

Abstract
Talin, an adaptor between integrin and the actin cytoskeleton at sites of cell adhesion, was recently found to be present at neuronal synapses, where its function remains unknown. Talin interacts with phosphatidylinositol-(4)-phosphate 5-kinase type Iγ, the major phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2]–synthesizing enzyme in brain. To gain insight into the synaptic role of talin, we microinjected into the large lamprey axons reagents that compete the talin–PIP kinase interaction and then examined their effects on synaptic structure. A dramatic decrease of synaptic actin and an impairment of clathrin-mediated synaptic vesicle endocytosis were observed. The endocytic defect included an accumulation of clathrin-coated pits with wide necks, as previously observed after perturbing actin at these synapses. Thus, the interaction of PIP kinase with talin in presynaptic compartments provides a mechanism to coordinate PI(4,5)P2 synthesis, actin dynamics, and endocytosis, and further supports a functional link between actin and clathrin-mediated endocytosis.
Introduction
In order for neurotransmitter release to continue reliably, neurotransmitter-containing synaptic vesicles must be rapidly and locally recycled. One predominant mechanism for their recycling involves clathrin-mediated endocytosis, a process that occurs at the periphery of active zones of secretion within an actin-rich region called the “periactive,” or endocytic, zone (Roos and Kelly, 1999; Teng and Wilkinson, 2000). Clathrin-mediated synaptic vesicle recycling requires intrinsic coat proteins and accessory factors, including actin regulatory proteins, as well as the interactions of these proteins with phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2], a phosphoinositide concentrated in the plasma membrane (Slepnev and De Camilli, 2000; Morgan et al., 2002).
Because PI(4,5)P2 participates in both clathrin coat and actin nucleation, it is important to identify how the presynaptic pool of PI(4,5)P2 is generated and maintained (Wenk and De Camilli, 2004). Two enzymes concentrated at synapses, phosphatidylinositol-(4)-phosphate 5-kinase type Iγ (PIPKIγ) and the polyphosphoinositide phosphatase synaptojanin, synthesize and degrade, respectively, a large fraction of the presynaptic pool of PI(4,5)P2 (McPherson et al., 1996; Cremona et al., 1999; Gad et al., 2000; Harris et al., 2000; Wenk et al., 2001; Verstreken et al., 2003; Di Paolo et al., 2004). Membrane recruitment and enzymatic activity of PIPKIγ are regulated by interactions with its membrane-localized binding partners, Rho family and Arf6 GTPases (Honda et al., 1999; Krauss et al., 2003). In addition, the predominant splice variants of PIPKIγ expressed in brain contain a unique 28-aa COOH-terminal extension that interacts with the focal adhesion protein talin (Di Paolo et al., 2002; Ling et al., 2002; Giudici et al., 2004). Although talin is present together with PIPKIγ at synapses, its function either pre- or post-synaptically is unknown. The interaction of talin with PIPKIγ is likely to be very important because it greatly up-regulates the catalytic activity of PIPKIγ in vitro (Di Paolo et al., 2002).
Talin is an adaptor between integrins and actin that mediates bi-directional integrin signaling at cell adhesion sites (Calderwood et al., 1999; Critchley et al., 1999; Calderwood and Ginsberg, 2003). The two highly homologous talin isoforms (talin 1 and 2) comprise a 47-kD NH2-terminal globular head and a 190-kD COOH-terminal rodlike tail. The tail has multiple binding sites for actin and vinculin, and the head contains a band 4.1/ezrin/radixin/moesin-like (FERM) domain that binds β-integrins, actin, and PI(4,5)P2 (Calderwood et al., 1999; Di Paolo et al., 2002; Ling et al., 2002). The FERM domain also contains the PIPKIγ binding site, which overlaps with the integrin binding site. These two interactions are mutually exclusive and competitive (Barsukov et al., 2003; Calderwood et al., 2004). Thus, a dynamic cycle has been proposed in which talin first recruits PIPKIγ to the membrane to generate PI(4,5)P2, and then upon binding PI(4,5)P2, shifts to integrin (Barsukov et al., 2003; Ling et al., 2003; Calderwood et al., 2004). Because talin exists as an antiparallel homodimer, another possibility is that the dimer simultaneously binds both PIPKIγ and integrin. Perturbation of these interactions in fibroblasts disrupts actin and causes cell detachment (Di Paolo et al., 2002; Ling et al., 2002).
At synapses, talin may participate in the recruitment of PIPKIγ to the membrane in order to generate the PI(4,5)P2 pool involved in clathrin coat and actin dynamics during vesicle recycling (Di Paolo et al., 2004). The lamprey reticulospinal synapses provide a tractable model to examine this question because of the prominence of the actin surrounding the large vesicle clusters (Gad et al., 2000; Shupliakov et al., 2002; Bloom et al., 2003). Here, we capitalize on the unique features of these synapses to demonstrate that perturbing talin function and, more specifically, perturbing its interactions at the PIP kinase binding site, drastically affects both actin dynamics and synaptic vesicle endocytosis. These results demonstrate that talin functions presynaptically within a protein network that links phosphoinositide metabolism to actin and clathrin coat dynamics.
Results and discussion
The talin interaction with a PIP kinase is conserved in lamprey
In Western blots of rat brain protein extracts, talin immunoreactivity migrates as a 230-kD band (Fig. 1 A) and a 190-kD proteolytic fragment lacking the head domain (Bolton et al., 1997; Fig. 1 A). Affinity chromatography using a GST fusion protein of the 28-aa COOH-terminal tail of human PIPKIγ (GST-PIPKIγ tail) resulted in the purification of the upper talin band, as expected (Fig. 1 A; Di Paolo et al., 2002). The proteolytic fragment, which lacks the PIPKIγ binding site, remained in the supernatant. Talin was the only major band retained by GST-PIPKIγ tail, as shown by Coomassie blue staining of the affinity-purified material (Fig. 1 B). Similarly, anti-talin antibodies recognized two proteins in lamprey spinal cord extracts of the appropriate size for talin and its proteolytic fragment (Fig. 1 C), and the larger protein, which corresponded to a major band visible by Coomassie blue (Fig. 1 D), was pulled down by GST-PIPKIγ tail. Western blots of lamprey extracts with an anti-PIPKIγ antibody raised against the 28-aa COOH-terminal tail revealed a 90-kD protein doublet that was affinity purified by a GST fusion protein of human talin head (GST-talin head; Fig. 1 E). Immunoprecipitates generated by this PIPKIγ antibody from lamprey extracts contained a much higher PIP2-synthesizing activity than did the control, as demonstrated by TLC separation of 32P-labeled phosphoinositides generated by in vitro incubation with brain lipids and γ-[32P] ATP (Fig. 1 F). Further, using anti-talin antibodies, PIPKIγ was coimmunoprecipitated from both rat brain (Fig. 1 G, middle lane) and lamprey extracts (Fig. 1 H, left lane) (Di Paolo et al., 2002). Thus, lamprey contains a PIP kinase that interacts with talin. Immunostaining of lamprey spinal cord cross sections demonstrated that talin is concentrated at synapses, including those of the large reticulospinal axons, as shown by its colocalization with the synaptic protein synapsin (Fig. 1 I) (De Camilli et al., 1983; Pieribone et al., 1995).
Figure 1.
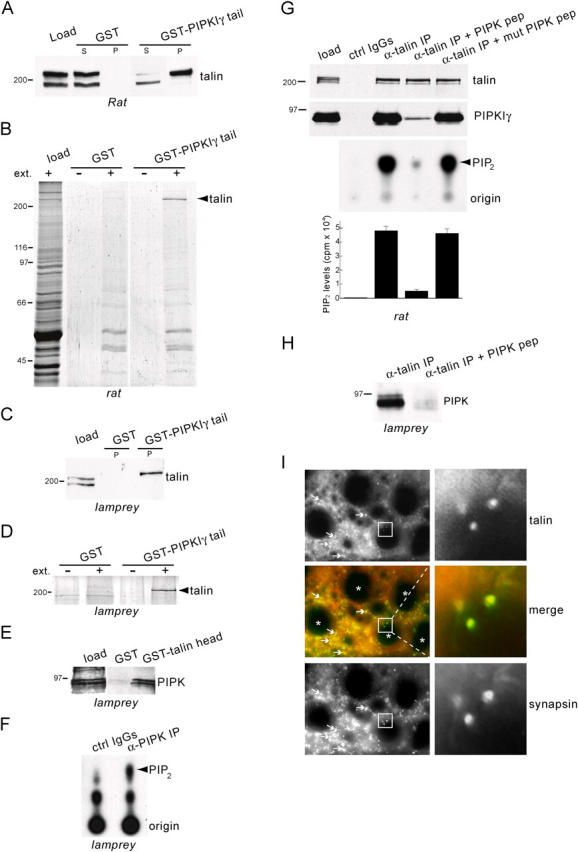
Interaction of PIPK with talin and synaptic localization of talin in the lamprey spinal cord. (A) Western blot with an anti-talin antibody of rat brain extract (load) and the material affinity purified from such extract by either GST or GST-PIPKIγ tail in pull-down experiments (S, supernatant; P, pellet). The top talin band is pulled down selectively by GST-PIPKIγ tail. (B) Coomassie blue–stained protein gel of a pull-down with GST-PIPKIγ tail from rat brain extracts (ext.) showing specificity of the talin–PIPKIγ interaction. (C) Western blot with an anti-talin antibody of lamprey extract (load) and of the material purified by either GST or GST-PIPKIγ tail. (D) Coomassie blue–stained protein gel of a pull-down with GST-PIPKIγ tail from lamprey spinal cord extracts. (E) Western blot with an anti-PIPKIγ antibody of lamprey extract (load) and of the material purified by either GST or GST-talin head. The 90-kD doublet is specifically affinity purified by GST-talin head. (F) TLC demonstrating the generation of phosphoinositides from lamprey extracts by immunoprecipitates of PIPKIγ antibody (α-PIPK IP) or control IgGs (ctrl IgGs). Note the prominent [32P]PIP2 band produced by the α-PIPK IP. (G, top panels) Western blots of anti-talin immunoprecipitates from rat brain extracts showing that coprecipitation of PIPKIγ (“α-talin IP” lane) is prevented by PIPK pep but not mutant PIPK pep (100 μM). (G, bottom panels) TLC and quantification of [32P]PIP2 showing correlation between PIPKIγ coprecipitation and PIP2-synthesizing activity. (cpm, counts per minute). (H) Western blot of anti-talin immunoprecipitates from lamprey spinal cord extracts showing that the PIPK pep (300 μM) prevents coprecipitation of PIPK. (I) Double immunofluorescence of cross sections of lamprey spinal cord stained for talin (red) and for the presynaptic marker, synapsin (green). Arrows indicate synapses in the neuropil. Asterisks indicate reticulospinal axons. Note the colocalization of talin with synapsin at the reticulospinal synapses (insets).
A PIPKIγ peptide inhibits the interaction of a PIP kinase with talin in lamprey
Next, we characterized a 14-mer PIPK peptide (PIPK pep) centered around the minimal talin-binding site in human PIPKIγ, WVYSPL (Fig. 2 A; Di Paolo et al., 2002). Pre-incubation of rat brain extracts with PIPK pep nearly completely abolished the ability of anti-talin antibodies to coprecipitate PIP kinase both from rat brain (Fig. 1 G, fourth lane) and lamprey spinal cord extracts (Fig. 1 H; right lane). In contrast, a mutant PIPK peptide (Mut PIPK pep; Fig. 2 A) in which the critical tryptophan was replaced by an alanine had negligible effects (Fig. 1 G, fifth lane). Isothermal titration calorimetry (ITC) revealed that PIPK pep binds to the head of talin in a saturable manner (Fig. 2, B and C) with a KD of 0.65 μM, as determined by fitting to a nonlinear least squares function. This value is roughly in agreement with previous measurements made by nuclear magnetic resonance spectroscopy using a slightly longer PIPKIγ peptide (Barsukov et al., 2003). Mut PIPK pep demonstrated only negligible binding to talin head (Fig. 2, D and E).
Figure 2.

Binding of a PIPK peptide to talin head. (A) Diagram showing the domain structure of human PIPKIγ and the sequences of the PIPK peptides. Underlined residues indicate the talin binding region. (B–E) ITC traces showing the responses when either PIPK pep (B) or Mutant PIPK pep (D) was injected (arrow) into a chamber containing talin head (Tal H). Decreased magnitude of the response indicates talin saturation by PIPK pep. Dose–response curves show that PIPK pep (C), but not Mutant PIPK pep (E), binds to talin head. Data points were derived from B and D and were fit with a nonlinear least squares function (solid line).
PIPK peptide inhibits the accumulation of actin at periactive zones
Lamprey reticulospinal axons form en passant synapses with motor neurons. The large vesicle clusters at these synapses are surrounded by a ring of actin at periactive zones that can be visualized by the microinjection of fluorescently labeled phalloidin (Fig. 3 A) (Shupliakov et al., 2002). To determine whether the talin–PIPK interaction regulates this pool of synaptic actin, individual lamprey axons were first microinjected with rhodamine-conjugated Mut PIPK pep or PIPK pep (red), and immediately after with Alexa 488–phalloidin (green). Although the Mut PIPK peptide did not affect the accumulation of phalloidin fluorescence at synapses, PIPK peptide strongly inhibited this accumulation (Fig. 3, B and C). The PIPK peptide caused an average 64% reduction in the synaptic phalloidin fluorescence at intra-axonal concentrations >10 μM, which is within the range expected to compete the talin–PIPK interaction (Fig. 3 D). This effect is likely an underestimation because the intensity of synaptic phalloidin fluorescence at sites of highest PIPK peptide concentration was often at the limit of detectability, and therefore was not measured. Even at concentrations as high as 200–700 μM, Mut PIPK peptide had no effect on synaptic phalloidin fluorescence (Fig. 3 D).
Figure 3.
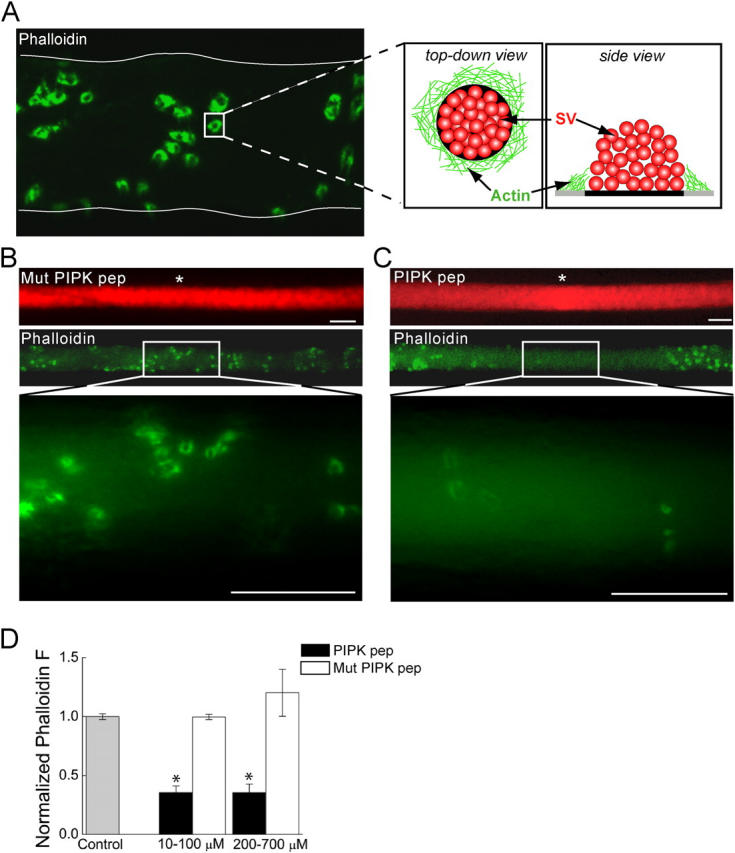
Microinjection of PIPK pep disrupts the actin cytoskeleton at synapses. (A) Confocal image of a lamprey reticulospinal axon injected with Alexa 488–phalloidin (green). Thin, white line denotes axonal border. Phalloidin labels rings of actin at periactive zones of synapses, as indicated by the cartoon (SV, synaptic vesicle). (B and C) Images of axons injected with rhodamine-conjugated (red) Mut PIPK pep or PIPK pep and then with Alexa 488–phalloidin at sites indicated by white asterisks. Although Mut PIPK pep had no effect (B), the fluorescence intensity of phalloidin rings is greatly diminished by PIPK pep (C). Bars, 20 μm. (D) Average fluorescence intensity of the phalloidin rings over several concentration ranges of PIPK peptides as compared with that in the absence of any peptides (Control). Data indicate mean values and SEM from synapses in 31 control, 15 PIPK pep–injected, and 14 Mut PIPK pep–injected axons. Asterisks indicate statistical significance as compared with control (for PIPK pep 10–100 μM, P < 0.05 × 10−13; for PIPK pep 200–700 μM, P < 0.05 × 10−8; t test).
The decreased accumulation of synaptic actin observed upon PIPK peptide injection may result from reduced PI(4,5)P2 levels as a consequence of impairing talin-mediated PIP kinase recruitment to the membrane. Additional effects of PIPK peptide on actin may be produced by a perturbation of the talin–integrin interaction, although this remains to be explored. Consistent with the results reported here, microinjection of antibodies directed against the PI(4,5)P2 phosphatase synaptojanin (e.g., a treatment that is expected to increase PI(4,5)P2) produced the opposite effect, which was a hypertrophy of perisynaptic actin at lamprey synapses (Gad et al., 2000).
PIPK peptide perturbs clathrin-mediated endocytosis
Next, we examined the effect of PIPK peptide on synaptic vesicle trafficking using EM. Axons were microinjected with either PIPK or mutant peptide, stimulated (20 Hz for 5 min) to induce exocytosis and compensatory synaptic vesicle recycling, and then fixed (Pieribone et al., 1995). Electron micrographs of synapses within mutant PIPK peptide–injected axons revealed the typical large synaptic vesicle clusters and very few clathrin-coated pits (Fig. 4 A). Under these stimulation conditions, synaptic vesicle recycling is very efficient in control synapses. In contrast, images of synapses from PIPK peptide–injected axons revealed numerous clathrin-coated pits and large folds of the plasma membrane at periactive zones that often extended toward the postsynaptic cell (Fig. 4, B and C). In addition, the average number of synaptic vesicles per synapse in PIPK peptide–injected axons was 33% smaller than in mutant PIPK peptide–injected control axons, indicating that synaptic vesicle recycling was perturbed (Fig. 4 D; P < 0.05; t test). A measurement of the plasma membrane cross-sectional profile within a 1-μm radial distance from the outer edge of the active zone revealed a twofold increase in length relative to mutant PIPK pep, denoting a striking expansion of the plasma membrane (Fig. 4 E; P < 0.05 × 10−6; t test). Further, the total number of clathrin-coated profiles per synapse dramatically increased 10-fold in the presence of PIPK pep (Fig. 4 F; P < 0.05 × 10−8; t test). When the coated profiles were staged according to state of maturation, the greatest increase was observed in unconstricted coated pits (Fig. 4 G).
Figure 4.
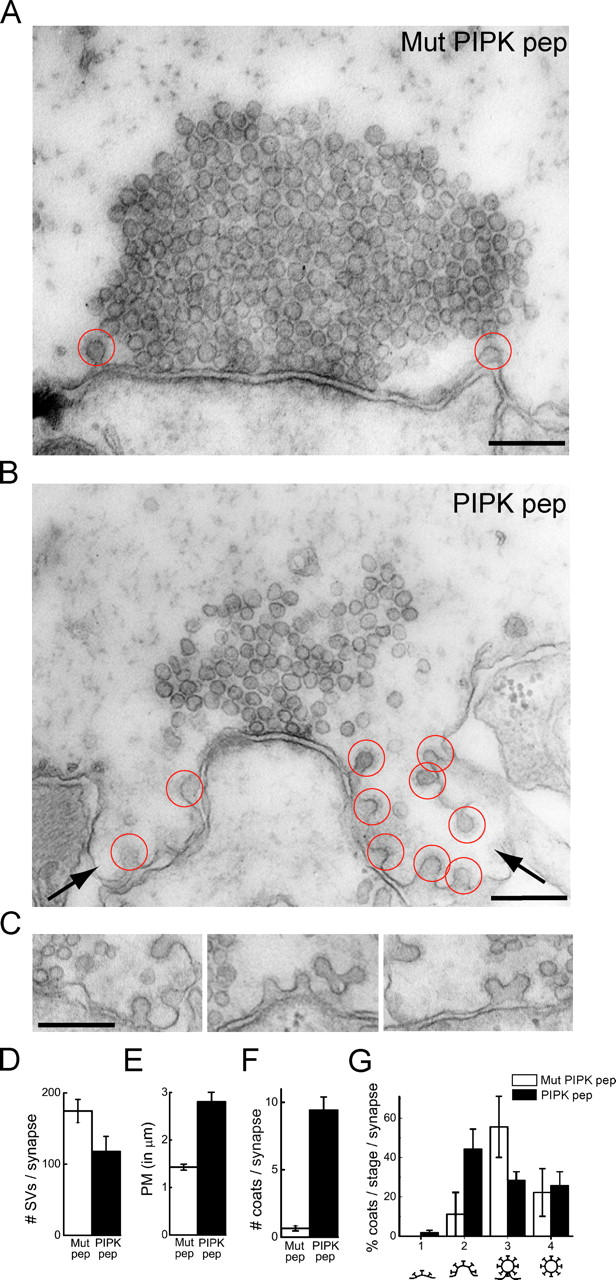
Ultrastructural changes produced by PIPK pep at lamprey synapses. (A and B) Electron micrographs of stimulated synapses after axonal injection of either Mut PIPK pep or PIPK pep. In the presence of the Mut PIPK pep (A), only few coated pits are observed. In contrast, numerous clathrin-coated pits (red circles) and large plasma membrane foldings (arrows) are observed in the presence of PIPK pep (B). (C) Gallery showing unconstricted clathrin-coated pits at periactive zones of synapses within PIPK pep–injected axons. Bars, 0.2 μm. (D–G) Quantification of the number of synaptic vesicles (D, SVs), the plasma membrane (PM) cross-sectional profile (E), the total number of clathrin-coated profiles (F), and the percentages of coated profiles at various stages of maturation (G) per synapse. Data represent mean values and SEM for 21 synapses from 2 axons injected with PIPK pep and 20 synapses from 2 axons injected with Mutant PIPK pep.
To further assess the specificity of the effects on synaptic vesicle recycling, we injected GST-talin head. This reagent is expected to compete the interactions of the FERM domain of endogenous talin with its binding partners. Electron micrographs of stimulated synapses within axons injected with GST-talin head revealed a perturbation of clathrin-mediated endocytosis at synapses similar to that observed with PIPK pep (Fig. 5). Compared with GST controls, GST-talin head caused an increase in the length of the plasma membrane cross-sectional profile around synapses (Fig. 5, A, B, and E; P < 0.05 t test), and a twofold increase in the number of clathrin-coated profiles (Fig. 5 F). Like the PIPK peptide, GST-talin head specifically increased the number of unconstricted coated pits (Fig. 5, B, C, and G; P < 0.05 t test), further supporting a presynaptic role for talin in coated vesicle maturation. Only a modest, nonstatistically significant decrease in the number of synaptic vesicles was observed in the presence of GST-talin head (Fig. 5 D). The less dramatic phenotype observed with GST-talin head, rather than with the PIPK peptide, likely reflects the lower axonal concentration that was achieved by microinjection of this fusion protein, which was estimated to be in the range of 0.5–2.5 μM (i.e., in the same range of the KD of the talin–PIPK interaction). However, the similarity to the PIPK peptide phenotype indicates that GST-talin head targeted the same process.
Figure 5.
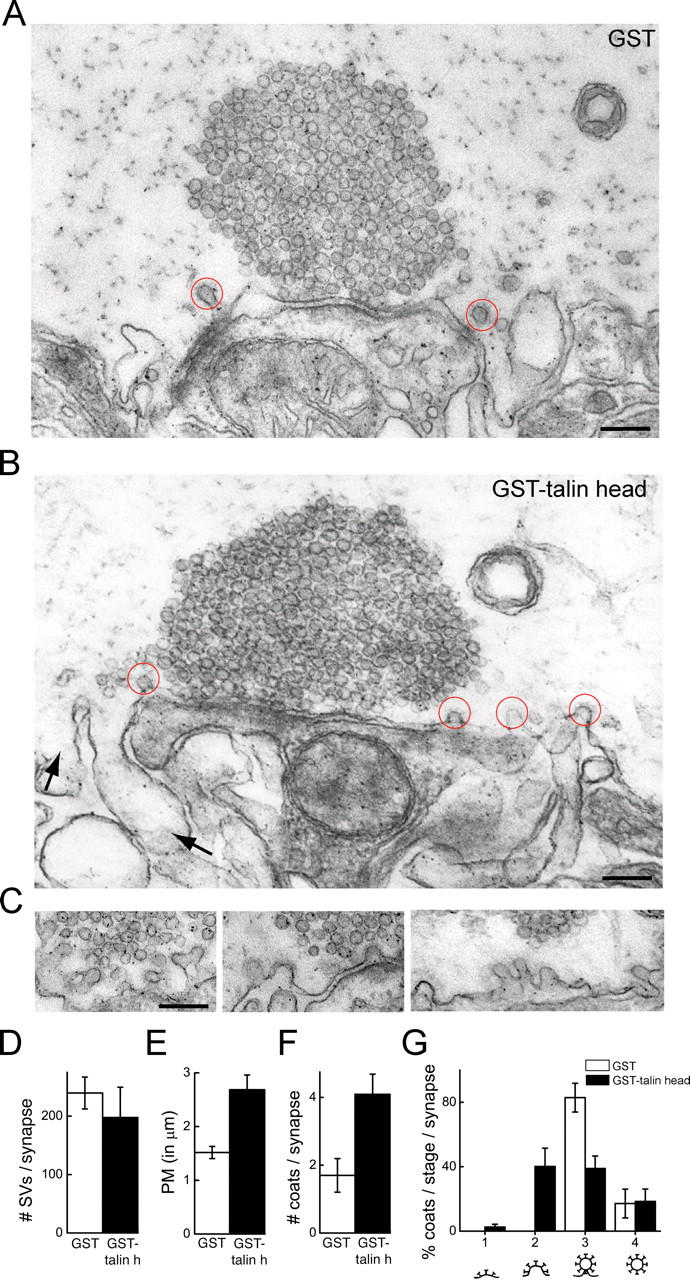
Ultrastructural changes produced by talin head at lamprey synapses. (A and B) Electron micrographs of stimulated synapses after axonal injection of either GST or GST-talin head. In the presence of GST (A), only few coated pits are observed. In contrast, more clathrin-coated pits (red circles) and large membrane foldings (arrows) are observed in the presence of GST-talin head (B). (C) Gallery showing unconstricted clathrin-coated pits in periactive zones within GST-talin head–injected axons. Bars, 0.2 μm. (D–G) Quantification of the number of SVs (D), the PM cross-sectional profile (E), the total number of clathrin-coated profiles (F), and percentages of coated profiles at various stages of maturation (G) per synapse cross section. Data represent mean values and SEM of 10 synapses from 4 axons injected with GST controls and 11 synapses from 4 axons injected with GST-talin head.
The ultrastructural changes produced by the microinjection of both the PIPK peptide and the GST-talin head indicate a kinetic delay or an arrest of the clathrin-mediated endocytic reaction. The increased number of coated pits may seem at odds with the role of PI(4,5)P2 in nucleating clathrin coat formation (Wenk and De Camilli, 2004). However, partial recruitment of PIPKIγ to the plasma membrane may occur by alternate mechanisms even when the interaction with talin is prevented. For example, PIPKIγ recruitment may occur via interactions with Rho family or Arf6 GTPases (Chong et al., 1994; Honda et al., 1999; Krauss et al., 2003). Given that the PIP kinase interactions with Arf6 and Rho occur outside the talin-binding region (because the interactions are conserved amongst all type I PIP kinase isoforms), it is unlikely that our reagents altered recruitment by these GTPases (Doughman et al., 2003). Thus, the recruitment of PIPKIγ by small GTPases may produce sufficient amounts of PI(4,5)P2 for clathrin coat nucleation by the clathrin adaptors. Other PI(4) 5-kinases may also come into play. Furthermore, accumulation of clathrin-coated pits could occur in the presence of lower PI(4,5)P2 levels if coat maturation was delayed.
Strikingly, the majority of clathrin-coated pits observed at PIPK peptide–treated synapses have a wide neck, reflecting a defect at an early stage of endocytosis. A similar accumulation of unconstricted clathrin-coated pits was observed at these synapses after treatment with actin-disrupting toxins (Shupliakov et al., 2002). Thus, our experiments seem to have perturbed regulatory mechanisms at the interface between endocytosis and actin (Qualmann and Kessels, 2002; Engqvist-Goldstein and Drubin, 2003). We note that dynamin, a GTPase critically implicated in the fission of endocytic vesicles, is thought to function as an actin regulatory protein (Lee and De Camilli, 2002; Schafer, 2004). Dynamin's recruitment to endocytic intermediates and its activity are regulated in part by its interaction with PI(4,5)P2 (Schmid et al., 1998). Talin may affect a PI(4,5)P2 pool specifically involved in an actin-dependent maturation of endocytic intermediates.
A conclusion of our work is that talin participates in the biology of periactive zones of synapses by regulating actin and clathrin coat dynamics. Although it cannot be completely excluded that some of the PIPK peptide effects may result from perturbing interactors other than talin, in vitro binding experiments have shown that talin is by far the major binding partner of PIPKIγ (Fig. 1 C; Di Paolo et al., 2002). Furthermore, the fact that talin head produces a phenotype on vesicle trafficking similar to that of the PIPK peptide supports the hypothesis that PIPKIγ acts primarily by disrupting talin function. The prominent effects of the PIPK peptide on both actin and clathrin-mediated endocytosis in our work provides further evidence for a close functional link between these processes (Merrifield et al., 2002; Shupliakov et al., 2002; Kaksonen et al., 2003). Accordingly, a number of possible molecular links between actin and clathrin-mediated endocytosis, which include Hip/Hip1R, dynamin, intersectin, PACSIN/syndapin, tuba, and cortactin, have been described previously (Qualmann and Kessels, 2002; Engqvist-Goldstein and Drubin, 2003).
These results extend our knowledge of the importance of phosphoinositide metabolism and actin polymerization during vesicle trafficking at the synapse and establish a role for talin in coordinating these processes. Further, our results suggest that synapses share mechanisms to coordinate membrane–cytoskeletal dynamics in common with other sites of cell adhesion.
Materials and methods
Protein and lipid biochemistry
Human PIPKIγ tail (aa 635–662) and human talin 2 head (aa 1–438) were expressed as GST fusion proteins and purified using a standard protocol. Affinity chromatography, immunoprecipitation, and TLC experiments were performed as described previously (Wenk et al., 2001; Di Paolo et al., 2002).
Rhodamine-conjugated PIPK peptide and mutant PIPK peptide were synthesized at the W.M. Keck Foundation Biotechnology Resource Laboratory (Yale University, New Haven, CT). The anti-talin antibody (TD77) used for detecting talin from GST pulldowns and with immunofluorescence was purchased from CHEMICON International, Inc. Anti-synapsin antibody was provided by Dr. Ona Bloom (Yale University, New Haven, CT). Anti–human talin 2 used for IPs and anti-PIPKIγ antibodies were generated in the De Camilli laboratory.
ITC
Peptides (300 μM) were repeatedly delivered (6 μl/injection; 23°C) into an ITC chamber containing 11 μM GST-talin head. The heat generated by each injection was measured and then analyzed with an ITC version of Microcal Origin 5.0 using embedded nonlinear least squares fitting routines. ITC experiments were performed at the Biophysics Resource of the W.M. Keck Foundation Biotechnology Resource Laboratory.
Imaging
Adult lampreys (Ichthyomyzon unicuspis) were anaesthetized with tricaine methanesulfonate (0.1 g/l tank water). A spinal cord segment was removed and transferred ventral side up to a chamber containing ice-cold, oxygenated ringer (mM): 91 NaCl, 2.1 KCl, 2.6 CaCl2, 1.8 MgCl2, 4.0 glucose, and 2.0 Hepes-KOH, pH 7.4. Ventral orientation permits easy access to the large diameter (20–100 μm) reticulospinal axons.
Fluorescent reagents were diluted with lamprey internal solution (180 mM KCl and 10 mM Hepes-KOH, pH 7.4), loaded into glass microelectrodes, and then injected into several reticulospinal axons maintained at 10–15°C using N2 pulses delivered by a General Valve Picospritzer (10–100 psi; 10–200 ms; 0.3–1.0 Hz). Fluorescence was visualized with a microscope (Axioskop 2FS; Carl Zeiss MicroImaging, Inc.) using 10 and 40× water-immersion objectives. Images were acquired with a CCD camera (Photometrics Cascade 650; Roper Scientific) and analyzed with Metamorph software (Universal Imaging Corp.).
To quantify the PIPK peptide effect on actin, images of Alexa 488–phalloidin (Molecular Probes, Inc.) rings were taken at 40×. The axonal background fluorescence was subtracted and then divided from the synaptic fluorescence value in order to normalize for differences in amounts of injected phalloidin. All synapses within a 60-μm segment centered at the injection site were analyzed and then an averaged for each axon. Peptide concentration was measured by comparing the average rhodamine intensity within the 60-μm segment to a calibration curve.
EM
Reagents were injected into single axons nearest the spinal cord central canal. Bright field and fluorescence images were made for later identification of the injected axon. The axon was then stimulated intracellularly by action potentials using depolarizing pulses (1 ms; 30–100 nA; 20 Hz, 5 min), which were delivered via an Axoclamp 2B amplifier (Axon Instruments). Preparations were fixed overnight (3% glutaraldehyde, 1% formaldehyde, and 0.1% phosphate buffer; pH 7.4), processed for EM as described in Pieribone et al. (1995), and imaged (28,500×) using a microscope (model CM10; Philips).
To measure the cross-sectional profile of the plasma membrane, a straight line was drawn between the outer edge of the active zone and a point on the plasma membrane located 1 μm away. The curved profile of the plasma membrane between these two points was measured and then divided by the straight distance.
Acknowledgments
The authors would like to thank Chrissie Horensavitz for EM assistance, Sergey Voronov for ITC data analysis, and Henry Tan for help with digital reproduction.
This work was supported by National Institutes of Health (NIH) grants (NS36251 and CA46128) to P. De Camilli, NIH NRSA (F32 MH067385) and Grass Foundation Fellowship to J.R. Morgan, a DFG fellowship to H. Werner, and a NIH grant (NS037823) to V.A. Pieribone.
Abbreviations used in this paper: FERM, band 4.1/ezrin/radixin/moesin-like; ITC, isothermal titration calorimetry; PI(4,5)P2, phosphatidylinositol-(4,5)-bisphosphate; PIPKIγ, phosphatidylinositol-(4)-phosphate 5-kinase type Iγ.
References
- Barsukov, I.L., A. Prescot, N. Bate, B. Patel, D.N. Floyd, N. Bhanji, C.R. Bagshaw, K. Letinic, G. Di Paolo, P. De Camilli, et al. 2003. Phosphatidylinositol phosphate kinase type 1γ and β1-integrin cytoplasmic domain bind to the same region in the talin FERM domain. J. Biol. Chem. 278:31202–31209. [DOI] [PubMed] [Google Scholar]
- Bloom, O., E. Evergren, N. Tomilin, O. Kjaerulff, P. Low, L. Brodin, V.A. Pieribone, P. Greengard, and O. Shupliakov. 2003. Colocalization of synapsin and actin during synaptic vesicle recycling. J. Cell Biol. 161:737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton, S.J., S.T. Barry, H. Mosley, B. Patel, B.M. Jockusch, J.M. Wilkinson, and D.R. Critchley. 1997. Monoclonal antibodies recognizing the N- and C-terminal regions of talin disrupt actin stress fibers when microinjected into human fibroblasts. Cell Motil. Cytoskeleton. 36:363–376. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., and M.H. Ginsberg. 2003. Talin forges the links between integrins and actin. Nat. Cell Biol. 5:694–697. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., R. Zent, R. Grant, D.J. Rees, R.O. Hynes, and M.H. Ginsberg. 1999. The Talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274:28071–28074. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., V. Tai, G. Di Paolo, P. De Camilli, and M.H. Ginsberg. 2004. Competition for talin results in trans-dominant inhibition of integrin activation. J. Biol Chem. 279:28889–28895. [DOI] [PubMed] [Google Scholar]
- Chong, L.D., A. Traynor-Kaplan, G.M. Bokoch, and M.A. Schwartz. 1994. The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell. 79:507–513. [DOI] [PubMed] [Google Scholar]
- Cremona, O., G. Di Paolo, M.R. Wenk, A. Luthi, W.T. Kim, K. Takei, L. Daniell, Y. Nemoto, S.B. Shears, R.A. Flavell, et al. 1999. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 99:179–188. [DOI] [PubMed] [Google Scholar]
- Critchley, D.R., M.R. Holt, S.T. Barry, H. Priddle, L. Hemmings, and J. Norman. 1999. Integrin-mediated cell adhesion: the cytoskeletal connection. Biochem. Soc. Symp. 65:79–99. [PubMed] [Google Scholar]
- De Camilli, P., R. Cameron, and P. Greengard. 1983. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. I. Its general distribution in synapses of the central and peripheral nervous system demonstrated by immunofluorescence in frozen and plastic sections. J. Cell Biol. 96:1337–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo, G., L. Pellegrini, K. Letinic, G. Cestra, R. Zoncu, S. Voronov, S. Chang, J. Guo, M.R. Wenk, and P. De Camilli. 2002. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1γ by the FERM domain of talin. Nature. 420:85–89. [DOI] [PubMed] [Google Scholar]
- Di Paolo, G., H.S. Moskowitz, K. Gipson, M.R. Wenk, S. Voronov, M. Obayashi, R. Flavell, R.M. Fitzsimonds, T.A. Ryan, and P. De Camilli. 2004. Impaired PI(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 431:415–422. [DOI] [PubMed] [Google Scholar]
- Doughman, R.L., A.J. Firestone, and R.A. Anderson. 2003. Phosphatidylinositol phosphate kinases put PI4,5P2 in its place. J. Membr. Biol. 194:77–89. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A.E., and D.G. Drubin. 2003. Actin assembly and endocytosis: from yeast to mammals. Annu. Rev. Cell Dev. Biol. 19:287–332. [DOI] [PubMed] [Google Scholar]
- Gad, H., N. Ringstad, P. Low, O. Kjaerulff, J. Gustafsson, M. Wenk, G. Di Paolo, Y. Nemoto, J. Crun, M.H. Ellisman, et al. 2000. Fission and uncoating of synaptic clathrin-coated vesicles are perturbed by disruption of interactions with the SH3 domain of endophilin. Neuron. 27:301–312. [DOI] [PubMed] [Google Scholar]
- Giudici, M.L., P.C. Emson, and R.F. Irvine. 2004. A novel neuronal-specific splice variant of Type I phosphatidylinositol 4-phosphate 5-kinase isoform γ. Biochem. J. 379:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, T.W., E. Hartwieg, H.R. Horvitz, and E.M. Jorgensen. 2000. Mutations in synaptojanin disrupt synaptic vesicle recycling. J. Cell Biol. 150:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda, A., M. Nogami, T. Yokozeki, M. Yamazaki, H. Nakamura, H. Watanabe, K. Kawamoto, K. Nakayama, A.J. Morris, M.A. Frohman, and Y. Kanaho. 1999. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 99:521–532. [DOI] [PubMed] [Google Scholar]
- Kaksonen, M., Y. Sun, and D.G. Drubin. 2003. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell. 115:475–487. [DOI] [PubMed] [Google Scholar]
- Krauss, M., M. Kinuta, M.R. Wenk, P. De Camilli, K. Takei, and V. Haucke. 2003. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Iγ. J. Cell Biol. 162:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, E., and P. De Camilli. 2002. Dynamin at actin tails. Proc. Natl. Acad. Sci. USA. 99:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, K., R.L. Doughman, A.J. Firestone, M.W. Bunce, and R.A. Anderson. 2002. Type Iγ phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 420:89–93. [DOI] [PubMed] [Google Scholar]
- Ling, K., R.L. Doughman, V.V. Iyer, A.J. Firestone, S.F. Bairstow, D.F. Mosher, M.D. Schaller, and R.A. Anderson. 2003. Tyrosine phosphorylation of type Iγ phosphatidylinositol phosphate kinase by Src regulates an integrin-talin switch. J. Cell Biol. 163:1339–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson, P.S., E.P. Garcia, V.I. Slepnev, C. David, X. Zhang, D. Grabs, W.S. Sossin, R. Bauerfeind, Y. Nemoto, and P. De Camilli. 1996. A presynaptic inositol-5-phosphatase. Nature. 379:353–357. [DOI] [PubMed] [Google Scholar]
- Merrifield, C.J., M.E. Feldman, L. Wan, and W. Almers. 2002. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 4:691–698. [DOI] [PubMed] [Google Scholar]
- Morgan, J.R., G.J. Augustine, and E.M. Lafer. 2002. Synaptic vesicle endocytosis: the races, places, and molecular faces. Neuromolecular Med. 2:101–114. [DOI] [PubMed] [Google Scholar]
- Pieribone, V.A., O. Shupliakov, L. Brodin, S. Hilfiker-Rothenfluh, A.J. Czernik, and P. Greengard. 1995. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 375:493–497. [DOI] [PubMed] [Google Scholar]
- Qualmann, B., and M.M. Kessels. 2002. Endocytosis and the cytoskeleton. Int. Rev. Cytol. 220:93–144. [DOI] [PubMed] [Google Scholar]
- Roos, J., and R.B. Kelly. 1999. The endocytic machinery in nerve terminals surrounds sites of exocytosis. Curr. Biol. 9:1411–1414. [DOI] [PubMed] [Google Scholar]
- Schafer, D.A. 2004. Regulating actin dynamics at membranes: a focus on dynamin. Traffic. 5:463–469. [DOI] [PubMed] [Google Scholar]
- Schmid, S.L., M.A. McNiven, and P. De Camilli. 1998. Dynamin and its partners: a progress report. Curr. Opin. Cell Biol. 10:504–512. [DOI] [PubMed] [Google Scholar]
- Shupliakov, O., O. Bloom, J.S. Gustafsson, O. Kjaerulff, P. Low, N. Tomilin, V.A. Pieribone, P. Greengard, and L. Brodin. 2002. Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc. Natl. Acad. Sci. USA. 99:14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepnev, V.I., and P. De Camilli. 2000. Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nat. Rev. Neurosci. 1:161–172. [DOI] [PubMed] [Google Scholar]
- Teng, H., and R.S. Wilkinson. 2000. Clathrin-mediated endocytosis near active zones in snake motor boutons. J. Neurosci. 20:7986–7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken, P., T.W. Koh, K.L. Schulze, R.G. Zhai, P.R. Hiesinger, Y. Zhou, S.Q. Mehta, Y. Cao, J. Roos, and H.J. Bellen. 2003. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron. 40:733–748. [DOI] [PubMed] [Google Scholar]
- Wenk, M., and P. De Camilli. 2004. Protein lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc. Natl. Acad. Sci. USA. 101:8262–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk, M.R., L. Pellegrini, V.A. Klenchin, G. Di Paolo, S. Chang, L. Daniell, M. Arioka, T.F. Martin, and P. De Camilli. 2001. PIP kinase Iγ is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron. 32:79–88. [DOI] [PubMed] [Google Scholar]