ras activation in living cells, we used the GFP-tagged RBD of Raf1, which binds to and thus should monitor Ras-GTP levels and its localization in cells (Chiu et al., 2002; Hancock, 2003). We investigated the GFP-RBD localization in randomly moving vegetative cells. As illustrated in Fig. 3 A, resting vegetative cells display an even cytoplasmic fluorescent signal with labeling of regions of membrane ruffling and circular, “crown-like” structures, which may be the precursors of macropinosomes. These are the regions in which F-actin and PI(3,4,5)P3 accumulate and to which PI3K specifically translocates (Parent et al., 1998; Funamoto et al., 2001, 2002). In aggregation-competent cells, which are differentiated by cAMP pulsing and competent to chemotax to cAMP, PH domain-containing proteins (e.g., CRAC, Akt/PKB, and PhdA) and PI3K rapidly translocate to the plasma membrane in response to global stimulation with a chemoattractant (Parent et al., 1998; Meili et al., 1999; Funamoto et al., 2002). We measured the kinetics of PH domain and PI3K cortical localizations using a faster and more sensitive camera than used previously, allowing us to see differences in the initial steps of the two responses (Funamoto et al., 2002). As shown in Fig. 3 C, the translocation of PI3K occurs faster than that of the PH domain, whereas the loss of PI3K from the plasma membrane occurs more slowly than that of the PH domain. For this work, we use the NH2-terminal domain of PI3K1 (N-PI3K1) that does not contain the RBD and is necessary and sufficient for PI3K localization. This reporter exhibits the same subcellular localization and kinetics of localization as GFP-PI3K (Funamoto et al., 2002). Interestingly, GFP-RBD also rapidly and transiently localizes to the plasma membrane in response to global stimulation (Fig. 3 B and Video 1, available at http:www.jcb.org/cgi/content/full/jcb.200406177/DC1). The signal intensity of GFP-RBD was significantly weaker than that of the PH domain-containing proteins, possibly because the potential binding sites for PH domains (PI(3,4,5)P3/PI(3,4)P2) are amplified relative to Ras-GTP by the catalytic activity of PI3K, thus providing an amplified signal, and/or because of our inability to obtain strains expressing high levels of RBD-GFP compared with the levels of expression of GFP fusions of PH domains and N-PI3K. The weaker signal could also be caused by the affinity of the Raf1 RBD for Ras-GTP in an in vivo cellular context.
Figure 3.
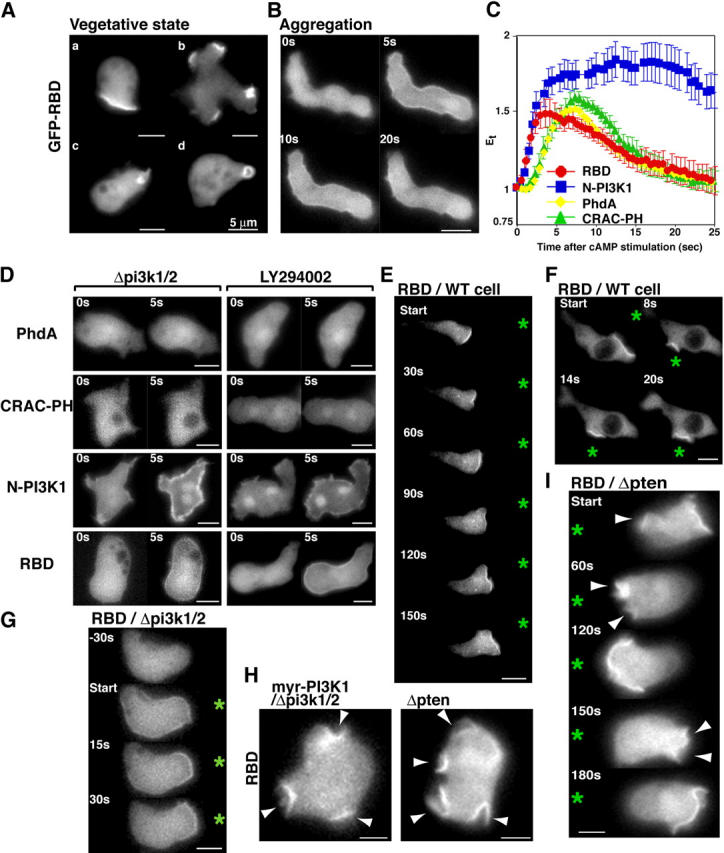
Spatial-temporal activation of Ras during chemotaxis. (A and B) The localization of GFP-RBD in wild-type vegetative cells (A) or aggregation-competent cells (B) was imaged. Translocation of GFP-RBD was imaged after stimulation with cAMP as described previously (Funamoto et al., 2001). (C) Translocation kinetics of GFP-tagged PhdA, CRAC-PH, N-PI3K1, and RBD in wild-type cells were obtained from time-lapse recordings. The graphs represent an average of data of movies taken from at least three separate experiments. The fluorescence intensity of membrane-localized GFP fusion protein was quantitated as Et as defined in Materials and methods. (D) Translocation of indicated GFP proteins in pi3k1/2 null cells or wild-type cells treated with 50 μM LY294002 for 20 min before cAMP stimulation. (E–I) Fluorescent images of GFP-RBD expressing wild-type cells (E and F), pi3k1/2 null cells (G), or pten null and myr-PI3K expressing cells (I) chemotaxing in a gradient of chemoattractant. An asterisk indicates the position of the micropipette. Fluorescent images of GFP-RBD and myr-PI3K1–expressing pi3k1/2 null cells (H, left) or GFP-RBD-expressing pten null cells (H, right) are shown. The sites producing multiple pseudopodia are marked with arrowheads.
Quantitation of the kinetics of GFP-RBD translocation indicates Ras is activated with levels peaking at ∼3–6 s, and this initial Ras activation occurs as early as N-PI3K1 translocation (Fig. 3 C). Thus, Ras activation (determined by GFP-RBD membrane localization) is contemporaneous with or possibly precedes that of N-PI3K1. We observe a delay in the initiation of PH domain-containing PhdA and CRAC translocation, the expected result if Ras-driven PI3K activation and PI(3,4,5)P3 production are required for PH domain localization (Fig. 3 C). The kinetics of PH domain localization correspond to the kinetics of Ras-GTP, suggesting Ras-GTP levels regulate the PI3K response. GFP-RBD and N-PI3K1 translocation occurred in both pi3k1/2 null cells and LY294002-treated cells, whereas PhdA and CRAC did not detectably translocate in either pi3k1/2 null cells or LY294002-treated cells (Fig. 3 D). These data suggest that initial Ras activation as well as PI3K translocation occur upstream of PI3K signaling, and Ras is at least one of the key intermediate molecules between the chemoattractant receptor and PI3K activation.
Ras is activated at the leading edge of chemotaxing cells
To study the spatial localization of signaling events in chemotaxing cells, we placed GFP-RBD–expressing cells in a chemoattractant gradient created by micropipette filled with chemoattractant. In chemotaxing cells, GFP-RBD exclusively localized to the leading edge (Fig. 3, E and F; and Video 2, available at http:www.jcb.org/cgi/content/full/jcb.200406177/DC1). To assess the qualitative dynamics and specificity of the GFP-RBD localization, we changed the pipette position to the opposite side of the cell (Fig. 3 F). The GFP-RBD signal was rapidly lost from the initial site and concomitantly reaccumulated at the new site on the membrane closest to the micropipette. GFP-RBD thus accumulated at the region of the future front of the cell, which then produced a pseudopod (Fig. 3 F). As shown in Fig. 3 G, this localized Ras activation also occurred in pi3k1/2 null cells, suggesting localized Ras activation does not require PI3K activity, as we had previously observed that cells expressing myristoyl (myr)-tagged PI3K1 produce multiple pseudopodia (Funamoto et al., 2002). In these cells, GFP-RBD accumulates at these sites of new functional pseudopodia formation (Fig. 3 H), providing a correlation between activated Ras and pseudopod formation. In addition, GFP-RBD localizes to the pseudopodia in randomly migrating wild-type cells (unpublished data). As pten null and PTEN hypomorphic cells exhibit a similar random localization of PH domain reporters (Funamoto et al., 2002; Iijima and Devreotes, 2002), we also examined GFP-RBD localization in pten null cells (Iijima and Devreotes, 2002). In pten null cells, GFP-RBD localized to the membrane in response to a global stimulation and accumulated at the multiple sites of new pseudopodia during random migration (Fig. 3 H and Video 3, available at http:www.jcb.org/cgi/content/full/jcb.200406177/DC1). Occasionally, membrane localization of GFP-RBD was observed at the rear of a cell, in which case the cell started to move “backward” relative to the chemoattractant gradient (Fig. 3 I and Video 3). From this series of observations, we conclude that activated Ras localizes to the leading edge and to membrane domains that will form a leading edge. We previously demonstrated that PI3K activation, as determined by Akt activation, requires a functional Ras binding domain, which is consistent with a requirement of Ras for mammalian PI3K activation (Funamoto et al., 2002). As myr-PI3K–expressing cells and pten null cells have high basal PI(3,4,5)P3 (Funamoto et al., 2001, 2002; Iijima and Devreotes, 2002), these observations also suggest the possible existence of PI(3,4,5)P3-mediated feedback Ras activation.
PI3K localization and Ras activation are differentially regulated
In response to chemoattractant stimulation, Dictyostelium and mammalian cell types exhibit a rapid burst of F-actin polymerization, which is followed by a rapid decrease to a near-basal level and a subsequent slower rise to a peak level ∼1/3 as high as the initial peak (Hall et al., 1988). The initial F-actin peak is similar in timing to Ras activation and PI3K translocation. Therefore, we examined if F-actin polymerization was needed to locally activate leading-edge signaling events using the F-actin polymerization inhibitor latrunculin A (LatA). After LatA treatment, cells became progressively rounder and stopped moving (Parent et al., 1998). Consistent with previous findings, under these conditions, there was a reduced amount of F-actin associated with the Triton X-100–insoluble cortical fraction and chemoattractant stimulation did not induce F-actin polymerization (Fig. 4 D). As in previous work (Parent et al., 1998), a global stimulation of LatA-treated cells resulted in a rapid cortical localization of the PH-domain-containing protein (Fig. 4 A).
Figure 4.
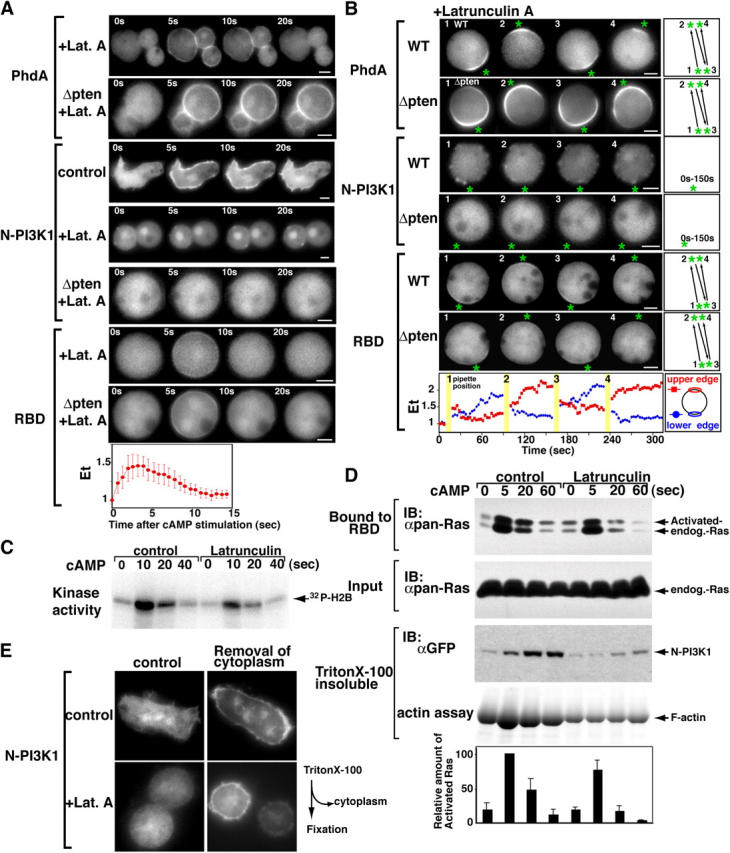
Differential regulation between PI3K translocation and Ras activation. (A and B) Effect of LatA on the translocation of GFP-tagged PhdA-, N-PI3K1–, and RBD-expressing wild-type cells and pten null cells. Asterisk indicates the position of the micropipette (B). These images were captured ∼30 s after changing the micropipette position. The behaviors were consistently observed over five independent sessions. Translocation kinetics of RBD were obtained from a time-lapse recording of GFP-RBD–expressing wild-type cells. Fluorescence intensities of the upper and lower plasma membranes were quantitated as Et (see Materials and methods). (C) Effect of LatA on Akt/PKB activation. Akt/PKB assays were performed as described in Fig. 1. Data are representative of at least three independent experiments. (D) N-PI3K1 is recruited into the Triton X-100–resistant cytoskeleton in response to chemoattractant stimulation. Cytoskeletal fractions (see Materials and methods) were subjected to SDS-PAGE. Actin and N-PI3K1 recruitment were assessed with Coomassie stain or anti-GFP antibody, respectively. Ras activation was assayed using an aliquoted lysate. Graph shows an average of the results that were obtained from four independent experiments. (E) Fluorescent images of GFP-tagged N-PI3K1–expressing wild-type cells. Cells were treated with (right) or without (left) 0.01% Triton X-100 before fixation by 3.7% formaldehyde.
We investigated the effect of the actin cytoskeleton on N-PI3K1 localization. We expected that, like PH domain-containing proteins, N-PI3K1 would translocate to the membrane in response to chemoattractant stimulation in the presence of LatA. However, after treatment with LatA, we observed a very low, basal level of cortically associated N-PI3K1, and this amount did not increase in response to chemoattractant stimulation (Fig. 4 A). In the presence of lower levels of LatA, which did not completely block F-actin polymerization, we observed a weak N-PI3K1 cortical localization in response to chemoattractant stimulation (unpublished data). When a micropipette containing a chemoattractant was placed near the cells, N-PI3K1 showed no directed cortical accumulation (Fig. 4 B). We have done parallel experiments with GFP fusions of the complete PI3K1 and PI3K2 and with the NH2-terminal localization domain of PI3K2 (Funamoto et al., 2002), and the chemoattractant-mediated translocation of these reporters is also blocked by LatA (unpublished data). This finding confirms that PI(3,4,5)P3 can accumulate at the side of the cell closest to the chemoattractant source without de novo asymmetric PI3K localization. These data suggest the presence of a basal level of PI3K on the plasma membrane in unstimulated cells. Unstimulated cells do not show obvious PI3K localized to the plasma membrane (Fig. 4 E); however, such a cortical PI3K pool may be occluded by a large cytoplasmic pool. To examine this possibility, we removed the cytoplasmic fraction by mild Triton X-100 treatment. Fig. 4 E shows the presence of some PI3K localized to the plasma membrane before chemoattractant stimulation. We suggest cells have a low, basal level of preexisting PI3K at the plasma membrane that is activated upon chemoattractant stimulation.
To verify the involvement of the actin cytoskeleton in PI3K distribution, we extracted the cytoskeletal fraction. In control cells (not treated with LatA), N-PI3K1 became associated with the cytoskeletal fraction after chemoattractant stimulation, whereas LatA strongly inhibited N-PI3K1 cytoskeleton association. Under the same conditions, Ras was activated by cAMP in the presence of LatA, although the level was decreased compared with that in control cells (Fig. 4 D). Consistent with the results from Fig. 4 (A and B), our observations suggest that chemoattractant-mediated N-PI3K1 localization, but not the initial Ras activation, requires F-actin polymerization. Our findings suggest that localized PI(3,4,5)P3 production is cooperatively regulated by localized Ras activation and F-actin–mediated recruitment of PI3K.
To elucidate the mechanisms of local PI(3,4,5)P3 production in the presence of LatA, we examined where Ras is activated in these cells. In response to global chemoattractant stimulation, GFP-RBD translocated uniformly to the whole plasma membrane. In response to a directional signal, GFP-RBD accumulated on the site of the plasma membrane closest to the micropipette (Fig. 4 B and Video 4, available at http:www.jcb.org/cgi/content/full/jcb.200406177/DC1). Upon changing the position of the micropipette, GFP-RBD redistributed toward the new position of the micropipette (Fig. 4 B). The change in membrane fluorescence with the change in the position of the micropipette is plotted in Fig. 4 B. These results demonstrate that eukaryotic cells can spatially sense a directional chemoattractant signal at a step downstream of G protein activation and upstream of Ras activation without an actin cytoskeleton. However, PI3K localization, and presumably the amplification of the PI3K signal, requires F-actin polymerization. Consistent with this hypothesis, we observe a reduction in Akt/PKB activation in LatA-treated cells (Fig. 4 C). Thus, in the absence of an F-actin cytoskeleton, chemoattractants induce a basal PI(3,4,5)P3 response, which is amplified by subsequent F-actin–mediated PI3K accumulation at the plasma membrane at the site closest to the chemoattractant source.
It was recently reported that PTEN accumulates at the posterior even in the presence of LatA, suggesting the possibility that PTEN regulates local PI(3,4,5)P3 levels in the presence of LatA (Iijima et al., 2004). To examine this hypothesis further, we tested whether or not GFP-N-PI3K1 localized to the front of pten null cells. Consistent with our findings in wild-type cells, we found that GFP-N-PI3K1 in LatA-treated pten null cells did not translocate upon chemoattractant stimulation (Fig. 4, A and B). We observed that in pten null cells in the presence of LatA, GFP-RBD, GFP-CRAC, and GFP-PhdA (Fig. 4 B and not depicted) accumulated toward the micropipette. When the position of the micropipette was moved, the reporters rapidly delocalized from the original site and immediately reaccumulated on the membrane at a site closest to the new position of the micropipette (Fig. 4 B). The only difference in the spatial localization of PhdA between wild-type and pten null cells was that the size of the arc was larger in pten null cells. This finding is consistent with PTEN localizing to the lateral sides and posterior of cells and being required to help spatially restrict the PI3K response (Funamoto et al., 2002; Iijima and Devreotes, 2002; Iijima et al., 2004). These studies on PI3K indicate that the presence or absence of PTEN does not affect PI3K localization and, as in wild-type cells, this localization is dependent on localized F-actin polymerization. These data also suggest that the localization of PTEN is not essential for the regulation of local PI(3,4,5)P3 levels in the presence of LatA; directional Ras activation occurs in the absence of F-actin, directional PI3K localization, and PTEN.
Ras, PI3K, and F-actin form a positive feedback loop
Although initial Ras activation requires neither PI3K activity nor F-actin polymerization, the Ras activation level was reduced in the presence of LY294002 or LatA (Figs. 3 and 4). Furthermore, Ras was spontaneously activated at multiple sites in randomly moving (no chemoattractant) pten null cells. Those observations suggest that Ras itself may be activated, in part, by events involving PI(3,4,5)P3 and F-actin polymerization. To examine this hypothesis, we treated randomly moving pten null cells with LY294002. After the drug treatment, both spontaneous PhdA accumulation at the membrane and the formation of multiple pseudopodia were completely abolished (Fig. 5 A). The result is consistent with high PI(3,4,5)P3 inducing F-actin polymerization in pten null cells PI3K (Iijima et al., 2004). Strikingly, spontaneous PI3K localization and Ras activation at multiple pseudopodia also completely disappeared after LY294002 treatment. The results suggest that, under some conditions such as “stochastic activation” during random movement, PI3K may regulate Ras activation and PI3K localization through PI(3,4,5)P3-mediated signaling. We further examined the possible involvement of F-actin polymerization in activating Ras and PI3K in randomly moving pten null cells. The “spontaneous” (no external stimulation) Ras activation, PIP3 accumulation, and PI3K localization gradually decreased in pten null cells after LatA addition. The loss of these responses coincided with an inability to make new F-actin, as judged by a complete smoothness of the cell surface. After removal of the LatA, cells regained the spontaneous Ras activation, PI(3,4,5)P3 accumulation, and PI3K localization at sites of F-actin accumulation (protrusions; Fig. 5 A). The findings suggest that F-actin is important for observable autonomous Ras and PI3K activation. As Ras is required for PI3K activation, we suggest that Ras and PI3K activation are tightly connected through a positive feedback loop.
Figure 5.

Feedback loop–mediated Ras/PI3K activation. (A) Fluorescent images of indicated GFP-protein expressing pten null cells before or 5 min after 50-μM LY294002 treatment or 25 min after 5 μM LatA treatment, and 20 min after the removal of LatA. (B) Fluorescent images of GFP-PhdA– and RasGQ61L-expressing rasG null cells.
As PI(3,4,5)P3 could induce F-actin–dependent Ras activation, we examined if constitutively activated Ras could induce persistent PI(3,4,5)P3 production. We coexpressed constitutively active RasGQ61L and PhdA in rasG null cells. After chemoattractant stimulation, we observe a rapid, transient, global accumulation of PI(3,4,5)P3, followed by persistent PI(3,4,5)P3 production at a localized site on the membrane (Fig. 5 B). When cells are pretreated with LatA, this persistent PI(3,4,5)P3 production is blocked (Fig. 5 B). These data are consistent with those in Fig. 5 A and suggest that F-actin and/or F-actin–mediated PI3K recruitment is required for extended PI3K activation. These findings are consistent with a model in which robust Ras/PI3K activation, as well as pseudopod formation, depends on a feedback loop comprising Ras, PI3K, and F-actin, although the initial Ras activation can occur without PI(3,4,5)P3 and F-actin. In support of this conclusion, we note that the level of Ras activation is reduced in the presence of LatA (Fig. 4, A and D).
Ras regulates cell polarity and directionality of the chemotaxing cells
Our previous findings that a functional RBD is required for Dictyostelium PI3K activation (Funamoto et al., 2002) and our observations here that Ras is activated at the leading edge suggest that RasG and Ras proteins of related function may be required for directional sensing. Further, several studies have implicated Ras in chemotaxis: rasG null cells have a slight loss of motility (Tuxworth et al., 1997; Lim et al., 2002), and a null mutation of the RasG-GTP binding protein RIP3 exhibits chemotaxis defects (Lee et al., 1999). Fig. 6 (A, B, and G; and Videos 5 and 6, available at http:www.jcb.org/cgi/content/full/jcb.200406177/DC1) extends these studies and shows that rasG null cells, compared with wild-type cells, exhibit reduced cell polarity and directionality (persistent movement toward the chemoattractant source).
Figure 6.
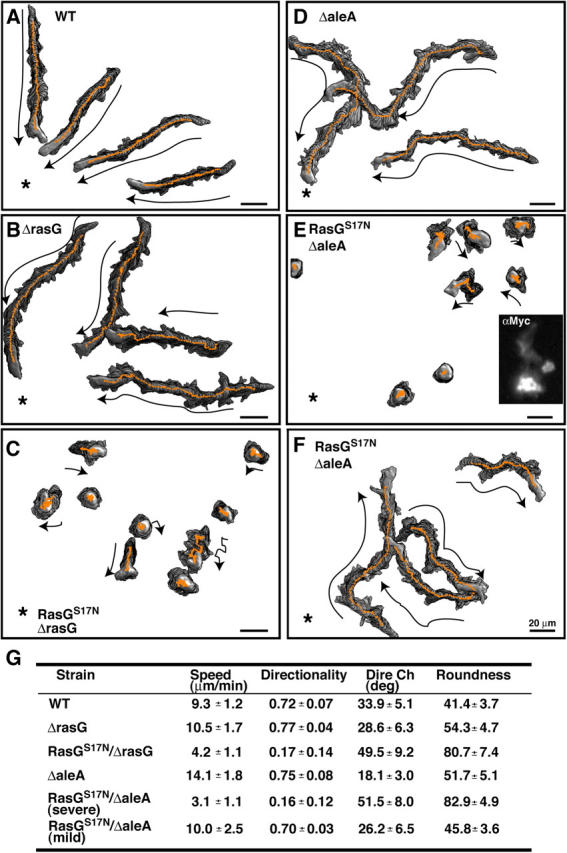
Ras signaling regulates proper chemotaxis. (A–F) Time-lapse recording of chemotaxis of indicated strains pulsed with cAMP for 6 h. An asterisk indicates the position of a micropipette containing 150 μM cAMP. Tracing of the chemotaxis of individual cells is shown. The expression level of myc-tagged RasGS17N in aleA null cells was monitored by immunofluorescence (E, inset). (G) Analysis of chemotaxis using DIAS software (Soll and Voss, 1998). Speed refers to the speed of the cell's centroid movement along the total path. Directionality is a measure of how straight the cells move. Direction change (Dire Ch) is a measure of the number and frequency of turns the cell makes. Roundness is an indication of the polarity of the cells.
Dominant-negative forms of small GTPases inhibit downstream function by blocking the activity of their cognate exchange factor (GDP/GTP exchange factor [GEF]). To further examine the roles of Ras in chemotaxis, we expressed dominant-negative RasGS17N in rasG null cells, which should block the activation of other Ras proteins that are regulated by GEFs sensitive to inhibition by RasGS17N. As expected if expression of RasGS17N were to inhibit Ras activation and thus decrease the activity of a downstream effector such as PI3K, we observed that activation of Ras, Akt/PKB, and PI(3,4,5)P3 production (as illustrated by PhdA membrane localization) in RasGS17N/rasG null cells was significantly reduced (but not eliminated) compared with rasG null cells (Fig. 7 A). Fig. 7 A also shows that as one titrates down Ras function (wild-type, rasG null, and RasGS17N/rasG null cells), there is a decrease in chemoattractant-mediated F-actin polymerization. When chemotaxis was assayed, RasGS17N/rasG null cells produced numerous small pseudopodia at right or oblique angles to the direction of the cAMP gradient and displayed severe polarization and migration defects with very little forward movement (Fig. 6 C and Video 7, available at http:www.jcb.org/cgi/content/full/jcb.200406177/DC1). These results suggest that, in addition to RasG, other Ras proteins play an important role in controlling the ability of cells to polarize and move. Our observation that these cells produce multiple pseudopodia on all sides when placed in a chemoattractant gradient suggests that the cells cannot effectively sense direction. These cells are able to polymerize F-actin, as illustrated by the ability to extend pseudopodia, but the absolute level of chemoattractant-mediated F-actin polymerization is reduced (Fig. 7 A).
Figure 7.

Activation of Akt/PKB and Ras. The effects of RasGS17N on Akt/PKB, Ras activation, and production of PI(3,4,5)P3 in rasG (A) and aleA (B) null cells were assayed as described in the legends to Figs. 1–4. The results were similar in five independent assays.
To examine the possibility that Ras signaling may control directional cell movement, we took advantage of aleA null cells, which carry a disruption of a gene encoding the putative Ras GEF Aimless and which were described as exhibiting aberrant chemotaxis (Insall et al., 1996). Fig. 7 B illustrates that the level of Ras activation is significantly reduced in aleA null cells compared with wild-type cells, suggesting that aberrant Ras signaling in aleA null cells might cause the aleA null chemotaxis defects. Next, we examined in greater detail the aleA null cell chemotaxis defects. Interestingly, aleA null cells were able to polarize and move well, although they displayed some defect in the directionality of movement toward the chemoattractant source (Fig. 6 D and Video 8, available at http:www.jcb.org/cgi/content/full/jcb.200406177/DC1), as previously suggested (Insall et al., 1996). This finding suggests Ras-mediated directional sensing may be independent from Ras-mediated cell polarization.
As there are >20 putative Ras GEFs in the Dictyostelium genome (Wilkins et al., 2000), we took advantage of the reduction in Ras activation in aleA null cells by expressing RasGS17N in these cells to potentially block other GEFs that activate RasG and other Ras proteins, using the same logic as when expressing RasGS17N in rasG null cells. RasGS17N/aleA null cells show a further reduction in Ras and Akt/PKB activation compared with aleA null cells (Fig. 7 B). These cells appear to produce very little PI(3,4,5)P3 in response to a chemoattractant (Fig. 7 B). When chemotaxis was examined, we observed two different types of behavior, both of which are more severe than aleA null cell phenotypes. One phenotype was very similar to RasGS17N/rasG null cells: a loss in cell polarity and cells did not move very far (Fig. 6 E and Video 9, available at http:www.jcb.org/cgi/content/full/jcb.200406177/DC1). The other phenotype was that cells polarized and moved but it took several minutes longer for these cells to polarize than aleA null cells. Once RasGS17N/aleA null cells polarized, they exhibited persistent movement, but the direction of movement was completely random relative to the direction of the chemoattractant source (Fig. 6 F and Video 9). The cells that exhibited this behavior were heterogenous in that different cells polarized with different kinetics (see Video 9). As might be expected, we observed a range of GFP-RasGS17N expression levels (Fig. 6 E, inset), suggesting that differences in kinetics or severity of the responses is the result of different expression levels. The results suggest Ras pathways are critical for regulating both cell polarity and directional movement.
Discussion
Ras plays an essential role in regulating directionality and cell polarity
We previously proposed that Ras is an essential regulator of Dictyostelium PI3Ks based on our observations that PI3K carrying a mutation in the Ras binding domain is unable to activate PI3K-dependent responses (Funamoto et al., 2002). Here, we demonstrate that Ras is rapidly and transiently activated upon global chemoattractant stimulation and that in chemotaxing cells, activated Ras, but not total Ras, is restricted to the cell's leading edge. This finding parallels that of Mochizuki et al. (2001), in which activated Ras was found at the peripheral edges of the plasma membrane in growth factor–stimulated COS-1 cells. A report that was published as we were submitting this manuscript also demonstrated that RasG is activated in response to chemoattractant stimulation (Kae et al., 2004). Their findings show a significantly more prolonged activation or the possibility of a bimodal activation profile. We are uncertain of the reason for this difference but the kinetics of RasG activation from our biochemical assay correspond to the kinetics of the cortical localization of the RBD-GFP probe and the kinetics of PI3K activation (Huang et al., 2003). As the Raf1 RBD does not effectively bind RasC-GTP (Kae et al., 2004), we do not think that our work examines RasC.
Our results implicate Ras as an essential part of the cell's compass that controls the ability of cells to sense a chemoattractant gradient. As previously reported (Insall et al., 1996), we find that aleA null cells exhibit reduced directionality without a significant loss in cell polarity or speed. Further depletion of Ras function through the expression of RasGS17N in aleA null cells causes severe directionality defects. A portion of these cells and rasG null/RasGS17N cells show a severe loss of movement, even though they produce cell protrusions in response to chemoattractant stimulation. However, the protrusions occur at random sites independent of the direction of the chemoattractant source. In the second phenotype, cells polarize in response to a directional signal, but do so much more slowly than wild-type cells. Most importantly, the direction of movement of these cells is essentially random relative to the direction of the chemoattractant source. As the level of Ras activation was reduced through a knockout of RasG and then expression of RasGS17N in these cells, we observed a sequential loss of chemoattractant-mediated F-actin polymerization.
We suggest that Ras may control chemotaxis through effectors in addition to PI3K. The directionality defect caused by depleting Ras function is significantly more severe than that of pi3k1/2 null cells or cells treated with 30 μM LY294002 (Funamoto et al., 2001). RasG-GTP binds to RIP3 (Ras interacting protein-3), an orthologue of the TOR complex 2 protein AVO1 in yeast, which has an orthologue in mammalian cells (Lee et al., 1999; Loewith et al., 2002). rip3 null cells are less polarized, migrate more slowly than wild-type cells, and produce pseudopodia at right or oblique angles to the direction of the chemoattractant source leading to a loss in directionality. We suggest the severe defect in cell polarization conferred by RasGS17N is due to its suppression of PI3K and RIP3, and likely other effectors. Although rasG null cells expressing RasGS17N have significantly reduced PI3K pathway activation (assayed by Akt activation), some activity remains, probably resulting from an inability to fully inhibit Ras activation through RasGS17N expression as shown by our data.
Local PI(3,4,5)P3 production is achieved cooperatively by local Ras activation, PI3K localization, and feedback loops
When a chemoattractant gradient is applied to cells, both PI(3,4,5)P3 accumulation and Ras activation occur locally at the site on the plasma membrane closest to the micropipette. We suggest that the localized increase of PI(3,4,5)P3 is the result of coordination between F-actin–independent local Ras activation and F-actin–driven PI3K localization (Fig. 8). The kinetics of Ras activation, F-actin polymerization, and PI3K translocation are similar, although our detailed analysis suggests that Ras activation (membrane localization of the GFP-RBD reporter) slightly precedes the localization of N-PI3K1. Strikingly, we found that translocation of N-PI3K1 (and full-length PI3K1 and PI3K2) is regulated by F-actin polymerization. We have shown that unstimulated cells have a low, basal level of PI3K on the plasma membrane.
Figure 8.
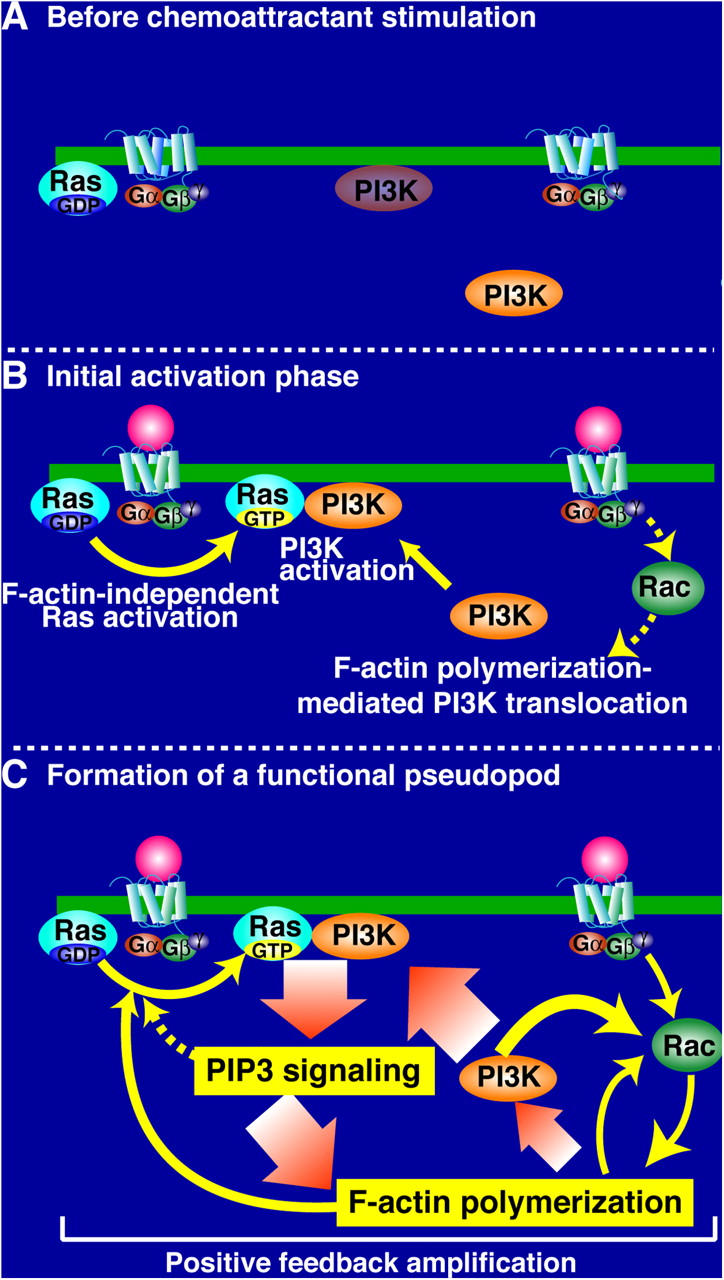
Model for the spatial and temporal regulation of Ras-induced chemotaxis. A model illustrating the intracellular signaling leading to local PI(3,4,5)P3 production is shown. (A) Resting cells with a basal level of PI3K at the plasma membrane. (B) The chemoattractant locally activates Ras at the presumptive leading edge (site closest to the chemoattractant source), where Ras then locally activates PI3K. There is a local polymerization of F-actin at the presumptive leading edge, which is partially Ras/PI3K independent, presumably controlled by Rho GTPase, WASP/SCAR, and Arp2/3. Our data suggest that the F-actin mediates PI3K translocation. (C) Locally produced PI(3,4,5)P3 induces further F-actin polymerization by activating downstream effectors, which would enhance the recruitment of PI3K to the membrane.
We suggest that F-actin cytoskeleton-independent Ras activation controls the initial PI3K response at the leading edge, when cells first respond to a chemoattractant gradient. This provides an initial amplification of the shallow extracellular gradient through the highly localized production of PIP3 and the localization of PH domain proteins. As PI3K-mediated PI(3,4,5)P3 production is required for full F-actin polymerization (Funamoto et al., 2001; Iijima and Devreotes, 2002; Chen et al., 2003), further amplification of the Ras-PI3K response results from a positive feedback loop comprising PI(3,4,5)P3-induced F-actin polymerization, PI3K localization, and Ras activation, which may help stabilize the nascent leading edge and aid persistent cell movement (Fig. 8). Consistent with this model, we show that concentrations of LatA that fully inhibit F-actin polymerization reduce the activity of the PI3K pathway (PKB activation), as might be expected if robust PI3K translocation to the membrane requires F-actin polymerization. A separate feedback loop whereby PI(3,4,5)P3 causes activation of PI3K has been proposed (Niggli, 2000; Wang et al., 2002).
The F-actin feedback loop we propose parallels a similar model proposed by the Bourne laboratory in which PI3K and F-actin are part of a feedback loop regulated by Rac GTPases that promotes leading edge function (Wang et al., 2002; Weiner et al., 2002). These findings parallel previous observations in Dictyostelium that PI3K is required for full F-actin polymerization and enhancement of the PI3K pathway through the deletion of PTEN, which causes an augmented F-actin response (Chung et al., 2001b; Funamoto et al., 2001; Iijima and Devreotes, 2002; Chen et al., 2003). In separate studies, we found that Dictyostelium RacB, which binds to the CRIB domains of Dictyostelium PAKa, PAKc, and WASP, has a biphasic activation profile paralleling that of F-actin. Disruption of RacB causes a >50% decrease in both peaks of chemoattractant-mediated F-actin polymerization (Park et al., 2004). RacB activation is partially dependent on PI3K and is enhanced in a PTEN hypomorphic strain. These findings provide a mechanism by which both PI3K-dependent and -independent pathways stimulate F-actin polymerization.
Spatially aberrant Ras activation and adventitious pseudopod formation are observed in pi3k1/2 null cells expressing myr-PI3K, which is uniformly localized around the plasma membrane. A similar response is seen in pten null cells, as the normal mechanisms that restrict PI(3,4,5)P3 levels and its localization to the leading edge are absent (Funamoto et al., 2002; Iijima and Devreotes, 2002). Aberrant lateral pseudopodia are also formed in both Dictyostelium cells and leukocytes lacking myosin II assembly in the cell's posterior (Wessels et al., 1988; Chung and Firtel, 1999; Worthylake and Burridge, 2003; Xu et al., 2003). We suggest that feedback loops exist in which cells, as they become more polarized and form a stable leading edge and posterior, are more resistant to adventitious pseudopod formation along the lateral sides and posterior of the cell. We expect that reduced Ras function limits the ability to form a single, stable pseudopod and concomitantly results in adventitious pseudopod formation, as the pathways that control the feedback loops controlling the actin cytoskeleton are not sufficiently robust (Fig. 8).
Materials and methods
Materials
We obtained LatA, LY294002, and anti-FLAG (M2) antibody from Sigma-Aldrich, monoclonal anti-Ras (Ab-3) antibody from Oncogene Research Products, and rabbit anti-GFP (FL) and anti-myc antibodies from Santa Cruz Biotechnology, Inc. The GFP-Raf Ras-binding domain (GST-RBD) was generated by PCR amplification of the coding region 1–146 and subcloned into the EcoRI–XhoI site of GFP-EXP4(+) or that of pGEX4T-3. All constructs were sequenced. We produced GST-RBD in BL21 bacteria (Stratagene) and purified it on glutathione-sepharose (Amersham Biosciences) as per the manufacturer's instructions. The pten null strain and CRAC-PH expression vector were obtained from P. Devreotes (Johns Hopkins School of Medicine, Baltimore, MD) and the aleA null cells were obtained from R. Insall (University of Birmingham, Birmingham, UK).
Biochemical assays
PKB activity was measured as described previously (Meili et al., 1999). The F-actin–enriched, Triton X-100–insoluble cytoskeleton was isolated as described previously (Steimle et al., 2001). For the Ras pull-down assay, the cell extract was incubated with 10 μg of GST-RBD on glutathione-agarose beads at 4°C for 30 min in the presence of 5 mg/ml BSA. The beads were washed three times. Ras proteins were separated on a 14% SDS-PAGE gel and immunoblotted with the appropriate antibody.
Chemotaxis and image acquisition
The analyses of chemotaxis toward cAMP and global responses to cAMP were performed as described previously (Chung and Firtel, 1999) and the data were analyzed with the DIAS program (Soll and Voss, 1998). Images were collected on a microscope (model TE300; Nikon) with DIC and fluorescence imaging and 40×/0.60 and 60×/1.40 objectives using a Coolsnap-HQ camera. Initial images were captured using Metamorph software. Quantitation of membrane or cortical localization of GFP fusion proteins and the analysis of chemotaxis of each strain represents the averages of at least five cells from at least three separate experiments. We measured the intensity of cortical GFP using NIH Image software. The level of peripheral GFP is defined as Et and is calculated by dividing the intensity before stimulation (E0) by the intensity at each time point (Et). E0 is set at 1.0. E is the intensity at the membrane minus the background intensity divided by the intensity of the cytosol minus the background intensity. For the LatA experiments, cells were pretreated with 3 μM of LatA (Sigma-Aldrich) for 20 min before assay.
Online supplemental material
Videos 1–3, which show the localization of the GFP-RBD in response to global stimulation (Video 1) and chemotaxing (Video 2) in wild-type cells and in pten null cells (Video 3), correspond to Fig. 3 in the text. Video 4 shows GFP-RBD translocation in the LatA-treated cells and corresponds to Fig. 4 B. Videos 5–9 show chemotaxis of wild-type (Video 5), rasG null (Video 6), RasGS17N/rasG null (Video 7), aleA null (Video 8), and RasGS17N/aleA null (Video 9) cells and correspond to Fig. 6 in the text. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200406177/DC1.
Acknowledgments
We thank Firtel laboratory members for their stimulating discussions and helpful suggestions, Susan Lee and Yung Duong for excellent technical assistance, and Jennifer Roth and Michelle Mendoza for help in preparing this manuscript. We thank A. Yoshimura for critical comments, M. Iijima and P. Devreotes for the pten null strain, and R. Insall for the aleA null strain.
A.T. Sasaki was supported, in part, by a Japanese Society for the Promotion of Science Research Fellowship for Research Abroad. This work was funded by research grants from the United States Public Health Service to R.A. Firtel.
Abbreviations used in this paper: GEF, GDP/GTP exchange factor; LatA, latrunculin A; myr, myristoyl; PI3K, phosphatidylinositol 3-kinase.
References
- Chan, T.O., U. Rodeck, A.M. Chan, A.C. Kimmelman, S.E. Rittenhouse, G. Panayotou, and P.N. Tsichlis. 2002. Small GTPases and tyrosine kinases coregulate a molecular switch in the phosphoinositide 3-kinase regulatory subunit. Cancer Cell. 1:181–191. [DOI] [PubMed] [Google Scholar]
- Chen, L., C. Janetopoulos, Y.E. Huang, M. Iijima, J. Borleis, and P.N. Devreotes. 2003. Two phases of actin polymerization display different dependencies on PI(3,4,5)P3 accumulation and have unique roles during chemotaxis. Mol. Biol. Cell. 14:5028–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, V., T. Bivona, A. Hach, J. Sajous, J. Silletti, H. Wiener, R.N. Johnson, A. Cox, and M. Philips. 2002. Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 4:343–350. [DOI] [PubMed] [Google Scholar]
- Chung, C.Y. and R.A. Firtel. 1999. PAKa, a putative PAK family member, is required for cytokinesis and the regulation of the cytoskeleton in Dictyostelium discoideum cells during chemotaxis. J. Cell Biol. 147:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, C., S. Funamoto, and R. Firtel. 2001. a. Signaling pathways controlling cell polarity and chemotaxis. Trends Biochem. Sci. 26:557–566. [DOI] [PubMed] [Google Scholar]
- Chung, C., G. Potikyan, and R. Firtel. 2001. b. Control of cell polarity and chemotaxis by Akt/PKB and PI3 kinase through the regulation of PAKa. Mol. Cell. 7:937–947. [DOI] [PubMed] [Google Scholar]
- Funamoto, S., K. Milan, R. Meili, and R. Firtel. 2001. Role of phosphatidylinositol 3′ kinase and a downstream pleckstrin homology domain–containing protein in controlling chemotaxis in Dictyostelium. J. Cell Biol. 153:795–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funamoto, S., R. Meili, S. Lee, L. Parry, and R. Firtel. 2002. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 109:611–623. [DOI] [PubMed] [Google Scholar]
- Hall, A.L., A. Schlein, and J. Condeelis. 1988. Relationship of pseudopod extension to chemotactic hormone-induced actin polymerization in amoeboid cells. J. Cell. Biochem. 37:285–299. [DOI] [PubMed] [Google Scholar]
- Hancock, J.F. 2003. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 4:373–384. [DOI] [PubMed] [Google Scholar]
- Huang, Y., M. Iijima, C. Parent, S. Funamoto, R. Firtel, and P. Devreotes. 2003. Receptor-mediated regulation of PI3Ks confines PI(3,4,5)P3 to the leading edge of chemotaxing cells. Mol. Biol. Cell. 14:1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima, M., and P. Devreotes. 2002. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 109:599–610. [DOI] [PubMed] [Google Scholar]
- Iijima, M., Y. Huang, and P. Devreotes. 2002. Temporal and spatial regulation of chemotaxis. Dev. Cell. 3:469–478. [DOI] [PubMed] [Google Scholar]
- Iijima, M., Y.E. Huang, H.R. Luo, F. Vazquez, and P.N. Devreotes. 2004. Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J. Biol. Chem. 279:16606–16613. [DOI] [PubMed] [Google Scholar]
- Insall, R.H., J. Borleis, and P.N. Devreotes. 1996. The aimless RasGEF is required for processing of chemotactic signals through G-protein-coupled receptors in Dictyostelium. Curr. Biol. 6:719–729. [DOI] [PubMed] [Google Scholar]
- Janetopoulos, C., T. Jin, and P. Devreotes. 2001. Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science. 291:2408–2411. [DOI] [PubMed] [Google Scholar]
- Jin, T., N. Zhang, Y. Long, C. Parent, and P. Devreotes. 2000. Localization of the G protein betagamma complex in living cells during chemotaxis. Science. 287:1034–1036. [DOI] [PubMed] [Google Scholar]
- Kae, H., C.J. Lim, G.B. Spiegelman, and G. Weeks. 2004. Chemoattractant-induced Ras activation during Dictyostelium aggregation. EMBO Rep. 5:602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S., C.A. Parent, R. Insall, and R.A. Firtel. 1999. A novel Ras-interacting protein required for chemotaxis and cyclic adenosine monophosphate signal relay in Dictyostelium. Mol. Biol. Cell. 10:2829–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., M. Hannigan, Z. Mo, B. Liu, W. Lu, Y. Wu, A.V. Smrcka, G. Wu, L. Li, M. Liu, et al. 2003. Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell. 114:215–227. [DOI] [PubMed] [Google Scholar]
- Lim, C., G. Spiegelman, and G. Weeks. 2001. RasC is required for optimal activation of adenylyl cyclase and Akt/PKB during aggregation. EMBO J. 20:4490–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, C.J., G.B. Spiegelman, and G. Weeks. 2002. Cytoskeletal regulation by Dictyostelium Ras subfamily proteins. J. Muscle Res. Cell Motil. 23:729–736. [DOI] [PubMed] [Google Scholar]
- Loewith, R., E. Jacinto, S. Wullschleger, A. Lorberg, J.L. Crespo, D. Bonenfant, W. Oppliger, P. Jenoe, and M.N. Hall. 2002. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell. 10:457–468. [DOI] [PubMed] [Google Scholar]
- Meili, R., C. Ellsworth, S. Lee, T. Reddy, H. Ma, and R. Firtel. 1999. Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J. 18:2092–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlot, S., and R. Firtel. 2003. Leading the way: Directional sensing through phosphatidylinositol 3-kinase and other signaling pathways. J. Cell Sci. 116:3471–3478. [DOI] [PubMed] [Google Scholar]
- Mochizuki, N., S. Yamashita, K. Kurokawa, Y. Ohba, T. Nagai, A. Miyawaki, and M. Matsuda. 2001. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 411:1065–1068. [DOI] [PubMed] [Google Scholar]
- Niggli, V. 2000. A membrane-permeant ester of phosphatidylinositol 3,4, 5-trisphosphate (PIP(3)) is an activator of human neutrophil migration. FEBS Lett. 473:217–221. [DOI] [PubMed] [Google Scholar]
- Pacold, M., S. Suire, O. Perisic, S. Lara-Gonzalez, C. Davis, E. Walker, P. Hawkins, L. Stephens, J. Eccleston, and R. Williams. 2000. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 103:931–943. [DOI] [PubMed] [Google Scholar]
- Park, K.C., F. Rivero, R. Meili, S. Lee, F. Apone, and R.A. Firtel. 2004. Rac regulation of chemotaxis and morphogenesis in Dictyostelium. EMBO J. 10.1038/sj.emboj.7600368. [DOI] [PMC free article] [PubMed]
- Parent, C., and P. Devreotes. 1999. A cell's sense of direction. Science. 284:765–770. [DOI] [PubMed] [Google Scholar]
- Parent, C., B. Blacklock, W. Froehlich, D. Murphy, and P. Devreotes. 1998. G protein signaling events are activated at the leading edge of chemotactic cells. Cell. 95:81–91. [DOI] [PubMed] [Google Scholar]
- Servant, G., O. Weiner, P. Herzmark, T. Balla, J. Sedat, and H. Bourne. 2000. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 287:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soll, D.R., and E. Voss. 1998. Two- and three-dimensional computer systems for analyzing how animal cells crawl. Motion Analysis of Living Cells. D.R. Soll and D. Wessels, editors. Wiley-Liss, New York. 25–52.
- Steimle, P.A., S. Yumura, G.P. Cote, Q.G. Medley, M.V. Polyakov, B. Leppert, and T.T. Egelhoff. 2001. Recruitment of a myosin heavy chain kinase to actin-rich protrusions in Dictyostelium. Curr. Biol. 11:708–713. [DOI] [PubMed] [Google Scholar]
- Suire, S., P. Hawkins, and L. Stephens. 2002. Activation of phosphoinositide 3-kinase gamma by Ras. Curr. Biol. 12:1068–1075. [DOI] [PubMed] [Google Scholar]
- Taylor, S., R. Resnick, and D. Shalloway. 2001. Nonradioactive determination of Ras-GTP levels using activated ras interaction assay. Methods Enzymol. 333:333–342. [DOI] [PubMed] [Google Scholar]
- Tuxworth, R., J. Cheetham, L. Machesky, G. Spiegelmann, G. Weeks, and R. Insall. 1997. Dictyostelium RasG is required for normal motility and cytokinesis, but not growth. J. Cell Biol. 138:605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F., P. Herzmark, O. Weiner, S. Srinivasan, G. Servant, and H. Bourne. 2002. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 4:513–518. [DOI] [PubMed] [Google Scholar]
- Weiner, O., P. Neilsen, G. Prestwich, M. Kirschner, L. Cantley, and H. Bourne. 2002. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 4:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels, D., D.R. Soll, D. Knecht, W.F. Loomis, A. De Lozanne, and J. Spudich. 1988. Cell motility and chemotaxis in Dictyostelium amebae lacking myosin heavy chain. Dev. Biol. 128:164–177. [DOI] [PubMed] [Google Scholar]
- Wilkins, A., J.R. Chubb, and R.H. Insall. 2000. A novel Dictyostelium RasGEF is required for normal endocytosis, cell motility and multicellular development. Curr. Biol. 10:1427–1437. [DOI] [PubMed] [Google Scholar]
- Worthylake, R.A., and K. Burridge. 2003. RhoA and ROCK promote migration by limiting membrane protrusions. J. Biol. Chem. 278:13578–13584. [DOI] [PubMed] [Google Scholar]
- Xu, J., F. Wang, A. Van Keymeulen, P. Herzmark, A. Straight, K. Kelly, Y. Takuwa, N. Sugimoto, T. Mitchison, and H. Bourne. 2003. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 114:201–214. [DOI] [PubMed] [Google Scholar]
- Yart, A., S. Roche, R. Wetzker, M. Laffargue, N. Tonks, P. Mayeux, H. Chap, and P. Raynal. 2002. A function for phosphoinositide 3-kinase beta lipid products in coupling beta gamma to Ras activation in response to lysophosphatidic acid. J. Biol. Chem. 277:21167–21178. [DOI] [PubMed] [Google Scholar]