

Abstract
The αvβ3 integrin participates in cell morphogenesis, growth factor signaling, and cell survival. Activation of the integrin is central to these processes and is influenced by specific ECM components, which engage both integrins and syndecans. This paper demonstrates that the αvβ3 integrin and syndecan-1 (S1) are functionally coupled. The integrin is dependent on the syndecan to become activated and to mediate signals required for MDA-MB-231 and MDA-MB-435 human mammary carcinoma cell spreading on vitronectin or S1-specific antibody. Coupling of the syndecan to αvβ3 requires the S1 ectodomain (ED), as ectopic expression of glycosylphosphatidylinositol-linked S1ED enhances αvβ3 recognition of vitronectin; and treatments that target this domain, including competition with recombinant S1ED protein or anti-S1ED antibodies, mutation of the S1ED, or down-regulation of S1 expression by small-interfering RNAs, disrupt αvβ3-dependent cell spreading and migration. Thus, S1 is likely to be a critical regulator of many cellular behaviors that depend on activated αvβ3 integrins.
Introduction
The αvβ3 integrin is a key regulator of adhesion and signaling in numerous biological processes, including tumor cell migration and metastasis, and angiogenesis. The activated form of this integrin participates in arrest of tumor cells in the blood stream (Pilch et al., 2002), enhancing their extravasation to target tissues, especially bone, where the activated integrin has further roles in tumor cell proliferation and survival (Brooks et al., 1994; Petitclerc et al., 1999; Eliceiri, 2001). In endothelial cells forming new blood vessels, the active integrin is linked not only to adhesion-dependent processes but also to signaling in response to FGF-2 (Eliceiri et al., 1998; Hood et al., 2003).
Although αvβ3 integrin expression in mammary epithelium is low, activated αvβ3 is expressed on most, if not all, successful mammary carcinoma metastases (Liapis et al., 1996; Felding-Habermann et al., 2001). We have reported previously that αvβ3 integrin on MDA-MB-231 mammary carcinoma cells appears to be functionally linked to syndecan-1 (S1); the cells spread when adherent to an artificial substratum comprised solely of S1-specific antibody, and although this spreading occurs in the absence of an αvβ3 ligand, the spreading requires activated αvβ3 integrin (Beauvais and Rapraeger, 2003). This finding suggests that even on a native ECM, anchorage of S1 to the matrix may serve as an important regulator of αvβ3 integrin activation and signaling.
Although classically defined as a vitronectin (VN) receptor, αvβ3 is promiscuous and binds many ECM components including fibronectin (FN), fibrinogen, von Willebrand Factor, proteolysed fragments of collagen (COL), laminin (LN), osteopontin, and others (van der Flier and Sonnenberg, 2001). Mechanisms leading to activation of this integrin are complex, including proteolytic cleavage (Ratnikov et al., 2002), conformational changes (affinity modulation), and clustering (avidity modulation; Carman and Springer, 2003). Activation is regulated by “inside-out” signals from the cell interior and is stabilized by ligand interactions that trigger “outside-in” signaling (Giancotti and Ruoslahti, 1999). Cell surface receptors known to modulate αvβ3 activity include CD87/uPAR and CD47/IAP, which associate with the β3 integrin subunit via their extracellular domains (Lindberg et al., 1996; Xue et al., 1997) and may also regulate αvβ3 function indirectly via a pertussis-toxin–sensitive G-protein signaling pathway (Gao et al., 1996; Degryse et al., 2001).
The syndecan family of cell surface heparan sulfate (HS) proteoglycans is comprised of four vertebrate members. These receptors are expressed on virtually all cell types, although their expression may be altered in disease states such as cancer (Beauvais and Rapraeger, 2004). The syndecan core proteins share a high degree of conservation in their short cytoplasmic and transmembrane (TM) domains; in contrast, their ectodomains (EDs) are divergent with the exception of attachment sites for HS glycosaminoglycans. Via their HS chains, syndecans regulate the signaling of growth factors, chemokines, and morphogens and engage components of the ECM including VN, FN, LN, tenascin, thrombospondin, and the fibrillar COLs (Bernfield et al., 1999).
In addition to the activities of their HS chains, the syndecan core proteins have roles in cell adhesion signaling (Rapraeger, 2000; Tumova et al., 2000). Conserved and variable regions of the syndecan cytoplasmic domains appear critical for binding interactions that lead to adhesion-mediated signaling and reorganization of the actin cytoskeleton (Couchman et al., 2001). Important roles for the TM domain have also been demonstrated for S1 and S4 (Tkachenko and Simons, 2002; McQuade and Rapraeger, 2003). Perhaps the least expected active protein domain is the syndecan ED, which bears the HS chains. Nonetheless, several emerging studies suggest that the syndecan ED may have important regulatory roles in cell adhesion signaling. Cell spreading and morphogenetic activities in COS-7 and Schwann cells trace in part to the S1ED (Carey et al., 1994; Adams et al., 2001). Raji cells require the S1 TM domain for initial spreading, but depend on a S1ED activity for cell polarization (McQuade and Rapraeger, 2003). Moreover, inhibition of ARH-77 myeloma and hepatocellular carcinoma cell invasion into a COL I matrix by S1 also traces to a region of its extracellular core protein domain (Liu et al., 1998; Ohtake et al., 1999).
The activities of other syndecans also trace to their EDs. Overexpression of syndecan-2 (S2) in COS-1 and Swiss 3T3 cells induces filipodial extension and deletion mutants of S2 map activity to the S2ED (Granes et al., 1999). Up-regulation of S2 expression in colon carcinoma cells leads to altered cell morphology and colony formation in soft agar; treatment with recombinant S2ED disrupts these behaviors (Park et al., 2002; Kim et al., 2003). Finally, activated B-lymphocytes, when seeded on S4ED antibodies, exhibit morphological changes and filipodial extensions. Intriguingly, only the S4ED is required for this response, indicating that it may interact with a TM partner to transmit a dendritic signal (Yamashita et al., 1999).
Our previous work in the MDA-MB-231 cells suggested that cell spreading induced upon anchorage of the cells to a S1 antibody relies on functional coupling of the syndecan to activated αvβ3 integrins (Beauvais and Rapraeger, 2003). This spreading response is rapid (∼15–30 min) and occurs even in the absence of an integrin ligand (i.e., spreading is not blocked by cycloheximide or EGTA treatment), so long as the cells are adherent via S1. Intriguingly, the αvβ3-dependent spreading mechanism is blocked by the addition of soluble, recombinant S1ED, suggesting that anchorage of S1 to a ligand provides a platform for αvβ3 integrin activation and adhesion signaling via binding interaction of its ED. These findings raised a fundamental question about the role of S1 in ECM signaling, in particular whether or not S1 is required for αvβ3 activation and signaling in response to a native matrix ligand. Here, we show that MDA-MB-231 and MDA-MB-435 human mammary carcinoma cells, which express αvβ3, require S1 engagement with the matrix for αvβ3 activity on VN. Addition of recombinant mouse S1 (mS1) ED or anti-S1ED polyclonal antibodies (pAbs) blocks integrin activation and disrupts αvβ3-dependent spreading and migration of MDA-MB-231 and -435 cells. In contrast, α5β1-dependent spreading and migration on FN is unaffected by these treatments. Furthermore, we show that down-regulation of human S1 (hS1) expression by small-interfering RNA (siRNA) disrupts MDA-MB-231 cell spreading and migration on VN, but not on FN. Expression of a mS1 construct containing the mS1ED alone tethered to the membrane by a glycosylphosphatidylinositol (GPI) tail, which is unaffected by the human-specific siRNA, rescues αvβ3-dependent spreading and migration on VN. These data suggest that S1 and the αvβ3 integrin are functionally coupled via the S1ED and that coupling is required for αvβ3 integrin activation and signaling.
Results
MDA-MB-231 cell spreading on VN is disrupted by soluble, recombinant S1ED
MDA-MB-231 human mammary carcinoma cells, plated in the absence of serum, adhere to and spread (∼15–20 min after plating) on wells coated with either 10 μg/ml VN or FN (Fig. 1). Cells treated with 30 μg/ml LM609 to block αvβ3 integrins fail to spread. LM609 has no effect on spreading in response to FN; instead, the cells rely on α5β1 integrin to respond to FN as spreading is blocked by either 25 μg/ml mAb 13 (Fig. 1; Mould et al., 1996) or mAb 16 (Akiyama et al., 1989; unpublished data). Our previous works have shown that αvβ3 integrins are essential for signaling when these cells are adherent to S1-specific antibody, suggesting a collaboration between these two adhesion receptors even in the absence of an integrin ligand; this collaboration between S1 and αvβ3 integrins is disrupted by the addition of recombinant GST-mS1ED (Beauvais and Rapraeger, 2003). To test whether or not a similar S1/αvβ3 collaboration is at work when cells are bound to matrix, cells were plated on either VN or FN in the presence of 20 μM of recombinant GST-mS1ED. This treatment blocks cell spreading on VN but has no effect on cell spreading on FN (Fig. 1). Treatment with either GST alone (unpublished data) or GST-mS4ED has no effect on spreading on either ligand (Fig. 1, insets).
Figure 1.
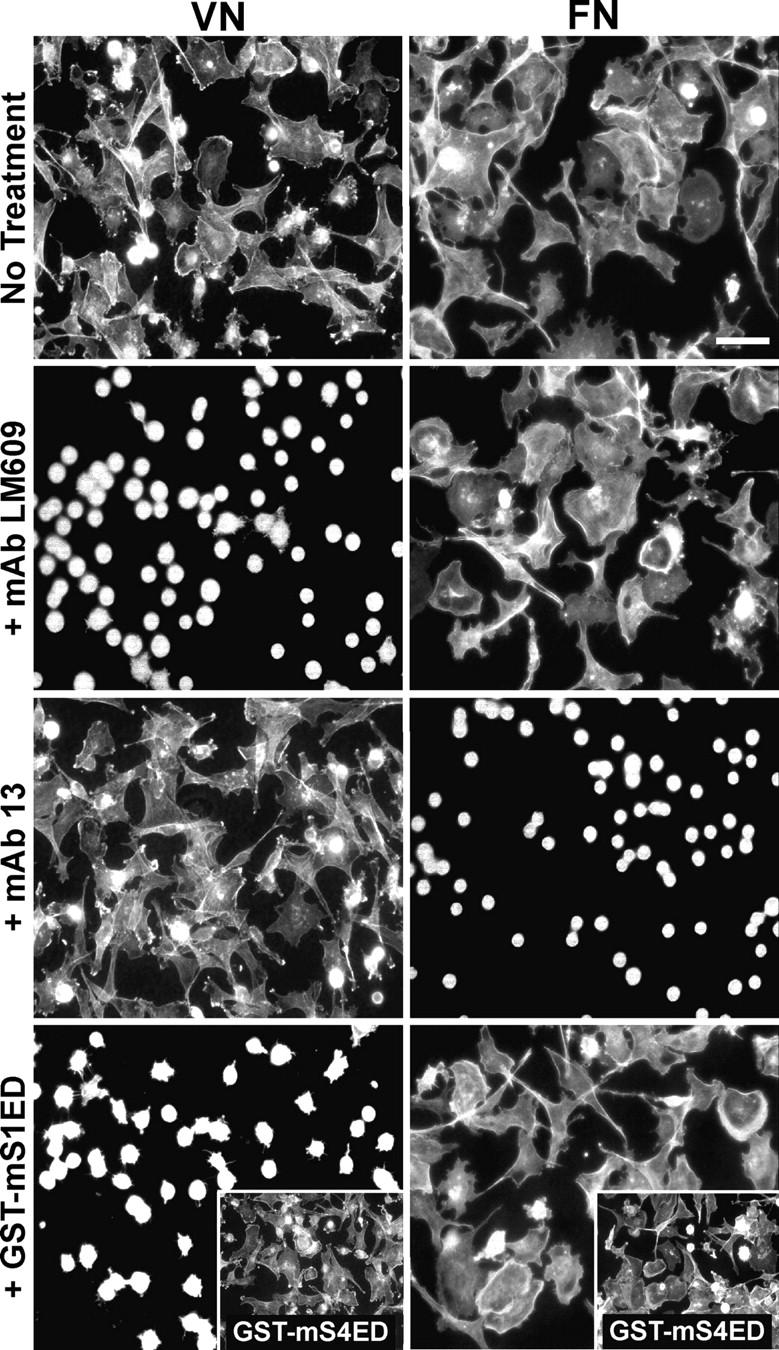
MDA-MB-231 human mammary carcinoma cell spreading on VN, but not FN, is disrupted by soluble, recombinant S1ED. Cells were plated on wells coated with 10 μg/ml VN or FN in plating medium alone or medium containing either 30 μg/ml mAb LM609, 25 μg/ml mAb 13, or 20 μM GST-mS1ED or -mS4ED. Cells were incubated at 37°C for 2 h, fixed, and stained with rhodamine-conjugated phalloidin. Bar, 50 μm.
S1 adhesion–mediated cell spreading correlates with αvβ3 integrin expression and activity
To test whether or not the αvβ3 integrin's dependence on S1 extends to other carcinoma cells, we screened a panel of human carcinoma cells with mAb LM609 using FACS analysis. MDA-MB-435 cells exhibit higher αvβ3 expression than MDA-MB-231 cells, whereas MCF-7 cells are negative for this integrin (Fig. 2 A). To test the collaboration between S1 and αvβ3 integrin when cells are adherent via S1, MDA-MB-435 and MCF-7 cells were plated on wells coated with hS1-specific mAb B-B4. Although αvβ3-positive MDA-MB-435 cells spread on this antibody substratum, their spreading is blocked (Beauvais and Rapraeger, 2003) by treatment with either mAb LM609 (added either before plating or 30 min after plating, when cells have already begun to spread) or GST-mS1ED protein (which is not recognized by mAb B-B4). MCF-7 (Fig. 2 B) and T47D (unpublished data) cells, which are αvβ3 negative, bind to mAb B-B4 but fail to spread.
Figure 2.
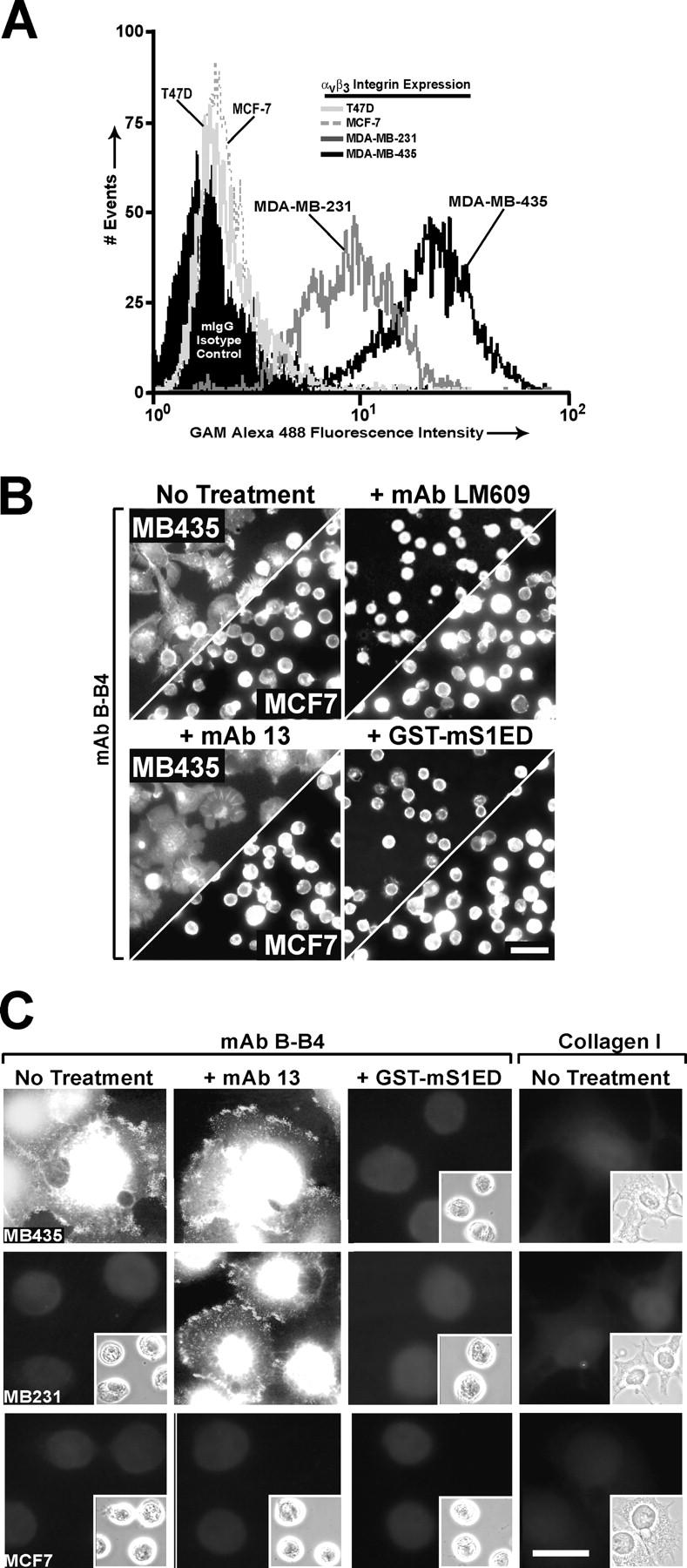
S1 adhesion–mediated cell spreading correlates with αvβ3 integrin expression and activity. (A) FACS analysis of αvβ3 integrin expression (mAb LM609) in human mammary carcinoma cells against an IgG isotype control. (B) Depicted on split panels are phalloidin-stained MDA-MB-435 (top) and MCF-7 (bottom) cells 2 h after plating on wells coated with mAb B-B4 in plating medium alone or medium containing either 30 μg/ml mAb LM609, 1 μg/ml mAb 13, or 20 μM GST-mS1ED. Bar, 50 μm. (C) Untreated MDA-MB-435, MDA-MB-231, and MCF-7 cells (first and last columns) and cells pretreated in suspension with 1 μg/ml mAb 13 (middle columns) were seeded on wells coated with either mAb B-B4 or COL I in plating medium alone (first, second, and last columns) or medium containing 20 μM GST-mS1ED (third column). Cells were incubated at 37°C for 2 h, fixed, permeabilized, and stained with WOW1 and an Alexa 488–conjugated secondary antibody. Panel insets are corresponding phase-contrast pictures. Bar, 20 μm.
To verify that cell spreading induced upon S1 ligation correlates with αvβ3 activation, we relied on binding of the ligand-mimetic WOW1 mouse Fab, a probe that detects activated but unligated αvβ3 integrin. MDA-MB-435 cells adherent to mAb B-B4 display strong binding of WOW1 both centrally and within puncta at the spreading margin of cells (Fig. 2 C). In contrast, WOW1 fails to bind to cells treated with 20 μM GST-mS1ED, and this correlates with the failure of the cells to spread (Fig. 2 C, phase insets). WOW1 also binds to MDA-MB-231 cells adherent to mAb B-B4 and pretreated with mAb 13 before plating; treatment with mAb 13 relieves a negative β1-β3 integrin cross-talk mechanism active in the MDA-MB-231 cells, thus allowing for αvβ3 integrin activation (Beauvais and Rapraeger, 2003). Like the MDA-MB-435 cells, WOW1 staining is also lost from the MDA-MB-231 cells when cells are treated with 20 μM GST-mS1ED. As a control, WOW1 fails to bind MDA-MB-435 or MB-231 cells adherent and spread on COL I, a β1 integrin–specific ligand. This finding agrees with growing evidence that activation of certain β1 integrins can down-regulate αvβ3 integrin activity in several cell lines (Kim et al., 2000; Kiosses et al., 2001; Gonzalez et al., 2002; Beauvais and Rapraeger, 2003). We do not see WOW1 binding to the αvβ3-negative MCF-7 cells.
MDA-MB-435, but not MCF-7, human carcinoma cells display functional coupling of S1 and αvβ3 integrin on VN
To test if αvβ3 integrin activation on an ECM ligand is also functionally coupled to S1, MDA-MB-435 and MCF-7 cells were plated on either VN or FN (Fig. 3). MDA-MB-435 cells spread on VN and require αvβ3 integrins for this activity as spreading is blocked by mAb LM609. Although the MCF-7 cells spread on VN, this spreading is unaffected by LM609; these cells rely instead on αvβ1 integrins as spreading is blocked by either mAb 13 (Fig. 3) or αv-specific mAb M9 (de Vries et al., 1986). Neither cell type uses αvβ3 to respond to FN, rather both use α5β1 integrins that are blocked by either mAb 13 (Fig. 3) or mAb 16 (unpublished data).
Figure 3.
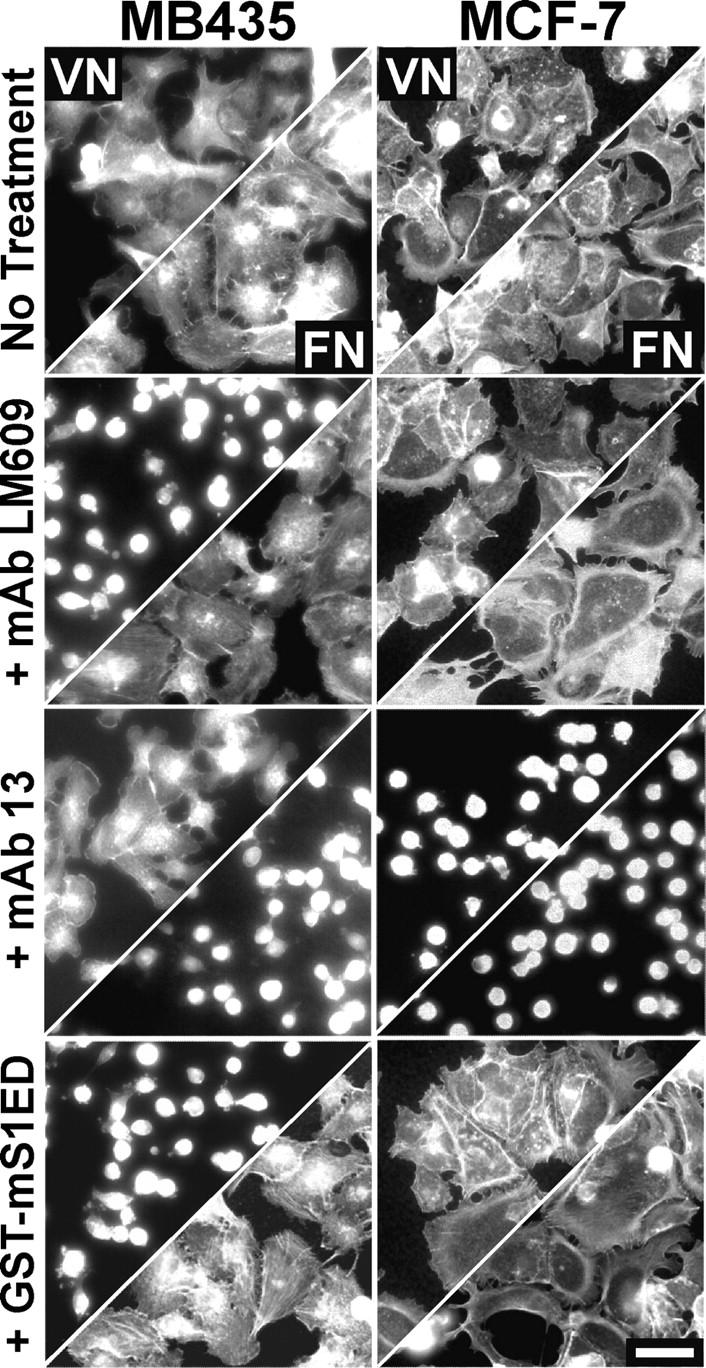
MDA-MB-435, but not MCF-7, human carcinoma cells display functional coupling of S1 and αvβ3 integrins on VN. Depicted are phalloidin-stained cells 2 h after plating on wells coated with 10 μg/ml VN (top half of panels) or FN (bottom half) in plating medium alone or medium containing either 30 μg/ml mAb LM609, 25 μg/ml mAb 13, or 20 μM GST-mS1ED or -mS4ED. Bar, 50 μm.
To test whether or not GST-mS1ED specifically blocks αvβ3 integrin–dependent spreading or acts as a general inhibitor to all αv integrin heterodimers, MDA-MB-435 and MCF-7 cells were plated on VN in the presence of 20 μM GST-mS1ED. Only the MDA-MB-435 cells fail to spread in response to VN after treatment with GST-mS1ED, although they retain their ability to spread on FN. This effect appears specific for S1ED as treatment with GST alone or GST-mS4ED (unpublished data) has no effect.
Polyclonal S1ED antibodies disrupt αvβ3 integrin–dependent cell spreading and migration on VN
To target the syndecan directly, MDA-MB-231 and MB-435 (αvβ3-positive) cells were treated with S1ED-specific pAb before plating. The pAb recognizes S1 on blots and live cells, but fails to recognize other syndecan family members (unpublished data). The cells display a dose-dependent inhibition in VN-dependent cell spreading over a pAb concentration range of 10–250 μg/ml (Fig. 4). The number of spread cells (diameter ≥20 μm) on VN was reduced from 92 ± 6% in the absence of pAb to 30 ± 4% at 100 μg/ml pAb with almost complete inhibition (≤7%) for both cell types at 250 μg/ml. Note that the treatment of either cell type with pAb does not alter their spreading in response to FN. The relatively high pAb concentration required to achieve full inhibition of spreading may indicate that the “blocking” antibody is a relatively minor fraction of the pAb mix. Treatment of cells with 250 μg/ml of anti-GST pAb has no effect on cell spreading on either matrix ligand (unpublished data).
Figure 4.
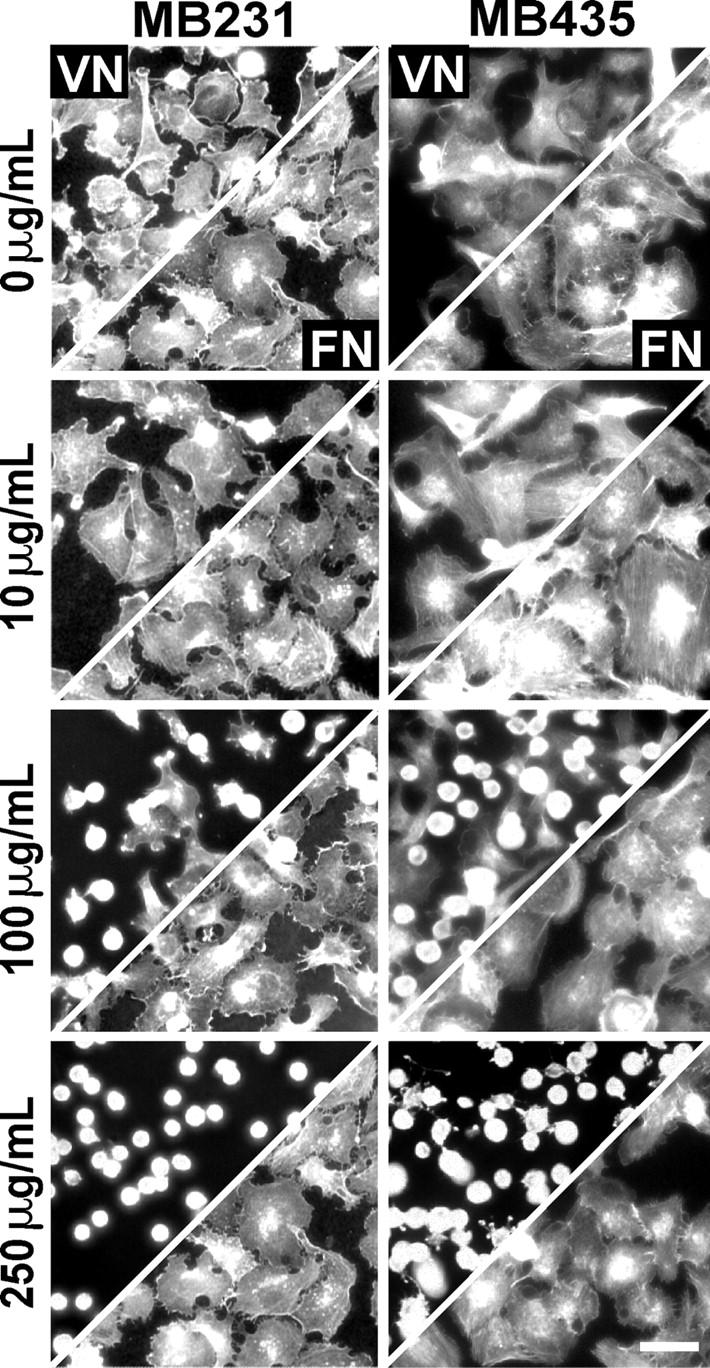
Polyclonal S1ED antibodies disrupt αvβ3 integrin–dependent cell spreading on VN. MDA-MB-231 and -435 cells were plated on wells coated with 10 μg/ml VN (top half) or FN (bottom half) in plating medium alone or medium containing 10, 100, or 250 μg/ml of anti-mS1ED pAb. Cells were incubated at 37°C for 2 h, fixed, and stained with rhodamine-conjugated phalloidin. Bar, 50 μm.
To test the role of the S1ED in αvβ3 integrin–dependent signaling in another functional assay, cells were examined for their ability to migrate across VN- or FN-coated filters in the presence or absence of S1ED pAb (250 μg/ml) or GST-mS1ED recombinant protein (20 μM). Migration of treated MDA-MB-231 and MB-435 cells across VN is suppressed two- to fourfold relative to untreated controls (Fig. 5 A). MCF-7 cell migration across VN, which is αvβ1 dependent, is unaffected by either treatment. Furthermore, neither inhibitor has any effect on cell migration across FN in any of the three cell types tested (Fig. 5 B).
Figure 5.
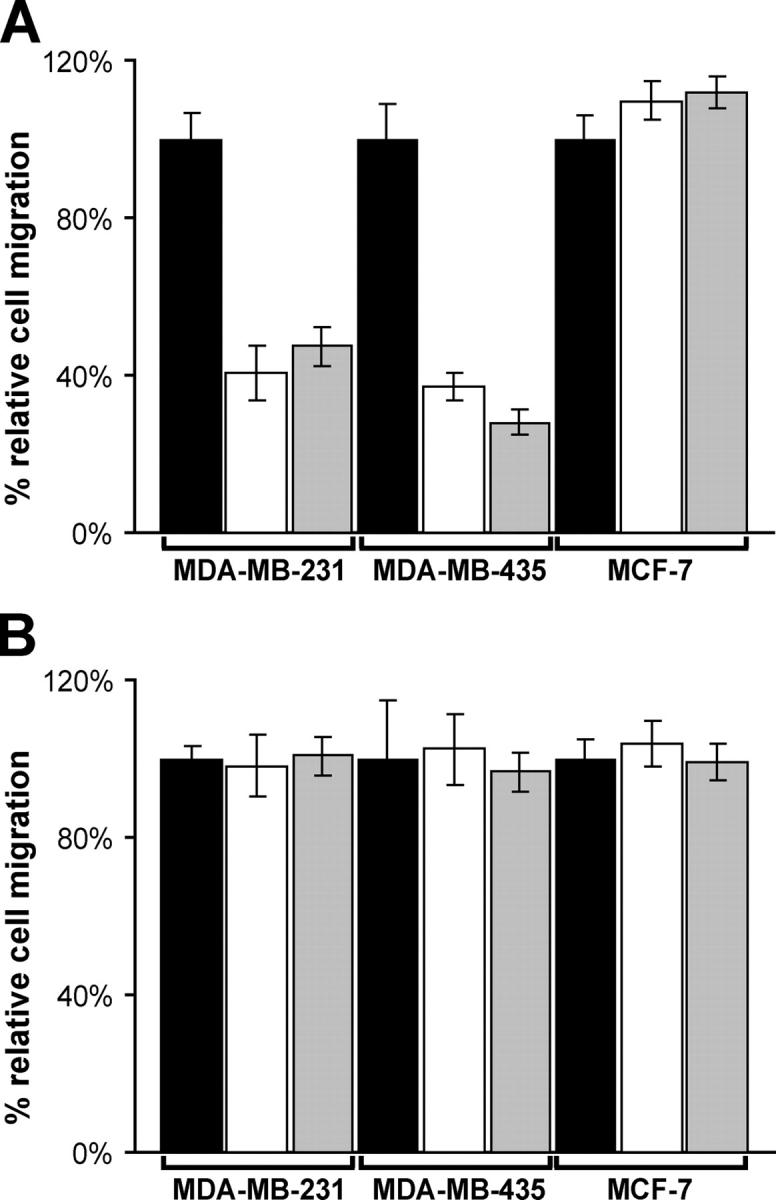
S1ED inhibitors disrupt αvβ3 integrin–dependent cell migration on VN. MDA-MB-231, MDA-MB-435, and MCF-7 cells in plating medium alone (black) or in plating medium containing either 20 μM GST-mS1ED (white) or 250 μg/ml mS1ED pAbs (gray) were seeded on polycarbonate filters coated with either 10 μg/ml VN (A) or FN (B) in a modified Boyden chamber. After 16 h, cells that migrated through the filter in response to 10% FBS in the lower chamber were quantified by colorimetric staining. The error bars represent the SEM from three independent experiments.
Activity resides within the ED of the S1 core protein
To confirm that the S1ED is necessary and sufficient for the activation of αvβ3 integrin–dependent cell spreading, MDA-MB-231 cells were transfected with mS1 expression constructs (Fig. 6 A). Populations of high-expressing clones were sorted by FACS analysis using mAb 281.2, an antibody that selectively recognizes mS1, to ensure comparable levels of expression. Cells were then plated on S1-specific antibodies to assess their ability to spread in response to S1 ligation.
Figure 6.
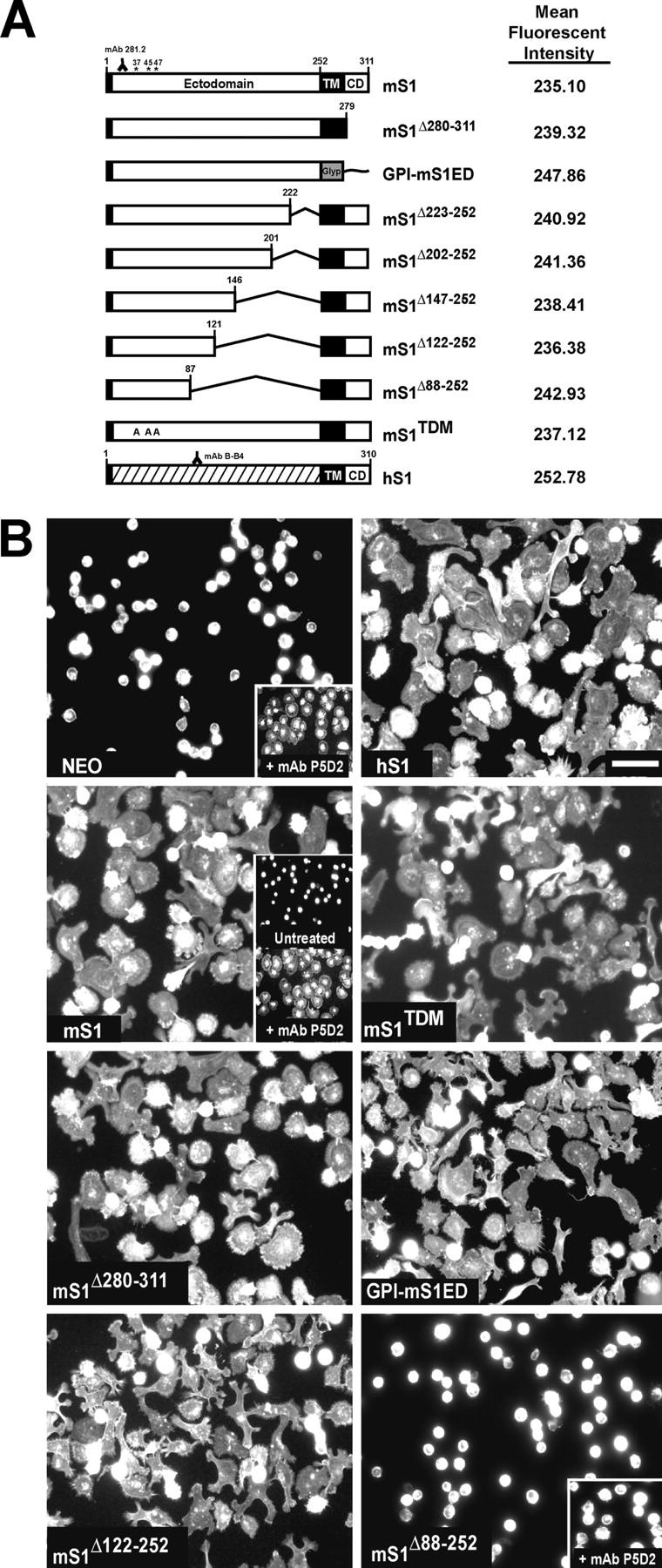
Deletion of a region of the S1ED blocks αvβ3-mediated cell spreading. (A) Graphic representation of S1 expression constructs transfected into MDA-MB-231 cells and their relative expression levels as detected by FACS (mean fluorescent intensity). Asterisks indicate the HS attachment sites. (B) Cells transfected with empty vector (NEO) or S1 constructs were seeded in plating medium on wells coated with either anti-hS1 mAb B-B4 (NEO + inset, hS1, and insets of mS1) or anti-mS1 mAb 281.2 (all others). Where noted, cells were pretreated in suspension with 1 μg/ml mAb P5D2 for 15 min before plating. Cells were incubated at 37°C for 2 h, fixed, and stained with rhodamine-conjugated phalloidin. Bar, 50 μm.
NEO (pcDNA3 empty vector) cells (Fig. 6 B) adherent to hS1-specific antibody mAb B-B4 fail to spread unless treated with mAb P5D2, a β1 integrin–neutralizing antibody (Fig. 6, NEO, inset), that relieves an α2β1-dependent repression of αvβ3 integrins (Beauvais and Rapraeger, 2003). To test the activity of the mS1 constructs, cells were plated on mAb 281.2. Interestingly, cells expressing full-length mS1 spread (Fig. 6 B, mS1) without prior inhibition of β1. This response is not unique to mS1, as hS1-overexpressors (Fig. 6 B, hS1), when plated on mAb B-B4, also spread in the absence of β1 integrin blockade, suggesting that overexpression of S1 overcomes the negative β1-β3 integrin cross-talk mechanism in these cells. Intriguingly, mS1-overexpressing cells fail to spread on mAb B-B4 unless cells are treated with the β1 blocker (Fig. 6, mS1, insets) mimicking the response of NEO cells plated on a similar substratum (Fig. 6 B, NEO). Thus, endogenous hS1 and ectopic mS1 appear to act independently of each other and the cells respond only to the ligated S1.
To identify the properties of S1 required to regulate αvβ3 integrin activity, cells expressing mS1 mutants were plated on a substratum of mAb 281.2. A mutant that lacks its HS chains (mS1TDM) retains its ability to spread. Cells expressing a S1 construct that lacks either its cytoplasmic domain (mS1Δ280-311) or both its TM and cytoplasmic domains (GPI-mS1ED) retain their ability to spread, confirming that activity resides in the S1ED. Mutants with progressively larger ED deletions (mS1Δ223-252, mS1Δ202-252, and mS1Δ147-252) all retain activity (unpublished data) as does a S1 construct (mS1Δ122-252) that lacks 131 amino acids located between the HS attachment sites and the TM domain. However, cells expressing a mutant (mS1Δ88-252) that lacks 34 additional amino acids, fail to spread; and spreading cannot be rescued by treatment with mAb P5D2 (Fig. 6, mS1Δ88-252, inset), a treatment that would have otherwise enhanced αvβ3 integrin activation (Fig. 6, NEO, inset).
Overexpression and ligation of S1 “primes” cells to spread in response to VN
To test if overexpression of S1 enhances VN recognition via the αvβ3 integrin, cell attachment and spreading were assessed on wells containing increasing concentrations of VN (1, 3, and 10 μg/ml; Fig. 7 A). NEO control cells largely fail to bind to wells coated with 1 μg/ml VN and display only modest spreading in response to 3 μg/ml. Full adhesion and spreading is not achieved until cells encounter high concentrations of VN (10 μg/ml). In contrast, cells overexpressing the S1ED (GPI-mS1ED) attach and spread on low VN concentrations and this response increases with higher concentrations of VN. However, this response is dependent on S1's engagement of the matrix as cells expressing mS1TDM (which lacks its HS chains) mimic the response of NEO control cells.
Figure 7.
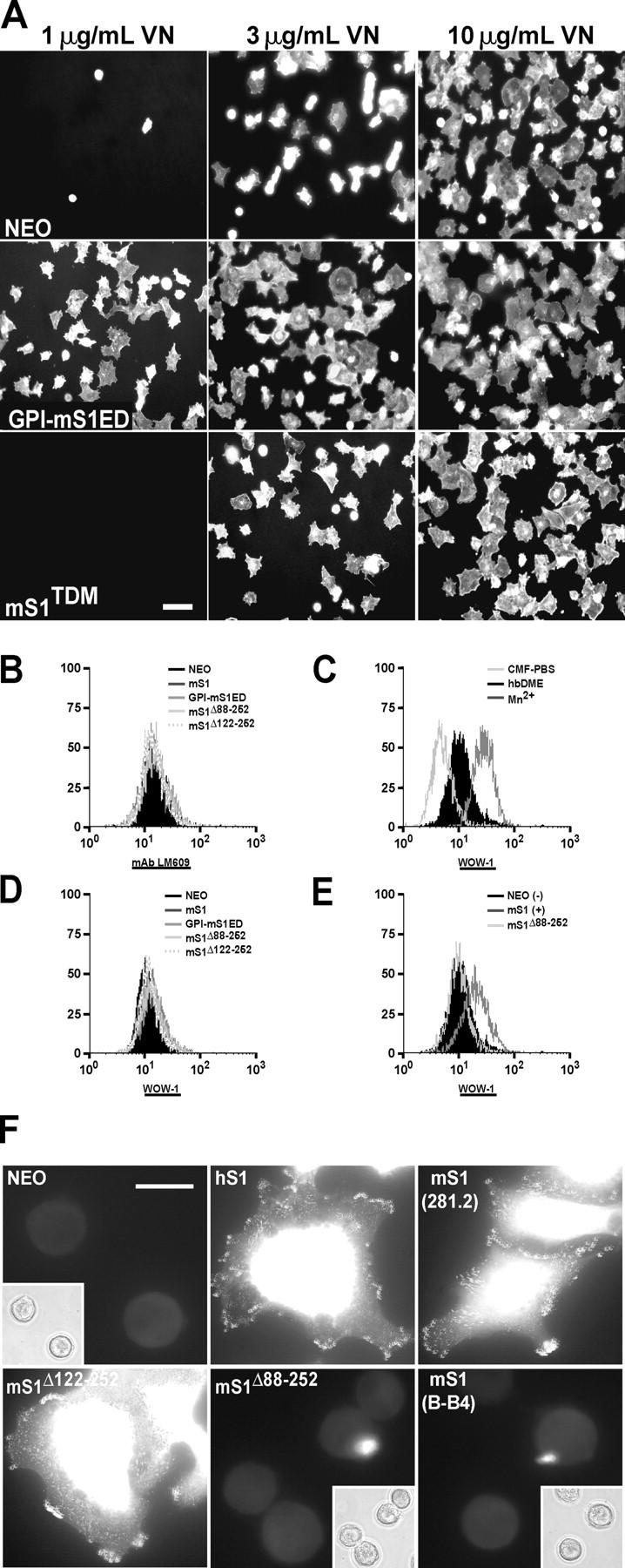
Overexpression and ligation of S1 activates αvβ3 integrins and primes cells to spread on VN. (A) MDA-MB-231 cells transfected with empty vector (NEO), GPI-mS1ED, or mS1TDM were seeded on wells coated with 1, 3, or 10 μg/ml VN. Cells were incubated at 37°C for 2 h, fixed, and stained with rhodamine-conjugated phalloidin. Bar, 50 μm. (B–E) Suspended cells in which mS1 was clustered (mAb 281.2, B and E) or not clustered (mAb KY8.2, C and D) were fixed and labeled with mAb LM609 (B) or WOW1 mouse Fab (C–E) followed by an Alexa 488–conjugated secondary antibody and analyzed by FACS. As controls for WOW1 staining (C), suspended cells were incubated with plating medium alone (black-filled histogram), plating medium containing 1 mM MnCl2 (right-shifted histogram), or divalent cation-free PBS (left-shifted histogram) before fixation and staining. (F) Cells were seeded on wells coated with either mAb B-B4 or 281.2, incubated at 37°C for 2 h, fixed, permeabilized, and stained with WOW1 and an Alexa 488–conjugated secondary antibody. Panel insets are corresponding phase-contrast pictures. Bar, 20 μm.
These results correlate with the induction in αvβ3 activation observed when cells are adherent to S1 antibody (Fig. 2 C) and suggest that αvβ3 activation depends on S1 being engaged by ligand. To test this, integrin activation and expression was examined in S1-overexpressing cells in suspension (± mS1 clustering) and when adherent to S1 antibody. On suspended cells when mS1 is not clustered, we detect no increase in αvβ3 activity (Fig. 7 D, WOW1). However, when mS1 is clustered using mAb 281.2, although we detect no change in αvβ3 expression (Fig. 7 B, mAb LM609), we do detect a demonstrable increase in αvβ3 activity with either mS1 or GPI-mS1ED (unpublished data), but not with mS1Δ88-252 (Fig. 7 E, WOW1). Activation of the integrin with 1.0 mM Mn2+ (Smith et al., 1994; Lin et al., 1997; Pampori et al., 1999) is shown for comparison (Fig. 7 C); this contrasts with the diminished WOW1 binding observed following inactivation of the integrin by the removal of divalent cations (Fig. 7 C, CMF-PBS).
Although enhanced S1 expression has no effect on αvβ3 activation levels on suspended cells (Fig. 7 D), a different result is obtained when the cells are assessed in antibody-based adhesion assays (Fig. 7 F). Cells overexpressing hS1 or mS1 stain positively with WOW1 when adherent to their respective S1-specific antibodies, whereas NEO cells bound to B-B4 display no significant WOW1 binding. However, cells overexpressing mS1 fail to bind WOW1 when adherent via their endogenous hS1 (B-B4), indicating again that the syndecan must be ligated to efficiently activate the αvβ3 integrin. Cells overexpressing mS1Δ122-252, a S1ED mutant that retains its ability to signal spreading in response to S1 ligation (Fig. 6 B), also display positive staining for WOW1, but cells overexpressing mS1Δ88-252, a mutant which fails to signal spreading, do not.
Down-regulation of S1 expression by siRNA disrupts cell spreading and migration in response to VN
To test the activity of the mS1 mutants on matrix ligands, the expression of the endogenous hS1 needs to be blocked. Thus, cells expressing mS1 constructs were transfected with siRNA designed to specifically target hS1 (Fig. 8 A). Transfection with siRNA efficiently silences hS1 (>90% reduction) in both NEO vector-control cells (Fig. 8 B) and in cells expressing mS1 constructs (GPI-mS1ED provided as a representative result; Fig. 8 C). Importantly, hS1 siRNA affects neither mS1 expression (Fig. 8 E) nor the expression of hS4 in either NEO vector control or mS1-expressing cells (Fig. 8 D). In addition, hS1 siRNA has no effect on the expression levels of either αvβ3 or β1 integrins as determined by FACS (unpublished data).
Figure 8.
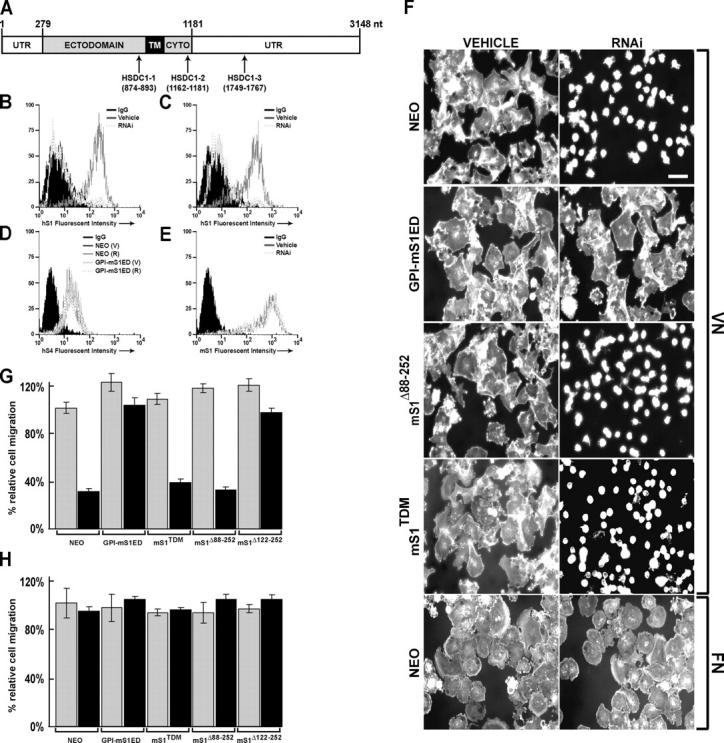
Down-regulation of S1 expression by siRNA disrupts cell spreading and migration on VN. (A) SiRNA targeting of hS1 mRNA. FACS analysis for (B and C) hS1 (mAb B-B4), (D) hS4 (mAb F94-8G3), and (E) mS1 (mAb 281.2) expression against IgG controls (black-filled histograms) in NEO- and GPI-mS1ED–expressing MDA-MB-231 cells 72 h after transfection with either lipid-vehicle alone (Vehicle or (V)) or 200 nM siRNA (RNAi or (R)). (F) NEOMDA-MB-231 cells and MDA-MB-231 cells expressing GPI-mS1ED, mS1Δ88-252, mS1Δ122-252, and mS1TDM were transfected with lipid-vehicle alone or 200 nM hS1-siRNA and seeded on wells coated with either 10 μg/ml VN or FN. Cells were incubated at 37°C for 2 h, fixed, and stained with rhodamine-conjugated phalloidin. Bar, 50 μm. (G and H) Lipid-vehicle (gray) or hS1-siRNA (black) transfected cells were also plated on polycarbonate filters coated with either 10 μg/ml VN (G) or FN (H) in a modified Boyden chamber. After 16 h, cells that migrated through the filter in response to 10% FBS in the lower chamber were quantified by colorimetric staining. The error bars represent the SEM from three independent experiments.
NEO vector control cells lacking hS1 fail to spread in response to VN, but are able to spread on FN. Cell spreading on VN is recovered by expression of GPI-mS1ED (Fig. 8 F) and mS1Δ122-252 (unpublished data). It is unlikely that the siRNA treatment has any nonspecific cellular effects because spreading is specifically rescued by expression of GPI-mS1ED. However, spreading is not recovered in cells expressing mS1Δ88-252, the S1ED mutant that fails to signal spreading in S1 antibody–based adhesion assays. Cells expressing mS1TDM, a mutant unable to engage the matrix, also fail to spread on VN, confirming that S1-mediated adhesion is required for full αvβ3 integrin activity.
To test the effects of hS1 silencing on cell migration, the migration of hS1 siRNA-transfected cells was examined on VN- or FN-coated filters. Migration of the NEO cells across VN is reduced approximately threefold by siRNA relative to untreated controls (Fig. 8 G). In addition, cell migration in response to VN is rescued in the siRNA-treated cells by expression of GPI-mS1ED or mS1Δ122-252, but not by mS1TDM or mS1Δ88-252. None of the cells display any defects in their ability to migrate in response to FN (Fig. 8 H).
Discussion
The current study demonstrates a functional coupling between the αvβ3 integrin and S1 that regulates the activation and signaling of the integrin during carcinoma cell spreading and migration. Whether this functional coupling is via a signaling pathway or via a direct interaction between these two receptors is not yet known; however, we do describe several features of this mechanism. First, S1-dependent activation of αvβ3 is contingent on the syndecan engaging a ligand. On antibody, ligation of S1 devoid of HS leads to integrin activation and cell spreading, the latter using integrin-mediated signaling in the absence of an integrin ligand. On VN, a ligand that engages both S1 and αvβ3, the integrin activation requires S1 bearing its HS chains, presumably to engage the heparin-binding domain of VN.
The finding that ligation of S1 is not only necessary but seemingly sufficient for integrin activation, as occurs on S1 antibody, is surprising. Most, if not all, ECM ligands have heparin binding domains that presumably engage S1; yet, the αvβ3 integrin is not always active on these matrices. An example is the cell behaviors that we observe on FN, where α5β1 signaling predominates; the αvβ3 integrin is inactive despite the fact that it (Charo et al., 1990) and S1 can engage the FN. Thus, unlike the antibody substratum, recognition of ECM components by other integrins and syndecans may disrupt the syndecan-αvβ3 coupling mechanism or target the αvβ3 directly, thus defeating S1 and inactivating the integrin.
In the MDA-MB-231 cells, the αvβ3 integrin is maintained in an inactive state by negative cross-talk, apparently from the α2β1 integrin (Beauvais and Rapraeger, 2003). When cells are plated on S1 antibody, this cross-talk mechanism prevents αvβ3 integrin activation; however, expression of higher S1 levels overrides the competing inhibition from the α2β1 integrin as long as S1 is ligated. Overexpression of mS1 will not lead to integrin activation if the cells are adherent only via their endogenous hS1, indicating that hS1 and mS1 act independently and are unlikely to multimerize. A similar “priming” of integrin activation due to enhanced S1 expression drives αvβ3-dependent adhesion and spreading on low concentrations of VN—levels at which parental cells cannot respond. It is worth noting that increased S1 expression in breast carcinomas and melanomas correlates with an aggressive metastatic phenotype and poor clinical prognosis (Timar et al., 1992; Barbareschi et al., 2003; Burbach et al., 2003); this finding may trace to up-regulation in αvβ3 activity.
A second feature of the coupling mechanism is its reliance on the S1ED. Regardless of whether S1 is engaged by antibody or VN, integrin activation is blocked by treatments that target this domain, including competition with anti-S1 antibodies or recombinant S1ED. Furthermore, inactivation of the integrin seen upon siRNA-dependent inhibition of hS1 expression is overcome by expression of GPI-linked mS1ED. Admittedly, the inhibitory effects of these reagents on cell spreading and migration on VN was a surprise. Unlike the S1 antibody–based adhesion assays, cells on VN are clearly provided an αvβ3 integrin ligand, yet even in the presence of this ligand, the integrin still requires S1 in order to signal, indicating a potentially important role for the S1ED that extends beyond the initial activation of the integrin. Although further experimentation will be necessary to identify the active site, a syndecan mutant lacking amino acids 121–252 of the ED retains activity, whereas one lacking an additional 34 amino acids (Δ88–252) does not. Importantly, within this 34–amino acid stretch mS1 and hS1 share 58% identity and 72% homology, indicating that activity of the S1ED is likely conserved between the species. This is also evidenced by the fact that overexpression of either S1 species is sufficient to confer enhanced αvβ3 activity.
Other studies have implicated the syndecan EDs in important protein interactions at cell surfaces. S1- and S4EDs mediate binding interactions with cultured fibroblasts and endothelial cells (McFall and Rapraeger, 1997, 1998). Antibodies that target the EDs of S1 and S3 block Schwann cell spreading on LN and FN (Carey et al., 1994) and FGF2-dependent proliferation of cultured chondrocytes (Kirsch et al., 2002), respectively. Competition with recombinant ED is effective in disrupting cell spreading and inducing cell cycle arrest in colon carcinoma cells that overexpress S2 (Park et al., 2002). Finally, polarization of S1-expressing Raji cells is dependent on the S1ED (McQuade and Rapraeger, 2003). In each of these cases, the exact mechanism of the extracellular core protein interaction remains unknown.
A third feature of the coupling mechanism is that it is specific for the αvβ3 integrin. MCF-7 cells spread and migrate on VN, but use the αvβ1 integrin. S1 is not required for the activity of this integrin, nor is it inactivated by any treatments that target S1. Similarly, cell spreading and migration on FN, which requires α5β1 integrin activity, appears to occur independent of S1, and vice-versa, coupling to the αvβ3 integrin appears to be specific for S1. Coupling is not observed in MDA-MB-231 cells adherent via S4 (i.e., treatment with mAb LM609 has no effect on cells adherent and spread on S4 antibody, mAb 150.9) nor in cells adherent to mAb RVS-10, an anti-CD71/transferrin receptor antibody (unpublished data). CD71-adherent cells fail to spread even in the presence of a function-blocking β1 integrin antibody that stimulates spreading in cells adherent via S1 (Beauvais and Rapraeger, 2003).
Multiple mechanisms, including affinity and avidity modulation, regulate integrin function. Affinity modulation of αvβ3 is complex and involves conformational changes within its extracellular domain (Beglova et al., 2002; Takagi et al., 2002). This process is regulated by inside-out signaling that impinges on the integrin's cytoplasmic domains either by stimulating proteolysis (Du et al., 1995; Smith, 1997), phosphorylation (Blystone et al., 1996; Jenkins et al., 1998), or binding of intracellular proteins such as talin (Calderwood et al., 1999, 2002) and β3-endonexin (Shattil et al., 1995; Kashiwagi et al., 1997). These intracellular events lead to the exposure of ligand-binding epitopes in the integrin's extracellular domains (Hughes et al., 1996). Studies on VN suggest that αvβ3 can assume two or more distinct activation states (Takagi et al., 2002), and distinct αvβ3 conformations have been detected for different matrix ligands (Boettiger et al., 2001). Ligand binding, in turn, stabilizes structural changes that initiate outside-in signaling that include tyrosine phosphorylation of the β3 cytoplasmic tail (Schaffner-Reckinger et al., 1998; Law et al., 1999) and association of the β3 subunit cytoplasmic tail with intracellular effectors.
Although interactions of αvβ3 with extracellular ligands stimulate outside-in signaling, signaling via unligated αvβ3 is important in a process known as “integrin-mediated death” (IMD; Stupack et al., 2001). In cells sensitive to IMD, αvβ3 may act as a sensor during cell invasion, inducing cell death when the cells encounter a nonpermissive ECM (Ilic et al., 1998). Until now, it has been unclear whether or not unligated αvβ3 integrins can participate in cell signaling processes other than IMD. However, in this work, the unligated αvβ3 is capable of transmitting signals that lead to cell spreading when S1 is engaged. It is appealing to speculate that S1 via its adhesion-dependent activation of αvβ3 may act as a negative regulator of IMD.
αvβ3 integrins interact with several cell surface receptors including PDGFR-β and VEGFR-2 (Borges et al., 2000), CD87/uPAR (Wei et al., 1996), and CD47/IAP (Lindberg et al., 1996; Fujimoto et al., 2003) via the β3 extracellular domain. It is possible that S1 also engages directly with the integrin or with one of these other receptors. Like CD47, we find that the S1ED, when expressed in cells and engaged with ligand, is sufficient to mediate a functional interaction with the β3 subunit that alters the conformation of the integrin to a high affinity ligand binding state. However, soluble recombinant S1ED, which is not tethered to the membrane and unable to sense the mechanical force imbued by an immobilized ligand, acts as a functional inhibitor of αvβ3. Intriguingly, soluble recombinant CD87 binds to the αvβ3 integrin (Degryse et al., 2001) and competitively inhibits the physical and functional coupling of CD87 to the integrin (Wei et al., 1996; Simon et al., 2000).
Why must the syndecan be anchored? Anchorage of the syndecan to an immobilized ligand may cluster the syndecan and/or induce conformational changes in the S1ED. These changes may induce clustering of the integrin itself or enhance signaling required for αvβ3 activation. Our data support a role in affinity modulation (e.g., WOW1 binding), although avidity modulation may take place as well.
In summary, this work highlights a novel mechanism in which the activity and function of the αvβ3 integrin is directly modulated by its physical or functional coupling to S1. As such, S1 is likely to be a critical regulator of αvβ3 integrin in the multiple cell behaviors that rely on this integrin.
Materials and methods
Reagents
Matrix ligands include human plasma FN (provided by D. Peters, University of Wisconsin, Madison, WI) and VN (Promega) and type I COL (BD Biosciences). Mouse mAbs B-B4 (Wijdenes et al., 1996), F94-8G3 (David et al., 1992), RVS-10 (Chemicon), rat mAbs 281.2 (Jalkanen et al., 1985), and KY 8.2 (Yamashita et al., 1999) recognize hS1, hS4, CD71/transferrin receptor, mS1, and mS4, respectively. Integrin antibodies include the following: β1 and α5β1 inhibitory mAb 13 and mAb 16 (provided by S. Akiyama, National Institute of Dental Research, Bethesda, MD), β1 inhibitory mAb P5D2, αv inhibitory mAb M9, and αvβ3 inhibitory mAb LM609 (Chemicon). The ligand-mimetic Fab WOW1 (provided by S. Shattil, Scripps Research Institute, La Jolla, CA) was used to detect activated αvβ3 integrin (Pampori et al., 1999).
Recombinant GST–fused mS1ED (GST-mS1ED) and S4ED (GST-mS4ED) protein was prepared as described previously (McFall and Rapraeger, 1998). GST-mS1ED was used as an antigen in New Zealand white rabbits, and antibodies were affinity-purified by sequential GST-mS1ED and GST columns (Harlow and Lane, 1988).
Cell culture and transfection
MDA-MB-231, MDA-MB-435, and MCF-7 human carcinoma cells were grown in DME (Life Technologies) supplemented with 10% FBS (Hyclone Laboratories), 4 mM l-glutamine (Sigma-Aldrich), and 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies) at 37°C and 92.5% air/7.5% CO2. Growth medium for MCF-7 cells was additionally supplemented with 10 μg/ml of bovine pancreatic insulin (Sigma-Aldrich).
Syndecan cDNA constructs (provided by R. Sanderson, University of Arkansas for Medical Sciences, Little Rock, AR) in vector pcDNA3 (Invitrogen) have been previously described (Langford et al., 1998; Liu et al., 1998; McQuade and Rapraeger, 2003). MDA-MB-231 cells were transfected using LipofectAMINE PLUS (Invitrogen) and 10 μg of plasmid in accordance with the manufacturer's instructions. Stable populations expressing high but equal levels of ectopic S1 were selected in 1.5 mg/ml G418 (GIBCO BRL) and sorted by FACS.
siRNA design and transfection
Three siRNAs (nucleotide annotation: 874AGGACTTCACCTTTGAAACC893, 1162AGGAGGAATTCTATGCCTGA1181, and 1749GGTAAGTTAAGTAAGTTGA1767 [GenBank/EMBL/DDBJ accession no. NM_002997]) specific for hS1 were designed by Ambion; each silences hS1 expression by ≥90%. For transfection, 200 nM siRNA was added to 2.0 × 105 cells in 35-mm wells using LipofectAMINE2000 and Opti-MEM I transfection medium (Invitrogen) lacking serum and antibiotics. Control cells were transfected with lipid-based vehicle alone. At 4 h after transfection, each well was supplemented with 3 ml of complete growth medium; at 24 h after transfection the cells were lifted in trypsin (0.25% wt/vol) and expanded in 100-mm tissue-culture plates. Cells were harvested 72 h after transfection and experimental cohorts subjected to two-color FACS analysis.
Cell spreading assays
Nitrocellulose-coated 10-well glass slides (Erie Scientific) were prepared as described previously (Lebakken and Rapraeger, 1996). Wells were coated with ligands at 37°C for 2 h. mAbs 281.2 and B-B4 (10 μg/ml), diluted in calcium and magnesium-free PBS (CMF-PBS, 135 mM NaCl, 2.7 mM KCl, 10.2 mM Na2HPO4 · 7H2O, and 1.75 mM KH2PO4, pH 7.4), were coated on wells directly or on wells precoated with 200 μg/ml of goat anti–rat or anti–mouse IgG, respectively (Jackson ImmunoResearch Laboratories). 10 μg/ml FN, 10 μg/ml COL I, or 1–10 μg/ml VN were heated to 37°C for 15 min, diluted in serum-free 15 mM Hepes-buffered (Hb) DME, pH 7.4, and coated directly to wells. Wells were blocked with serum-free Hb-DME containing 1.0% heat-denatured BSA (plating medium) for 1 h at 37°C. Cells were lifted in Tris-EDTA-saline (20 mM Tris, pH 7.5, 165 mM NaCl, and 5 mM EDTA), washed with plating medium, and plated on wells (50 μl per well) in the same medium (± inhibitors) at a cell density of 4.0 × 105 cells per milliliter. Where syndecan mAb was used as a substratum, the plating medium was supplemented with 0.2 U/ml of heparinase mix (heparinase I, II, and III) and 0.05 U/ml chondroitin ABC lyase (Seikagaku America) to enzymatically remove glycosaminoglycans. Cells were allowed to adhere and spread for 2 h at 37°C, followed by washing in CMF-PBS and fixation for 2 h in 2% PFA at 4°C.
Cell staining and quantification of spreading
Fixed cells were stained with rhodamine-conjugated phalloidin as described previously (Lebakken et al., 2000). Alternatively, cells were stained with WOW-1 Fab (1:4 dilution) diluted in Hepes tyrode buffer (Beauvais and Rapraeger, 2003) for 1 h at 37°C followed by Alexa-488–conjugated goat anti–mouse IgG (H+L) F(ab')2 secondary antibody (Molecular Probes). Slides were mounted with a coverslip in aqueous, non-fluorescing mounting medium (Immu-mount; Thermo Shandon). All images were acquired at RT with a 20× Fluor objective (0.75 NA; Nikon), with the exception of a 63× Planapo objective (1.4 NA; Carl Zeiss MicroImaging, Inc.) for Fig. 7 F, on a microscope (model Microphot-FX; Nikon) and attached Image Point Scientific cooled CCD camera (Photometrics), using Image-Pro Plus version 1.3 (Nikon). Images were processed (cropping, contrast, and size adjustments) in Adobe Photoshop version 7.0. All images represent results from triplicate wells and three independent experiments.
Migration assays
Migration assays were performed in 48-well modified Boyden chambers (Neuroprobe) using 8-μm polycarbonate filters (Osmonics Inc.) coated on both sides O/N with 10 μg/ml of either VN or FN. 2.0 × 104 cells were plated in serum-free Hb-DME containing 0.2% fatty acid–free BSA in quadruplicate wells and allowed to migrate in response to 10% FBS in the lower chamber for 16 h. The upper side of the filter was scraped to remove nonmotile cells, fixed and stained with Diff-Quik® (Dade-Behring) for scanning, and quantification by densitometry using NIH Image software.
Flow cytometry
Suspended cells were incubated for 1 h on ice with 1 μg of primary antibody per 3 × 105 cells, washed, and counterstained with Alexa-488 and/or R-PE–conjugated secondary antibodies (Molecular Probes). To cluster mS1, cells in suspension were incubated with 1 μg/ml mAb 281.2 (or mAb KY8.2 as an IgG control) for 15 min at 37°C, washed, and incubated with a 5 μg/ml of goat anti–rat IgG secondary (Jackson ImmunoResearch Laboratories) for an additional 15 min before staining and scanning on a FACSCalibur benchtop cytometer (BD Biosciences). Cell scatter and propidium iodide (Sigma-Aldrich; 1 μg/sample) staining profiles were used to gate live, single-cell events for data analysis. Cells were sorted under sterile conditions on a triple-laser FACSVantage SE equipped with the FACSDiVa digital electronics analyzer.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants HD21881 and GM48850 to A. Rapraeger, training grant stipend T32-GM08688 to D. Beauvais, and the core facilities of the University of Wisconsin Comprehensive Cancer Center, supported by NIH grant P30-CA14520.
Abbreviations used in this paper: COL, collagen; ED, ectodomain; FN, fibronectin; GPI, glycosylphosphatidylinositol; Hb, Hepes-buffered; HS, heparan sulfate; hS1, human S1; IMD, integrin-mediated death; LN, laminin; mS1, mouse S1; pAb, polyclonal antibody; S1, syndecan-1; siRNA, small-interfering RNA; TM, transmembrane; VN, vitronectin.
References
- Adams, J.C., N. Kureishy, and A.L. Taylor. 2001. A role for syndecan-1 in coupling fascin spike formation by thrombospondin-1. J. Cell Biol. 152:1169–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama, S.K., S.S. Yamada, W.T. Chen, and K.M. Yamada. 1989. Analysis of fibronectin receptor function with monoclonal antibodies: roles in cell adhesion, migration, matrix assembly, and cytoskeletal organization. J. Cell Biol. 109:863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbareschi, M., P. Maisonneuve, D. Aldovini, M.G. Cangi, L. Pecciarini, F. Angelo Mauri, S. Veronese, O. Caffo, A. Lucenti, P.D. Palma, et al. 2003. High syndecan-1 expression in breast carcinoma is related to an aggressive phenotype and to poorer prognosis. Cancer. 98:474–483. [DOI] [PubMed] [Google Scholar]
- Beauvais, D.M., and A.C. Rapraeger. 2003. Syndecan-1-mediated cell spreading requires signaling by αvβ3 integrins in human breast carcinoma cells. Exp. Cell Res. 286:219–232. [DOI] [PubMed] [Google Scholar]
- Beauvais, D.M., and A.C. Rapraeger. 2004. Syndecans in tumor cell adhesion and signaling. Reprod. Biol. Endocrinol. 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglova, N., S.C. Blacklow, J. Takagi, and T.A. Springer. 2002. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 9:282–287. [DOI] [PubMed] [Google Scholar]
- Bernfield, M., M. Gotte, P.W. Park, O. Reizes, M.L. Fitzgerald, J. Lincecum, and M. Zako. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 68:729–777. [DOI] [PubMed] [Google Scholar]
- Blystone, S.D., F.P. Lindberg, M.P. Williams, K.P. McHugh, and E.J. Brown. 1996. Inducible tyrosine phosphorylation of the β3 integrin requires the αV integrin cytoplasmic tail. J. Biol. Chem. 271:31458–31462. [DOI] [PubMed] [Google Scholar]
- Boettiger, D., L. Lynch, S. Blystone, and F. Huber. 2001. Distinct ligand-binding modes for integrin αvβ3-mediated adhesion to fibronectin versus vitronectin. J. Biol. Chem. 276:31684–31690. [DOI] [PubMed] [Google Scholar]
- Borges, E., Y. Jan, and E. Ruoslahti. 2000. Platelet-derived growth factor receptor β and vascular endothelial growth factor receptor 2 bind to the β3 integrin through its extracellular domain. J. Biol. Chem. 275:39867–39873. [DOI] [PubMed] [Google Scholar]
- Brooks, P.C., A.M. Montgomery, M. Rosenfeld, R.A. Reisfeld, T. Hu, G. Klier, and D.A. Cheresh. 1994. Integrin αvβ3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 79:1157–1164. [DOI] [PubMed] [Google Scholar]
- Burbach, B.J., A. Friedl, C. Mundhenke, and A.C. Rapraeger. 2003. Syndecan-1 accumulates in lysosomes of poorly differentiated breast carcinoma cells. Matrix Biol. 22:163–177. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., R. Zent, R. Grant, D.J. Rees, R.O. Hynes, and M.H. Ginsberg. 1999. The Talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274:28071–28074. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., B. Yan, J.M. de Pereda, B.G. Alvarez, Y. Fujioka, R.C. Liddington, and M.H. Ginsberg. 2002. The phosphotyrosine binding-like domain of talin activates integrins. J. Biol. Chem. 277:21749–21758. [DOI] [PubMed] [Google Scholar]
- Carey, D.J., R.C. Stahl, G. Cizmeci-Smith, and V.K. Asundi. 1994. Syndecan-1 expressed in Schwann cells causes morphological transformation and cytoskeletal reorganization and associates with actin during cell spreading. J. Cell Biol. 124:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman, C.V., and T.A. Springer. 2003. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr. Opin. Cell Biol. 15:547–556. [DOI] [PubMed] [Google Scholar]
- Charo, I.F., L. Nannizzi, J.W. Smith, and D.A. Cheresh. 1990. The vitronectin receptor αvβ3 binds fibronectin and acts in concert with α5β1 in promoting cellular attachment and spreading on fibronectin. J. Cell Biol. 111:2795–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couchman, J.R., L. Chen, and A. Woods. 2001. Syndecans and cell adhesion. Int. Rev. Cytol. 207:113–150. [DOI] [PubMed] [Google Scholar]
- David, G., B. van der Schueren, P. Marynen, J.J. Cassiman, and H. van den Berghe. 1992. Molecular cloning of amphiglycan, a novel integral membrane heparan sulfate proteoglycan expressed by epithelial and fibroblastic cells. J. Cell Biol. 118:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries, J.E., G.D. Keizer, A.A. te Velde, A. Voordouw, D. Ruiter, P. Rumke, H. Spits, and C.G. Figdor. 1986. Characterization of melanoma-associated surface antigens involved in the adhesion and motility of human melanoma cells. Int. J. Cancer. 38:465–473. [DOI] [PubMed] [Google Scholar]
- Degryse, B., S. Orlando, M. Resnati, S.A. Rabbani, and F. Blasi. 2001. Urokinase/urokinase receptor and vitronectin/αvβ3 integrin induce chemotaxis and cytoskeleton reorganization through different signaling pathways. Oncogene. 20:2032–2043. [DOI] [PubMed] [Google Scholar]
- Du, X., T.C. Saido, S. Tsubuki, F.E. Indig, M.J. Williams, and M.H. Ginsberg. 1995. Calpain cleavage of the cytoplasmic domain of the integrin β3 subunit. J. Biol. Chem. 270:26146–26151. [DOI] [PubMed] [Google Scholar]
- Eliceiri, B.P. 2001. Integrin and growth factor receptor crosstalk. Circ. Res. 89:1104–1110. [DOI] [PubMed] [Google Scholar]
- Eliceiri, B.P., R. Klemke, S. Stromblad, and D.A. Cheresh. 1998. Integrin αvβ3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 140:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felding-Habermann, B., T.E. O'Toole, J.W. Smith, E. Fransvea, Z.M. Ruggeri, M.H. Ginsberg, P.E. Hughes, N. Pampori, S.J. Shattil, A. Saven, and B.M. Mueller. 2001. Integrin activation controls metastasis in human breast cancer. Proc. Natl. Acad. Sci. USA. 98:1853–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto, T.T., S. Katsutani, T. Shimomura, and K. Fujimura. 2003. Thrombospondin-bound integrin-associated protein (CD47) physically and functionally modifies integrin αIIbβ3 by its extracellular domain. J. Biol. Chem. 278:26655–26665. [DOI] [PubMed] [Google Scholar]
- Gao, A.G., F.P. Lindberg, J.M. Dimitry, E.J. Brown, and W.A. Frazier. 1996. Thrombospondin modulates αvβ3 function through integrin-associated protein. J. Cell Biol. 135:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Gonzalez, A.M., M. Gonzales, G.S. Herron, U. Nagavarapu, S.B. Hopkinson, D. Tsuruta, and J.C. Jones. 2002. Complex interactions between the laminin α4 subunit and integrins regulate endothelial cell behavior in vitro and angiogenesis in vivo. Proc. Natl. Acad. Sci. USA. 99:16075–16080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granes, F., R. Garcia, R.P. Casaroli-Marano, S. Castel, N. Rocamora, M. Reina, J.M. Urena, and S. Vilaro. 1999. Syndecan-2 induces filopodia by active cdc42Hs. Exp. Cell Res. 248:439–456. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 726 pp.
- Hood, J.D., R. Frausto, W.B. Kiosses, M.A. Schwartz, and D.A. Cheresh. 2003. Differential αv integrin–mediated Ras-ERK signaling during two pathways of angiogenesis. J. Cell Biol. 162:933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, P.E., F. Diaz-Gonzalez, L. Leong, C. Wu, J.A. McDonald, S.J. Shattil, and M.H. Ginsberg. 1996. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J. Biol. Chem. 271:6571–6574. [DOI] [PubMed] [Google Scholar]
- Ilic, D., E.A. Almeida, D.D. Schlaepfer, P. Dazin, S. Aizawa, and C.H. Damsky. 1998. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 143:547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalkanen, M., H. Nguyen, A. Rapraeger, N. Kurn, and M. Bernfield. 1985. Heparan sulfate proteoglycans from mouse mammary epithelial cells: localization on the cell surface with a monoclonal antibody. J. Cell Biol. 101:976–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins, A.L., L. Nannizzi-Alaimo, D. Silver, J.R. Sellers, M.H. Ginsberg, D.A. Law, and D.R. Phillips. 1998. Tyrosine phosphorylation of the β3 cytoplasmic domain mediates integrin-cytoskeletal interactions. J. Biol. Chem. 273:13878–13885. [DOI] [PubMed] [Google Scholar]
- Kashiwagi, H., M.A. Schwartz, M. Eigenthaler, K.A. Davis, M.H. Ginsberg, and S.J. Shattil. 1997. Affinity modulation of platelet integrin αIIbβ3 by β3-endonexin, a selective binding partner of the β3 integrin cytoplasmic tail. J. Cell Biol. 137:1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S., M. Harris, and J.A. Varner. 2000. Regulation of integrin αvβ3-mediated endothelial cell migration and angiogenesis by integrin α5β1 and protein kinase A. J. Biol. Chem. 275:33920–33928. [DOI] [PubMed] [Google Scholar]
- Kim, Y., H. Park, Y. Lim, I. Han, H.J. Kwon, A. Woods, and E.S. Oh. 2003. Decreased syndecan-2 expression correlates with trichostatin-A induced-morphological changes and reduced tumorigenic activity in colon carcinoma cells. Oncogene. 22:826–830. [DOI] [PubMed] [Google Scholar]
- Kiosses, W.B., S.J. Shattil, N. Pampori, and M.A. Schwartz. 2001. Rac recruits high-affinity integrin αvβ3 to lamellipodia in endothelial cell migration. Nat. Cell Biol. 3:316–320. [DOI] [PubMed] [Google Scholar]
- Kirsch, T., E. Koyama, M. Liu, E.E. Golub, and M. Pacifici. 2002. Syndecan-3 is a selective regulator of chondrocyte proliferation. J. Biol. Chem. 277:42171–42177. [DOI] [PubMed] [Google Scholar]
- Langford, J.K., M.J. Stanley, D. Cao, and R.D. Sanderson. 1998. Multiple heparan sulfate chains are required for optimal syndecan-1 function. J. Biol. Chem. 273:29965–29971. [DOI] [PubMed] [Google Scholar]
- Law, D.A., F.R. DeGuzman, P. Heiser, K. Ministri-Madrid, N. Killeen, and D.R. Phillips. 1999. Integrin cytoplasmic tyrosine motif is required for outside-in αIIbβ3 signalling and platelet function. Nature. 401:808–811. [DOI] [PubMed] [Google Scholar]
- Lebakken, C.S., and A.C. Rapraeger. 1996. Syndecan-1 mediates cell spreading in transfected human lymphoblastoid (Raji) cells. J. Cell Biol. 132:1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebakken, C.S., K.J. McQuade, and A.C. Rapraeger. 2000. Syndecan-1 signals independently of β1 integrins during Raji cell spreading. Exp. Cell Res. 259:315–325. [DOI] [PubMed] [Google Scholar]
- Liapis, H., A. Flath, and S. Kitazawa. 1996. Integrin αvβ3 expression by bone-residing breast cancer metastases. Diagn. Mol. Pathol. 5:127–135. [DOI] [PubMed] [Google Scholar]
- Lin, E.C., B.I. Ratnikov, P.M. Tsai, E.R. Gonzalez, S. McDonald, A.J. Pelletier, and J.W. Smith. 1997. Evidence that the integrin β3 and β5 subunits contain a metal ion-dependent adhesion site-like motif but lack an I domain. J. Biol. Chem. 272:14236–14243. [DOI] [PubMed] [Google Scholar]
- Lindberg, F.P., H.D. Gresham, M.I. Reinhold, and E.J. Brown. 1996. Integrin-associated protein immunoglobulin domain is necessary for efficient vitronectin bead binding. J. Cell Biol. 134:1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W., E.D. Litwack, M.J. Stanley, J.K. Langford, A.D. Lander, and R.D. Sanderson. 1998. Heparan sulfate proteoglycans as adhesive and anti-invasive molecules. Syndecans and glypican have distinct functions. J. Biol. Chem. 273:22825–22832. [DOI] [PubMed] [Google Scholar]
- McFall, A.J., and A.C. Rapraeger. 1997. Identification of an adhesion site within the syndecan-4 extracellular protein domain. J. Biol. Chem. 272:12901–12904. [DOI] [PubMed] [Google Scholar]
- McFall, A.J., and A.C. Rapraeger. 1998. Characterization of the high affinity cell-binding domain in the cell surface proteoglycan syndecan-4. J. Biol. Chem. 273:28270–28276. [DOI] [PubMed] [Google Scholar]
- McQuade, K.J., and A.C. Rapraeger. 2003. Syndecan-1 transmembrane and extracellular domains have unique and distinct roles in cell spreading. J. Biol. Chem. 278:46607–46615. [DOI] [PubMed] [Google Scholar]
- Mould, A.P., S.K. Akiyama, and M.J. Humphries. 1996. The inhibitory anti-β1 integrin monoclonal antibody 13 recognizes an epitope that is attenuated by ligand occupancy. Evidence for allosteric inhibition of integrin function. J. Biol. Chem. 271:20365–20374. [DOI] [PubMed] [Google Scholar]
- Ohtake, T., Y. Fujimoto, K. Ikuta, H. Saito, M. Ohhira, M. Ono, and Y. Kohgo. 1999. Proline-rich antimicrobial peptide, PR-39 gene transduction altered invasive activity and actin structure in human hepatocellular carcinoma cells. Br. J. Cancer. 81:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampori, N., T. Hato, D.G. Stupack, S. Aidoudi, D.A. Cheresh, G.R. Nemerow, and S.J. Shattil. 1999. Mechanisms and consequences of affinity modulation of integrin αvβ3 detected with a novel patch-engineered monovalent ligand. J. Biol. Chem. 274:21609–21616. [DOI] [PubMed] [Google Scholar]
- Park, H., Y. Kim, Y. Lim, I. Han, and E.S. Oh. 2002. Syndecan-2 mediates adhesion and proliferation of colon carcinoma cells. J. Biol. Chem. 277:29730–29736. [DOI] [PubMed] [Google Scholar]
- Petitclerc, E., S. Stromblad, T.L. von Schalscha, F. Mitjans, J. Piulats, A.M. Montgomery, D.A. Cheresh, and P.C. Brooks. 1999. Integrin αvβ3 promotes M21 melanoma growth in human skin by regulating tumor cell survival. Cancer Res. 59:2724–2730. [PubMed] [Google Scholar]
- Pilch, J., R. Habermann, and B. Felding-Habermann. 2002. Unique ability of integrin αvβ3 to support tumor cell arrest under dynamic flow conditions. J. Biol. Chem. 277:21930–21938. [DOI] [PubMed] [Google Scholar]
- Rapraeger, A.C. 2000. Syndecan-regulated receptor signaling. J. Cell Biol. 149:995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnikov, B.I., D.V. Rozanov, T.I. Postnova, P.G. Baciu, H. Zhang, R.G. DiScipio, G.G. Chestukhina, J.W. Smith, E.I. Deryugina, and A.Y. Strongin. 2002. An alternative processing of integrin αv subunit in tumor cells by membrane type-1 matrix metalloproteinase. J. Biol. Chem. 277:7377–7385. [DOI] [PubMed] [Google Scholar]
- Schaffner-Reckinger, E., V. Gouon, C. Melchior, S. Plancon, and N. Kieffer. 1998. Distinct involvement of β3 integrin cytoplasmic domain tyrosine residues 747 and 759 in integrin-mediated cytoskeletal assembly and phosphotyrosine signaling. J. Biol. Chem. 273:12623–12632. [DOI] [PubMed] [Google Scholar]
- Shattil, S.J., T. O'Toole, M. Eigenthaler, V. Thon, M. Williams, B.M. Babior, and M.H. Ginsberg. 1995. β3-endonexin, a novel polypeptide that interacts specifically with the cytoplasmic tail of the integrin β3 subunit. J. Cell Biol. 131:807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, D.I., Y. Wei, L. Zhang, N.K. Rao, H. Xu, Z. Chen, Q. Liu, S. Rosenberg, and H.A. Chapman. 2000. Identification of a urokinase receptor-integrin interaction site. Promiscuous regulator of integrin function. J. Biol. Chem. 275:10228–10234. [DOI] [PubMed] [Google Scholar]
- Smith, J.W. 1997. Allostery and proteolysis: two novel modes of regulating integrin function. Matrix Biol. 16:173–178. [DOI] [PubMed] [Google Scholar]
- Smith, J.W., R.S. Piotrowicz, and D. Mathis. 1994. A mechanism for divalent cation regulation of β3-integrins. J. Biol. Chem. 269:960–967. [PubMed] [Google Scholar]
- Stupack, D.G., X.S. Puente, S. Boutsaboualoy, C.M. Storgard, and D.A. Cheresh. 2001. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J. Cell Biol. 155:459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi, J., B.M. Petre, T. Walz, and T.A. Springer. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 110:599–611. [DOI] [PubMed] [Google Scholar]
- Timar, J., A. Ladanyi, K. Lapis, and M. Moczar. 1992. Differential expression of proteoglycans on the surface of human melanoma cells characterized by altered experimental metastatic potential. Am. J. Pathol. 141:467–474. [PMC free article] [PubMed] [Google Scholar]
- Tkachenko, E., and M. Simons. 2002. Clustering induces redistribution of syndecan-4 core protein into raft membrane domains. J. Biol. Chem. 277:19946–19951. [DOI] [PubMed] [Google Scholar]
- Tumova, S., A. Woods, and J.R. Couchman. 2000. Heparan sulfate chains from glypican and syndecans bind the Hep II domain of fibronectin similarly despite minor structural differences. J. Biol. Chem. 275:9410–9417. [DOI] [PubMed] [Google Scholar]
- van der Flier, A., and A. Sonnenberg. 2001. Function and interactions of integrins. Cell Tissue Res. 305:285–298. [DOI] [PubMed] [Google Scholar]
- Wei, Y., M. Lukashev, D.I. Simon, S.C. Bodary, S. Rosenberg, M.V. Doyle, and H.A. Chapman. 1996. Regulation of integrin function by the urokinase receptor. Science. 273:1551–1555. [DOI] [PubMed] [Google Scholar]
- Wijdenes, J., W.C. Vooijs, C. Clement, J. Post, F. Morard, N. Vita, P. Laurent, R.X. Sun, B. Klein, and J.M. Dore. 1996. A plasmocyte selective monoclonal antibody (B-B4) recognizes syndecan-1. Br. J. Haematol. 94:318–323. [DOI] [PubMed] [Google Scholar]
- Xue, W., I. Mizukami, R.F. Todd III, and H.R. Petty. 1997. Urokinase-type plasminogen activator receptors associate with β1 and β3 integrins of fibrosarcoma cells: dependence on extracellular matrix components. Cancer Res. 57:1682–1689. [PubMed] [Google Scholar]
- Yamashita, Y., K. Oritani, E.K. Miyoshi, R. Wall, M. Bernfield, and P.W. Kincade. 1999. Syndecan-4 is expressed by B lineage lymphocytes and can transmit a signal for formation of dendritic processes. J. Immunol. 162:5940–5948. [PubMed] [Google Scholar]