

Abstract
HLA-DR2 is associated with susceptibility to multiple sclerosis (MS). A peptide from human myelin basic protein (MBP, residues 85–99) was previously found to bind to purified HLA-DR2 (DRA, DRB1*1501) and to be recognized by human MBP-specific T cell clones. Soluble HLA-DR2 was expressed in the baculovirus system by replacing the hydrophobic transmembrane regions and cytoplasmic segments of DRα and DRβ with leucine zipper dimerization domains from the transcription factors Fos and Jun. In the expression construct, the MBP(85–99) sequence was covalently linked to the N terminus of the mature DRβ chain. The recombinant protein was secreted by Sf9 cells infected with the recombinant baculovirus and purified by affinity chromatography. The leucine zipper dimerization domains were then cleaved from the assembled HLA-DR2/MBP peptide complex with V8 protease, and the protein was further purified by anion-exchange HPLC. Analysis by HPLC gel filtration indicated that the HLA-DR2/MBP peptide complex did not have a tendency to aggregate. The purified HLA-DR2/MBP peptide complex readily crystallized by the hanging drop method in 15–18% polyethylene glycol 6000/100 mM glycine, pH 3.5. At a synchrotron radiation source, a crystal with a tetragonal space group diffracted to a resolution of 2.6 Å. The expression of such homogenous HLA-DR/peptide complexes may facilitate cocrystallization with T cell receptors as well as other molecules involved in T cell receptor recognition and signaling.
Multiple sclerosis (MS) is a polygenic disease, and the major histocompatibility complex (MHC) class II region on chromosome 6 represents an important susceptibility locus. Many population-based studies have demonstrated an increased frequency of the HLA-DR2 (DRB1*1501) haplotype in MS patients. Since HLA-DR and HLA-DQ genes are in linkage disequilibrium, both DR and DQ alleles of this haplotype may account for the increased susceptibility to MS (refs. 1–4, reviewed in refs. 5 and 6). The HLA-DR2 haplotype (DRB1*1501) is also associated with susceptibility to Goodpasture’s syndrome, an autoimmune disease characterized by antibodies against glomerular and alveolar basement membranes (7).
An immunodominant peptide of human myelin basic protein (MBP) was previously shown to bind to purified HLA-DR2 (DRA, DRB1*1501) and to be recognized by human MBP-specific T cell clones (8–10). The interaction of the MBP(85–99) peptide with HLA-DR2 as well as T cell receptor (TCR) recognition of the DR2/MBP peptide complex has been studied in detail. On the basis of the HLA-DR2-binding and TCR-recognition motif, microbial peptides were identified that activate MBP-specific T cell clones; these peptides were quite distinct in their primary sequence from the MBP peptide (11, 12). To further study the interaction of the MBP peptide with HLA-DR2 as well as the recognition of HLA-DR2/peptide complexes by MBP-specific TCRs, efforts were made to produce soluble HLA-DR2/peptide complexes that would be suitable for crystallization.
Human MHC class I molecules can be efficiently expressed by the refolding of MHC class I heavy chains in the presence of β2-microglobulin and appropriate peptides (13). The refolding of MHC class II molecules has proven to be more difficult (14). Soluble HLA-DR1 and HLA-DR4 were generated in insect cells by coexpressing the DRα and DRβ ectodomains; these molecules were then loaded with single peptides and crystallized (15–17). HLA-DR2 failed to assemble when this approach was attempted; however, assembly and secretion of soluble molecules could be achieved by addition of leucine zipper dimerization domains as a functional replacement for the hydrophobic transmembrane segments (18). Leucine zipper dimerization domains were also reported to promote the efficient assembly of murine I-A molecules (19). For crystallization of a defined HLA-DR2/peptide complex, the MBP peptide sequence was attached to the N terminus of the mature DRβ chain, as described previously for murine MHC class II molecules (20). The purification and crystallization of this HLA-DR2/MBP peptide complex are reported here.
MATERIALS AND METHODS
Expression Construct.
The construct with the covalently linked MBP peptide was generated by PCR such that the 5′ end (DRβ signal peptide–MBP peptide–linker) was made first and then joined with the larger DRβ-Jun segment by overlapping PCR. The DRβ signal peptide was amplified by using a 60-bp reverse primer that encoded the MBP(85–99) peptide up to residue 97 (5′-TGT CAC GAT ATT TTT GAA GAA ATG CAC CAC AGG GTT CTC TAG ACC AGA CAA AGC CAG TGG-3′). This fragment was extended at the 3′ end in a second round of PCR with a 45-bp primer that encoded the remaining sequence of the MBP peptide and eight amino acids of the linker (5′-TGG CAC TAG TGA GCC ACC ACC AGA TCT TGG TGT CAC GAT ATT TTT-3′). The mature DRβ chain-Jun segment was amplified with a 54-bp 5′ oligonucleotide that created a 15-bp overlap with the first PCR product in the linker region (5′-GGC TCA CTA GTG CCA CGG GGC TCT GGA GGA GGT GGG TCC GGG GAC ACC CGA CCA-3′). These two fragments were joined by overlapping PCR using primers for the 5′ end of the DRβ signal peptide and the 3′ end of the construct.
The construct in which only two amino acids (Gly-Leu, whose codons made up part of an XbaI site) were present between the DRβ signal peptide and the MBP peptide sequence did not result in secretion of the protein. The segment between the signal peptide and the MBP peptide sequence was therefore extended to five amino acids (GDTGL, with the first three amino acids representing residues 1–3 of the mature DRβ sequence) in a second round of overlapping PCR (sense primer for GDTGL segment, 5′-TCT GGT GAT ACA GGT CTA GAG AAC CCT GTG-3′; antisense primer for GDTGL segment, 5′-TAG ACC TGT ATC ACC AGA CAA AGC CAG TGG-3′). This construct was cloned into a eukaryotic expression vector (pcDNA I; Invitrogen), sequenced, and amplified with the following oligonucleotides for subcloning into pAcAB3: DRβ-forward, 5′-AAA AAA GGA TCCATG GTG TGT CTG AAG CTC CC-3′; DRβ-reverse, 5′-AAA AAA GGA TCC TCA ATG GTT CAT GAC TTT CT-3′, BamHI sites underlined. The DRα-Fos construct was excised with BamHI from a eukaryotic expression vector (pRSV); these constructs had been generated by amplification of the 5′ end of the DRα cDNA (representing the signal sequence and the ectodomain) and the Fos dimerization domain, which were joined by a SalI restriction site in the seven amino acid linker.
The constructs were cloned in baculovirus vector pAcAB3 (PharMingen) which carries two p10 promoters in opposite orientation. The DRα-Fos construct was cloned in the BglII site; the BamHI site was used for the DRβ-Jun construct with the covalently linked MBP peptide. Sequences were confirmed by dideoxynucleotide sequencing. The vector was cotransfected with linearized baculovirus DNA (BaculoGold; PharMingen) into Sf9 cells according to the supplier’s recommendations; this viral DNA contained a lethal deletion that was rescued by the pAcAB3 transfer vector. Recombinant viruses were plaque purified, tested for expression, and amplified.
Purification of the HLA-DR2/MBP Peptide Complex.
Sf9 cells were grown in serum-free medium, either in roller bottles (Sf-900 II serum-free medium; Life Technologies) or in spinner flasks (HyQ-CCM3 medium; HyClone). Large-scale spinner cultures were prepared by the National Cell Culture Center, Minneapolis, MN, which has a cooperative agreement award from the National Center for Research Resources, National Institutes of Health. Five days after infection, supernatants were clarified by centrifugation and concentrated ≈3- to 10-fold by ultrafiltration in an Amicon CH2 ultrafiltration system with a spiral membrane cartridge (molecular mass cutoff of 30 kDa). The protein was purified with mAb L243 (American Type Culture Collection no. HB55) conjugated to cyanogen bromide-activated Sepharose CL4B beads (Pharmacia Biotech). Samples were passed over a Sepharose CL4B precolumn column and an L243 affinity column at a flow rate not exceeding 30 ml/hr; the L243 column was then washed with 40 ml of 20 mM sodium phosphate/0.01% sodium azide, pH 7.0, followed by elution with three bed volumes (15 ml) of 50 mM glycine, pH 11.5. Fractions (1 ml) were collected and neutralized by adding 70 μl of 2 M Tris⋅HCl, pH 6.8. The protein content of these fractions was determined with the Coomassie Plus protein assay reagent (Pierce). Positive fractions were pooled and dialyzed twice against 2.5 liters of 50 mM sodium phosphate, pH 7.8 at 4°C. Protein purity was monitored by SDS/PAGE.
Cleavage of Leucine Zipper Dimerization Domains.
Cleavage of the leucine zipper was tested with Staphylococcus aureus V8 protease and endoproteinase Asp-N (Boehringer Mannheim). Conditions were tested using 1 μg of DR2/MBP peptide complex, 50 ng of V8 protease, and 20–50 ng of endoproteinase Asp-N in 50 mM sodium phosphate, pH 7.7 for 16 hr. Products were separated by SDS/PAGE under nonreducing conditions and transferred to a poly(vinylidene difluoride) (PVDF) membrane. Western blots were incubated with polyclonal antisera specific for DRα or DRβ (1:20,000 dilution) in 5% nonfat dry milk/50 mM Tris⋅HCl, pH 7.5/150 mM NaCl/0.2% Tween 20 at 4°C overnight. Blots were then washed and incubated with a horseradish peroxidase-conjugated anti-rabbit IgG antibody (1:3,000), followed by detection with enhanced chemiluminescence (ECL; Amersham).
Large-scale digestions were done with DR2/MBP at 0.5 mg/ml and V8 protease at 2.5 μg/ml in 50 mM sodium phosphate, pH 7.8, for 16 hr at room temperature. After digestion, the DR2/MBP peptide complex was separated by anion-exchange HPLC (Mono Q, Pharmacia). Separations were done by using a 20-min linear gradient made with buffer A (50 mM ethanolamine, pH 9.0/0.02% sodium azide) and buffer B (50 mM ethanolamine, pH 9.0/1 M NaCl/0.02% sodium azide) at a flow rate of 1 ml/min. The major peak that eluted at ≈55–60% B was concentrated by ultrafiltration in a Centricon 10 unit (Amicon) to a final concentration of 10 mg/ml in 25 mM Tris⋅HCl, pH 7.5/1 mM EDTA/1 mM phenylmethylsulfonyl fluoride/1 μM leupeptin. The protein concentration was determined by using the BCA (bicinchoninic acid) protein assay reagent (Pierce).
Crystallization of the HLA-DR2/MBP Peptide Complex.
Crystals were grown by the hanging drop method, by placing 1 μl of well solution and 1 μl of protein (10 mg/ml) on silane-treated coverslips. Optimal conditions for crystal growth were 15–18% (wt/vol) polyethylene glycol (PEG) 6000/100 mM glycine, pH 3.5 at 18°C. For seeding experiments, serial dilutions of microcrystals with a tetragonal morphology were made in 30% PEG 6000/100 mM glycine, pH 3.5 (starting at 1:100, steps of 5-fold dilution). These were streaked with a dog whisker into preequilibrated 2-μl drops of 15–18% PEG 6000/100 mM glycine, pH 3.5, containing 10 μg of protein. Crystals were harvested into 25–30% PEG 6000/100 mM glycine, pH 3.5, and allowed to equilibrate for 12–24 hr. Stepwise transfers (12–24 hr) were performed into 30% PEG 6000/100 mM glycine, pH 3.5, containing increasing concentrations of ethylene glycol (5%, 10%, and 15%, vol/vol) as a cryoprotectant. Crystals were then mounted on 0.1- to 0.4-mm-diameter loops and cryocooled in liquid nitrogen.
Low-resolution diffraction data (15.0 Å to 3.5 Å) were collected by using an Elliot GX-13 rotating anode x-ray source with the beam collimated by double mirrors, and diffraction recorded on an imaging plate detector (Mar 345, Mar Research). High-resolution data (15.0 Å to 2.5 Å) were collected on an ADSC 1K charge-coupled device detector at the A1 beamline (λ = 0.91 Å) at CHESS (Cornell High Energy Synchrotron Source) and data were processed by using denzo (21).
RESULTS AND DISCUSSION
Approaches Used to Generate Defined HLA-DR2/Peptide Complexes.
A number of different approaches were attempted to generate soluble HLA-DR2/peptide complexes that would be suitable for crystallization. Initially, HLA-DR2 molecules were purified from B cell lines homozygous for the HLA-DR2 haplotype, as described by Gorga et al. (22). Since the DR2 haplotype carries two DRβ chain genes, these B cell lines coexpressed DR2a (encoded by DRA, DRB5*0101) and DR2b (encoded by DRA, DRB1*1501) (23). Separation of DR molecules with mAbs available at the time did not yield pure preparations. Additional difficulties were related to the fact that the majority of these molecules were already loaded with high-affinity peptides and that the hydrophobic transmembrane domains had to be cleaved by limited proteolysis with papain.
To overcome these problems, the expression of recombinant HLA-DR2 (DRA, DRB1*1501) was attempted. No assembly of the DRα and DRβ ectodomains of HLA-DR2 was observed in the baculovirus system, when materials with stop codons inserted before the transmembrane region were used, an approach that had allowed the expression of soluble HLA-DR1 (15). However, addition of leucine zipper dimerization domains from the transcription factors Fos and Jun resulted in the assembly and secretion of soluble DR2 molecules (18). These constructs were first expressed in a yeast expression system (Pichia pastoris, yield of ≈0.3 mg/liter of culture) and then in an insect cell expression system (Drosophila Schneider cells, yield of ≈1 mg/liter of culture prior to peptide loading). Soluble DR2 molecules that were purified from supernatants of Drosophila Schneider cell transfectants were loaded with the MBP(85–99) peptide. Crystallization was attempted with and without proteolytic cleavage of the leucine zipper dimerization domains, but only small crystals of poor quality were obtained. We then decided to express DR2 with a covalently linked MBP(85–99) peptide to improve the final yield and homogeneity of HLA-DR2/peptide complexes.
Expression and Purification of Soluble HLA-DR2 with a Covalently Linked MBP Peptide.
The DRα and DRβ chain constructs (Fig. 1) were cloned in a baculovirus vector (pAcAB3) with dual p10 promoters, which allowed both chains to be expressed under the control of a promoter that is expressed early after infection. Generation of a single recombinant baculovirus obviated the need for matching titers of two recombinant baculoviruses and greatly facilitated large-scale protein production. In the cDNA constructs, the leucine zipper dimerization domains of Fos and Jun were attached to the 3′ end of the DRα and DRβ ectodomains, respectively, through a seven amino acid linker (VDGGGGG) that contained a SalI restriction site. The 5′ end of the DRβ chain construct constituted the DRβ chain signal peptide, five residues (GDTGL) that represented the first three residues of the mature DRβ chain for cleavage of the signal peptide as well as part of a XbaI site, followed by the sequence of the MBP(85–99) peptide, a 16 amino acid linker, and the mature DRβ chain sequence. The linker was changed only at the first position (Gly to Ser) from that reported previously (20), to create a BglII site for exchange of the covalently linked peptide. Even though cleavage of the linker was not required for T cell recognition of I-A or I-E molecules with covalently linked peptides (20), the thrombin cleavage site in the linker was left in case the linker would interfere with crystallization or TCR binding. The murine I-Ad molecule was recently crystallized with covalently linked peptides from ovalbumin and influenza virus hemagglutinin; in these constructs, the linker was shortened to six residues (24, 25).
Figure 1.

Construct for the expression of the HLA-DR2/MBP peptide complex. In the DRβ chain construct, the MBP(85–99) sequence was attached through a 16-amino acid linker to the N terminus of the mature DRβ chain sequence. A 5-amino acid segment (GDTGL) was placed between the DRβ chain signal peptide and the MBP sequence to allow for cleavage of the signal peptide and to introduce an XbaI site. The linker between the MBP peptide sequence and the mature DRβ chain sequence encoded a thrombin cleavage site (LVPRGS) for optional cleavage of the linker. Leucine zipper dimerization domains of the transcription factors Fos and Jun were attached to the 3′ end of DRα and DRβ ectodomains through short flexible linkers. These constructs were cloned in the baculovirus vector pAcAB3, which carried two p10 promoters in opposite orientation.
The construct was cotransfected with viral DNA into Sf9 cells, recombinant viruses were cloned, and-high titer virus stocks were generated. Western blot analysis of supernatants from infected Sf9 cells indicated that DRα and DRβ chains were secreted into the supernatant. For large-scale expression, Sf9 cells were infected in roller bottles or in spinner flasks and the protein was purified by affinity chromatography using antibody L243. N-terminal sequence analysis confirmed the predicted sequences for the mature DRα chain and the DRβ chain with the covalently linked peptide. The yield of protein from the affinity column was ≈2.3 mg/liter from cultures grown in roller bottles and ≈1.4 mg/liter from spinner flasks. The yield after cleavage of the leucine zipper and anion-exchange chromatography was 0.83 and 0.73 mg/liter of initial culture.
Cleavage of Leucine Zipper Dimerization Domains from the Assembled HLA-DR2/MBP Peptide Complex.
Proteolytic cleavage of the leucine zippers was performed because the leucine zipper dimerization domains and the linkers could interfere with crystallization. Because the flexible linker region [sequence: Val-Asp-(Gly)5] was likely to be accessible, two proteases that cleaved N- or C-terminal to Asp were tested. Endoproteinase Asp-N cleaves peptide bonds N-terminal to Asp and cysteic acid at pH 6.0 to pH 8.5. The specificity of staphylococcal V8 protease is pH dependent; at pH 7.8 it specifically cleaves C-terminal to Glu as well as Asp-Gly bonds (present in the linker). Since the C-terminal amino acid in the DRα ectodomain was Glu, a second potential site for cleavage by V8 protease was present between the DRα ectodomain and the Fos segment.
After digestion with different quantities of these enzymes, products were analyzed by SDS/PAGE and Western blot analysis using antisera specific for DRα or DRβ (Fig. 2). Soluble DR1 that was expressed in the baculovirus system was included for comparison (lane 5). For both α and β chains of DR2, the products obtained by cleavage with V8 protease migrated at a slightly lower apparent molecular mass than those resulting from cleavage with endoproteinase Asp-N. The α chain of cleaved DR2 migrated at a molecular mass similar to that of the DR1 α chain; the DR2 β chain migrated at a higher apparent molecular mass than the DR1 β chain, because of the covalently linked MBP peptide. Only small quantities of V8 protease were required for large-scale digestion (5 ng of enzyme per μg of DR2), indicating that cleavage of both chains was efficient. No breakdown products of DRα or DRβ were detected by Western blot analysis with a 10-fold larger amount of V8 protease (50 ng of enzyme for 1 μg of DR2/MBP), indicating that other potential cleavage sites for the enzyme were not accessible on the native protein. Because of the lower cost of V8 protease compared with endoproteinase Asp-N, this enzyme was used for preparative work.
Figure 2.

Cleavage of leucine zipper dimerization domains from the assembled HLA-DR/MBP peptide complex. Western blot analysis of DR2/MBP peptide complexes was performed after cleavage of leucine zipper dimerization domains with V8 protease or endoproteinase Asp-N. Western blots were probed with antisera specific for DRα (A) or DRβ (B). After cleavage, the α chain of DR2 migrated at a position similar to that of the α chain of soluble DR1; the β chain of DR2 had a higher apparent molecular mass than the β chain of soluble DR1, because of the covalently linked MBP peptide. Lanes 1–4, 1 μg of DR2/MBP; lane 2, + 50 ng of endoproteinase Asp-N; lane 3, + 50 ng of V8 protease; lane 4, + 20 ng of endoproteinase Asp-N and 50 ng of V8 protease; lane 5, 0.5 μg of soluble DR1 (no protease digestion). Digestions were done for 16 hr at room temperature and proteins were separated on an SDS/12% polyacrylamide gel under nonreducing conditions, followed by transfer to a poly(vinylidene difluoride) membrane. The membrane was probed with polyclonal antisera specific for DRα or DRβ, followed by a horseradish peroxidase-conjugated anti-rabbit IgG antibody and detection by enhanced chemiluminescence. Positions of markers (kDa) are shown on the left.
Solubility of the HLA-DR2/MBP Peptide Complex.
After digestion with V8 protease, the DR2/MBP peptide complex was further purified by anion-exchange HPLC (Fig. 3). The protein was eluted from a DEAE column (Mono Q, Pharmacia) with a linear salt gradient (0 to 1 M NaCl) in 50 mM ethanolamine, pH 9.0. The major peak, which eluted at 55–60% B, represented the DR2/MBP peptide complex; this material was used for crystallization. A second, small, peak that eluted at a higher salt concentration also contained DR2, but this material was not used. SDS/PAGE demonstrated a doublet for DRα (higher molecular mass band) and a single band for the DRβ chain with the covalently linked peptide before and after cleavage of the leucine zipper (Fig. 4, lanes 1 and 2, respectively).
Figure 3.

Purification of the HLA-DR2/MBP peptide complex by anion-exchange HPLC after cleavage of leucine zipper dimerization domains. After affinity purification using mAb L243, the leucine zipper dimerization domains were cleaved with V8 protease (using 5 μg of enzyme per mg of DR2/MBP peptide complex). The HLA-DR2/MBP peptide complex was further purified by using a DEAE column (Mono Q, Pharmacia). Six milligrams of protein was loaded onto the column followed by elution with a 20-min linear NaCl gradient (buffer A, 50 mM ethanolamine, pH 9.0/0.02% sodium azide; buffer B, 50 mM ethanolamine, pH 9.0/0.02% sodium azide/1 M NaCl). The major peak that eluted between ≈55% and 60% B was used for crystallization. Horizontal axis, time (min); left vertical axis, absorbance at 280 nm, expressed in μV.
Figure 4.
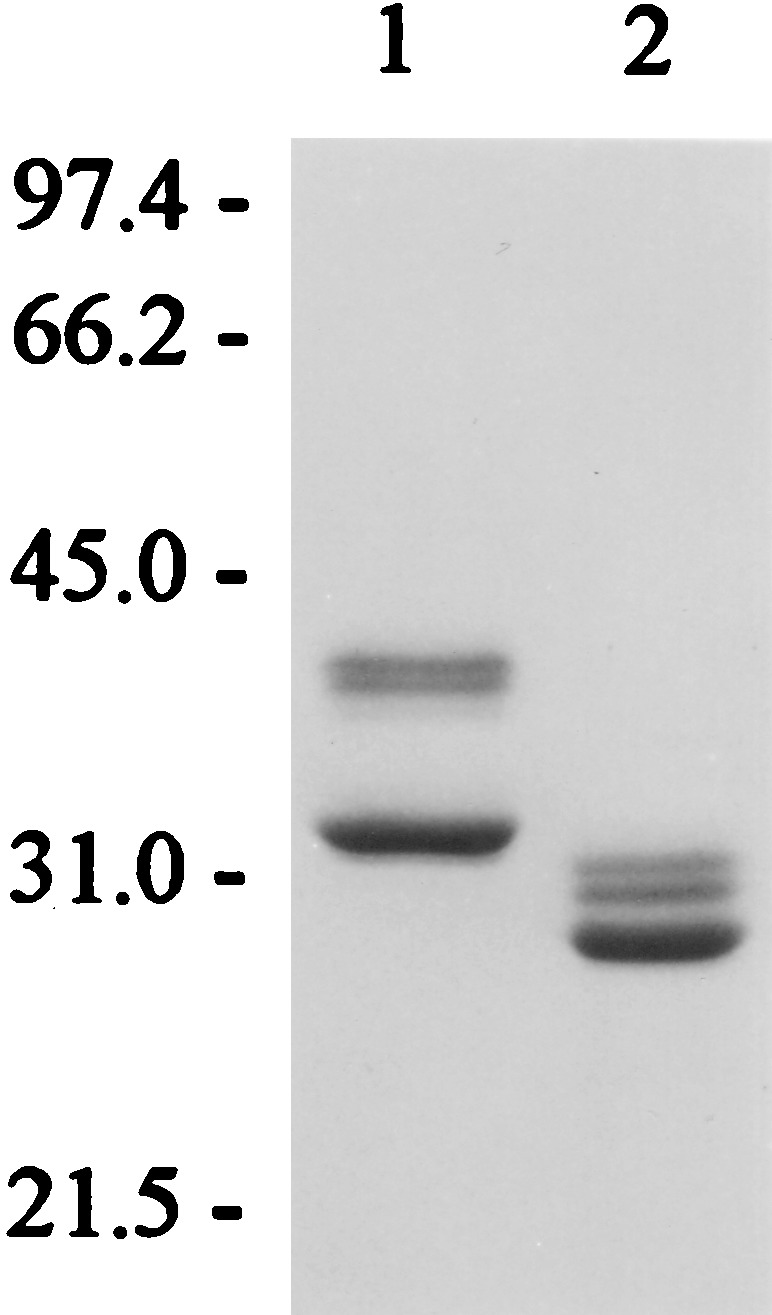
SDS/PAGE of purified HLA-DR2/MBP peptide complex. Lane 1, 5 μg of DR2/MBP prior to cleavage of leucine zipper dimerization domains; lane 2, 3.6 μg of DR2/MBP after cleavage with V8 protease. The lower band represents the DRβ chain with the covalently linked MBP peptide; the higher molecular mass doublet, the DRα chain. The SDS/12% polyacrylamide gel was stained with Coomassie blue.
In HPLC gel filtration, the DR2/MBP peptide complex eluted as a single symmetrical peak of the appropriate molecular mass (Fig. 5). Importantly, there was no evidence of protein aggregates. Empty DR1 molecules were found to aggregate unless they were loaded with a high-affinity peptide (15). The absence of protein aggregates in the DR2/MBP peptide preparation indicated that the covalently bound peptide protected these molecules from aggregation. This may have been an important factor in the successful crystallization of the DR2/MBP peptide complex.
Figure 5.

HPLC gel filtration analysis of the HLA-DR2/MBP peptide complex. Because empty MHC class II molecules have a tendency to aggregate, the purified HLA-DR2/MBP peptide complex was examined by gel filtration chromatography (Bio-Sil SEC 250 column; Bio-Rad). Ten micrograms of DR2/MBP peptide complex was injected at time 0; the mobile phase was PBS, pH 6.8, the flow rate 1 ml/min, and the total run time 15 min. IgM, IgG, BSA, and β-lactoglobulin (molecular masses of >900, 150, 66.2, and 18.4 kDa) were used as standards (marked by arrows). The HLA-DR2/MBP peptide complex eluted at a molecular mass slightly higher than that of BSA. There was no evidence for aggregation of the HLA-DR2/MBP peptide complex. Axes as in Fig. 3.
Crystallization of the HLA-DR2/MBP Peptide Complex.
For crystallization, the protein was concentrated to 10 mg/ml by ultrafiltration. Crystallization was performed by the hanging drop method using 1 μl of protein (at 10 mg/ml) and 1 μl of well solution. PEG 6000 was evaluated as a precipitant because it had been successfully used to crystallize other HLA-DR molecules (16, 17, 26–28). The protein readily crystallized in 15–18% PEG 6000 at a pH of 3.0 to 4.0. Several different crystal morphologies were observed, including tetragonal crystals (Fig. 6). These crystals were found to diffract to a resolution of ≈3.5Å under room temperature conditions on an Elliot GX-13 rotating anode x-ray source.
Figure 6.

Crystal of the HLA-DR2/MBP peptide complex. The HLA-DR2/MBP peptide complex was crystallized by the hanging drop method, using 1 μl of HLA-DR2/MBP peptide complex (10 mg/ml) and 1 μl of well solution. This crystal was grown in 15% PEG 6000/100 mM glycine, pH 3.5 at 18°C. Dimensions of the crystal were ≈0.7 × 0.3 mm.
Appropriate harvesting/cryocooling conditions were difficult to establish because crystals had a tendency to dissolve or crack. However, the crystals could be stabilized by harvesting into a buffer with a higher concentration of PEG 6000 (25–30%). After harvesting in buffers with a high PEG concentration, crystals were transferred to buffers containing increasing concentrations (5%, 10%, and 15%) of different cryoprotectants (ethylene glycol, glycerol, 2-methyl-2,4-pentanediol, PEG 450) and flash-cooled in liquid nitrogen; best results were obtained with ethylene glycol. Seeding was used to increase the yield of crystals with a tetragonal morphology. Serial dilution of microcrystals and streak seeding yielded large numbers of tetragonal crystals, ranging is size from microcrystals to crystals that were >0.5 mm in one dimension. These crystals were harvested in 30% PEG/100 mM glycine, pH 3.5, transferred to buffers containing increasing amounts of ethylene glycol (5%, 10%, and 15%) and flash-cooled in liquid nitrogen. Several of these crystals were analyzed at a synchrotron radiation source (CHESS, Cornell University), and one of these crystals diffracted to a resolution of 2.6 Å.
Summary.
Leucine zipper dimerization domains allowed the assembly and secretion of HLA-DR2; these dimerization domains could be cleaved from assembled HLA-DR2/MBP peptide complexes by using small quantities of V8 protease. The covalently linked MBP peptide improved the yield and homogeneity of defined HLA-DR2/peptide complexes. Such defined HLA-DR/peptide complexes may facilitate cocrystallization with TCRs by allowing crystallization in the presence of stoichiometric amounts of TCRs and the appropriate MHC class II/peptide complex.
Acknowledgments
We thank Dr. Hidde Ploegh for providing polyclonal DRα and DRβ antisera. This work was supported by grants from the National Institutes of Health (AI 42316 to K.W.W. and HD-17461 to D.C.W.), the Howard Hughes Medical Institute, the National Multiple Sclerosis Society (to K.W.W.), and the Howard Hughes Medical Institute. K.W.W. is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society. K.J.S. is supported by a Wellcome Trust Travelling Fellowship. D.C.W. is an investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- MHC
major histocompatibility complex
- TCR
T cell receptor
- MBP
myelin basic protein
References
- 1.The Multiple Sclerosis Study Group. Nat Genet. 1996;13:469–471. doi: 10.1038/ng0896-469. [DOI] [PubMed] [Google Scholar]
- 2.Sawcer S, Jones H B, Feakes R, Gray J, Smaldon N, Chataway J, Robertson N, Clayton D, Goodfellow P N, Compston A. Nat Genet. 1996;13:464–468. doi: 10.1038/ng0896-464. [DOI] [PubMed] [Google Scholar]
- 3.Ebers G C, Kukay K, Bulman D E, Sadovnick A D, Rice G, Anderson C, Armstrong H, Cousin K, Bell R B, Hader W, et al. Nat Genet. 1996;13:472–476. doi: 10.1038/ng0896-472. [DOI] [PubMed] [Google Scholar]
- 4.Hillert J, Kall T, Vrethem M, Fredrikson S, Ohlson M, Olerup O. J Neuroimmunol. 1994;50:95–100. doi: 10.1016/0165-5728(94)90219-4. [DOI] [PubMed] [Google Scholar]
- 5.Spielman R S, Nathenson N. Epidemiol Rev. 1982;4:45–65. doi: 10.1093/oxfordjournals.epirev.a036251. [DOI] [PubMed] [Google Scholar]
- 6.Oksenberg J R, Seboun E, Hauser S L. Brain Pathol. 1996;6:289–302. doi: 10.1111/j.1750-3639.1996.tb00856.x. [DOI] [PubMed] [Google Scholar]
- 7.Kalluri R, Wilson C B, Weber M, Gunwar S, Chonko A M, Neilson E G, Hudson B G. J Am Soc Nephrol. 1995;6:1178–1185. doi: 10.1681/ASN.V641178. [DOI] [PubMed] [Google Scholar]
- 8.Wucherpfennig K W, Sette A, Southwood S, Oseroff C, Matsui M, Strominger J L, Hafler D A. J Exp Med. 1994;179:279–290. doi: 10.1084/jem.179.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valli A, Sette A, Kappos L, Oseroff C, Sidney J, Miescher G, Hochberger M, Albert E D, Adorini L. J Clin Invest. 1993;91:616–628. doi: 10.1172/JCI116242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogt A B, Kropshofer H, Kalbacher H, Kalbus M, Rammensee H-G, Coligan J E, Martin R. J Immunol. 1994;151:1665–1673. [PubMed] [Google Scholar]
- 11.Wucherpfennig K W, Strominger J L. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hemmer B, Fleckenstein B T, Vergelli M, Jung G, McFarland H, Martin R, Wiesmüller K-H. J Exp Med. 1997;185:1651–1659. doi: 10.1084/jem.185.9.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garboczi D N, Hung D T, Wiley D C. Proc Natl Acad Sci USA. 1992;89:3429–3433. doi: 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altman J D, Reay P A, Davis M M. Proc Natl Acad Sci USA. 1993;90:10330–10334. doi: 10.1073/pnas.90.21.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stern L J, Wiley D C. Cell. 1992;68:465–477. doi: 10.1016/0092-8674(92)90184-e. [DOI] [PubMed] [Google Scholar]
- 16.Stern L J, Brown J H, Jardetzky T S, Urban R, Strominger J L, Wiley D C. Nature (London) 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 17.Dessen A, Lawrence C M, Cupo S, Zaller D M, Wiley D C. Immunity. 1997;7:473–481. doi: 10.1016/s1074-7613(00)80369-6. [DOI] [PubMed] [Google Scholar]
- 18.Kalandadze A, Galleno M, Foncerrado L, Strominger J L, Wucherpfennig K W. J Biol Chem. 1996;271:20156–20162. doi: 10.1074/jbc.271.33.20156. [DOI] [PubMed] [Google Scholar]
- 19.Scott C A, Garcia K C, Carbone F R, Wilson I A, Teyton L. J Exp Med. 1996;183:2087–2095. doi: 10.1084/jem.183.5.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozono H, White J, Clements J, Marrack P, Kappler J W. Nature (London) 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski Z. In: Proceedings of CCP4 Study Weekend: Data Collection and Processing. Sawyer L, Isaacs N, Bailey S, editors. Warrington, U.K.: SERC Daresbury Lab.; 1993. pp. 56–62. [Google Scholar]
- 22.Gorga J C, Horejsi V, Johnson D R, Raghupathy R, Strominger J L. J Biol Chem. 1987;262:16087–16094. [PubMed] [Google Scholar]
- 23.Sone T, Tsukamoto K, Hirayama K, Nishimura Y, Takenouchi T, Aizawa M, Sasazuki T. J Immunol. 1985;135:1288–1298. [PubMed] [Google Scholar]
- 24.Scott C A, Garcia K C, Peterson P A, Wilson I A, Teyton L. Protein Sci. 1998;7:413–418. doi: 10.1002/pro.5560070222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott C A, Peterson P A, Teyton L, Wilson I A. Immunity. 1998;8:319–329. doi: 10.1016/s1074-7613(00)80537-3. [DOI] [PubMed] [Google Scholar]
- 26.Gorga J C, Brown J H, Jardetzky T, Wiley D C, Strominger J L. Res Immunol. 1991;142:401–407. doi: 10.1016/0923-2494(91)90038-k. [DOI] [PubMed] [Google Scholar]
- 27.Brown J H, Jardetzky T S, Gorga J C, Stern L J, Urban R G, Strominger J L, Wiley D C. Nature (London) 1993;364:33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh P, Amaya M, Mellins E, Wiley D C. Nature (London) 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]