

Abstract
The Ras-related GTPase Rap1 stimulates integrin-mediated adhesion and spreading in various mammalian cell types. Here, we demonstrate that Rap1 regulates cell spreading by localizing guanine nucleotide exchange factors (GEFs) that act via the Rho family GTPase Rac1. Rap1a activates Rac1 and requires Rac1 to enhance spreading, whereas Rac1 induces spreading independently of Rap1. Active Rap1a binds to a subset of Rac GEFs, including VAV2 and Tiam1 but not others such as SWAP-70 or COOL-1. Overexpressed VAV2 and Tiam1 specifically require Rap1 to promote spreading, even though Rac1 is activated independently of Rap1. Rap1 is necessary for the accumulation of VAV2 in membrane protrusions at the cell periphery. In addition, if VAV2 is artificially localized to the cell edge with the subcellular targeting domain of Rap1a, it increases cell spreading independently of Rap1. These results lead us to propose that Rap1 promotes cell spreading by localizing a subset of Rac GEFs to sites of active lamellipodia extension.
Introduction
Rap1 is a member of the Ras subfamily of small GTPases. GTPases act as molecular switches by cycling between inactive GDP-bound and active GTP-bound conformations (Takai et al., 2001). The GTP-bound form binds to proteins that function as downstream effectors. Transition between the GDP- and GTP-bound states is tightly regulated by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). GEFs promote release of bound GDP and stabilize the nucleotide-free state until displaced by GTP. GAPs promote the hydrolysis of bound GTP and hence return GTPases to the inactive state. The distinct functions of GTPases depend on which effectors they bind and where in the cell they are localized. Localization is primarily governed by the COOH terminus, which contains a hypervariable (HV)/polybasic domain and a prenylated CAAX box for association with membranes. The HV sequence and nature of the prenylation can specify which type of cell membrane is targeted (Hancock, 2003). Identifying proteins that either regulate the activity of GTPases or relay their downstream biochemical signals is essential for understanding how these molecules regulate cellular functions.
Mammalian Rap1 was identified as a cDNA that reverts the loss of adhesion accompanying cellular transformation by an oncogenic mutant of K-Ras (Kitayama et al., 1989). Given that Rap1 and Ras have similar effector-binding regions, Rap1 was initially thought to simply act as an antagonist by sequestering Ras effectors in nonproductive complexes (Zwartkruis and Bos, 1999; Stork, 2003). Subsequent studies have concluded that Rap1 overcomes the effects of active K-Ras largely by increasing the function of the integrin family of heterodimeric cell adhesion molecules (Bos et al., 2001). Rap1 is activated by numerous extracellular stimuli via integrins, receptor tyrosine kinases, G protein–coupled receptors, and other transmembrane proteins (Quilliam et al., 2002). Signaling from Rap1 leads to downstream effects on MAP kinases, transcription, differentiation, and cell morphology, mediated by a variety of identified and unidentified effector molecules (Bos et al., 2001). Rap1 plays a central role in basic cellular events such as adhesion and spreading, as well as in more complicated processes including migration, thrombosis, phagocytosis, inflammation, extravasation, and differentiation (Bos et al., 2001; Caron, 2003). The importance of Rap1 in development was shown by the early embryonic lethality of mutation of a Rap1-specific GEF (Ohba et al., 2001; Voss et al., 2003) as well as deletion of the Rap1 orthologue in Drosophila melanogaster (Hariharan et al., 1991; Asha et al., 1999; Knox and Brown, 2002).
Although it is evident that Rap1 regulates cell adhesion and spreading, little is known about how this occurs. One candidate Rap1 effector, RAPL, increases cell adhesion and colocalizes with clustered integrins (Katagiri et al., 2003). However, RAPL expression is limited to lymphoid tissues, whereas Rap1 regulates adhesion in numerous cell types. Signaling proteins that mediate the morphological effects of Rap1 in nonlymphoid cells have not been identified.
In budding yeast, the Rap1 orthologue Bud1/Rsr1p acts via a GEF, Cdc24p, that acts on a Rho protein, Cdc42p, at sites of bud emergence (Gulli and Peter, 2001). In animal cells, the Rho proteins RhoA, Cdc42, and Rac1 regulate many aspects of cell morphology in response to appropriate signals (Bishop and Hall, 2000). RhoA induces stress fibers and focal adhesions, Rac1 drives the formation of lamellipodia, and Cdc42 stimulates the formation of filopodia. Thus, it is plausible that the effects of Rap1 on animal cell morphology are mediated by Rho proteins.
Here, we have investigated the contribution of Rho family GTPases in cell spreading stimulated by Rap1. We show that Rac1, but not RhoA or Cdc42, is required downstream of Rap1 for cell spreading. Rap1 interacts with a subset of Rac GEFs that includes VAV2 and Tiam1, but not SWAP-70 or COOL-1. Overexpressed VAV2 and Tiam1 require Rap1 activity to enhance cell spreading even though they increase Rac1.GTP levels equally in the absence or presence of Rap1 activity. Moreover, the active form of Rap1 localizes VAV2 to the plasma membrane at sites where protrusive structures are actively engaging the extracellular matrix. Our findings suggest that Rap1 promotes cell spreading through Rac1 by targeting a specific subset of Rac GEFs to sites of cell–matrix contact. Thus, only when properly localized can Rac GEFs give rise to changes in cell morphology.
Results
Rap1a functions upstream of Rac
Rap1 regulates cell spreading and adhesion through an unknown mechanism. Given that Rap1 and Rho family GTPases lead to similar alterations in cell morphology, Rho proteins may function downstream of Rap1. To investigate this possibility, we made use of adherent HeLa cells transiently transfected with various constructs. Cells were suspended with EDTA, held in suspension in serum-free medium for 1 h, plated on fibronectin-coated surfaces, and examined 30 to 60 min later. Under these conditions, Rac1a and Rap1 are both activated, which is consistent with a possible functional relationship (unpublished data). Moreover, HeLa cells expressing activated (63E mutant) Rap1a adopted a flat, isotropic morphology (Fig. 1 A, 2) and resembled cells overexpressing activated Rac1 (Fig. 1 B, d) or Rac GEFs (see Fig. 4).
Figure 1.

Rac1 acts downstream of Rap1 in spreading cells. (A) Rac1 is necessary for cell spreading by active Rap1. HeLa cells were transfected with vectors encoding GFP or constitutively active GFP-63E Rap1a alone or together with inhibitory effector fragments that block signaling downstream from Rho family proteins. Myc-RBD POSH, Myc-GBD N-WASP, and Myc-RBD Rhotekin specifically inhibit signaling from Rac1, Cdc42, and RhoA, respectively. Rac1, Cdc42, and likely other Rho proteins are inhibited by Myc-PBD PAK1. Transfected cells were suspended, plated on fibronectin for 30 min, fixed, and labeled with Myc antibodies. (B) Decreased spreading by Rap1 inactivation is rescued by active Rac1. HeLa cells were transfected with vectors encoding GFP or Flag-Rap1GAP alone or together with constitutively active GFP-61L Rac1, and then allowed to spread on fibronectin for 1 h. (C) Histogram showing the percentage (average plus the SD) of flat, well-spread cells in the experiment described in A. 1, GFP (white bar); 2, GFP-63E Rap1a; 3, GFP-63E Rap1a + Myc-PBD PAK1; 4, GFP-63E Rap1a + Myc-RBD POSH; 5, GFP-63E Rap1a + Myc-GBD N-WASP; 6, GFP-63E Rap1a + Myc-RBD Rhotekin. (D) Histogram showing the percentage (average plus the SD) of refractile, poorly spread cells in the experiment described in B. a, GFP (white bar); b, Flag-Rap1GAP; c, Flag-Rap1GAP + GFP-61L Rac1; d, GFP-61L Rac1.
Figure 4.

Rap1 activity is necessary for cell spreading promoted by VAV2 and Tiam1, but not SWAP-70 or COOL-1. HeLa cells were transiently transfected with vectors encoding GFP, GFP-Rap1GAP, HA-DH-PH-CRD VAV2, or Myc-C1199 Tiam1, or cotransfected with vectors encoding GFP-Rap1GAP and HA-DH-PH-CRD VAV2, Myc-C1199 Tiam1, HA-SWAP-70, or Myc-COOL-1. Transfected cells were suspended, plated on fibronectin for 1 h, fixed, and labeled with the HA or Myc antibodies. The histogram shows the percentage (average plus the SD) of refractile, poorly spread cells for each condition in the absence (open bars) or presence (closed bars) of GFP-Rap1GAP.
To investigate the possible involvement of Rac1 or other Rho-related GTPases in Rap1-induced spreading, we made use of fragments from various effector proteins to sequester Rac1, Cdc42, or RhoA and prevent them from binding to endogenous effector molecules. The Rac-binding domain of POSH (RBD POSH) binds specifically to Rac1, the GTPase-binding domain (GBD) of N-WASP binds to Cdc42, the RhoA-binding domain of Rhotekin (RBD Rhotekin) binds to RhoA, and the p21-binding domain (PBD) of PAK1 binds to Rac1, Cdc42, and other Rho GTPases (Wennerberg et al., 2002). The increase in flat, well-spread cells caused by GFP-63E Rap1a was significantly antagonized by inhibiting Rac1 with RBD POSH or the PBD of PAK1, but not by the Cdc42 or RhoA antagonists (Fig. 1, A and C). Dominant-negative 17N Rac1 also inhibited spreading induced by 63E Rap1a (unpublished data). These data suggest that signaling from Rac1, but not Cdc42 or RhoA, is required for Rap1-induced cell spreading.
Inactivating Rap1 inhibits HeLa cell spreading, leading to a refractile, poorly-spread morphology (Fig. 1 B, b; and Fig. 1 D). If Rap1 induces spreading through Rac1, then activating Rac1 by other means may overcome the effects of Rap1 inactivation. Indeed, coexpression of constitutively active (61L mutant) Rac1 significantly rescued the loss of spreading caused by Rap1 inhibition. Cells expressing 61L Rac1 alone or together with the Rap1 inactivator Flag-Rap1GAP were well spread (Fig. 1 B, c and d). Rap1GAP fully inactivates Rap1a under these conditions (unpublished data). Thus, Rac1 is sufficient for cell spreading in the absence of Rap1 activity.
To test whether Rap1 regulates Rac1 GTP levels, we measured the effect of activated Rap1a on Rac1.GTP levels in cells that had been plated on fibronectin or poly-l-lysine for 3 h (Fig. 2). Rac1 activity was measured by precipitating active GTP-bound Rac1 with a GST fusion protein containing the PBD of PAK1 (del Pozo et al., 2000). Rac1 was activated by 63E Rap1a, on both fibronectin and poly-l-lysine surfaces. This finding suggests that Rap1 activates Rac1, and such activation is not secondary to Rap1-induced increases in outside-in signaling through integrins.
Figure 2.

Rap1 activates Rac1. HeLa cells were transfected with an empty vector or a vector encoding constitutively active HA-63E Rap1a. Cells were suspended, plated on fibronectin (FN) or poly-l-lysine (PLL) for 3 h, and lysed, and then active GTP-bound Rac1 was precipitated with GST-PBD PAK1. Precipitates (active) and total cell lysates (total) were then immunoblotted with Rac1 or HA antibodies.
Rap1a functions upstream of Rac by controlling specific Rac GEFs
Rap1 may induce cell spreading through Rac1 by any of several possible direct or indirect mechanisms involving parallel or sequential pathways. One possibility is that Rap1 may interact with a Rac1 GEF. Therefore, we tested if activated Rap1 might bind to one or more Rac GEFs.
We first tested whether or not Rap1a associates with VAV2. VAV2 is a widely expressed GEF that reportedly acts on Rac1, RhoA, and Cdc42 in vitro, although overexpression of VAV2 leads to a predominantly Rac1-like morphology (Schuebel et al., 1998; Liu and Burridge, 2000; Marignani and Carpenter, 2001). VAV2 contains NH2-terminal Calponin homology and acidic domains and central Dbl homology (DH) and Pleckstrin homology (PH) domains (Fig. 3 B). The DH domain mediates catalysis of GDP-GTP exchange on Rho proteins, whereas the PH domain binds phospholipids and participates in DH domain regulation (Han et al., 1998; Ma et al., 1998). The VAV2 COOH-terminal region is made up of a cysteine-rich domain (CRD) followed by a Src homology 2 domain flanked by two Src homology 3 domains. The CRD was of particular interest to this work because it is a motif that is also found in Raf and facilitates Ras-Raf binding (Hu et al., 1995). Additionally, the Calponin homology and DH-PH module have been separately implicated in Bud1 binding to Cdc24 (Park et al., 1997; Gulli and Peter, 2001). We conducted coimmunoprecipitation experiments using serum-starved cells expressing HA-tagged wild-type Rap1a (mostly GDP-bound) or constitutively active 63E Rap1a (GTP-bound) to determine if Rap1 binds to VAV2 (Fig. 3 A). We found that endogenous VAV2 coimmunoprecipitated with 63E Rap1a, and to a lesser extent with wild-type Rap1a. Another HA-tagged protein, HA-CRD VAV2, did not coimmunoprecipitate endogenous VAV2. Similar results were obtained in reciprocal immunoprecipitations using VAV2 antibodies. We also found that bacterial GST-Rap1a, but not Rab5 or H-Ras, precipitated bacterially expressed and purified His-DH-PH-CRD VAV2, suggesting that these proteins interact directly (Fig. 3 B).
Figure 3.

Rap1 binds to a subset of Rac GEFs. (A) Rap1 and VAV2 coimmunoprecipitate. HA (left) and VAV2 (right) antibodies were used for immunoprecipitation from serum-starved HeLa cells electroporated with vectors encoding HA-CRD VAV2 (negative control), HA-wild-type Rap1a (mostly GDP bound), or HA-63E Rapla (GTP bound). Immunoprecipitates (IP) and total cell lysates (TCL) were then immunoblotted with HA or VAV2 antibodies. (B) Rap1 binds to VAV2 directly. GTP-loaded GST-Rab5, GST-H-Ras, and GST-Rap1a on glutathione beads were incubated in the absence (−) or presence (+) of bacterially expressed His-DH-PH-CRD VAV2 (input). Beads were washed, and the precipitates were immunoblotted with an anti-His antibody (His-VAV2) or stained with Coomassie blue (GST-GTPases). (C) Rap1 interacts with the DH-PH module of VAV2. GST-63E Rapla was used to pulldown HA-DH-PH-CRD VAV2 (truncation a), HA-DH-PH-CRD W503L VAV2 (b), HA-DH-PH VAV2 (c), HA-DH VAV2 (d), and HA-CRD VAV2 (e) from transiently transfected HeLa cells. Pulldowns (GST-63E Rapla) and total cell lysates were immunoblotted with HA antibodies. (D) Rap1 interacts with a subset of Rac GEFs. GST-63E Rapla was used to pulldown Myc-COOL-1, Myc-C1199 Tiam1, HA-DH-PH-CRD VAV2, HA-SWAP-70, and HA-DH-PH Tiam1 from transiently transfected HeLa cells. Pulldowns and total cell lysates were immunoblotted with Myc (left) or HA (right) antibodies.
We tested which domains of VAV2 bind to Rap1 by incubating GST-63E Rap1a with cell lysates containing transiently expressed HA-tagged truncation mutants of VAV2. All proteins containing the DH and PH domains of VAV2 bound efficiently to GST-63E Rap1a (Fig. 3 C) and GST-Rap1a.GTP, but not GST-Rab5.GTP (not depicted). A W503L mutation in the PH domain of DH-PH-CRD VAV2 abolishes phospholipid binding (Booden et al., 2002) but not Rap1a binding. The DH domain alone also bound to Rap1a but at reduced levels. Unexpectedly, the CRD of VAV2 was neither necessary nor sufficient for binding to 63E Rap1a. These findings suggest that Rap1 binds to the DH-PH module of VAV2 and that the contribution of the PH domain is independent of phospholipid binding. Despite VAV2 binding to Rap1 via its catalytic DH-PH module, VAV2 does not stimulate nucleotide exchange on Rap1 in vitro or in cells (unpublished data).
Nearly all of the more than 60 Dbl family Rho GEFs contain a DH-PH module (Schmidt and Hall, 2002). Our finding that Rap1 binds to the DH-PH module of VAV2 led us to consider that Rap1 may bind to other Rac GEFs. Therefore, GST-63E Rap1a binding experiments were performed with several transiently expressed Rac GEFs (Fig. 3 D). We found that truncated, activated mutant C1199 Tiam1 and the DH-PH region of Tiam1 bound to 63E Rap1a, whereas COOL-1/βPIX and SWAP-70 did not. These assays suggest that Rap1 binds to a subset of Rac GEFs that includes VAV2 and Tiam1 but not SWAP-70 or COOL-1.
We tested whether or not Rac GEFs that can bind Rap1 require Rap1 to induce cell spreading. We examined the morphology of cells transiently expressing various full-length or activated Rac GEFs in the presence or absence of Rap1GAP to inactivate Rap1 (Fig. 4). As expected, GFP-Rap1GAP inhibited spreading relative to control cells expressing only GFP, whereas SWAP-70, COOL-1, full-length VAV2, the DH-PH-CRD module of VAV2, and C1199 Tiam1 each enhanced spreading (Fig. 4 and not depicted). Coexpression of SWAP-70 and COOL-1 with Rap1GAP decreased the incidence of poorly spread cells relative to cells expressing Rap1GAP alone. Strikingly, however, full-length VAV2, DH-PH-CRD VAV2, and C1199 Tiam1 were not able to rescue the decrease in spreading caused by Rap1GAP overexpression (Fig. 4 and not depicted). This finding was surprising because the mutant forms of VAV2 and Tiam1 are constitutively active. These data indicate that the Rap1-binding Rac GEFs, VAV2 and Tiam1, require Rap1.GTP to stimulate cell spreading. In contrast, SWAP-70 and COOL-1 function in a Rap1.GTP-independent manner.
Rap1 is necessary for the proper localization of VAV2
Several potential mechanisms may explain why VAV2 and Tiam1 require Rap1 activity to regulate cell morphology. First, Rap1 could stimulate the GEF activities of VAV2 and Tiam1. Second, Rap1 could localize VAV2 and Tiam1 to specific sites where Rac1 activation is required for cell spreading. We tested whether or not Rap1 regulates Rac1 activation by exogenous VAV2 and Tiam1 (Fig. 5). Expression of truncated, constitutively active variants of DH-PH-CRD VAV2 or C1199 Tiam1 increased Rac1 GTP content even when endogenous Rap1 was inactivated by Rap1GAP. Furthermore, the addition of Rap1.GTP to in vitro exchange reactions did not augment GTP loading of Rac by VAV2 (unpublished data). These data show that Rap1.GTP is dispensable for Rac1 activation by constitutively active VAV2 and Tiam1, but do not exclude the possibility that Rap1 regulates to the catalytic activity of full-length GEFs in physiological conditions. These findings (Figs. 4 and 5) further suggest that increased total cellular levels of Rac1.GTP are not sufficient for cell spreading in the absence of Rap1 activity. Activation of a specific subpopulation of Rac1 may be required for spreading.
Figure 5.

Rap1 activity is dispensable for Rac1 activation by VAV2 and Tiam1. (left) HeLa cells were transfected with an empty vector (vector) or a vector encoding constitutively active HA-DH-PH-CRD VAV2 (VAV2), FLAG-Rap1GAP (Rap1GAP), or both. (right) In separate experiments, cells were transfected with an empty vector or a vector encoding constitutively active Myc-C1199 Tiam1 (Tiam1), FLAG-Rap1GAP, or both. The cells were lysed, and active GTP-bound Rac was precipitated with GST-PBD PAK1. Precipitates (active) and total cell lysates (total Rac1, HA/Myc, FLAG) were immunoblotted with Rac1, HA, Myc, or FLAG antibodies.
We used two approaches to test whether or not Rap1 can regulate the subcellular localization of VAV2. First, Cho and Klemke (2002) have developed a technique for identifying proteins that are enriched in pseudopodia engaging a fibronectin-coated substrate. We prepared pseudopodial extracts and total extracts from cells with normal or attenuated Rap1 activity and used Western blotting to examine the distribution of endogenous proteins (Fig. 6). As reported previously (Cho and Klemke, 2002), Paxillin was concentrated in pseudopodia and ERK was distributed equally in the pseudopodia and total extracts. Relative to ERK, Rap1 was also concentrated in pseudopodia. However, Rap1 localization was not affected by Rap1GAP overexpression. Like Rap1, VAV2 accumulated in pseudopodia under control conditions. However, the targeting of VAV2 to pseudopodia was blocked by expression of Rap1GAP. These data demonstrate that both GDP- and GTP-bound forms of Rap1 are concentrated in matrix-associated membrane protrusions and suggest that the active form of Rap1 mediates the localization of VAV2 to these structures.
Figure 6.
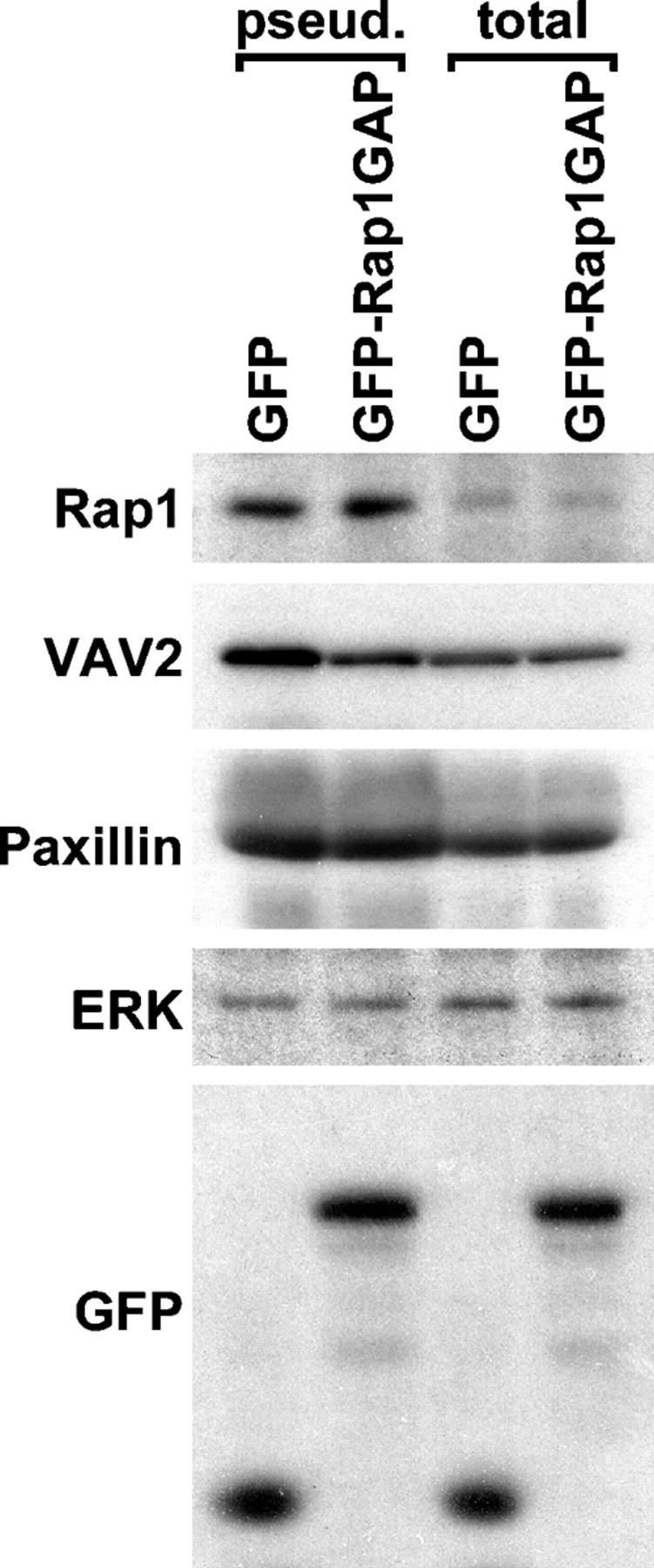
Rap1 targets VAV2 to matrix-associated membrane protrusions. Pseudopodia and total cell extracts were prepared with Transwell filters from HeLa cells electrotransfected with vectors encoding GFP or GFP-Rap1GAP. Extracts were adjusted for equal content of ERK, a protein found at equal levels in pseudopodia and total cell extracts (Cho and Klemke, 2002). Pseudopodial (pseud.) and total cell (total) extracts were immunoblotted with antibodies for Rap1, VAV2, Paxillin, ERK, or GFP.
As a second test for VAV2 localization, we examined the subcellular distribution of Rap1 and VAV2 in spreading cells using immunofluorescence (Fig. 7). Consistent with a recent paper (Bivona et al., 2004), a fraction of activated 63E Rap1a, but not dominant-negative 17N Rap1a, was found in membrane protrusions around the circumference of transiently transfected cells (Fig. 7 A, left). The absence of 17N Rap1a at the cell periphery conflicted with our pseudopodia fractionation studies and could be a secondary effect due to the lack of membrane protrusions around the circumference of cells in which Rap1 is inhibited. Accordingly, the Rac GEF SWAP-70, which induces cell spreading independently of Rap1, was expressed with 63E or 17N Rap1a. Consistent with the pseudopodia fractionation results (Fig. 6), both 63E Rap1a and 17N Rap1a were enriched in circumferential protrusions in SWAP-70–expressing cells (Fig. 7 A). These data suggest that both active and inactive Rap1 localize to Rac-dependent membrane protrusions. We examined the localization of the Rap1-dependent and -independent Rac GEFs VAV2 and COOL-1 in cells with normal or attenuated Rap1 activity. Overexpressed VAV2 and COOL-1 were both in peripheral membrane ruffles in cells with normal Rap1 activity. However, when Rap1 was inhibited with Rap1GAP, VAV2 but not COOL-1 was displaced from the cell periphery (unpublished data). Because the cells expressing VAV2 and Rap1GAP were poorly spread (Fig. 4), we again used SWAP-70 to induce Rap1-independent spreading (Fig. 7 B). Under these conditions, the cells were well-spread regardless of Rap1 activity, but VAV2 was displaced from circumferential membrane protrusions when Rap1 was inactivated. Together, these experiments suggest that active Rap1 relocalizes Rac GEFs that bind to Rap1, such as VAV2, to the periphery of spreading cells. In contrast, Rac GEFs that do not bind Rap1, such as COOL-1, accumulate in the periphery of spreading cells independently of Rap1 activity.
Figure 7.

Rap1 targets VAV2 to circumferential membrane protrusions. (A) Both active and inactive Rap1a localize to membrane protrusions. HeLa cells were transiently transfected with vectors encoding GFP-63E Rapla (63E) or GFP-17N Rap1a (17N) alone (left) or cotransfected with a HA-SWAP-70–encoding vector (right four panels). Transfected cells were suspended, plated on fibronectin for 1 h, fixed, and labeled with HA antibodies. Note that cells expressing 17N Rap1a do not spread, but when spreading is induced with SWAP-70, then 17N Rap1a is detected at the cell periphery. Arrowheads indicate localization of the GFP-Rap1a variants at the cell edge. (B) VAV2, but not COOL-1, requires Rap1 activity to localize to membrane protrusions. HeLa cells were transiently cotransfected with vectors encoding Myc-VAV2 or Myc-COOL-1 and GFP or GFP-Rap1GAP together with HA-SWAP-70. HA-SWAP-70 was cotransfected with the Rac GEFs and GFP vectors to allow Rap1-independent spreading. Transfected cells were treated as in A, labeled with Myc antibodies, and only well-spread cells were analyzed. Arrowheads indicate localization of the GEFs at the cell edge.
Artificial localization of VAV2 bypasses the Rap1 requirement for cell spreading
Collectively, our data suggest that VAV2 requires Rap1.GTP for accumulation at the cell periphery and that this relocalization is required for VAV2 to stimulate cell spreading. A prediction of this model is that Rap1.GTP-dependent Rac GEFs would be able to promote cell spreading in the absence of Rap1 activity if they were targeted to the vicinity of Rap1 by other means. To test this idea, we created fusion proteins of VAV2 and 63E Rap1a, 17N Rap1a, or fragments of Rap1a. The fragments used for these fusions consisted of the 20-residue membrane-targeting domain at the COOH terminus of Rap1a. This domain includes the prenylated (geranylgeranyl modified) COOH-terminal CAAX box that allows for membrane insertion and 16 adjacent residues NH2-terminal to the CAAX box that make up the HV/polybasic domain and dictate what type of membrane is targeted (Hancock, 2003). As a control, a C/S mutation was generated in the CAAX box to prevent prenylation and thus abolish membrane targeting. The ability of the Rap1a-targeted VAV2 fusions to stimulate cell spreading was analyzed in cells in which Rap1 was inhibited with Rap1GAP (Fig. 8). As in Fig. 4, expression of Rap1GAP led to poor spreading, and this phenotype was not reverted by coexpression of the DH-PH-CRD module of VAV2. In contrast, the refractile, poorly spread phenotype was reverted to a highly spread morphology when a fusion of the DH-PH-CRD to 63E Rap1a was coexpressed with Rap1GAP. Because 63E Rap1a is resistant to Rap1GAP, this result could be either because the DH-PH-CRD is correctly localized or because Rap1a is not inactivated. However, a fusion of DH-PH-CRD to inactive 17N Rap1a or to just the HV-CAAX region of Rap1a also reverted cells expressing Rap1GAP from a poorly spread morphology. In contrast, a fusion of the DH-PH-CRD to the nonprenylated HV-SAAX region of Rap1a did not rescue. Thus, fusing the localization signals present in the HV-CAAX region of Rap1a onto VAV2 is sufficient to bypass the requirement for Rap1.GTP for cell spreading. The effect of the Rap1a CAAX region was specific because fusing VAV2 DH-PH-CRD to the K-Ras HV-CAAX region did not induce Rap1-independent spreading (unpublished data). These findings are consistent with a model in which Rap1.GTP relocalizes VAV2 and, likely, other Rac1GEFs including Tiam1 to the cell periphery where it stimulates Rac1-dependent cell spreading.
Figure 8.
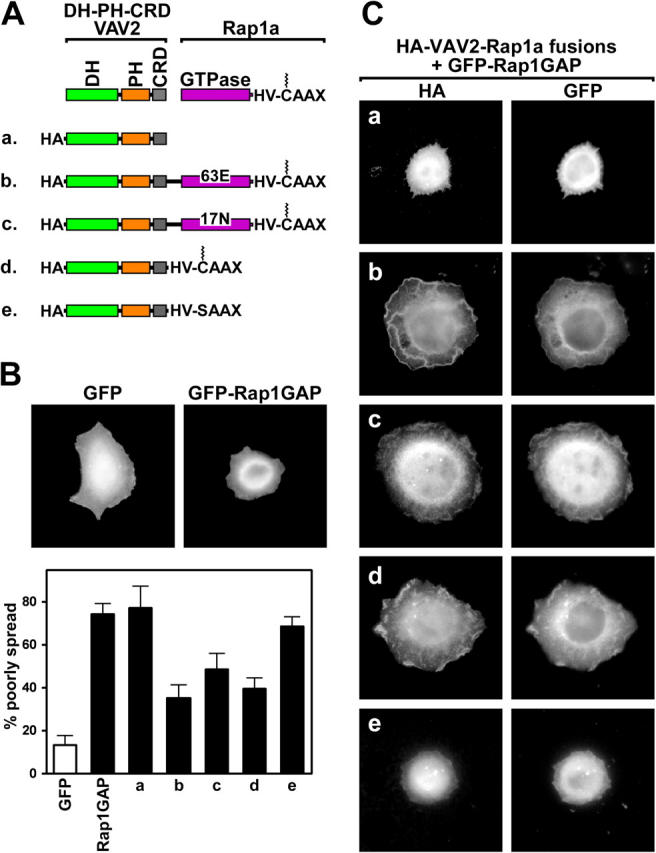
Rap1-targeted VAV2 fusion proteins function independently of Rap1. (A) Schematic representation of HA epitope-tagged DH-PH-CRD VAV2 (VAV2) fused to full-length 63E Rap1a or 17N Rap1a, consisting of the GTPase domain (GTPase), hypervariable domain (HV), and CAAX box. Also shown are VAV2 fused to the COOH terminus of Rap1a encoding the HV and the prenylated (jagged line) CAAX box. The SAAX mutation abolished membrane targeting by preventing prenylation. HeLa cells were transfected with expression vectors for GFP or GFP-Rap1GAP alone (B) or GFP-Rap1GAP together with HA-VAV2 (a), HA-VAV2-63E Rapla (b), HA-VAV2-17N Rapla (c), HA-VAV2-HV-CAAX (d), or HA-VAV2-HV-SAAX (e) vectors (C). Transfected cells were suspended, plated on fibronectin for 1 h, fixed, and labeled with HA epitope tag antibodies. The histogram shows the percentage (average plus the SD) of refractile, poorly spread cells for each condition.
Discussion
Rap1 regulates the interaction of a variety of cell types with the extracellular matrix or adhesion molecules on adjacent cells (Bos et al., 2001; Caron, 2003). Loss of Rap1 activity in D. melanogaster disrupts adherens junctions and cell–cell adhesion in the embryo, eye disc, and ovary (Hariharan et al., 1991; Asha et al., 1999; Knox and Brown, 2002). Rap1 is activated in animal cells in response to numerous stimuli and is important for cell adhesion, spreading, and migration (Bos et al., 2001). In this work, we examined the mechanism by which Rap1 enhances spreading of HeLa cells on the extracellular matrix protein fibronectin in the absence of serum. We found that Rap1 acts through the Rho family GTPase Rac1 (Fig. 1). Both active and inactive Rap1 are present in pseudopods and at the periphery of spreading cells, where plasma membrane is actively engaging the extracellular matrix (Figs. 6 and 7). When associated with GTP, Rap1 binds to specific Rac GEFs, including VAV2 and Tiam1, via their DH-PH regions (Fig. 3). Rap1 is required to recruit VAV2, and likely other GEFs, to sites of membrane protrusion (Fig. 7). Localization of VAV2 by Rap1 is required to induce Rac1-dependent protrusive activity (Figs. 4 and 8). In addition to localizing VAV2, Rap1 has the potential to also activate VAV2 locally at the cell periphery, but our experiments did not address this possibility. After Rac-dependent protrusive activity, new sites for Rap1 localization are created (Fig. 7 A), continuing the cycle. Rac activation has recently been reported to regulate the localization of a Rap1 GEF, GRP2, to sites of actin polymerization (Caloca et al., 2004). Therefore, Rap1 and Rac may promote membrane protrusion in a highly cooperative manner (Fig. 9 A).
Figure 9.

Model: Rap1 regulates morphology by targeting a subset of Rac GEFs to the edge of spreading cells. (A) Only when Rac GEFs are properly targeted do they locally activate Rac, resulting in cell spreading by the formation of productive membrane protrusions adjacent to the ECM. COOL-1 and SWAP-70, two Rac GEFs that do not interact with Rap, are targeted to the protrusive structures by Rap1-independent mechanisms. In contrast, the Rac GEFs VAV2 and Tiam1 are targeted to ECM-associated plasma membranes by binding to active Rap1 (Rap1.GTP) but not inactive Rap1 (Rap1.GDP), following Rap1 activation by adhesion signals. (B) Our proposed model of Rap1 regulation of spreading through Rac GEFs is comparable to the mechanism by which the yeast protein Bud1/Rsr1 controls budding via Cdc24, a Cdc42 GEF.
An implication of this paper is that other Rac GEFs such as SWAP-70 and COOL-1, which stimulate cell spreading independently of Rap1 when overexpressed, are either absent from HeLa cells or inactive under the serum-free conditions we used for assay. These GEFs may relocalize to the cell periphery and stimulate spreading in response to other signals in a Rap1-independent manner. For example, SWAP-70 localizes to membrane ruffles by binding to phosphatidylinositol-3, 4, 5-triphosphate (Shinohara et al., 2002) and PKL links COOL-1 to Paxillin, a cytoskeletal protein found in nascent protrusions and adhesions (Tumbarello et al., 2002).
Cell spreading on extracellular matrix is a complex, multistep process. Spreading requires attachment, the initial interaction between integrins and specific extracellular matrix proteins. Attachment is known to be regulated by Rap1 and can be augmented by soluble factors that promote inside-out signaling to integrins (Bos et al., 2001). Rap1 increases binding to extracellular ligands by regulating integrin affinity, avidity, or both. Affinity regulation involves conformational changes that result in increased binding of individual integrin heterodimers to their extracellular ligands (Hynes, 2002). Avidity modulation occurs when integrins cluster such that overall adhesion is increased without noticeable changes in affinity. After the initial integrin–matrix attachment is established, outside-in signaling by integrins leads to inside-out signals through Rap1 that further modify integrin affinity or avidity and promote adhesion (Bos et al., 2001). Concurrent with these signaling events, integrins trigger rearrangements in the actin cytoskeleton through Rho family proteins. Activation of Rac1 and Cdc42 as well as RhoA inhibition are essential for cytoskeletal dynamics that give rise to efficient cell spreading. Interestingly, we found that spreading can occur in cells overexpressing SWAP-70 or COOL-1 when Rap1 is inactive and presumably unable to up-regulate integrin function. Under these conditions, some GTPase other than Rap1 may up-regulate integrins. In this regard, we found that R-Ras promotes spreading independently from Rap1 (unpublished data), likely through integrin activation (Kinbara et al., 2003).
Our work does not show which steps in spreading are promoted by Rap1-mediated Rac GEF recruitment. Other papers using dominant-negative GEFs and cells from knockout mice have implicated VAV2, the VAV2-related GEF VAV1, and Rac1 in adhesion, spreading, and integrin clustering (Marignani and Carpenter, 2001; Krawczyk et al., 2002; Ardouin et al., 2003; del Pozo et al., 2003). Indeed, dominant-negative VAV2 reduces cell spreading in our assay (unpublished data). However, whether inside-out signaling downstream of integrins depends on, follows from, or lies in parallel to cytoskeletal rearrangements is not clear. Some recent models suggest that the functional activation of integrins by Rap1 is mediated by cytoskeletal dynamics and cytoskeleton-associated proteins (Caron, 2003). In contrast, because VAV1 interacts with talin, a cytoskeletal protein that regulates integrin conformation (Fischer et al., 1998; Calderwood, 2004; Nayal et al., 2004), VAV1 has the potential to directly affect integrins independently of Rac1. Thus, VAV2 may independently or cooperatively regulate both integrin affinity and Rac1-dependent cytoskeletal rearrangements when it is recruited to the cell periphery by Rap1.
Our data suggest that Rap1-mediated recruitment of Rac GEFs is necessary and sufficient for cell spreading, but active Rap1 binds to a variety of effectors and it is likely that other mechanisms downstream from Rap1 also regulate cell–matrix interactions. Katagiri and colleagues (2003) have reported that the Rap1 effector RAPL colocalizes with integrins and increases adhesion. The Rap1 effector AF-6 has been implicated in adherens junction regulation by Rap1 and may contribute to cell–matrix interactions through Profilin (Boettner et al., 2000). Thus, Rap1-mediated recruitment of Rac GEFs may not be necessary for cell spreading by physiological stimuli that activate Rap1 if other effectors, such as RAPL, are present.
Recently, Bivona et al. (2004) reported that Rap1 is absent from the plasma membrane of serum-starved COS cells and is dynamically transported via an exocytic pathway from endomembranes to the plasma membrane after growth factor stimulation. This movement coincides with activation of Rap1, and an activated mutant of Rap1 is enriched on the plasma membrane whereas an inactive mutant is restricted to internal membranes. This suggests that the activity state of Rap1 influences localization to the plasma membrane. However, we found both active and inactive Rap1 in pseudopods and at the ruffling edge of HeLa cells actively spreading on fibronectin. This finding implies that Rap1 may be present at sites of active integrin engagement independent of its activity state. However, to detect 17N Rap1a at the cell edge we found it necessary to enhance protrusion with a Rap1-independent Rac GEF. Consequently, we cannot rule out the possibility that inactive Rap1 is absent from the cell circumference under normal conditions. Another possibility is that various Rap1 GEFs activate different pools of Rap1. If Rap1 is activated by a GEF that is restricted to internal membranes, then the exocytic pathway (Bivona et al., 2004) may be essential to relocate Rap1 to the cell periphery.
In addition to targeting Rac GEFs to the cell periphery, we and others have found that Rap1 stimulates GTP loading of Rac1 (Maillet et al., 2003). Because increased Rac1 GTP loading by GEFs is not sufficient to cause cell spreading (e.g., in cells that overexpress VAV2 but in which Rap1 is inhibited by Rap1GAP), the subpopulation of Rac1 that induces spreading may be small. Significantly, the COOH-terminal targeting domains and distributions of Rap1 and Rac1 are very similar (Quinn et al., 1992; Pizon et al., 1994; Sato et al., 1994; D'Silva et al., 1997; Radhakrishna et al., 1999; Boettner et al., 2000; Michaelson et al., 2001; Knox and Brown, 2002; Bivona et al., 2004). Thus, Rap1 may increase GTP loading on Rac1 present in endomembranes and on the plasma membrane by localizing Rac GEFs to both compartments.
Ras protein interaction is becoming a common mechanism of GEF regulation (Schmidt and Hall, 2002). In addition to the classic regulation of the GEF RalGDS by Ras, several Ras family GEFs contain Ras-binding domains that may enable their subcellular localization by Rap or M-Ras (Quilliam et al., 2002). Additionally, Ras regulates the Rac GEFs Tiam1 and SOS (Lambert et al., 2002; Margarit et al., 2003). In Saccharomyces cerevisiae, the Rap1 orthologue Bud1/Rsr1p directs actin polymerization at the site of bud formation (Gulli and Peter, 2001; Park et al., 2002). Active Bud1 at the incipient bud recruits the Rho family GEF Cdc24. Cdc24 then locally catalyzes nucleotide exchange on Cdc42 and thereby activates the PAK orthologue Cla4. Clearly, parallels can be made between the Bud1-Cdc24-Cdc42 system and the Rap1-Rac GEF-Rac1 mechanism described in this paper (Fig. 9 B). The concept that Bud1/Rap1 GTPases can regulate the function of Rho family GEFs by dictating their subcellular localization is conserved across distantly related species. Given the evolutionary conservation of the pathway, the localization of many more of the over 60 Dbl family Rho GEFs (Schmidt and Hall, 2002) may be determined by Rap or Ras family GTPases. In this work, we focused on Rac GEFs due to the finding that Rac and not Cdc42 or RhoA was essential for spreading by Rap1 (Fig. 1 A.). However, we identified several Cdc42 and RhoA GEFs that also interact with Rap1 (unpublished data). Similarly, recent studies have demonstrated that Rap1 colocalizes with and acts upstream of the Cdc42 GEF FRG in the context of cell–cell adhesion (Fukuyama, T., H. Ogita, T. Sato, T. Kawakatsu, T. Fukuhara, T. Yamada, K. Shimizu, T. Nakamura, M. Matsuda, and Y. Takai, personal communication). Thus, it is evident that GEFs for multiple Rho proteins participate in Rap1-mediated biological processes.
Materials and methods
Reagents
The following vectors were provided by the indicated investigators: pCMV-Myc-PBD PAK, -RBD POSH, -GBD N-WASP, -RBD Rhotekin, pEGFP-Q61L Rac1, pGEX-PBD PAK1, pZip-Myc-C1199 Tiam1, pCGN-HA-DH-PH Tiam1, and pCGN-HA-DH-PH-CRD VAV2 mutants and truncations (K. Wennerberg, M. Booden, C. Der, and K. Burridge, University of North Carolina, Chapel Hill, NC), pcDNA3-Myc-COOL-1/βPIX (M. Brown and C. Turner, State University of New York, Syracuse, NY), pEFBos-HA-SWAP-70 (R. Jessberger, Mount Sinai School of Medicine, New York, NY), and pCMV5-Myc-VAV2 (C. Carpenter, Harvard Medical School, Boston, MA). His-DH-PH-CRD VAV2 was provided by S. Ellerbroek (University of North Carolina, Chapel Hill, NC). pCGN-HA WT, 17N, and 63E Rap1A mutants were made as described previously (Tsygankova et al., 2001). pFLAG CMV2-Rap1GAP (Castro et al., 2003) was generated by cloning a BglII–BamHI fragment of Rap1GAP (Rubinfeld et al., 1991) into pFLAG-CMV2 (Sigma-Aldrich). pEGFP-17N Rap1a and pEGFP-63E Rap1a were made by subcloning BamHI fragments from pCGN-HA-Rap1a into pEGFP-C1 (CLONTECH Laboratories, Inc.) linearized with BamHI. pEGFP-Rap1GAP was made by subcloning a BglII–BamHI fragment from pFLAG CMV2-Rap1GAP into BglII linearized pEGFP-C3. pCGN-HA-DH-PH-CRD VAV2 fusion with HV-CAAX, HV-SAAX, 63E Rap1a, or 17N Rap1a were constructed by replacing the pCGN-HA-DH-PH-CRD VAV2 stop codon with a BglII site. A BamHI VAV2 insert was then subcloned into pBluescript II KS+. BamHI fragments of 17N, 63E, HV-CAAX, or HV-SAAX of Rapla were ligated into the BglII site of the DH-PH-CRD VAV2 in pBluescript II KS+. The HV-CAAX fragment encodes the amino acid sequence INRKTPVEKKKPKKKSCLLL. BamHI fragments of DH-PH-CRD VAV2-Rap1a variants from pBluescript II KS+ were subcloned into BamHI-digested pCGN-HA. The following antibodies were obtained: rabbit anti-Rap1 (Santa Cruz Biotechnology, Inc.), mouse anti-Rac1 and -Paxillin (BD Biosciences), VAV2 rabbit anti-sera (provided by B. Liu and K. Burridge, University of North Carolina, Chapel Hill, NC), mouse anti-GFP (Roche), mouse anti-FLAG (Sigma-Aldrich), mouse anti-Myc (Invitrogen), and mouse anti-HA.11(Covance). The rabbit anti-ERK was generated in our laboratory.
Cell culture, transfection, and cell stimulation
HeLa cells were maintained in DME supplemented with 10% FBS and antibiotics. Cells were transfected with the indicated mammalian expression vectors using LipofectaminePLUS (Invitrogen) or Nucleofector Electroporation Kit R (Amaxa Biosystems) according to the manufacturer's instructions. Transfected cells were detached using 0.5 mM EDTA in Hepes-buffered saline, pH 7.4, and placed in suspension for 1 h in DME, 0.1% BSA. Cells were plated in the presence of DME, 0.1% BSA, on glass coverslips or tissue culture plates that had been coated overnight with 4 μg/ml of fibronectin or 10 μg/ml of poly-l-lysine (Sigma-Aldrich) and blocked with 0.5% BSA. Cells were fixed or lysed after spreading for 30 min (Fig. 1 A) or 1 h.
Immunofluorescence and spreading assay
Cells on glass coverslips were fixed for 15 min at RT in 3.7% formaldehyde, and then permeabilized for 4 min in 0.2% Triton X-100. Cells expressing epitope-tagged proteins were labeled by incubation with the relevant mouse mAbs followed by incubation with Texas red–conjugated goat anti–mouse antibodies (Jackson ImmunoResearch Laboratories). All images were collected at RT by 40× magnification epifluorescence on a microscope (model Axiophot; Carl Zeiss MicroImaging, Inc.) with a cooled CCD digital camera (model Photometrics SenSys; Roper Scientific) using MetaMorph software (Universal Imaging Corp.). To quantitate spreading, the total number of cells expressing the relevant constructs (70–140 cells per coverslip) and the number of flat, well-spread cells (Fig. 1 A only) or refractile, poorly-spread cells were counted in each condition. Histograms are from representative experiments and show the average percent of well- or poorly spread cells from three coverslips plus the SD. Data was considered statistically significant if the p-value was <0.05 as determined by two-tailed t tests.
Rac activity assay
The levels of activated, GTP-bound Rac1 from cells plated for 3 h on fibronectin or poly-l-lysine was measured using a technique similar to previously described methods (del Pozo et al., 2000). In brief, cells were lysed in 400 μl of 50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, and protease inhibitors. 500–750 μg of lysates were cleared at 16,000 g for 5 min and the supernatant was rotated for 30 min with 30 μg of GST-PBD PAK1 bound to glutathione-Sepharose 4B beads (Amersham Biosciences), and then washed three times in lysis buffer. GST-PBD PAK1 pulldowns (active Rac1) and lysates (total Rac1) were then immunoblotted with Rac1 antibodies.
Pulldown and coimmunoprecipitation assays
For GST-Rap1a pulldowns, transfected cells were lysed in 1% Triton X-100, 20 mM Hepes, pH 7.4, 150 mM NaCl, 5 mM MgCl2, and protease inhibitors. Lysates were centrifuged at 16,000 g for 10 min. 250-μg samples were incubated at 4°C for 1 h with 2 μg of GST or GST Rap1a mutants bound to glutathione-Sepharose 4B beads. The beads were washed four times with lysis buffer, and then subjected to immunoblot analysis with antibodies for Myc or HA. Membranes were stained with Ponceau S before immunoblot analysis or with Coomassie blue after immunoblot analysis and indicated that similar amounts of fusion proteins were used in all experiments. In vitro binding assays with GTP-loaded GST-GTPases and 1 μg of bacterially expressed His-DH-PH-CRD VAV2 were conducted in a similar manner except that the concentration of Triton X-100 was reduce to 0.1%. For Rap and VAV2 coimmunoprecipitations, starved cells expressing Rap1a mutants were lysed in 1% NP-40, 0.5% deoxycholate, 50 mM Tris, pH 7.4, 137 mM NaCl, 0.5 mM MgCl2, and protease inhibitors and precleared with rabbit anti–mouse–conjugated agarose beads (Sigma-Aldrich) or protein A–Sepharose beads (Amersham Biosciences). Samples were cleared at 16,000 g for 10 min. Lysates were incubated overnight with 0.5 μg of mAb HA.11 or with 1 μl of rabbit VAV2 antisera, followed by incubation for 2 h with 10 μl of bed volume of anti-mouse–conjugated agarose beads or protein A–Sepharose beads. Sample were washed six to eight times in lysis buffer, run on 12.5% SDS-PAGE gels, transferred to PVDF, and immunoblotted with antibodies for VAV2 or the HA epitope tag.
Pseudopodia purification
Pseudopodia were purified from electrotransfected cells expressing GFP or GFP-Rap1GAP using a technique based on a method described by Cho and Klemke (2002). In brief, the bottom surfaces of 75-mm diameter polycarbonate Transwell inserts with 3-μm pores (Corning Inc.) were coated overnight with 4 μg/ml of fibronectin (Sigma-Aldrich). The upper surfaces were coated for 1 h with 0.1 μg/ml of fibronectin. Both surfaces were blocked with 0.5% BSA in PBS, washed with PBS, and placed in DME, 0.1% BSA. Transfected cells that had been held in suspension for 1 h in DME, 0.1% BSA, were seeded to confluency on the upper surfaces and allowed to incubate at 37°C for 3 h. The Transwells were fixed in ice-cold 100% methanol and cell bodies on the upper surfaces were removed with a cotton swab and Kimwipes. The Transwells were washed briefly with ice-cold Hepes-buffered saline and the remaining pseudopodia on the lower surfaces were lysed in boiling sample buffer (pseudopodial extracts). Cells that had been suspended for the duration of the experiment were lysed in sample buffer and used as a source of total cell extracts. Total cell and pseudopodial extracts containing equal amounts of ERK, a protein found at equal levels in total cell and pseudopodia extracts (Cho and Klemke, 2002), were run on 12.5% SDS-PAGE gels, transferred to PVDF, and immunoblotted with antibodies for Paxillin, ERK, VAV2, Rap1, and GFP.
Acknowledgments
The authors are very grateful to the members of the Cooper laboratory, Krister Wennerberg, Shawn Ellerbroek, and Susan Parkhurst for helpful discussions, and Michelle Booden, Channing Der, Keith Burridge, Michael Brown, Christopher Turner, Rolf Jessberger, and Christopher Carpenter for providing reagents. We thank Cecilia Moens for suggesting a modification to the protocol used in Fig. 7 B.
This work was supported by Ruth L. Kirschstein National Research Service Award F32 NS046492-01 (W.T. Arthur), National Institutes of Health research grant R37-CA41072 (J.A. Cooper), and an Indiana University Biomedical Research grant (L.A. Quilliam).
Abbreviations used in this paper: CRD, cysteine-rich domain; DH, Dbl homology; GAP, GTPase activating protein; GBD, GTPase-binding domain; GEF, guanine nucleotide exchange factor; HV, hypervariable; PBD, p21-binding domain; PH, Pleckstrin homology; RBD POSH, Rac-binding domain of POSH; RBD Rhotekin, RhoA-binding domain of Rhotekin.
References
- Ardouin, L., M. Bracke, A. Mathiot, S.N. Pagakis, T. Norton, N. Hogg, and V.L. Tybulewicz. 2003. Vav1 transduces TCR signals required for LFA-1 function and cell polarization at the immunological synapse. Eur. J. Immunol. 33:790–797. [DOI] [PubMed] [Google Scholar]
- Asha, H., N.D. de Ruiter, M.G. Wang, and I.K. Hariharan. 1999. The Rap1 GTPase functions as a regulator of morphogenesis in vivo. EMBO J. 18:605–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, A.L., and A. Hall. 2000. Rho GTPases and their effector proteins. Biochem. J. 348:241–255. [PMC free article] [PubMed] [Google Scholar]
- Bivona, T.G., H.H. Wiener, I.M. Ahearn, J. Silletti, V.K. Chiu, and M.R. Philips. 2004. Rap1 up-regulation and activation on plasma membrane regulates T cell adhesion. J. Cell Biol. 164:461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettner, B., E.E. Govek, J. Cross, and L. Van Aelst. 2000. The junctional multidomain protein AF-6 is a binding partner of the Rap1A GTPase and associates with the actin cytoskeletal regulator profilin. Proc. Natl. Acad. Sci. USA. 97:9064–9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booden, M.A., S.L. Campbell, and C.J. Der. 2002. Critical but distinct roles for the pleckstrin homology and cysteine-rich domains as positive modulators of Vav2 signaling and transformation. Mol. Cell. Biol. 22:2487–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos, J.L., J. de Rooij, and K.A. Reedquist. 2001. Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell Biol. 2:369–377. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A. 2004. Integrin activation. J. Cell Sci. 117:657–666. [DOI] [PubMed] [Google Scholar]
- Caloca, M.J., J.L. Zugaza, M. Vicente-Manzanares, F. Sanchez-Madrid, and X.R. Bustelo. 2004. F-Actin-dependent translocation of the Rap1 GDP/GTP exchange factor RasGRP2. J Biol Chem. 279:20435–20446. [DOI] [PubMed] [Google Scholar]
- Caron, E. 2003. Cellular functions of the Rap1 GTP-binding protein: a pattern emerges. J. Cell Sci. 116:435–440. [DOI] [PubMed] [Google Scholar]
- Castro, A.F., J.F. Rebhun, G.J. Clark, and L.A. Quilliam. 2003. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem. 278:32493–32496. [DOI] [PubMed] [Google Scholar]
- Cho, S.Y., and R.L. Klemke. 2002. Purification of pseudopodia from polarized cells reveals redistribution and activation of Rac through assembly of a CAS/Crk scaffold. J. Cell Biol. 156:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo, M.A., L.S. Price, N.B. Alderson, X.D. Ren, and M.A. Schwartz. 2000. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 19:2008–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo, M.A., M.A. Schwartz, J. Hu, W.B. Kiosses, A. Altman, and M. Villalba. 2003. Guanine exchange-dependent and -independent effects of Vav1 on integrin-induced T cell spreading. J. Immunol. 170:41–47. [DOI] [PubMed] [Google Scholar]
- D'Silva, N.J., D.H. DiJulio, C.M. Belton, K.L. Jacobson, and E.L. Watson. 1997. Immunolocalization of rap1 in the rat parotid gland: detection on secretory granule membranes. J. Histochem. Cytochem. 45:965–973. [DOI] [PubMed] [Google Scholar]
- Fischer, K.D., Y.Y. Kong, H. Nishina, K. Tedford, L.E. Marengere, I. Kozieradzki, T. Sasaki, M. Starr, G. Chan, S. Gardener, et al. 1998. Vav is a regulator of cytoskeletal reorganization mediated by the T-cell receptor. Curr. Biol. 8:554–562. [DOI] [PubMed] [Google Scholar]
- Gulli, M.P., and M. Peter. 2001. Temporal and spatial regulation of Rho-type guanine-nucleotide exchange factors: the yeast perspective. Genes Dev. 15:365–379. [DOI] [PubMed] [Google Scholar]
- Han, J., K. Luby-Phelps, B. Das, X. Shu, Y. Xia, R.D. Mosteller, U.M. Krishna, J.R. Falck, M.A. White, and D. Broek. 1998. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 279:558–560. [DOI] [PubMed] [Google Scholar]
- Hancock, J.F. 2003. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 4:373–384. [DOI] [PubMed] [Google Scholar]
- Hariharan, I.K., R.W. Carthew, and G.M. Rubin. 1991. The Drosophila roughened mutation: activation of a rap homolog disrupts eye development and interferes with cell determination. Cell. 67:717–722. [DOI] [PubMed] [Google Scholar]
- Hu, C.D., K. Kariya, M. Tamada, K. Akasaka, M. Shirouzu, S. Yokoyama, and T. Kataoka. 1995. Cysteine-rich region of Raf-1 interacts with activator domain of post-translationally modified Ha-Ras. J. Biol. Chem. 270:30274–30277. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687. [DOI] [PubMed] [Google Scholar]
- Katagiri, K., A. Maeda, M. Shimonaka, and T. Kinashi. 2003. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 4:741–748. [DOI] [PubMed] [Google Scholar]
- Kinbara, K., L.E. Goldfinger, M. Hansen, F.L. Chou, and M.H. Ginsberg. 2003. Ras GTPases: integrins' friends or foes? Nat. Rev. Mol. Cell Biol. 4:767–776. [DOI] [PubMed] [Google Scholar]
- Kitayama, H., Y. Sugimoto, T. Matsuzaki, Y. Ikawa, and M. Noda. 1989. A ras-related gene with transformation suppressor activity. Cell. 56:77–84. [DOI] [PubMed] [Google Scholar]
- Knox, A.L., and N.H. Brown. 2002. Rap1 GTPase regulation of adherens junction positioning and cell adhesion. Science. 295:1285–1288. [DOI] [PubMed] [Google Scholar]
- Krawczyk, C., A. Oliveira-dos-Santos, T. Sasaki, E. Griffiths, P.S. Ohashi, S. Snapper, F. Alt, and J.M. Penninger. 2002. Vav1 controls integrin clustering and MHC/peptide-specific cell adhesion to antigen-presenting cells. Immunity. 16:331–343. [DOI] [PubMed] [Google Scholar]
- Lambert, J.M., Q.T. Lambert, G.W. Reuther, A. Malliri, D.P. Siderovski, J. Sondek, J.G. Collard, and C.J. Der. 2002. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 4:621–625. [DOI] [PubMed] [Google Scholar]
- Liu, B.P., and K. Burridge. 2000. Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not β1 integrins. Mol. Cell. Biol. 20:7160–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, A.D., A. Metjian, S. Bagrodia, S. Taylor, and C.S. Abrams. 1998. Cytoskeletal reorganization by G protein-coupled receptors is dependent on phosphoinositide 3-kinase γ, a Rac guanosine exchange factor, and Rac. Mol. Cell. Biol. 18:4744–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet, M., S.J. Robert, M. Cacquevel, M. Gastineau, D. Vivien, J. Bertoglio, J.L. Zugaza, R. Fischmeister, and F. Lezoualc'h. 2003. Crosstalk between Rap1 and Rac regulates secretion of sAPPα. Nat. Cell Biol. 5:633–639. [DOI] [PubMed] [Google Scholar]
- Margarit, S.M., H. Sondermann, B.E. Hall, B. Nagar, A. Hoelz, M. Pirruccello, D. Bar-Sagi, and J. Kuriyan. 2003. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 112:685–695. [DOI] [PubMed] [Google Scholar]
- Marignani, P.A., and C.L. Carpenter. 2001. Vav2 is required for cell spreading. J. Cell Biol. 154:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson, D., J. Silletti, G. Murphy, P. D'Eustachio, M. Rush, and M.R. Philips. 2001. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 152:111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayal, A., D.J. Webb, and A.F. Horwitz. 2004. Talin: an emerging focal point of adhesion dynamics. Curr. Opin. Cell Biol. 16:94–98. [DOI] [PubMed] [Google Scholar]
- Ohba, Y., K. Ikuta, A. Ogura, J. Matsuda, N. Mochizuki, K. Nagashima, K. Kurokawa, B.J. Mayer, K. Maki, J. Miyazaki, and M. Matsuda. 2001. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. EMBO J. 20:3333–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, H.O., E. Bi, J.R. Pringle, and I. Herskowitz. 1997. Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc. Natl. Acad. Sci. USA. 94:4463–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, H.O., P.J. Kang, and A.W. Rachfal. 2002. Localization of the Rsr1/Bud1 GTPase involved in selection of a proper growth site in yeast. J. Biol. Chem. 277:26721–26724. [DOI] [PubMed] [Google Scholar]
- Pizon, V., M. Desjardins, C. Bucci, R.G. Parton, and M. Zerial. 1994. Association of Rap1a and Rap1b proteins with late endocytic/phagocytic compartments and Rap2a with the Golgi complex. J. Cell Sci. 107:1661–1670. [DOI] [PubMed] [Google Scholar]
- Quilliam, L.A., J.F. Rebhun, and A.F. Castro. 2002. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. Prog. Nucleic Acid Res. Mol. Biol. 71:391–444. [DOI] [PubMed] [Google Scholar]
- Quinn, M.T., M.L. Mullen, A.J. Jesaitis, and J.G. Linner. 1992. Subcellular distribution of the Rap1A protein in human neutrophils: colocalization and cotranslocation with cytochrome b559. Blood. 79:1563–1573. [PubMed] [Google Scholar]
- Radhakrishna, H., O. Al-Awar, Z. Khachikian, and J.G. Donaldson. 1999. ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J. Cell Sci. 112:855–866. [DOI] [PubMed] [Google Scholar]
- Rubinfeld, B., S. Munemitsu, R. Clark, L. Conroy, K. Watt, W.J. Crosier, F. McCormick, and P. Polakis. 1991. Molecular cloning of a GTPase activating protein specific for the Krev-1 protein p21rap1. Cell. 65:1033–1042. [DOI] [PubMed] [Google Scholar]
- Sato, K.Y., P.G. Polakis, H. Haubruck, C.L. Fasching, F. McCormick, and E.J. Stanbridge. 1994. Analysis of the tumor suppressor activity of the K-rev-1 gene in human tumor cell lines. Cancer Res. 54:552–559. [PubMed] [Google Scholar]
- Schmidt, A., and A. Hall. 2002. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16:1587–1609. [DOI] [PubMed] [Google Scholar]
- Schuebel, K.E., N. Movilla, J.L. Rosa, and X.R. Bustelo. 1998. Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J. 17:6608–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara, M., Y. Terada, A. Iwamatsu, A. Shinohara, N. Mochizuki, M. Higuchi, Y. Gotoh, S. Ihara, S. Nagata, H. Itoh, et al. 2002. SWAP-70 is a guanine-nucleotide-exchange factor that mediates signalling of membrane ruffling. Nature. 416:759–763. [DOI] [PubMed] [Google Scholar]
- Stork, P.J. 2003. Does Rap1 deserve a bad Rap? Trends Biochem. Sci. 28:267–275. [DOI] [PubMed] [Google Scholar]
- Takai, Y., T. Sasaki, and T. Matozaki. 2001. Small GTP-binding proteins. Physiol. Rev. 81:153–208. [DOI] [PubMed] [Google Scholar]
- Tsygankova, O.M., A. Saavedra, J.F. Rebhun, L.A. Quilliam, and J.L. Meinkoth. 2001. Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A. Mol. Cell. Biol. 21:1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbarello, D.A., M.C. Brown, and C.E. Turner. 2002. The paxillin LD motifs. FEBS Lett. 513:114–118. [DOI] [PubMed] [Google Scholar]
- Voss, A.K., P. Gruss, and T. Thomas. 2003. The guanine nucleotide exchange factor C3G is necessary for the formation of focal adhesions and vascular maturation. Development. 130:355–367. [DOI] [PubMed] [Google Scholar]
- Wennerberg, K., S.M. Ellerbroek, R.Y. Liu, A.E. Karnoub, K. Burridge, and C.J. Der. 2002. RhoG signals in parallel with Rac1 and Cdc42. J. Biol. Chem. 277:47810–47817. [DOI] [PubMed] [Google Scholar]
- Zwartkruis, F.J., and J.L. Bos. 1999. Ras and Rap1: two highly related small GTPases with distinct function. Exp. Cell Res. 253:157–165. [DOI] [PubMed] [Google Scholar]