

Abstract
Upon completion of mitosis, daughter nuclei assemble all of the organelles necessary for the implementation of nuclear functions. We found that upon entry into daughter nuclei, snRNPs and SR proteins do not immediately colocalize in nuclear speckles. SR proteins accumulated in patches around active nucleolar organizing regions (NORs) that we refer to as NOR-associated patches (NAPs), whereas snRNPs were enriched at other nuclear regions. NAPs formed transiently, persisting for 15–20 min before dissipating as nuclear speckles began to form in G1. In the absence of RNA polymerase II transcription, NAPs increased in size and persisted for at least 2 h, with delayed localization of SR proteins to nuclear speckles. In addition, SR proteins in NAPs are hypophosphorylated, and the SR protein kinase Clk/STY colocalizes with SR proteins in NAPs, suggesting that phosphorylation releases SR proteins from NAPs and their initial target is transcription sites. This work demonstrates a previously unrecognized role of NAPs in splicing factor trafficking and nuclear speckle biogenesis.
Introduction
At the onset of mitosis, mammalian cell nuclei undergo dramatic structural and functional alterations such as chromatin condensation, inactivation of the transcription machinery, nuclear envelope breakdown, and disassembly of nuclear compartments such as nucleoli and interchromatin granule clusters (IGCs). The constituents of nuclear compartments become diffusely distributed throughout the cytosol, and some of them assemble into various cytoplasmic structures as mitosis progresses (Ferriera et al., 1994; Jimenez-Garcia et al., 1994; Thiry, 1995; Dundr et al., 1997; Dundr and Olson, 1998; Everett et al., 1999). Although the function of the cytoplasmic bodies is largely unknown, the segregation of components from distinct nuclear compartments into distinct cytoplasmic bodies suggests that they maintain multiprotein complexes for the various nuclear bodies throughout mitosis (for review see Hernandez-Verdun et al., 2002). The nuclear entry of many nuclear body constituents is likely to occur by active transport of single macromolecules or macromolecular complexes, not by direct uptake or fusion of the entire cytoplasmic structures with forming nuclei. Organization of the nuclear compartments begins at mitotic exit. Interestingly, assembly of the nuclear envelope is followed in telophase by sequential nuclear entry of RNA polymerase II and transcription factors, followed by entry of pre-mRNA splicing factors and other pre-mRNA processing factors (Prasanth et al., 2003).
As cells enter G1, chromatin decondenses, the nuclei expand, global transcription is reactivated, and functional nuclear compartments such as the nucleoli, Cajal bodies (CBs), PML bodies, and nuclear speckles are reestablished. Little is known about nuclear body reformation aside from the nucleolus, for which a multistep process is initiated by rRNA transcription at nucleolar organizing regions (NORs) and is followed by recruitment of rRNA-processing factors and other nucleolar proteins. The nucleolar protein fibrillarin coats the surfaces of chromosomes during mitosis, and then decorates NORs when they become transcriptionally active and is involved in early rRNA processing events (for review see Hernandez-Verdun et al., 2002). Nucleolar proteins that are involved in late rRNA processing, such as nucleolin, remain cytosolic during mitosis, and then enter daughter nuclei and form structures called prenucleolar bodies or nucleolar-derived foci before recruitment to nucleoli (for reviews see Dundr et al., 1997; Dundr and Olson, 1998; Sleeman and Lamond, 1999; Hernandez-Verdun et al., 2002).
IGCs, also called nuclear speckles, are storage, assembly, and/or modification sites for pre-mRNA processing factors in interphase nuclei (for review see Lamond and Spector, 2003). Nuclear speckles disassemble during mitosis, and their constituents are diffusely distributed throughout the cytoplasm, later organizing into cytoplasmic structures called mitotic IGCs (MIGs; Verheijen et al., 1986; Leser et al., 1989; Ferriera et al., 1994). Early studies on the behavior of nuclear speckles through the cell cycle described MIGs as the mitotic equivalent of interphase nuclear speckles based on their similar granular structure and composition (Spector and Smith, 1986; Leser et al., 1989; Thiry, 1995; see Fig. 1, a–c). MIGs disappear concomitant with nuclear entry of pre-mRNA processing factors at telophase (Ferriera et al., 1994; Prasanth et al., 2003), and then nuclear speckles reassemble in G1 and are maintained through interphase.
Figure 1.

Distribution of SR splicing factors during telophase. Endogenous SR splicing factor SF2/ASF (green) and DNA (pseudocolored red) localization during the HeLa cell cycle (a–e). Throughout mitosis, nuclear speckle constituents reside in the cytosol and in MIGs (a–c, arrowheads). At telophase, SF2/ASF enters daughter nuclei and localizes in NAPs (d, arrows) that correspond to DAPI-negative zones (d, inset, arrows) before it is localized in nuclear speckles in G1 (e). YFP-SF2/ASF localization was followed by confocal microscopy during telophase in living cells (f–k), where it initially accumulates in NAPs (f–j, arrows) and later localizes to nuclear speckles (k, arrowheads; see Video 1 for the entire time course, available at http://www.jcb.org/cgi/content/full/jcb.200404120/DC1). Images in f–k are 2-μm optical sections. Projections of deconvolved z-stacks (l–o) show that SR splicing factor SC35-CFP (l, arrow, pseudocolored green) colocalizes in NAPs with SF2/ASF (m and n, arrows). The SR protein kinase Clk/STY (q, arrow) colocalizes with endogenous SF2/ASF in NAPs (p and r, arrows). DNA was stained with DAPI to monitor cell cycle phase (a–e, o, and s; arrows in o and s indicate NAP position). Bars, 5 μm.
To understand in more detail how the nuclear speckle pattern is established in daughter nuclei, we studied the localization and dynamics of nuclear speckle constituents at mitotic exit. Here, we show that in telophase splicing factors do not immediately localize to nuclear speckles upon nuclear entry. Surprisingly, different families of splicing factors initially target different subnuclear regions. In telophase, SR splicing factors initially localize around NORs before the establishment of nuclear speckles, whereas snRNPs localize in polar regions of daughter nuclei. We propose that the concentration of SR proteins in a region of the nucleus away from other splicing factors could increase the likelihood for arginine-serine rich (RS) domain–RS domain interactions, thus facilitating the intermolecular associations important for subsequent association with transcription sites and nuclear speckles.
Results
SR proteins accumulate in NOR-associated patches (NAPs) upon entering telophase nuclei
Splicing factors reside in the cytosol during mitosis and assemble into MIGs as mitosis progresses (Fig. 1, a–c). MIGs begin to appear during metaphase, when two or three MIGs form near the periphery of the metaphase plate (Fig. 1 a, arrowheads; Ferriera et al., 1994). During anaphase, additional MIGs form (Fig. 1 b, arrowheads), increasing in number and size until telophase (Fig. 1 c, arrowheads; Ferriera et al., 1994), when splicing factors are imported into daughter nuclei resulting in the formation of nuclear speckles in G1 (Fig. 1 e; Prasanth et al., 2003). To our surprise, we observed a novel nuclear localization of the SR splicing factor SF2/ASF at the earliest time point of SF2/ASF nuclear entry. In telophase, several bright patches (NAPs) of SF2/ASF were found inside daughter nuclei (Fig. 1 d, arrows). There was a reduction of DAPI staining (Fig. 1 d, inset) and histone H2B localization (not depicted) in these regions, suggesting that DNA is decondensed or absent in the areas where NAPs form.
To study the nuclear entry of SF2/ASF in living daughter nuclei with regard to NAPs, we generated a HeLa cell line stably expressing YFP-SF2/ASF. During telophase, YFP-SF2/ASF transiently accumulated in NAPs that increased in size over 15 min (Fig. 1, f–i) and then began to disappear after ∼20 min (Fig. 1, i and j) as the cells entered G1 and nuclear speckles formed (Fig. 1 k, arrowheads). The complete time-lapse video of this sequence is available online (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200404120/DC1). Repeated analysis of YFP-SF2/ASF localization in NAPs in living cells showed that accumulation in NAPs was transient, and it typically persisted for 15–20 min in HeLa telophase cells before NAPs disappeared and speckles formed (Fig. S1, a–d; and Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200404120/DC1). In cases when NAP formation was staggered by several minutes between two daughter nuclei (e.g., compare the timing of NAP assembly/disassembly between the two daughter nuclei in Fig. 1, f–j), NAP formation and dissipation was always coordinated among multiple NAPs within the same daughter nucleus, and again NAPs remained intact for ∼15–20 min in each nucleus.
Next, we were interested in determining if additional SR proteins localize in NAPs during telophase. We found that the SR splicing factor SC35 tagged with CFP (Fig. 1 l, arrows) colocalized with SF2/ASF in NAPs (Fig. 1 m, arrows). Additionally, some endogenous SR protein kinase Clk/STY (Fig. 1 q, arrows) that regulates associations among SR proteins (Colwill et al., 1996b) colocalized with endogenous SF2/ASF in NAPs (Fig. 1 p, arrows). The presence of Clk/STY in NAPs suggests that modulation of SR protein phosphorylation could play a role in SR protein complex assembly and/or release of SR proteins from NAPs, which is similar to the role Clk/STY plays in releasing SR proteins from interphase speckles (Colwill et al., 1996a,b; Sacco-Bubulya and Spector, 2002).
Immunolocalization studies allowed us to determine the phosphorylation state of SR proteins in NAPs. An antibody that recognizes the phosphorylated form of SC35 (Fu and Maniatis, 1990) labeled cytoplasmic MIGs and did not recognize any nuclear SC35 during telophase (Fig. 2 b). This antibody does not recognize SC35 in NAPs (Fig. 2 b, arrow), which is consistent with the possibility that the SC35 in NAPs must be in a hypophosphorylated state (Prasanth et al., 2003). In fact, hyperphosphorylated SR proteins in general are not found in NAPs, as antibodies 3C5 (Fig. 2 f, arrow; Turner and Franchi, 1987) and mAb104 (Roth et al., 1990), which recognize phosphoepitopes on a subset of SR proteins, also did not label NAPs or recognize nuclear SR proteins until G1. However, antibody SCf11 that preferentially recognizes hypophosphorylated SC35 (Fig. 2 j, arrows; Cavaloc et al., 1999) colocalized with YFP-SF2/ASF in NAPs (Fig. 2 i, arrows). An immunoblot of HeLa extract confirmed that SCf11 recognizes SC35 only in cell extract treated with phosphatase (Fig. 2 m, lane 2), preferentially recognizing hypophosphorylated SC35. Therefore, SR proteins in NAPs are predominantly hypophosphorylated, a characteristic that would promote SR protein–SR protein association in NAPs.
Figure 2.

SR proteins in NAPs are hypophosphorylated. SC35 antibody (b, arrow), which recognizes hyperphosphorylated SC35, does not recognize the SC35 in NAPs (a, arrow) and is absent from the nucleus (c and d, arrows). 3C5 antibody that recognizes a family of hyperphosphorylated SR proteins (f, arrow) also does not recognize SR proteins in NAPs (e, arrow) and is absent from the nucleus (g and h, arrows). Projections of deconvolved image stacks illustrate the strictly cytoplasmic localization of hyperphosphorylated SR proteins as well as exclusion from nuclei (a–h). SCf11 (j, arrows) colocalizes with YFP-SF2/ASF in NAPs (i and k, arrows) and recognizes SC35 by immunoblot only when cell extract is treated with phosphatase (m, lane 2). Arrows in d, h, and l indicate NAP position. Bar, 5 μm.
snRNPs are not enriched at NAPs
We expected that all nuclear speckle components would simultaneously reassemble into nuclear speckles upon entry into daughter nuclei, and we initially suspected that NAPs represent the first nuclear speckles in daughter nuclei. However, although SR proteins and snRNPs colocalize in interphase speckles (Huang and Spector, 1992) and in MIGs (Ferriera et al., 1994), NAPs are not enriched in snRNPs as shown by using an antibody against the U2 snRNP protein B′′ (Fig. 3, compare arrows in a and b). Interestingly, the majority of B′′ accumulated in regions of telophase daughter nuclei (Fig. 3 b, arrowheads) distinct from NAPs. DAPI staining indicates that DNA is decondensed or absent in these regions (Fig. 3 d, arrowheads). In addition, an antibody that recognizes the 5′ m3G cap of the snRNAs also showed snRNA accumulation in regions of telophase daughter nuclei distinct from NAPs (unpublished data). Because snRNP maturation is thought to occur in CBs (Sleeman and Lamond, 1999), we examined the localization of the CB protein p80 coilin in telophase cells. Coilin was found in regions of telophase nuclei (Fig. 3 f, arrowheads), distinct from NAPs (Fig. 3 e, arrows). In fact, coilin and U2 snRNP protein B′′ were colocalized in telophase nuclei (Fig. 3, j, k, and m, arrowheads) in areas separate from NAPs (Fig. 3, i–n, arrows). The colocalization of snRNPs and coilin suggests that in addition to NAPs there are regions of daughter nuclei that could be initial sites for snRNP assembly and/or modification (see Discussion).
Figure 3.

snRNPs and coilin are not enriched in NAPs. snRNPs (b, arrowheads) are enriched in regions of daughter nuclei away from SF2/ASF NAPs (a, arrows). Coilin is also found in regions of daughter nuclei (f, arrowheads; 5P10 antibody), away from SF2/ASF NAPs (e, arrow). The U2snRNP protein B′′ (j, arrowheads) colocalized with coilin (k, arrowheads; R228 antibody; merge shown in m, arrowheads) in regions separate from NAPs, which are indicated by arrows (i–n). The two regions are clearly distinct in a merged image of YFP-SF2/ASF and B′′ (l). DNA was stained with DAPI (d, h, and n). Arrows indicate NAP position (a–n). Arrowheads indicate “polar” localization of U2-B′′ (b–d), coilin (f–h), or both U2-B′′ and coilin (j–n). DNA was stained with DAPI (d, h, and n). Bars, 5 μm.
NAPs surround transcriptionally active NORs
Because NORs are activated in mitosis, we were interested to determine if there is a relationship between NORs and NAPs. Immunofluorescence analysis verified that SR proteins initially surround NORs upon nuclear entry. Endogenous SF2/ASF was localized around foci of fibrillarin in telophase daughter nuclei in HeLa cells (Fig. 4, a–c, arrows), in nontransformed human fibroblast IMR90 cells (Fig. 4, d–g, arrows), and in human osteosarcoma U2OS cells (Fig. 4, h–k, arrows). Some SF2/ASF is still present in MIGs at this stage (Fig. 4, a, d, and h, arrowheads). As there are multiple NORs in each nucleus (see Discussion), multiple NAPs were seen in these cell lines in telophase (Fig. 4, a, d, and h). To determine if the NORs are transcriptionally active when NAPs are formed, we looked for evidence of RNA polymerase I activity in the NORs. Foci of the RNA polymerase I transcription factor upstream binding factor (Fig. 4 m, arrow) that is localized in transcriptionally active NORs (Jordan et al., 1996; Roussel et al., 1996), foci of nascent transcripts (presumably RNA polymerase I transcripts; Fig. S1, i and j), and pre-ribosomal RNAs detected by RNA-FISH (Fig. 4 q, arrows) were surrounded by NAPs (Fig. 4 p, arrows).
Figure 4.
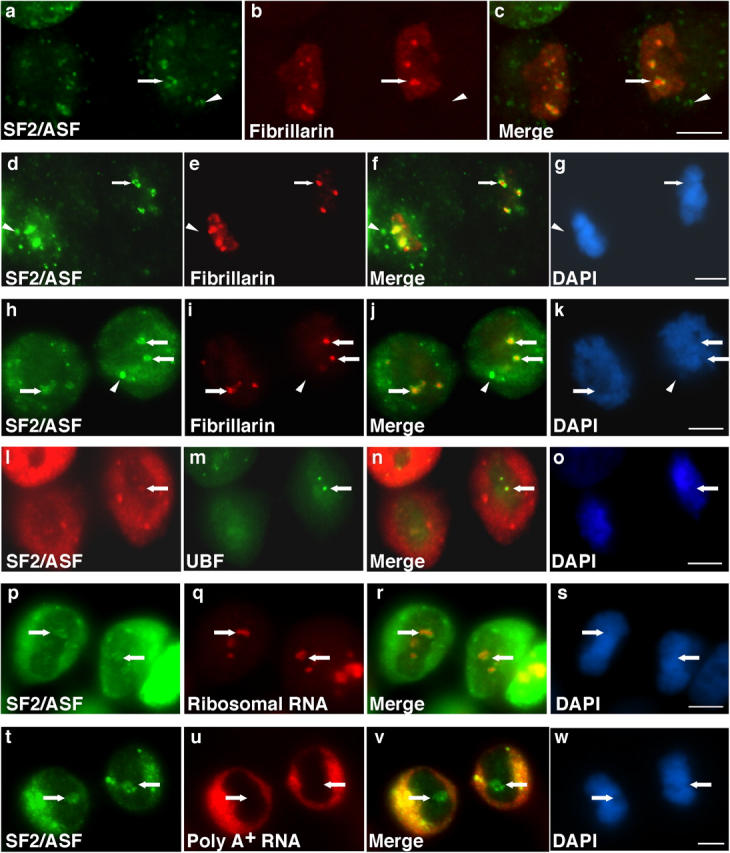
NAPs surround transcriptionally active NORs during telophase. During telophase, endogenous SF2/ASF localizes in NAPs (a, arrow) surrounding foci of fibrillarin (b, arrow) in HeLa cells. Endogenous SF2/ASF (d, arrow) is localized in NAPs surrounding foci of fibrillarin (e, arrow) in the nontransformed cell line IMR90. Endogenous SF2/ASF (h, arrow) is localized in NAPs surrounding foci of fibrillarin (i, arrow) in U2OS cells. SF2/ASF is also in MIGs at this stage (a–k, arrowheads). Endogenous SF2/ASF (l, arrow) surrounds foci of the RNA pol I transcription factor upstream binding factor (m, arrow) in HeLa cells. RNA-FISH for ribosomal RNA (q, arrows) showed that SF2/ASF NAPs (p, arrows) surrounded transcriptionally active NORs. RNA-FISH using oligo dT probes demonstrated that polyadenylated RNA (u, arrows) was absent from SF2/ASF NAPs (t, arrows). DNA was stained with DAPI (g, k, o, s, and w). NAP position is indicated by arrows (a–w). Bars, 5 μm.
SR splicing factors colocalize with pre-mRNAs in interphase nuclei (Misteli et al., 1997, 1998; Misteli and Spector, 1999; Shopland et al., 2003); therefore, it was important to rule out the possibility that SR protein accumulation in NAPs is a result of RNA polymerase II transcription or pre-mRNA processing in these regions. RNA-FISH using an oligo-dT probe showed that polyA+ RNA is not present in NAPs (Fig. 4 u, arrows) or elsewhere in the daughter nuclei. In addition, NAPs did not contain nascent transcripts labeled by incorporation of 5-fluoro-UTP (Fig. S1, i and j). Finally, we ruled out the possibility that NAPs correspond to other nucleolar-associated nuclear bodies that could be in the early stages of assembly during telophase. Proteins such as nucleolin (not depicted) and fibrillarin (Fig. 4) were not present in NAPs, ruling out the possibility that these are prenucleolar bodies (Ochs et al., 1985; Hernandez-Verdun et al., 2002). NAPs also do not represent initial assembly sites for the perinucleolar compartment (PNC), a transcriptionally active domain that contains pre-mRNA processing factors (Matera et al., 1995; Huang et al., 1998). The initial nuclear accumulations of the PNC component polypyrimidine tract-binding protein (PTB) were directly adjacent to NAPs as would be expected for a compartment that is on the nucleolar periphery, but the PNC did not show significant overlap with NAPs (Fig. S1, e–h).
NAPs form at established NORs
To examine the relationship between the association of fibrillarin with NORs and the assembly of NAPs, both structures were simultaneously examined in living cells. HeLa cells stably expressing CFP-fibrillarin were transiently transfected with YFP-SF2/ASF, and dual color four-dimensional imaging was performed in living mitotic cells (Fig. 5, a–l). The NORs were visible as distinct CFP-fibrillarin foci in early telophase (Fig. 5 a, arrow). YFP-SF2/ASF was not yet detectable in daughter nuclei at this time (Fig. 5 g). Clearly, the NAPs are the first sites where YFP-SF2/ASF is enriched in daughter nuclei (Fig. 5 i), and they appeared a few minutes after CFP-fibrillarin first accumulated in NORs. We conclude that fibrillarin associates with NORs before the accumulation of YFP-SF2/ASF in NAPs.
Figure 5.

NAPs form at established NORs. Dual-color four-dimensional imaging revealed the temporal sequence of nuclear domain establishment (a–l). Cells stably expressing CFP-fibrillarin (a–f) were transiently transfected with a cDNA construct encoding YFP-SF2/ASF (g–l). CFP-fibrillarin (a, arrow) had clearly accumulated in NORs for several minutes before YFP-SF2/ASF first accumulated in NAPs (i, arrows). Images are projections of confocal z-stacks collected every 2 min. Bar, 5 μm.
Ultrastructure of the NAPs
To investigate the ultrastructural relationship between the active NORs and NAPs, immunoelectron microscopy was performed on telophase HeLa cells. The early telophase state of the cell in Fig. 6 a is evident by the abundance of SF2/ASF in cytoplasmic MIGs (Fig. 6 a, arrowheads). Also, although there are several regions of dense heterochromatin throughout the nuclei, there is no indication of granular IGCs, confirming the absence of nuclear speckles at this stage (Fig. 6 a). In the daughter nuclei, endogenous SF2/ASF was localized in several electron-dense granular patches around NORs, and it was clearly excluded from the interior of the NOR (Fig. 6, b and c; enlarged NAPs from the top and bottom nuclei, respectively). Furthermore, the nuclear envelope appeared intact in nuclei where NAPs were observed.
Figure 6.
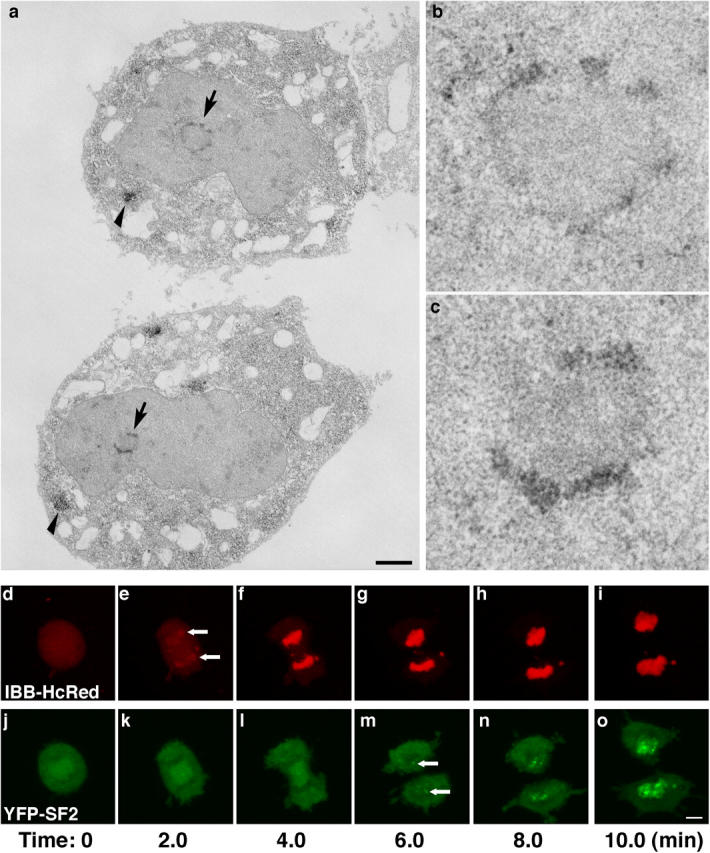
Ultrastructure of NAPs and time-lapse of nuclear envelope assembly. Ultrastructure of the NAPs was determined during early telophase (a–c) by using immunoelectron microscopy. Electron-dense material was deposited where SF2/ASF antibody was localized around forming nucleoli (a, arrows) and in MIGs (a, arrowheads). Enlarged images from the top (b) and bottom (c) cells show that SF2/ASF is excluded from the NOR interior. (d–o) Dual four-dimensional imaging of NAPs and nuclear import in living HeLa cells. A functional nuclear envelope was established as indicated by import of IBB-HcRed (e, arrows), before YFP-SF2/ASF began to accumulate in NAPs (m, arrows). Images are projections of confocal z-stacks collected every 2 min. Bars: (a) 1 μm; (d–o) 5 μm.
Organization of a functional nuclear envelope precedes NAP formation
SR protein accumulation in NAPs occurs earlier than bulk SR protein nuclear entry but after NORs are activated. Because rDNA becomes transcriptionally active during anaphase, we wanted to be certain that the nuclear membrane is established before SR proteins associate with NAPs. We performed a functional assay in living cells to assess whether or not the nuclear membrane is competent for import when NAPs first appear (Fig. 6, d–o). HeLa cells stably expressing YFP-SF2/ASF were transiently transfected with the importin-β binding domain (IBB) of importin α fused to HcRed. The IBB-HcRed entered nuclei in late anaphase (Fig. 6 e, arrows), whereas the initial accumulation of YFP-SF2/ASF in NAPs occurred minutes later (Fig. 6 m, arrows). This experiment confirmed that the nuclear membrane is fully assembled and functional for nuclear import before the formation of NAPs in daughter nuclei and indicates that YFP-SF2/ASF enters nuclei via active transport.
SF2/ASF rapidly exchanges between NAPs and the nucleoplasm
To determine if SF2/ASF traffics through NAPs, we examined the exchange of SF2/ASF at NAPs by FRAP (Fig. 7, a–f). A representative fluorescence recovery curve is shown in Fig. 7 f for the region bleached in Fig. 7 a (boxed). FRAP analysis of a typical NAP indicated that SF2/ASF had a half time of fluorescence recovery of ∼1.8 s (± 0.6). The average fluorescence recovery curve from 14 experiments is shown in Fig. S2 (available at http://www.jcb.org/cgi/content/full/jcb.200404120/DC1). As we also wanted to know how SR protein transit through NAPs compared with transit through the nucleoplasm, half of a nucleus was bleached such that the bleached region included a NAP (Fig. S2). Interestingly, the recovery of YFP-SF2/ASF in the NAPs occurred at the same rate as the YFP-SF2/ASF in the nucleoplasm, suggesting that diffusion is the limiting determinant for recovery.
Figure 7.

Dynamics and quantification of YFP-SF2/ASF in NAPs. FRAP analysis indicates that turnover of YFP-SF2/ASF at NORs is rapid. A representative of 14 NAP photobleaching experiments is shown in panels a–f. Photobleaching of NAPs (a, boxed area) revealed that the average half time of FRAP of YFP-SF2/ASF in NAPs was 1.8 s (± 0.6; recovery curve, f; see also Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200404120/DC1). High-resolution dual color four-dimensional imaging data set is shown in panels g–v. Intensity sums of fluorescence of YFP-SF2/ASF in different cellular compartments during the early stages of daughter nuclei reformation (g–v) revealed that ∼1.17% of YFP-SF2/ASF is found at NAPs during early telophase in the example shown (m, NAPs; v, right y-axis). Data shown in g–v is representative of three experiments. Bars: (a–e) 2.5 μm; (g–u) 5 μm.
Quantitation of YFP-SF2/ASF flux through NAPs
Next, we wanted to know if all SF2/ASF could potentially transit through NAPs upon nuclear entry. The amount of YFP-SF2/ASF in NAPs in living telophase cells was quantified by dual high-resolution four-dimensional imaging of YFP-SF2/ASF (Fig. 7, g–v). Hoechst staining allowed imaging of the chromatin for calculation of nuclear volume (Fig. 7, g–k). The amount of YFP-SF2/ASF in NAPs (Fig. 7 m) was quantified relative to nucleoplasmic and cytoplasmic pools of YFP-SF2/ASF (Fig. S3 for procedure and Table S1 for intensity sums, available at http://www.jcb.org/cgi/content/full/jcb.200404120/DC1). As expected, the cytoplasmic pool decreased as the nucleoplasmic pool increased (Fig. 7 v), and the total fluorescence was maintained throughout the experiment (Table S1). The average amount of YFP-SF2/ASF in NAPs in the example shown was ∼1.17% (Fig. 7 m; and Fig. 7 v, right y-axis) and it averaged 1.02% (± 0.146) of the total cellular signal in three independent experiments. Although there is no difference in recovery time of SF2/ASF in NAPs versus the nucleoplasm (Table S1), YFP-SF2/ASF is 2.7 times more concentrated in NAPs than in the nucleoplasm, further demonstrating the importance of these domains in the early stages of subnuclear SR protein organization. Combining the facts that ∼1% of cellular SF2/ASF resides in NAPs and that 50% of SF2/ASF in NAPs exchanges in <2 s allows the estimate that it would require <6 min to shuttle all cellular SF2/ASF through NAPs (100%/0.51% × 1.8 s = 353 s). As NAPs typically persist for 15–20 min, all SF2/ASF could feasibly visit NAPs at least once during a normal telophase.
NAP integrity is modulated by transcriptional activity
Because SR proteins are present in NAPs before entry into nuclear speckles, we were interested in determining the effect of NAP formation and maintenance under conditions where RNA polymerase II transcription was inhibited as cells exited mitosis. Interestingly, inhibition of RNA polymerase II transcription during telophase, by α-amanitin treatment, caused NAPs to become larger and brighter and to persist longer. Endogenous SF2/ASF accumulation at NAPs was exaggerated under these conditions (Fig. 8, a–d), suggesting that SR proteins preferentially target NAPs when transcription sites are absent. This result was confirmed in living cells, as YFP-SF2/ASF accumulation in NAPs continued for at least 2 h (Fig. 8, e–h), during which time the NAPs increased in size over the first 65 min and eventually diminished but were not completely absent by 2 h (Fig. 8 h). This is a dramatically longer association of SF2/ASF with NAPs, compared with SF2/ASF in untreated cells, in which NAPs disappear after ∼20 min (Fig. 1 j). Furthermore, SF2/ASF entry into nuclear speckles was delayed (i.e., speckles were just beginning to accumulate SF2/ASF by 2 h) compared with untreated cells in which nuclear speckles accumulate SF2/ASF immediately (Fig. 1 k). Interestingly, endogenous SF2/ASF (Fig. S4, a–h, available at http://www.jcb.org/cgi/content/full/jcb.200404120/DC1) and YFP-SF2/ASF (Fig. 8 i, arrows) in interphase cells treated with α-amanitin were redistributed from nuclear speckles to the peripheral nucleolar region. In the same cells, snRNPs remained in rounded, enlarged nuclear speckles and were not targeted to the nucleolar periphery (Fig. 8 j, arrowhead), consistent with the segregation of different families of splicing factors at telophase. SC35 tagged with RFP also redistributed from nuclear speckles to the peripheral nucleolar region (Fig. S4, i–p). Identical results were obtained with other RNA polymerase II inhibitors (e.g., actinomycin D and DRB; unpublished data). These results implicate RNA polymerase II transcription in appropriate subnuclear targeting of SR proteins after mitosis, and they demonstrate a previously unrecognized role of NAPs in the pathway of SR splicing factor trafficking and biogenesis of nuclear speckles.
Figure 8.

Inhibition of RNA polymerase II during telophase delays targeting of YFP-SF2/ASF to nuclear speckles. In mitotic cells treated with the specific RNA polymerase II inhibitor α-amanitin, endogenous SF2/ASF (a, arrow) accumulated much more extensively around NORs (labeled with fibrillarin; b and c, arrow) than in untreated cells (Fig. 4). In living mitotic cells treated with α-amanitin (e–h), YFP-SF2/ASF continued to accumulate in NAPs for at least 2 h (h, arrow indicates NAP remnant), ∼100 min longer than it persists in NAPs in untreated cells. Even after 2 h, accumulation of SF2/ASF nuclear speckles was not abundant (h, compare with Fig. 1, j and k). Arrows in a–h indicate NAP position. Confocal images are representative projections from a typical sequence in which z-stacks were collected every 5 min. Immunofluorescence of interphase cells treated with α-amanitin showed that similar to the localization of SR proteins during telophase, a significant amount of YFP-SF2/ASF was redistributed to the periphery of nucleoli (i, arrows). snRNPs remained in rounded, enlarged nuclear speckles and were not targeted to the nucleolar periphery (j, arrowhead). Arrows (i–l) indicate nucleolar periphery. Arrowheads (i–l) indicate rounded-up nuclear speckles. DNA was stained with DAPI (d and l). Bars, 5 μm.
Discussion
We have characterized a transient nuclear domain (NAPs) where SR proteins accumulate during a limited temporal window in telophase. The NAPs form around active NORs after nuclear envelope assembly, but before the onset of nuclear speckle formation. The results of this paper are key to building a time line of nuclear compartment assembly following mitosis. They also bring to light the fact that, in general, constituents of nuclear bodies, including prerRNA processing factors, components of CBs and PML bodies, and pre-mRNA processing factors, do not immediately localize to and/or assemble their resident bodies in daughter nuclei. Instead, there is a lag phase after mitosis as nuclear proteins first enter daughter nuclei and accumulate temporarily with proteins of similar function(s) before nuclear bodies (i.e., nuclear speckles and CBs) are established in G1. These transient associations of subsets of proteins may be necessary for modification/maturation or partial assembly of multimolecular subcomplexes before localization to nuclear bodies in G1. The subsequent organization of nuclear compartments such as nuclear speckles would then provide further modification/maturation and recycling/maintenance of protein complexes during interphase.
We expected that all pre-mRNA processing factors would simultaneously reassemble into speckles in telophase daughter cells. Instead, we found a distinct localization of different families of pre-mRNA processing factors in different regions of telophase nuclei. The U2 snRNP protein B′′, snRNAs (m3G), and the CB resident protein p80 coilin were all found at polar regions of daughter nuclei. CBs are discrete nuclear compartments that can associate with U1, U2, and U3 snRNA gene loci and histone gene loci (Frey and Matera, 1995; Smith et al., 1995; Gao et al., 1997; Frey et al., 1999) and have been implicated in snRNP maturation and assembly (Sleeman and Lamond, 1999; Darzacq et al., 2002) and in snoRNA posttranscriptional modification and targeting to the nucleolus (Narayanan et al., 1999; Verheggen et al., 2002). CBs do not acquire their distinctive morphology until late telophase/G1 (Sleeman and Lamond, 1999; unpublished data). Therefore, the observed localization of snRNPs and p80 coilin at polar regions of daughter nuclei may be important for assembling the initial nuclear populations of snRNPs before CB formation.
During interphase, snRNPs may follow a nonrandom path as a short pulse of YFP-snRNP protein expression allowed tracking of the accumulation of snRNPs from the CB to the nucleoli, and then to the speckles (Sleeman and Lamond, 1999). With the entry of snRNPs into the next compartment, there was clear depletion of snRNPs from the previous compartment (Sleeman and Lamond, 1999). Sleeman and Lamond (1999) concluded that the snRNPs are directly enriched in “speckles” during telophase without first passing through CBs, suggesting that snRNPs do not require modification/maturation at CBs upon entry into daughter nuclei. However, colocalization with SR proteins was not examined to confirm that these sites of snRNP enrichment are the equivalent of interphase speckles. As we have found a polar distribution of snRNPs in daughter nuclei, what was interpreted as a “speckle” during telophase (Sleeman and Lamond, 1999) is more likely equivalent to what we have shown here as polar localization of snRNPs. Furthermore, the accumulation of both snRNPs and coilin in the same nuclear region is more suggestive of the early stages of CB organization than of nuclear speckle organization.
The initial localization of SR proteins around NORs in telophase implicates this region in SR protein modification/maturation. The fact that α-amanitin blocks trafficking of SR proteins from the perinucleolar region to transcription sites and/or nuclear speckles during interphase implicates a role for the nucleolar periphery specifically in SR protein modification/maturation during interphase as well. Interestingly, telophase NAPs increased in size and were maintained for a longer period of time when RNA polymerase II transcription was turned off, and SR proteins were left without a pre-mRNA target. Transit through the nucleolar periphery in interphase may be very rapid, similar to the rapid exchange in NAPs observed in FRAP experiments during telophase. Thereby, the steady-state level of SR proteins at the nucleolar periphery may be below the detection limits or indistinguishable from the nuclear speckle pattern (e.g., speckles on the periphery of the nucleoli), and could therefore be observed only upon RNA polymerase II inhibition when a build up occurs due to the depletion of a downstream target. A similar scenario was observed for hnRNP proteins when it was first discovered that they shuttle continuously between the nucleus and the cytoplasm (Pinol-Roma and Dreyfuss, 1993). The hnRNP proteins are entirely nuclear when observed by immunofluorescence, and no hnRNP is detectable in the cytoplasm. However, upon inhibition of RNA polymerase II transcription, some hnRNP proteins (e.g., A1 and K) accumulated in the cytosol, revealing that these proteins traffic between the nucleus and cytoplasm during interphase (Pinol-Roma and Dreyfuss, 1993). Here, we have shown interruption in the trafficking pathway of SR proteins, with preferential targeting to the nucleolar periphery under conditions where RNA polymerase II is inhibited, suggesting that this region is important for continuous SR protein modification/maturation/trafficking. This interpretation is strengthened by similar observations made for snRNP trafficking during interphase, as snRNPs were shown to proceed from the cytoplasm to the CBs, and then into nucleoli before accumulation in nuclear speckles (Sleeman and Lamond, 1999). Interestingly, SR proteins such as SF2/ASF have been identified in proteomic analysis of nucleoli (Leung et al., 2003). Our results support a model in which SR proteins traffic through the nucleolar periphery in interphase cells, in contrast to snRNPs, which were not detected at the nucleolar periphery, and is consistent with what is seen at mitotic exit as the two families of splicing factors spatially separated in the daughter nuclei. The initial accumulations of SR proteins in NAPs during telophase may reflect the beginning of SR protein modification/maturation/trafficking in the new cell cycle, a time when global transcriptional activity is more reduced than during interphase and much less pre-mRNA target is available for the splicing machinery.
The state of chromatin condensation and nuclear speckle integrity are inversely correlated through the cell cycle (Smith et al., 1985). In our work, we found that SR proteins did not directly accumulate in nuclear speckles in telophase, but initially accumulated around NORs, which contain repetitive rDNA and are the first extensive regions of chromatin that become highly decondensed and transcribed following mitosis. Human nuclei contain 10 NORs located on the short arms of acrocentric chromosomes 13, 14, 15, 21, and 22 (Kaplan and O'Connor, 1995), six of which are associated with transcription factors in HeLa cells during mitosis and are the sites where rRNA is synthesized and nucleoli form (Roussel et al., 1996). We considered the possibility that NORs may be the initial targets for SR proteins because they serve some transient function in nucleolar biogenesis. However, this is unlikely because SF2/ASF is always found in regions directly surrounding NORs, not coincident with NORs. It is also unlikely that the SR proteins are involved in splicing pre-mRNAs originating from transcriptionally active genes that flank NORs or from active chromatin domains in the vicinity of NORs because large regions of heterochromatin isolate the rDNA repeats (Sylvester et al., 1986; Worton et al., 1988). Also, during the temporal window that NAPs were observed, polyA+ RNA was absent from nuclei. Furthermore, our data presented here do not support this possibility because the snRNPs, which are enriched elsewhere in the telophase nucleus, would also be expected to accumulate on such transcripts. We also addressed the possibility that SR proteins may be targeted to NAPs because they are directly involved in modulating the condensation state of rDNA. Recent reports indicated that topoisomerase I is found in fibrillar centers of nucleoli and NORs (Christensen et al., 2002) and that SF2/ASF interacts with topoisomerase I to inhibit its DNA relaxation activity (Andersen et al., 2002). However, we confirmed that GFP-topoisomerase I is found in fibrillar centers, but does not colocalize with SF2/ASF in NAPs (unpublished data), suggesting that NAPs are not directly involved in these processes.
Perhaps SF2/ASF accumulation in NAPs is necessary for modification of SR proteins required for their targeting to nuclear speckles. Phosphorylation by SR protein kinases regulates the release of SR proteins from nuclear speckles (Colwill et al., 1996b) as well as their splicing activity (Prasad et al., 1999). Our earlier studies showed that hyperphosphorylation of SR proteins in vivo does not simply release SR proteins from speckles, but leads to complete disassembly of nuclear speckles (Sacco-Bubulya and Spector, 2002). On the contrary, hypophosphorylation of SR proteins in vivo causes bright foci to form on the speckle periphery, consistent with inhibition of SR protein release from speckles (Sacco-Bubulya and Spector, 2002). We concluded that the SR protein–SR protein interactions are the basis for nuclear speckle organization during interphase and would therefore play a critical role in nuclear speckle assembly in daughter nuclei. NAPs provide the only example during the cell cycle in which SR proteins are sequestered from other pre-mRNA processing factors. We have now demonstrated in this paper that SR proteins in NAPs are hypophosphorylated. The sequestration and hypophosphorylated state of the SR proteins at NAPs would favor RS domain–RS domain interactions for establishing NAPs. As the recovery time of SR proteins in NAPs is rapid, a simple mechanism such as this for the establishment of NAPs is the most logical. The presence of Clk/STY at NAPs could be a result of the RS repeats found in the amino terminus of the kinase; however, it also suggests phosphorylation as a means for SR protein activation for subsequent participation in pre-mRNA splicing. Alternatively, the function of Clk/STY at NAPs could be to release SR proteins from NAPs, or both activities could result from the same phosphorylation event. Although it would potentially be informative to examine the localization of SR protein deletion mutants and substitutions with regard to their localization to NAPs during telophase, this is not possible due to the delayed entry of such proteins into daughter nuclei (unpublished data).
If SR protein interactions are the basis for nuclear speckle organization, nuclear speckles could be assembled/disassembled during the cell cycle simply by regulating the level of phosphorylation of RS domains. Phosphorylation of different serine residues within the RS domain may confer different activities or functions upon SR proteins or interactions with different partners and/or subnuclear compartments. This possibility is also supported by the finding that Schizosaccharomyces pombe contains only two SR proteins and nuclear speckles have not been observed in these cells (Potashkin et al., 1990), and no SR proteins are encoded in the Saccharomyces cerevisiae genome and once again no speckles have been identified in these cells (for review see Graveley, 2000). In mammalian cells, it is not clear how pre-mRNA processing factors are activated to a splicing-competent state in newly forming nuclei that have not yet assembled nuclear speckles. Our current observations suggest that regions surrounding NORs are important sites for initially segregating SR proteins away from other splicing factors, to modify them or to allow them to interact before association with pre-mRNA transcripts or nuclear speckles. This finding raises questions for future study regarding how SR proteins and snRNPs follow two separate pathways to eventually become targeted to the same speckle regions within daughter nuclei. It will be important to determine if SR proteins and snRNPs first meet at transcription sites before recycling of complexes through nuclear speckles. It also raises questions about how different nuclear compartments assemble/disassemble and communicate with each other as nuclear domains are established after mitosis.
Materials and methods
cDNA constructs
PCR was used to generate a restriction site at the stop codon of human SC35 cDNA for convenient subcloning into pEYFP-N1 (CLONTECH Laboratories, Inc.) or a vector encoding monomeric RFP. SF2/ASF was subcloned from GFP-SF2/ASF vector (Misteli et al., 1997) into pEYFP-C1. Fibrillarin (provided by S. Huang, Northwestern University Medical School, Chicago, IL) was subcloned from pEGFP-fibrillarin vector into pECFP. IBB-Hc-Red was provided by E. Zanin (EMBL, Heidelberg, Germany).
Cell culture and transfection
Cells were grown in Dulbecco's MEM (Life Technologies) supplemented with penicillin-streptomycin and 10% FBS (Hyclone). For transient transfection of HeLa cells, electroporation (240 V; 950 μF) was performed on cells resuspended in 250 μl of Dulbecco's MEM and transferred to cuvettes containing 2 μg of plasmid plus 20 μg of salmon sperm DNA. Cells were seeded onto acid-washed coverslips and processed for immunofluorescence localization of proteins 2 d after transfection. Stable HeLa cell lines expressing CFP-fibrillarin or YFP-SF2/ASF were selected in 1 mg/ml G-418. 50 μg/ml α-amanitin was added to culture medium for 6 h before fixation or 30 min before live cell imaging. For immunoblotting, HeLa cells were subjected to microfractionation (Mendez and Stillman, 2000), and 8 μl of soluble nuclear extract was incubated in a total volume of 10 μl with 2 U of calf intestinal phosphatase (New England Biolabs, Inc.) for 30 min at 37°C. Extract was applied to SDS PAGE followed by immunoblot using anti-SC35 (SCf11; 1:20) antibody.
Immunofluorescence
Cells were rinsed in PBS and fixed for 15 min in 2% formaldehyde in PBS, pH 7.4. Cells were permeabilized in PBS + 0.2% Triton X-100 + 0.5% goat serum. Nascent transcripts were labeled by adding 2 mM 5-fluoro-UTP to culture medium. The following primary antibodies were added for 1 h at RT: anti-(hyperphosphorylated)SC35 (1:1,000), SCf11 anti-(hypophosphorylated)SC35 (1:20; provided by J. Stevenin, Institut de Genetique et de Biologie, Moleculaire et Cellulaire, Illkirch, France); anti-B′′ (1:200; provided by W. van Venrooij, University of Nijmegen, Nijmegen, Netherlands), mAb103 anti-SF2/ASF (1:100; provided by A. Krainer, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY); anti-snRNA m3G (1:40); ANA-N (fibrillarin, 1:10); anti-UBF (1:100); anti-BrdU (1:500; Sigma-Aldrich); anti-nucleolin (1:100); anti-PTB (1:200); mouse anti-coilin 5P10 (1:100; provided by M. Carmo-Fonseca, University of Lisbon, Lisbon, Portugal); rabbit anti-coilin R228 (1:400; provided by E. Chan, The Scripps Research Institute, La Jolla, CA); and anti-Clk/STY (1:100; BD Biosciences). Cells were rinsed in PBS + 0.5% goat serum, and secondary anti-species–specific antibodies (Jackson ImmunoResearch Laboratories) were added for 1 h at RT. RNA-FISH was performed as described previously (Sacco-Bubulya and Spector, 2002) with probes synthesized from rDNA template (provided by M. Dundr, National Cancer Institute, Bethesda, MD) using a nick translation reagent kit (VYSIS) to incorporate spectrum red-dUTP (VYSIS). Fixed cells were examined using a fluorescence microscope (model Axioplan 2i; Carl Zeiss MicroImaging, Inc.) equipped with Chroma filters (Chroma Technology Corp.), and OpenLab software (Improvision) was used to collect digital images. Alternatively, fixed cells were observed on a Deltavision microscope (Applied Precision) and images were deconvolved using SoftWorx.
Live cell microscopy
HeLa cells stably expressing YFP-SF2/ASF were seeded onto coverslips and grown for 2 d. The cells were directly imaged in a LabTekII chamber slide (Lab Tek) or transferred to an FCS2 live-cell chamber (Bioptechs), which was mounted onto the stage of a microscope (model LSM 510; Carl Zeiss MicroImaging, Inc.) and maintained at 37°C. For time-lapse observations of NAPs, Z-stacks (2-μm optical sections) were acquired every minute for ∼1 h. Timing of appearance/disappearance of NAPs was confirmed by monitoring all sections in each stack to rule out shift in focus, movements of NAPs, or mitotic cell movements. Photobleaching experiments were done using a 63× 1.4 NA Plan-Apochromat oil immersion lens and 50 iterations of the 488-nm laser set at 100% power in the area of the NAP. Acquisition imaging was done at 1.2% laser power with an open pinhole and a pixel time of 1.6 μs; acquisition bleaching was negligible in all FRAP experiments. The average half time of recovery of YFP-SF2/ASF in NAPs was calculated based on 14 independent bleaching experiments using the same geometry, laser intensity, and bleaching length. Fluorescence of NAPs was normalized with the whole nucleus intensity (Fig. 7 a). To compare recovery in NAPs versus the nucleoplasm, half of the nucleus was photobleached and fluorescence recovery was measured in a NAP and in the nucleoplasm at the same distance from the bleach front. For quantification of SF2/ASF in NAPs, DNA was visualized in living cells by adding 10 μg/ml of Hoechst 33342 30 min before imaging. 1-μm optical sections were acquired through the entire mitotic cell (∼22–24 μm) every 5 min. Three high-resolution four-dimensional data sets were averaged for quantification of SF2/ASF in NAPs. For quantitation, images were preprocessed using an anisotropic diffusion filtering (Gerlich et al., 2001). Cellular compartments were defined by single thresholds that were applied to whole four-dimensional data sets (Fig. S3). Two different thresholds were interactively determined in the YFP-SF2/ASF channel to identify whole cellular regions (lower value) and NAP regions (higher value). An additional threshold was applied on the Hoechst channel to find nuclear regions. Cytoplasmic regions were determined by subtracting nuclear and NAP regions from whole cellular regions. Using these region definitions, total fluorescence intensity was calculated in the unprocessed YFP channel.
EM
HeLa cells were seeded onto gridded Mat Tek dishes and processed for immunolocalization of SF2/ASF. Cells were rinsed briefly in PBS, fixed in 2% formaldehyde for 15 min, washed in blocking buffer (PBS 0.5% NGS 0.3 M glycine) permeabilized in PBS 2% saponin 0.5% NGS 0.3M glycine, and washed in blocking buffer. Cells were incubated in anti-SF2/ASF (1:100) overnight at 4°C, washed in blocking buffer, incubated in biotinylated secondary antibody for 1 h, washed in buffer, and incubated in ABC complex (Vector Laboratories) for an additional hour. After washing, the cells were reacted for 6 min in 0.5 mg/ml DAB (Sigma-Aldrich) with 0.2 μL/ml of hydrogen peroxide, rinsed in buffer, and post-fixed in 1% osmium tetroxide for 1 h. Cells were rinsed in double distilled water, dehydrated in an ethanol series, and infiltrated with Durcupan ACM resin (Electron Microscopy Sciences). Electron micrographs were recorded from 80-nm-thick sections at 80 kV with a transmission electron microscope (model 1200 EX; Jeol Ltd.).
Online supplemental material
Supplemental Videos 1 and 2 show time-lapse of NAPs in HeLa cells. Fig. S1 shows images from a time-lapse of NAPs in HeLa cells that is presented in Video 2 and exclusion of PTB and nascent transcripts from NAPs. Fig. S2 shows FRAP recovery curves for SF2/ASF in NAPs. Fig. S3 illustrates selection of cellular compartments for quantification of fluorescence intensity. Table S1 shows fluorescence intensity values in different cellular compartments. Fig. S4 shows localization of endogenous SF2/ASF and SC35-RFP around nucleoli after α-amanitin treatment of HeLa cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200404120/DC1.
Acknowledgments
The authors wish to thank E. Zanin, M. Dundr, S. Huang, M. Carmo-Fonseca, E. Chan, A. Krainer, J. Stevenin, and W. van Venrooij for reagents. We also thank A. Bubulya and S.G. Prasanth for technical assistance and members of the Spector and Ellenberg laboratories for helpful discussions.
D. Gerlich is supported by a European Molecular Biology Organization long-term fellowship. J. Ellenberg acknowledges support by the Human Frontiers Science Program (grant RGP0031/2001-M). P. Bubulya obtained travel support from The Journal of Cell Science Traveling Fellowship for travel to the J. Ellenberg laboratory. D.L. Spector is funded by the National Institutes of Health/National Institute of General Medical Sciences (grant 42694).
Abbreviations used in this paper: CB, Cajal body; IBB, importin-β binding domain; IGC, interchromatin granule cluster; MIG, mitotic IGC; NAP, NOR-associated patch; NOR, nucleolar organizing region; PNC, perinucleolar compartment; PTB, polypyrimidine tract-binding protein; RS, arginine-serine rich.
References
- Andersen, F.F., T.O. Tange, T. Sinnathamby, J.R. Olesen, K.E. Andersen, O. Westergaard, J. Kjems, and B.R. Knudsen. 2002. The RNA splicing factor ASF/SF2 inhibits human topoisomerase I mediated DNA relaxation. J. Mol. Biol. 322:677–686. [DOI] [PubMed] [Google Scholar]
- Cavaloc, Y., C.F. Bourgeois, L. Kister, and J. Stevenin. 1999. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA. 5:468–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, M.O., H.U. Barthelmes, F. Boege, and C. Mielke. 2002. The N-terminal domain anchors human topoisomerase I at fibrillar centers of nucleoli and nucleolar organizer regions of mitotic chromosomes. J. Biol. Chem. 277:35932–35938. [DOI] [PubMed] [Google Scholar]
- Colwill, K., L.L. Feng, J.M. Yeakley, G.D. Gish, J.F. Caceres, T. Pawson, and X.D. Fu. 1996. a. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J. Biol. Chem. 271:24569–24575. [DOI] [PubMed] [Google Scholar]
- Colwill, K., T. Pawson, B. Andrews, J. Prasad, J.L. Manley, J.C. Bell, and P.I. Duncan. 1996. b. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 15:265–275. [PMC free article] [PubMed] [Google Scholar]
- Darzacq, X., B.E. Jady, C. Verheggen, A.M. Kiss, E. Bertrand, and T. Kiss. 2002. Cajal body-specific small nuclear RNAs: a novel class of 2'-O-methylation and pseudouridylation guide RNAs. EMBO J. 21:2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr, M., and M.O. Olson. 1998. Partially processed pre-rRNA is preserved in association with processing components in nucleolus-derived foci during mitosis. Mol. Biol. Cell. 9:2407–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr, M., U.T. Meier, N. Lewis, D. Rekosh, M.L. Hammarskjold, and M.O. Olson. 1997. A class of nonribosomal nucleolar components is located in chromosome periphery and in nucleolus-derived foci during anaphase and telophase. Chromosoma. 105:407–417. [DOI] [PubMed] [Google Scholar]
- Everett, R., P. Lomonte, T. Sternsdorf, R. van Driel, and A. Orr. 1999. Cell cycle regulation of PML modification and ND10 composition. J. Cell Sci. 112:4581–4588. [DOI] [PubMed] [Google Scholar]
- Ferriera, J.A., M. Carmo-Fonseca, and A.I. Lamond. 1994. Differential interaction of splicing snRNPs with coiled bodies and interchromatin granules during mitosis and assembly of daughter nuclei. J. Cell Biol. 126:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey, M.R., and A.G. Matera. 1995. Coiled bodies contain U7 small nuclear RNA and associate with specific DNA sequences in interphase human cells. Proc. Natl. Acad. Sci. USA. 92:5915–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey, M.R., A.D. Bailey, A.M. Weiner, and A.G. Matera. 1999. Association of snRNA genes with coiled bodies is mediated by snRNA transcripts. Curr. Biol. 9:126–135. [DOI] [PubMed] [Google Scholar]
- Fu, X.D., and T. Maniatis. 1990. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature. 343:437–441. [DOI] [PubMed] [Google Scholar]
- Gao, L., M.R. Frey, and A.G. Matera. 1997. Human genes encoding U3 snRNA associate with coiled bodies in interphase cells and are clustered on chromosome 17p11.2 in a complex inverted repeat structure. Nucleic Acids Res. 25:4740–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlich, D., J. Beaudouin, M. Gebhard, J. Ellenberg, and R. Eils. 2001. Four-dimensional imaging and quantitative reconstruction to analyse complex spatiotemporal processes in live cells. Nat. Cell Biol. 3:852–855. [DOI] [PubMed] [Google Scholar]
- Graveley, B.R. 2000. Sorting out the complexity of SR protein functions. RNA. 6:1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Verdun, D., P. Roussel, and J. Gebrane-Younes. 2002. Emerging concepts of nucleolar assembly. J. Cell Sci. 115:2265–2270. [DOI] [PubMed] [Google Scholar]
- Huang, S., and D.L. Spector. 1992. U1 and U2 small nuclear RNAs are present in nuclear speckles. Proc. Natl. Acad. Sci. USA. 89:305–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S., T.J. Deerinck, M.H. Ellisman, and D.L. Spector. 1998. The perinucleolar compartment and transcription. J. Cell Biol. 143:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Garcia, L.F., M.L. Segura-Valdez, R.L. Ochs, L.I. Rothblum, R. Hannan, and D.L. Spector. 1994. Nucleologenesis: U3 snRNA-containing prenucleolar bodies move to sites of active pre-rRNA transcription after mitosis. Mol. Biol. Cell. 5:955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, P., M. Mannervik, L. Tora, and M. Carmo-Fonseca. 1996. In vivo evidence that TATA-binding protein/SL1 colocalizes with UBF and RNA polymerase I when rRNA synthesis is either active or inactive. J. Cell Biol. 133:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, F.S., and J.P. O'Connor. 1995. Topographic changes in a heterochromatic chromosome block in humans (15P) during formation of the nucleolus. Chromosome Res. 3:309–314. [DOI] [PubMed] [Google Scholar]
- Lamond, A.I., and D.L. Spector. 2003. Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4:605–612. [DOI] [PubMed] [Google Scholar]
- Leser, G.P., S. Fakan, and T.E. Martin. 1989. Ultrastructural distribution of ribonucleoprotein complexes during mitosis. snRNP antigens are contained in mitotic granule clusters. Eur. J. Cell Biol. 50:376–389. [PubMed] [Google Scholar]
- Leung, A.K., J.S. Andersen, M. Mann, and A.I. Lamond. 2003. Bioinformatic analysis of the nucleolus. Biochem. J. 376:553–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera, A.G., M.R. Frey, K. Marelot, and S. Wolin. 1995. A perinucleolar compartment contains several RNA polymerase III transcripts as well as the polypyrimidine tract-binding protein, hnRNP I. J. Cell Biol. 129:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez, J. and B. Stillman. 2000. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell Biol. 20:8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T., and D.L. Spector. 1999. RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol. Cell. 3:697–705. [DOI] [PubMed] [Google Scholar]
- Misteli, T., J.F. Caceres, and D.L. Spector. 1997. The dynamics of a pre-mRNA splicing factor in living cells. Nature. 387:523–527. [DOI] [PubMed] [Google Scholar]
- Misteli, T., J.F. Caceres, J.Q. Clement, A.R. Krainer, M.F. Wilkinson, and D.L. Spector. 1998. Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J. Cell Biol. 143:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, A., W. Speckmann, R. Terns, and M.P. Terns. 1999. Role of the box C/D motif in localization of small nucleolar RNAs to coiled bodies and nucleoli. Mol. Biol. Cell. 10:2131–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs, R.L., M.A. Lischwe, E. Shen, R.E. Caroll, and H. Busch. 1985. Nucleologenesis: composition and fate of prenucleolar bodies. Chromosoma. 92:330–336. [DOI] [PubMed] [Google Scholar]
- Pinol-Roma, S., and G. Dreyfuss. 1993. hnRNP proteins: localization and transport between the nucleus and the cytoplasm. Trends Cell Biol. 3:151–155. [DOI] [PubMed] [Google Scholar]
- Potashkin, J.A., R.J. Derby, and D.L. Spector. 1990. Differential distribution of factors involved in pre-mRNA processing in the yeast cell nucleus. Mol. Cell. Biol. 10:3524–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad, J., K. Colwill, T. Pawson, and J.L. Manley. 1999. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol. 19:6991–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth, K.V., P.A. Sacco-Bubulya, S.G. Prasanth, and D.L. Spector. 2003. Sequential entry of components of gene expression machinery into daughter nuclei. Mol. Biol. Cell. 14:1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, M.B., C. Murphy, and J.G. Gall. 1990. A monoclonal antibody that recognizes a phosphorylated epitope stains lampbrush chromosomes and small granules in the amphibian germinal vesicle. J. Cell Biol. 111:2217–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel, P., C. Andre, L. Comai, and D. Hernandez-Verdun. 1996. The rDNA transcription machinery is assembled during mitosis in active NORs and absent in inactive NORs. J. Cell Biol. 133:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco-Bubulya, P., and D.L. Spector. 2002. Disassembly of interchromatin granule clusters alters the coordination of transcription and pre-mRNA splicing. J. Cell Biol. 156:425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopland, L.S., C.V. Johnson, M. Byron, J. McNeil, and J.B. Lawrence. 2003. Clustering of multiple specific genes and gene-rich R-bands around SC-35 domains: evidence for local euchromatic neighborhoods. J. Cell Biol. 162:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman, J.E., and A.I. Lamond. 1999. Newly assembled snRNPs associate with coiled bodies before speckles, suggesting a nuclear snRNP maturation pathway. Curr. Biol. 9:1065–1074. [DOI] [PubMed] [Google Scholar]
- Smith, H.C., D. L. Spector, C.L. Woodcock, R. L. Ochs and J. Bhorjee. 1985. Alterations in chromatin conformation are accompanied by reorganization of nonchromatin domains that contain U-snRNP protein p28 and nuclear protein p107. J. Cell Biol. 101:560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, K.P., K.C. Carter, C.V. Johnson, and J.B. Lawrence. 1995. U2 and U1 snRNA gene loci associate with coiled bodies. J. Cell. Biochem. 59:473–485. [DOI] [PubMed] [Google Scholar]
- Spector, D.L., and H.C. Smith. 1986. Redistribution of U-snRNPs during mitosis. Exp. Cell Res. 163:87–94. [DOI] [PubMed] [Google Scholar]
- Sylvester, J.E., D.A. Whiteman, R. Podolsky, J.M. Poszgay, J. Respess, and R.D. Schmickel. 1986. The human ribosomal RNA genes: structure and organization of the complete repeating unit. Hum. Genet. 73:193–198. [DOI] [PubMed] [Google Scholar]
- Thiry, M. 1995. Behavior of interchromatin granule clusters during the cell cycle. Eur. J. Cell Biol. 68:14–24. [PubMed] [Google Scholar]
- Turner, B.M., and L. Franchi. 1987. Identification of protein antigens associated with the nuclear matrix and with clusters of interchromatin granules in both interphase and mitotic cells. J. Cell Sci. 87:269–282. [DOI] [PubMed] [Google Scholar]
- Verheggen, C., D.L. Lafontaine, D. Samarsky, J. Mouaikel, J.M. Blanchard, R. Bordonne, and E. Bertrand. 2002. Mammalian and yeast U3 snoRNPs are matured in specific and related nuclear compartments. EMBO J. 21:2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen, R., H. Kuijpers, P. Vooijs, W. Van Venrooij, and F. Ramaekers. 1986. Distribution of the 70K U1 RNA-associated protein during interphase and mitosis. Correlation with other U RNP particles and proteins of the nuclear matrix. J. Cell Sci. 86:173–190. [DOI] [PubMed] [Google Scholar]
- Worton, R.G., J. Sutherland, J.E. Sylvester, H.F. Willard, S. Bodrug, I. Dube, C. Duff, V. Kean, P.N. Ray, and R.D. Schmickel. 1988. Human ribosomal RNA genes: orientation of the tandem array and conservation of the 5′ end. Science. 239:64–68. [DOI] [PubMed] [Google Scholar]