

Abstract
The integrin α9β1 is expressed on migrating cells, such as leukocytes, and binds to multiple ligands that are present at sites of tissue injury and inflammation. α9β1, like the structurally related integrin α4β1, mediates accelerated cell migration, an effect that depends on the α9 cytoplasmic domain. α4β1 enhances migration through reversible binding to the adapter protein, paxillin, but α9β1-dependent migration is paxillin independent. Using yeast two-hybrid screening, we identified the polyamine catabolizing enzyme spermidine/spermine N 1-acetyltransferase (SSAT) as a specific binding partner of the α9 cytoplasmic domain. Overexpression of SSAT increased α9β1-mediated migration, and small interfering RNA knockdown of SSAT inhibited this migration without affecting cell adhesion or migration that was mediated by other integrin cytoplasmic domains. The enzyme activity of SSAT is critical for this effect, because a catalytically inactive version did not enhance migration. We conclude that SSAT directly binds to the α9 cytoplasmic domain and mediates α9-dependent enhancement of cell migration, presumably by localized effects on acetylation of polyamines or of unidentified substrates.
Introduction
Cell migration is central to many biological and pathological processes, including embryogenesis, inflammation, tissue repair and regeneration, and cancer metastasis (Lauffenburger and Horwitz, 1996). For cells to migrate on a substrate, they must adhere and de-adhere in a coordinated manner and generate tensile force in the direction of migration. The force required to promote migration is generated by the actin cytoskeleton and the integrin-dependent protein complexes that anchor actin to specific sites on the cell membrane (Lauffenburger and Horwitz, 1996).
Although several members of the integrin family can mediate cell migration, two structurally related integrin α subunits, α4 and α9, appear to play specialized roles in accelerated migration. Several studies using chimeric integrin α subunits have demonstrated that this enhancement of migration depends on distinct sequences within the cytoplasmic domains of α4 (Chan et al., 1992; Kassner and Hemler, 1993; Kassner et al., 1995) and α9 (Young et al., 2001). Integrin α4–mediated cell migration depends on the specific interaction of the α4 cytoplasmic domain with the adapter protein paxillin (Liu et al., 1999), and it requires the dissociation of paxillin from integrin at the leading edge of migrating cells upon phosphorylation of a specific serine at position 988 in the α4 cytoplasmic domain (Han et al., 2001; Goldfinger et al., 2003). The α9 cytoplasmic domain is 47% identical to the α4 cytoplasmic domain, and also binds paxillin (Young et al., 2001). However, α9 does not contain a homologous phosphorylation site; although the interaction between paxillin and α9 is critical for the effects of the integrin α9β1 on cell spreading, this interaction is completely dispensable for α9-dependent enhancement of cell migration (Young et al., 2001). Therefore, we used yeast two-hybrid screening to identify proteins that interact with the α9 cytoplasmic domain to mediate enhanced cell migration.
Results
Identification of SSAT as a specific binding partner of the integrin α9 subunit cytoplasmic domain
Proteins that interact with the α9 cytoplasmic domain were identified by yeast two-hybrid screening of a human leukocyte cDNA library, using the entire cytoplasmic domain of human α9 (residues 974–1006) as bait. Several candidate cDNAs were isolated and sequenced. These candidates were rescreened by cotransformation with bait vectors containing the α9, α5, α4, α2, or β1 cytoplasmic domains. Only a single candidate, human spermidine/spermine N 1-acetyltransferase (SSAT), was found to specifically interact with α9 and not with α5, α4, α2, or β1 (Fig. 1 a).
Figure 1.

SSAT specifically binds to the α9 cytoplasmic domain. (a) Plasmid DNA isolated from positive colonies from α9 yeast two-hybrid library screening were cotransformed into yeast with pAS2-1–containing integrin α2, α4, α5, α9, or β1 cytoplasmic domain cDNA. 72 h after the transformation, yeast growth on YPD (-Ade/-His/-Leu/-Trp/α-X-gal) plate was shown. (b) GST pull-down assays with the α9, α2, α4, and α5 cytoplasmic domains fused to GST and immobilized on Glutathione Sepharose 4B. Wild-type SSAT protein was produced by in vitro transcription and translation (IVTT) in the presence of [35S]methionine. Bound proteins were separated by 15% SDS-PAGE and analyzed by autoradiography. 25% of IVTT products were also analyzed and showed the same amount of input. (c) Wild-type and full-length SSAT, NH2-terminal deletion SSAT (21–171), COOH-terminal deletion mutants (1–161) and (1–141), and enzymatically inactive (dominant negative [DN]) mutant SSAT (R101A/E152K) proteins were produced by IVTT, pulled down by GST-α9, and analyzed as in b.
To confirm the results of the two-hybrid screen, and to determine precisely which region(s) of SSAT interacted with the α9 cytoplasmic domain, we evaluated the binding of GST fusion proteins containing the α9, α5, α4, and α2 cytoplasmic domains (GST-α9, GST-α5, GST-α4, and GST-α2) to [35S]Met-labeled SSAT generated by in vitro transcription and translation. SSAT specifically bound to GST-α9, but not to α2, α4, α5, or GST alone (Fig. 1 b). To map the binding site in SSAT, we generated a series of NH2-terminal and COOH-terminal truncation mutants and found that the COOH-terminal 30 residues (142aa–171aa), including the catalytic site (Coleman et al., 1996), were not required for binding, but the 20 amino acids at the NH2 terminus were critical (Fig. 1 c). A double mutant that had previously been shown to be catalytically inactive (R101A/E152K; Coleman et al., 1996) could also bind the α9 cytoplasmic domain (Fig. 1 c).
SSAT binds to cell surface α9
To determine whether SSAT associates with α9 in cells, CHO cells stably expressing either wild-type α9 or chimeric α subunits containing the α9 extracellular and transmembrane domains, fused to the cytoplasmic domains of either α4 or α5 (Young et al., 2001), were transfected to stably express SSAT-Myc. Flow cytometry demonstrated that similar levels of α9, α9α4, and α9α5 were expressed on the cell surface (Fig. 2 a). Lysates from these cell lines were immunoprecipitated with the anti-α9β1 antibody Y9A2 (Wang et al., 1996) and immunoblotted with an anti-Myc antibody. Because endogenously and heterologously expressed SSAT have been shown to be rapidly degraded in CHO cells (McCloskey et al., 1999), the concentration of Myc-tagged SSAT was increased by treatment with the spermine analogue N 1,N 11-bis(ethyl)norspermine tetrahydrochloride (BE-3-3-3), which prevents proteosome-mediated SSAT degradation (Coleman and Pegg, 2001). Western blotting of immunoprecipitates with antiserum against the cytoplasmic domain of the integrin β1 subunit confirmed that similar amounts of α9β1 were precipitated from each lysate. SSAT was coimmunoprecipitated with full-length α9β1 in cells treated with BE-3-3-3, but no association with the α9α5 or α9α4 chimeras could be detected (Fig. 2 b).
Figure 2.


Coimmunoprecipitation of α9β1 and SSAT. (a) Flow cytometric evaluation of cell surface expression of α9 integrin on α9-, α9α5-, and α9α4-expressing CHO cells. Open peaks represent fluorescence (FL) of unstained CHO cells, and shaded peaks represent fluorescence of CHO stained with the anti-α9β1 antibody Y9A2. (b) CHO cells transfected with Myc-tagged SSAT were treated with 50 μM BE-3-3-3 and immunoprecipitated by protein G–Sepharose coated with the anti-α9β1 antibody Y9A2. Precipitated proteins (or cell lysates) were separated by SDS-PAGE and detected with anti-Myc mAb or anti–integrin β1 cytoplasmic domain antiserum. (c) Cell surface proteins were labeled with thiol-cleavable biotin, captured with streptavidin agarose, and eluted by reduction. Eluted proteins were then immunoprecipitated and analyzed as in b.
To determine whether there is physical interaction between SSAT and α9 at the cell surface, we labeled cell surface proteins with a thiol-cleavable amine-reactive biotinylation reagent, Sulfo-NHS-SS-Biotin. Biotin-labeled cells were lysed, and labeled proteins were captured by streptavidin agarose. Cell surface proteins were eluted from agarose by reduction and used for coimmunoprecipitation. SSAT was coimmunoprecipitated with full-length cell surface α9β1 from cells treated with BE-3-3-3, but no association with the α9α5 or α9α4 chimeras could be detected (Fig. 2 c). Thus, SSAT associates with α9β1 at the cell surface.
Increasing SSAT protein concentration specifically enhances cell migration mediated by the α9 subunit cytoplasmic domain
To determine whether the interaction between SSAT and the α9 cytoplasmic domain is critical for the enhancement of cell migration, we used CHO cells stably expressing wild-type or chimeric α9 subunits (Young et al., 2001) for cell adhesion and migration assays on an α9β1-specific substrate in the presence of a range of concentrations of BE-3-3-3 to increase endogenous levels of SSAT, and in the presence or absence of the α9β1-blocking antibody Y9A2. The α9β1-specific substrate we used (TNfn3RAA) was a recombinant form of the third fibronectin type III repeat in chicken tenascin C, in which the normal arginine-glycine-aspartic acid sequence (RGD) had been mutated to arginine-alanine-alanine (RAA) to prevent interaction with other integrins (Prieto et al., 1993; Yokosaki et al., 1994, 1998). The expression of SSAT was induced by BE-3-3-3 in a dose-dependent manner (Fig. 3 a). In the absence of BE-3-3-3, both the α9 and the α4 cytoplasmic domains caused similar enhancement of cell migration compared with the cytoplasmic domain of α5. However, BE-3-3-3 caused concentration-dependent enhancement of migration only in cells expressing wild-type α9 (Fig. 3 b). As we have previously reported (Young et al., 2001), all three versions of the α9 subunit supported equivalent adhesion to TNfn3RAA, and adhesion was unaffected by BE-3-3-3 treatment in all three cell lines (Fig. 3 c). As expected, adhesion and migration of all α9-expressing cells were inhibited by the anti-α9β1 antibody Y9A2. Migration on the irrelevant substrate, plasma fibronectin, was similar among all three cell lines and was unaffected by treatment with BE-3-3-3 (Fig. 3 d). Adhesion to fibronectin was also similar among all three cell lines and was unaffected by BE-3-3-3 (unpublished data). These results indicate that the effects of BE-3-3-3 are specific for α9β1-mediated cell migration and are dependent on the presence of the α9 cytoplasmic domain.
Figure 3.
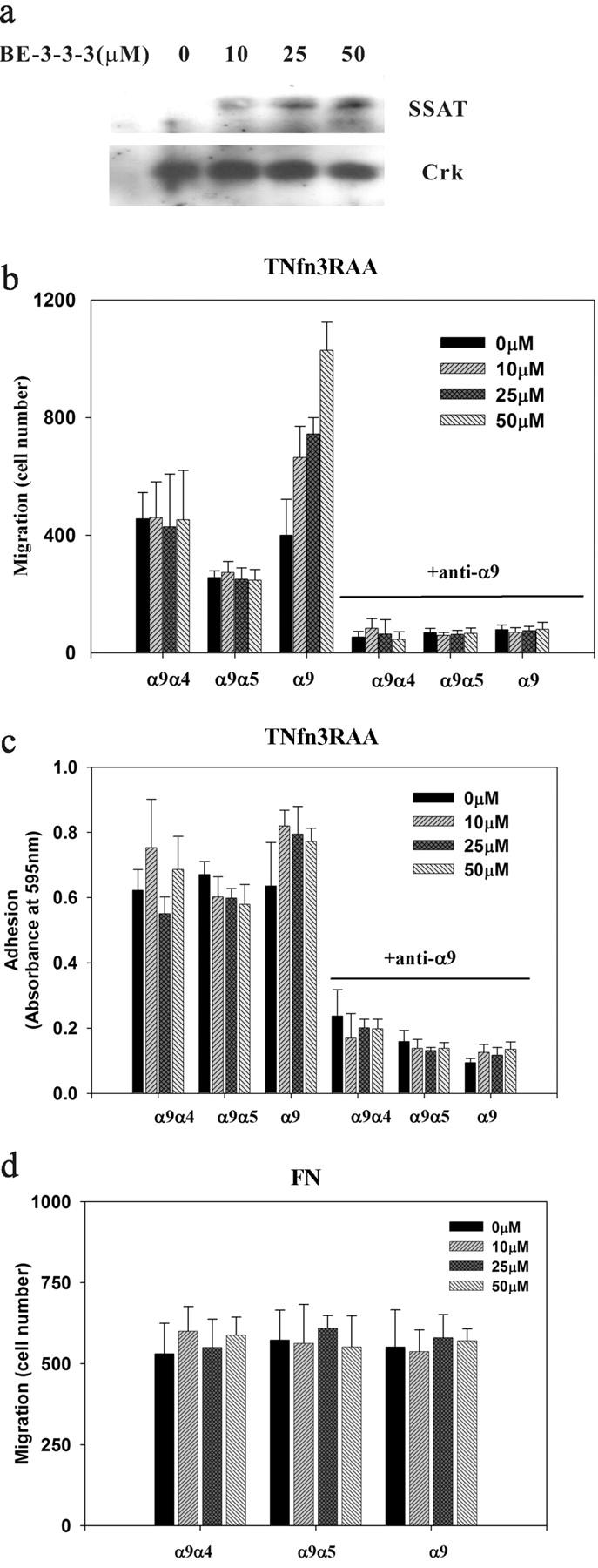
Effects of SSAT induction by BE-3-3-3 on adhesion and migration of α9-expressing CHO cells. (a) CHO cells were treated with BE-3-3-3. SSAT was detected by Western blotting with rabbit polyclonal anti-SSAT. Western blot for Crk was used as a control for equal protein loading. (b and c) α9-, α9α4-, and α9α5-expressing CHO cells were treated with a range of concentrations of BE-3-3-3 for 24 h and were suspended in serum-free medium. (b) Cells were seeded onto membranes coated with 5 μg/ml TNfn3RAA in the top well of 24-well plates in the presence or absence of the anti-α9β1 mAb Y9A2. After a 2-h incubation in the presence of 1% FCS in the bottom well, nonmigrated cells on the top side of the membrane were removed, and migrated cells on the bottom side were fixed, stained, and counted. (c) Cells were added to 96-well plates coated with 5 μg/ml TNfn3RAA in the presence or absence of the anti-α9β1 mAb Y9A2. They were allowed to attach for 60 min, and nonadherent cells were removed by centrifugation. Adherent cells were stained with crystal violet and quantified by measurement of absorbance at 595 nm. (d) Migration of α9-, α9α4-, and α9α5-expressing CHO cells was analyzed on plasma fibronectin (FN; 5 μg/ml). All data represent the means (±SEM) of triplicate experiments.
Confluent CHO cells expressing wild-type α9 or the α9α5 chimera were plated on TNfn3RAA and scratch wounded. Closure of the wound was quantified by phase-contrast microscopy as a measure of directed cell migration. Cells expressing the α9α5 chimera showed markedly slower migration and wound closure than cells expressing wild-type α9. Wound closure was accelerated by BE-3-3-3 in cells expressing wild-type α9, but not in cells expressing the α9α5 chimera (Fig. 4 a). Photomicrographs of the wound edge did not reveal any systematic differences in cell morphology for cells expressing α9 in the presence or absence of treatment with BE-3-3-3 (Fig. 4 b).
Figure 4.

The induction of SSAT by BE-3-3-3 enhances α9-mediated cell migration. (a) CHO cells stably expressing wild-type α9 or the α9α5 chimera were plated onto dishes coated with 5 μg/ml TNfn3RAA, scratch wounded at confluence in the presence or absence of BE-3-3-3, and allowed to migrate into the wound space for 8 h. The ratio of the final wound area to the area immediately after scratching is indicated as percent closure. (b) Cells transfected with wild-type α9 were incubated with or without BE-3-3-3 and fixed 8 h after scratch wounding.
SSAT-induced enhancement of α9-dependent cell migration requires the catalytic activity of SSAT
Endogenous levels of SSAT are below the level of detection by existing anti-SSAT antibodies. To confirm the relationship between the SSAT expression level and α9β1-dependent cell migration, and to determine whether the effects of SSAT depend on catalytic activity, we also performed adhesion and migration assays in CHO cells stably transfected to express either wild-type or catalytically inactive (R101A/E152K mutant) c-Myc–tagged SSAT. In the absence of BE-3-3-3, cells coexpressing wild-type α9 and exogenous wild-type SSAT demonstrated enhancement of cell migration on TNfn3RAA, compared with cells expressing wild-type α9 alone (Fig. 5 a); cell migration was further enhanced by treatment with BE-3-3-3. In contrast, neither treatment with BE-3-3-3 nor overexpression of wild-type SSAT had any effect on migration in cells expressing the chimeric subunits composed of the extracellular and transmembrane domains of α9 fused to the cytoplasmic domains of α4 (α9α4) or α5 (α9α5). The relative SSAT protein concentrations in mock- and SSAT-transfected cells in the presence and absence of treatment with BE-3-3-3–treated cells are shown in Fig. 5 d. Expression of catalytically inactive SSAT (Coleman et al., 1996) did not affect the baseline enhancement of cell migration in cells expressing wild-type α9, perhaps because the level of expression was not adequate to effectively compete with that of endogenous SSAT. However, catalytically inactive SSAT did not enhance cell migration, and it prevented even the increase in migration caused by BE-3-3-3 in cells expressing wild-type α9 alone, a result consistent with previous reports that this construct can function as a dominant negative inhibitor of wild-type SSAT (Coleman et al., 1996). Similar to the previous experimental results, overexpression of either form of SSAT had no effect on cell adhesion on TNfn3RAA or on migration on plasma fibronectin in the presence or absence of BE-3-3-3 (Fig. 5, b and c). These results confirm the specificity of SSAT for enhancement of α9-dependent cell migration and suggest that this effect depends on the catalytic activity of SSAT.
Figure 5.

Adhesion and migration of α9-expressing CHO cells transfected with SSAT. α9-expressing CHO cells were transfected with empty plasmid (Mock), wild-type SSAT (WT), or enzymatically inactive mutant SSAT (dominant negative [DN]). Cells were treated with or without 50 μM BE-3-3-3 for 24 h. (a) Cell migration on 5 μg/ml TNfn3RAA, (b) cell adhesion on 5 μg/ml TNfn3RAA, and (c) cell migration on 5 μg/ml fibronectin were assayed and analyzed as described in the Fig. 3 legend. All data represent the means (±SEM) of triplicate experiments. (d) Western blot showing level of SSAT protein detection in mock- and SSAT-transfected α9-expressing CHO cells in the presence or absence of treatment with BE-3-3-3. Western blot for Crk was used as a control for equal protein loading.
Knockdown of endogenous SSAT specifically inhibits cell migration mediated by the α9 subunit cytoplasmic domain
To further investigate the importance of SSAT for α9β1-dependent enhancement of cell migration, we used RNA interference to suppress the expression of SSAT in α9-expressing mouse embryonic fibroblasts (MEFs). MEFs were chosen because the availability of complete genomic sequence data in the mouse allows improved design of specific small interfering RNA (siRNA), and because mouse and human SSAT are virtually identical. Four 21-nucleotide siRNA segments directed against the middle (siRNA-1 and siRNA-3) or the 3′ end (siRNA-2 and siRNA-4) of the SSAT transcript were transfected into MEFs that had been stably transfected to express either wild-type α9, α9α4, or α9α5, and the cells were examined 24 h later (Young et al., 2001). Real-time PCR showed that SSAT mRNA was reduced by >75% by siRNA-2, siRNA-3, and siRNA-4, compared with untransfected cells or cells transfected with siRNA-1 (Fig. 6 a). Because endogenous SSAT cannot be detected by a polyclonal antibody against SSAT, BE-3-3-3 was used to induce the expression of SSAT 24 h after siRNA transfection. The SSAT protein level was decreased by siRNA-2, siRNA-3, and siRNA-4, compared with untransfected cells or cells transfected with siRNA-1 (Fig. 6 b). Transfection with siRNA-2, siRNA-3, and siRNA-4 decreased the cell migration on TNfn3RAA of cells expressing wild-type α9 to the level seen for cells transfected with the α9α5 chimera, but had no effect on the migration of cells expressing α9α4 or α9α5 (Fig. 6 c). The enhancement of α9-dependent cell migration by BE-3-3-3 was also decreased by siRNA transfection (Fig. 6 d). None of these treatments had any effect on cell adhesion on TNfn3RAA or on cell migration on fibronectin in any cell type (unpublished data). These results further support the concept that endogenous levels of SSAT play an important role in α9-dependent enhancement of cell migration.
Figure 6.
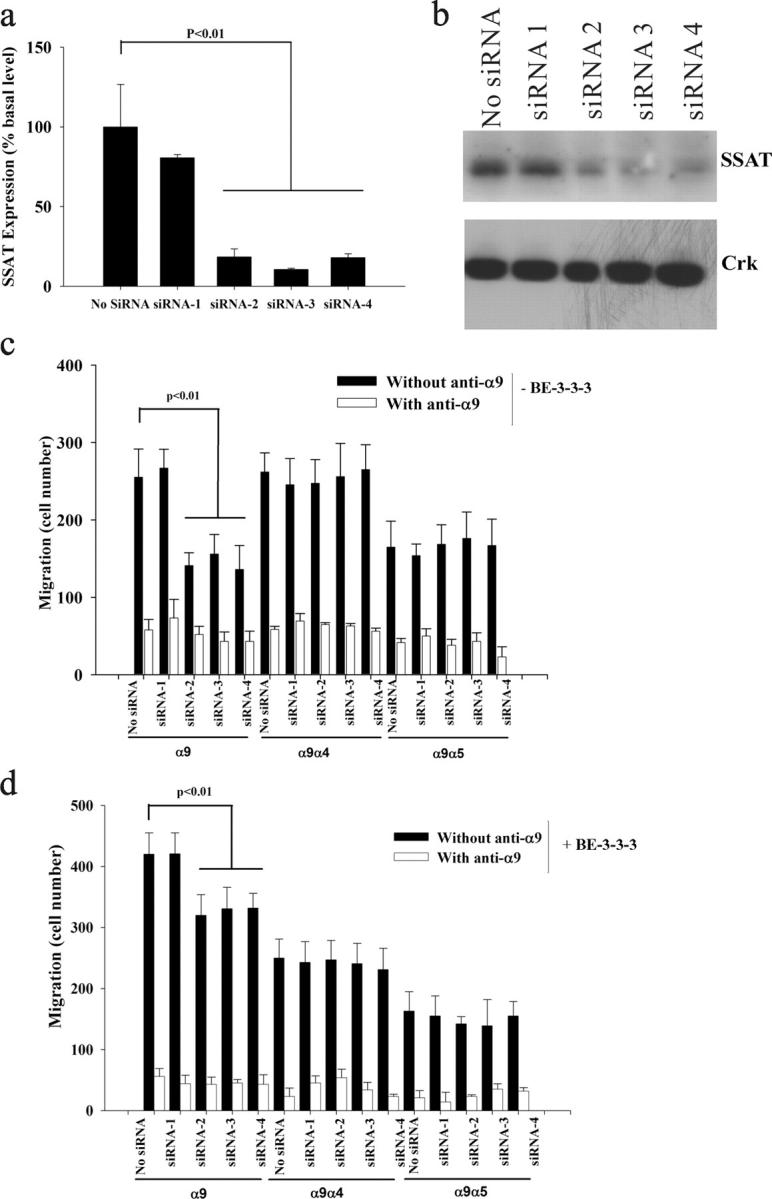
Effects of siRNA knockdown on α9-dependent enhancement of cell migration. (a) α9-expressing MEFs were transfected with siRNA-1, siRNA-2, siRNA-3, siRNA-4, and no siRNA control in 6-well plates. 24 h later, SSAT mRNA was assayed by quantitative PCR and expressed as percent basal level (normalized to no siRNA). (b) 50 μM BE-3-3-3 was used to induce the expression of SSAT 24 h after siRNA transfection, for 24 h. SSAT protein was detected by Western blot. Western blot for Crk was used as a control for equal protein loading. (c and d) α9-, α9α4-, and α9α5-expressing MEFs were transfected with siRNA-1, siRNA-2, siRNA-3, siRNA-4, and no siRNA; 24 h later, cells suspended in serum-free medium were used for migration assays without BE-3-3-3 (c) and with 50 μM BE-3-3-3 (d). The cells were assayed and counted as described in the Fig. 3 legend.
The target sequences of the functional siRNA-3 and siRNA-4 are the same in mouse and hamster SSAT cDNA. Therefore, we used these two siRNAs to inhibit the expression of endogenous SSAT in CHO cells. Both mRNA and protein concentrations of hamster SSAT were substantially reduced by each siRNA (Fig. 7, a and c). However, the sequence of human SSAT is different from the mouse and hamster sequences in the regions targeted by each of these siRNAs. Thus, neither siRNA-3 nor siRNA-4 affects the mRNA or protein concentration in cells transfected to express human SSAT (Fig. 7, b and c). In CHO cells that only express endogenous hamster SSAT, as in MEFs, the enhancement of α9-dependent cell migration by BE-3-3-3 was decreased by siRNA transfection (Fig. 7 d). However, α9-dependent enhancement of cell migration was not inhibited by siRNA-targeting mouse (and hamster) SSAT in CHO cells overexpressing human SSAT (Fig. 7 d). This reconstitution of α9-dependent enhancement of cell migration in siRNA-treated CHO cells with human SSAT that is resistant to these siRNAs suggests that the effects of the siRNAs tested are likely specifically due to the knockdown of SSAT.
Figure 7.

Rescue of siRNA knockdown of hamster SSAT by overexpression of human wild-type SSAT cDNA in CHO cells. (a and b) α9-expressing CHO cells stably expressing either empty vector or wild-type human SSAT (SSAT(WT)) were transfected with siRNA-1 (nonfunctional), siRNA-3, and siRNA-4 in 6-well plates. (a) 24 h later, hamster SSAT mRNA was assayed by quantitative PCR and expressed as percent basal level (normalized to siRNA-1). (b) Human SSAT mRNA was assayed by quantitative PCR and expressed as percent basal level (normalized to siRNA-1). (c) BE-3-3-3 was used to stabilize SSAT 24 h after transfection with nonfunctional siRNA (1) or two functional siRNAs (3 and 4) in CHO cells stably transfected with empty plasmid (CHO-α9-Mock) or wild-type human SSAT (CHO-α9-SSAT(WT)). SSAT protein was detected by Western blot. Western blot for Crk was used as a control for equal protein loading. (d) α9- or α9α5-expressing CHO cells with or without human SSAT expression were transfected with siRNA-1, siRNA-3, and siRNA-4; 24 h later, cells were suspended in serum-free medium and used for migration assays without BE-3-3-3 (0 μM) or with BE-3-3-3 (50 μM), as described in the Fig. 3 legend.
SSAT specifically enhances α9-mediated cell migration in HMVEC that express endogenous α9β1
To further investigate the importance of SSAT for endogenous α9β1-dependent enhancement of cell migration in untransfected cells, we tested the expression of integrin α9β1 on the cell surface of human microvascular endothelial cells (HMVEC) by flow cytometry (Fig. 8 a). The expression of SSAT in these cells was induced by BE-3-3-3 and inhibited by siRNA directed against human SSAT. Two siRNAs were evaluated, and only one (siRNA-A) consistently inhibited SSAT mRNA concentrations in untreated cells (Fig. 8 b) and dramatically reduced SSAT protein levels in BE-3-3-3–treated cells (Fig. 8 c). Induction of SSAT increased HMVEC migration on the α9β1-specific ligand, TNfn3RAA. Inhibition of SSAT by functional siRNA specifically decreased cell migration on TNfn3RAA (Fig. 8 d). Neither BE-3-3-3 nor siRNA against human SSAT had any effect on cell migration on the α9β1-irrelevant ligand, plasma fibronectin (Fig. 8 e). These results demonstrate that the expression level of SSAT specifically affects α9β1-dependent cell migration in cells that endogenously express α9β1.
Figure 8.

Effects of siRNA knockdown and BE-3-3-3 on endogenous α9-dependent HMVEC migration. (a) Flow cytometric evaluation of cell surface expression of α9 integrin on HMVEC. Open peaks represent unstained cells, and shaded peaks represent cells stained with the anti-α9β1 antibody Y9A2. (b) HMVEC were transfected with siRNA-A, siRNA-B, and no siRNA control in 6-well plates. 24 h later, SSAT mRNA was assayed by quantitative PCR and expressed as percent basal levels (normalized to no siRNA). (c) 50 μM BE-3-3-3 was used to induce the expression of SSAT 24 h after siRNA transfection, for 24 h. SSAT protein concentration was assessed by Western blot. Western blot for Crk was used as a control for equal protein loading. (d and e) HMVEC were transfected with siRNA-A, siRNA-B, and no siRNA, and 24 h later, cells were or were not treated with BE-3-3-3 (50 μM) for 24 h. After treatment with BE-3-3-3, cells were suspended in serum-free medium and were used for migration assays on 5 μg/ml TNfn3RAA (d) or on 5 μg/ml fibronectin (FN; e), as described in the Fig. 3 legend. All data represent the means (±SEM) of triplicate experiments.
Discussion
We have previously demonstrated that the integrin α9β1 mediates enhancement of cell migration (Taooka et al., 1999; Young et al., 2001). As with the closely related integrin α4β1, this mediation depends on sequences within the α subunit cytoplasmic domain. However, in contrast with the mechanism used by α4, α9-dependent enhancement of migration does not require the adapter protein paxillin (Young et al., 2001). We now show that the acetyltransferase SSAT specifically and directly binds to the α9 cytoplasmic domain, that increased levels of SSAT further accelerate cell migration only in cells expressing wild-type α9 and only on α9β1-specific substrate, and that decreased levels of SSAT only decrease cell migration under the same circumstances. Furthermore, these effects of SSAT appear to require catalytic activity. Together, these results strongly suggest that catalytically active SSAT is a critical component of the pathway by which the integrin α9β1 enhances cell migration, and that this pathway is highly specific for this integrin.
As we have previously reported (Young et al., 2001), α9β1 (containing the complete α9 cytoplasmic domain) is diffusely expressed on the cell surface and does not localize to focal adhesion structures, even in cells plated on specific ligands (e.g., TNfn3RAA). Although we are unable to detect endogenous SSAT with available reagents, heterologously expressed Myc- or RFP-tagged SSAT is diffusely expressed throughout the cytoplasm and nucleus (unpublished data). Thus, although both proteins can be seen along the cell membrane, we have not been able to infer “colocalization” from immunostaining experiments. Nonetheless, the broad expression pattern of each protein at least suggests that it is feasible for them to make contact in intact cells. In vivo, SSAT is ubiquitously expressed, whereas α9β1 expression is restricted to subsets of epithelial cells, muscle cells (Palmer et al., 1993), leukocytes (Taooka et al., 1999), and endothelial cells. Thus, SSAT is clearly expressed in cells that do not express α9β1, but all α9β1-expressing cells are likely to express SSAT.
Over the past several years, much has been learned about the common pathways by which ligated integrins activate cytoplasmic signals (outside-in signaling) and by which cytoplasmic signals affect the affinity and avidity of integrins (inside-out signaling). However, the divergent phenotypes of mice expressing null mutations of individual integrin subunits, and the divergent effects of specific integrins on cell behavior, make it clear that the signaling pathways used by integrins can be highly specific. In this paper, we describe a novel example of integrin specificity that appears to be critical for the best-characterized cellular function of the α9β1 integrin, acceleration of cell migration.
The oxidative catabolism of the higher order polyamines, spermidine and spermine, is accomplished by the concerted action of two different enzymes, SSAT and polyamine oxidase (PAO). Cytosolic SSAT N1 acetylates both spermidine and spermine, which then serve as substrates for peroxisomal PAO (Holtta, 1977). Because PAO strongly prefers acetylated polyamines to unmodified polyamines as its substrates, SSAT is generally considered the rate-controlling enzyme in the back conversion of the higher order polyamines, spermidine and spermine, to the lower order polyamine, putrescine (Casero and Pegg, 1993). Thus far, spermine and spermidine are the only physiological substrates that have been identified for SSAT. Therefore, it is tempting to speculate that SSAT enhances α9β1-dependent cell migration by local effects on polyamines. Indeed, previous studies have implicated polyamines in the regulation of cell migration, although polyamines' precise mechanisms of action in this process have been obscured (Johnson et al., 2002; Ray et al., 2003). Depletion of polyamines by treatment with DFMO, a drug that blocks the enzyme ornithine decarboxylase responsible for conversion of ornithine to putrescine, has been shown to globally inhibit cell migration and reduce cellular activity of the small GTPase rac1, a well-established mediator of cell migration (Johnson et al., 2002; Ray et al., 2003). Thus, it is conceivable that SSAT-mediated modulation of local polyamine concentrations enhances local rac1 activity to enhance cell migration. However, an equally plausible hypothesis is that SSAT is involved in acetylating other unknown substrates whose acetylation affects their role in enhancing cell migration.
The α9β1 integrin is expressed on several cells for which migration is critical for function. For example, this integrin is expressed on neutrophils and monocytes and has been shown to play an important role in neutrophil emigration across activated endothelial cells (Taooka et al., 1999). Transendothelial migration appears to depend on binding of α9β1 to VCAM-1, an inducible ligand expressed on endothelial cells at sites of infection, tissue injury, and inflammation. Several other α9β1 ligands have been identified; many of them, including tenascin C (Yokosaki et al., 1994), osteopontin (Smith et al., 1996; Yokosaki et al., 1999), tissue fibronectin (Liao et al., 2002), and the coagulation proteins von Willebrand factor and factor XIII (Takahashi et al., 2000), are highly enriched in injured and inflamed tissues. Thus, α9β1 is likely to play an important role in both leukocyte emigration from the vasculature and leukocyte migration through the extravascular space at sites of injury, infection, or chronic inflammation. Furthermore, mice expressing a null mutation of the α9 subunit demonstrate defects in development of lymphatic vessels (Huang et al., 2000), suggesting a possible role for α9β1 in the migration of lymphatic endothelial cells required for normal lymphatic development. Thus, our identification of SSAT as a critical effector molecule in α9β1-dependent cell migration provides a new target for intervention in disorders that are characterized by excessive leukocyte emigration as well as for modulation of the migration of other α9β1-expressing cells.
Materials and methods
Reagents
Mouse mAb (Y9A2) raised against human α9β1 (Wang et al., 1996) was prepared as described previously. mAb 9B11 against the Myc tag was purchased from Cell Signaling Technology. BE-3-3-3 was purchased from Tocris Cookson Inc. Polyclonal antibody against integrin β1 was purchased from CHEMICON International. Anti-Crk was purchased from Upstate Biotechnology. Polyclonal anti-SSAT antibody was prepared as described previously (Casero et al., 1991).
Yeast two-hybrid screening
The yeast MATCHMAKER two-hybrid system (CLONTECH Laboratories, Inc.) was used for a library screen. In brief, α9, α2, α4, α5, and β1 cytoplasmic domains (aa 2883–3053 for α9) were amplified by PCR and cloned into the Gal4-DNA–binding domain (Gal4-DB) vector (pAS2-1) to screen a library of human leukocyte cDNAs fused to the Gal4-DNA activation domain (Gal4-DA) vector (pACT2). AH109 were used as the host strain. Yeast two-hybrid library screening was performed according to the MATCHMAKER manual on plates lacking adenine, histidine, leucine, and tryptophan. Positive interactions were confirmed by β-galactosidase expression on α-X-Gal.
Expression and purification of GST fusion proteins
GST fusion proteins containing the α9, α2, α4, and α5 cytoplasmic domains were generated by cloning the PCR-amplified sequences into the bacterial expression plasmid pGEX-4T-1 (Amersham Biosciences) and transforming BL21-Gold bacteria (Stratagene). Transformed cells were cultured in 300 ml of YT medium in the presence of 0.1 mM isopropyl-β-d-1-thiogalactopyranoside (Sigma-Aldrich), and GST fusion proteins were purified on glutathione-Sepharose by B-PER GST Fusion Protein Purification Kit (Pierce Chemical Co.).
In vitro transcription and translation and protein binding assays
In vitro transcription and translation experiments were done with the T7 RNA polymerase transcription/translation systems, using rabbit reticulocyte lysate (Promega) and l-[35S]methionine (AG1094; Amersham Biosciences) to produce 35S-labeled proteins according to the manufacturer's recommendations. Plasmids containing full-length, truncated, or mutant forms of SSAT in pBluescript SK were used as templates. 0.8 nmol of the appropriate GST fusion protein was mixed with 40 μl of the in vitro translation reaction above and 50 μl of glutathione-Sepharose beads, and then incubated in binding buffer (PBS with 1% Triton X-100) at 4°C for 2 h. The beads were washed five times in 1 ml of binding buffer and resuspended in 25 μl Laemmli sample buffer and heated at 95°C for 5 min. Bound proteins were separated by SDS-PAGE (15%) under reducing conditions. Gels were fixed in 50% methanol and 10% acetic acid for 30 min, soaked in a fluorographic reagent (Amplify; Amersham Biosciences), dried, and developed by autoradiography.
Generation of stable cell lines
Integrin α9-, α9α4-, and α9α5-expressing CHO cells and MEFs were generated in our lab previously (Young et al., 2001). Transfected cells were analyzed for expression of α9, α9α4, and α9α5 integrins by flow cytometry with Y9A2. Fluorescence-activated cell sorting was performed to isolate heterogeneous populations of cells expressing high levels of α9β1 integrin on their cell surface (Yokosaki et al., 1998). All cell lines continuously expressed high surface levels of α9β1 as determined by flow cytometry with Y9A2.
Full-length SSAT cDNAs were amplified by forward primer (GCAGCGGATCCCCGCCATGGCTAAATTCGTGATCCGCCCAGCCACTGCGCCGACT) and reverse primer (CGTACCTCGAGTCAATTCAGATCCTCTTCTGAGATGAGTTTTTGTTCCTCCTCTGTTGCCATTTTTA) to add a c-Myc tag to the COOH terminus using either wild-type SSAT or the double mutant (R101A/E152K) in pBluescript SK as template (Coleman et al., 1995). The PCR products were digested by EcoRI and XhoI and cloned into pcDNA3.1/Hygro(+) (Invitrogen). Expression constructs were confirmed by restriction digestion and sequencing. Integrin α9-, α9α4-, and α9α5-expressing CHO cells were used for transfection. Stable clones were obtained by dilution subcloning and characterized by Western blotting. These double transfectants were maintained in medium containing 250 μg/ml G418 and 50 μg/ml hygromycin.
Coimmunoprecipitation and Western blot analysis
CHO cell lines expressing different chimeric α9β1 integrins were lysed on ice for 30 min in an immunoprecipitation buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM EDTA, 10 mM benzamidine-HCI, 0.02% sodium azide, 1% Triton X-100, 0.05% Tween 20, 2 mM PMSF, 5 μg/ml aprotinin, and 5 μg/ml leupeptin). Lysates were clarified by centrifuging at 16,000 g for 20 min at 4°C, and then were incubated with protein G–Sepharose coated with Y9A2 at 4°C overnight. The beads were washed with the same buffer five times, and precipitated polypeptides were extracted in Laemmli sample buffer, separated by SDS-PAGE under reducing conditions, probed with anti-Myc mAb, and detected by ECL (Amersham Biosciences).
Cell surface protein biotinylation and coimmunoprecipitation
To determine whether there is interaction between SSAT and cell surface α9β1, labeling of surface proteins was performed using the lysine-directed, membrane-impermeant biotinylating reagent sulfo-NHS-SS-biotin (Pierce Chemical Co.). Cells were washed four times with PBS at 4°C and incubated with 1.5 mg/ml sulfo-NHS-SS-biotin in PBS for 30 min at 4°C. After the sulfo-NHS-SS-biotin incubation, cells were washed twice with 100 mM glycine in PBS at 4°C and incubated for 20 min at 4°C in glycine/PBS. The glycine buffer was removed, lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM EDTA, 1% Triton X-100, 0.05% Tween 20, 1× protease inhibitor cocktail) was applied, and the mixture was shaken for 30 min at 4°C. The supernatants were cleared of insoluble material by pelleting at 15,000 rpm for 20 min at 4°C. ImmunoPure-immobilized streptavidin beads (Pierce Chemical Co.) were washed three times with 100 mM glycine/PBS and four times with lysis buffer, and finally were resuspended in lysis buffer. 210 μl of bead/lysis buffer slurry was added to 750 μl of supernatant, and the mixture was gently mixed for 1 h at room temperature. After incubation, the streptavidin beads were washed four times with lysis buffer and the biotinylated proteins were eluted from the beads with lysis buffer containing 50 mM DTT. Eluted samples were used for coimmunoprecipitation as described in the previous paragraph.
siRNA construction and transfection
siRNAs to mouse SSAT were designed with 3′-overhanging thymidine dimers as described previously (Elbashir et al., 2001). Target sequences were aligned to the mouse genome database in a BLAST search (www.ncbi.nlm.nih.gov/blast) to eliminate those with significant similarity to other genes. Web-based siRNA design software from Ambion (www.ambion.com/techlib/misc/siRNA_finder.html) was used for selecting siRNA sequences. Four siRNAs (siRNA-1, UCUAAGCCAGGUUGCCAUGTT; siRNA-2, AAGAAGAGGUGCUUCGGAUTT; siRNA-3, CACCCCUUCUACCACUGCCTT; and siRNA-4, AAAUGGCAGCAGAGG-AGUGTT) for mouse SSAT and two siRNAs (siRNA-A, UGGCUAAAUUCGUGAUCCGTT; and siRNA-B, GAUGGUUUUGGAGAGCACCTT) for human SSAT were synthesized (Proligo) and used for transfection with the siPORT Amine Transfection Agent (Ambion). In brief, MEF cells or HMVEC were grown to 50–70% confluence in complete medium without antibiotics in 6-well plates. Cells were washed with serum-free medium. 10 μl SiPORT Amine was added to Opti-MEM I (Invitrogen) medium for a final complexing volume of 200 μl, and the mixture was vortexed and then incubated at room temperature for 10–30 min. 1.25 μl of 20 μM siRNA was added, mixed gently, and incubated for 15 min. This mixture was added dropwise to cells in a volume of 800 μl Opti-MEM I.
Flow cytometry
Cultured cells were harvested by trypsinization and rinsed with PBS. Nonspecific binding was blocked with normal goat serum at 4°C for 10 min. Cells were then incubated with a primary antibody (Y9A2) for 20 min at 4°C, followed by a secondary goat anti–mouse antibody conjugated with phycoerythrin (CHEMICON International). Between incubations, cells were washed twice with PBS. The stained cells were resuspended in 100 μl PBS, and fluorescence was quantified on 5,000 cells with a FACScan flow cytometer (Becton Dickinson).
Cell adhesion assays
The wells of nontissue culture–treated 96-well microliter plates (Nunc) were coated by incubation with 100 μl TNfn3RAA for 1 h at 37°C. After incubation, wells were washed with PBS and then blocked with 1% BSA in DME at 37°C for 30 min. Control wells were filled with 1% BSA in DME. The cells were detached with 2.5 ml of trypsin solution (Sigma-Aldrich) followed by 2.5 ml of trypsin-neutralizing solution (Sigma-Aldrich), washed once in DME, and resuspended in DME at 5 × 105 cells/ml in the presence or absence of 50 μg/ml Y9A2 for 20 min at 4°C before plating. Plates were centrifuged (top side up) at 10 g for 5 min before incubation for 1 h at 37°C in humidified 5% CO2. Nonadherent cells were then removed by centrifugation (top side down) at 48 g for 5 min. Attached cells were fixed and stained in 40 μl of a 1% formaldehyde, 0.5% crystal violet, and 20% methanol solution for 30 min; then, the wells were washed three times with PBS. The relative number of cells in each well was evaluated after solubilization in 40 μl of 2% Triton X-100 by measuring the absorbance at 595 nm in a microplate reader (Bio-Rad Laboratories). All determinations were performed in triplicate, and the data represent the means ± SEM for a minimum of three experiments.
Cell migration assays
For chemotactic migration assays, 24-well Transwell plates (Costar) were used. The lower sides of the Transwell filters (6.5-mm diam, pore size 8.0 μm) were coated with TNfn3RAA dissolved in 250 μl DME for 60 min at 37°C. After incubation with TNfn3RAA, the filters were washed by adding 100 μl PBS to the top well and 500 μl PBS to the bottom well. After being washed twice, the filters were blocked with 1% BSA in DME for 30 min and washed again in PBS. Cells were detached as described in the previous paragraph and resuspended in DME at 5 × 105 cells/ml. Migration and adhesion assays were performed simultaneously, and the cells from the same dishes were used for both assays. Cells were incubated for 20 min on ice with or without 50 μg/ml of Y9A2, and then 100 μl (50,000 cells) were loaded in each chamber. Each chamber was inserted in a well containing 600 μl DME supplemented with 1% FCS to serve as a chemoattractant and incubated at 37°C in humidified 5% CO2 for 2 h (CHO cells) or 3 h (MEF cells). The medium was then aspirated and the filters were washed once with PBS. Cells on the bottom of the filters were fixed for 20 min in 500 μl of DifQuik fixative (Fisher Scientific), and the nonmigrated cells on the top of the filter were gently removed. Filters were allowed to completely dry, stained with DifQuik, washed in running distilled H2O, and allowed to destain in distilled H2O for 1 h. Filters were air dried (for ≥3 h), removed from the chamber with a scalpel, and mounted on glass slides with a Permamount/xylene solution (Fisher Scientific). Migrated cells were counted under a 25× objective with the use of a gridded eyepiece (reticule). 10 high-powered fields (HPF) per slide were counted, the average was taken, and the number of migrated cells was expressed as migrated cells per 10 HPF. The data represent the means ± SEM for a minimum of three experiments.
Scratch wound assay
Tissue culture dishes were coated with 5 μg/ml GST-TNfn3RAA overnight at 4°C. Cells were suspended with trypsin/EDTA and plated onto coated dishes at a density of 106 cells/35-mm dish. After 2 h, cell cultures were scratched with a single pass of a pipette tip and incubated at 37°C for 8 h. In some cases, culture media were supplemented with 50 μM BE-3-3-3. Cultures were washed twice with PBS, fixed in 3.7% formaldehyde for 10 min, and photographed. Cells were viewed on an inverted microscope (model PE300; Nikon) using Nikon objective lenses (10×/0.30; 20×/0.45). Images were processed by Openlab 2.2.5 software (Improvision) and an ICCD digital camera (model C4742-95; Hamamatsu Photonics) at room temperature, and were arranged and labeled using Adobe Photoshop.
Acknowledgments
The authors thank Nicholas Vlahakis and Gisli Jenkins for helpful suggestions on the manuscript.
This work was supported by grants HL56385, HL64353, HL53949, and HL66600 from the National Heart, Lung, and Blood Institute (to D. Sheppard) and by grant GM26290 from the National Institute of General Medical Sciences (to A.E. Pegg).
Abbreviations used in this paper: BE-3-3-3, N 1,N 11-bis(ethyl)norspermine tetrahydrochloride; HMVEC, human microvascular endothelial cells; MEF, mouse embryonic fibroblast; PAO, polyamine oxidase; siRNA, small interfering RNA; SSAT, spermidine/spermine N 1-acetyltransferase.
References
- Casero, R.A., Jr., and A.E. Pegg. 1993. Spermidine/spermine N1-acetyltransferase–the turning point in polyamine metabolism. FASEB J. 7:653–661. [PubMed] [Google Scholar]
- Casero, R.A., Jr., P. Celano, S.J. Ervin, N.B. Applegren, L. Wiest, and A.E. Pegg. 1991. Isolation and characterization of a cDNA clone that codes for human spermidine/spermine N1-acetyltransferase. J. Biol. Chem. 266:810–814. [PubMed] [Google Scholar]
- Chan, B.M., P.D. Kassner, J.A. Schiro, H.R. Byers, T.S. Kupper, and M.E. Hemler. 1992. Distinct cellular functions mediated by different VLA integrin α subunit cytoplasmic domains. Cell. 68:1051–1060. [DOI] [PubMed] [Google Scholar]
- Coleman, C.S., and A.E. Pegg. 2001. Polyamine analogues inhibit the ubiquitination of spermidine/spermine N1-acetyltransferase and prevent its targeting to the proteasome for degradation. Biochem. J. 358:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, C.S., H. Huang, and A.E. Pegg. 1995. Role of the carboxyl terminal MATEE sequence of spermidine/spermine N1-acetyltransferase in the activity and stabilization by the polyamine analog N1,N12-bis(ethyl)spermine. Biochemistry. 34:13423–13430. [DOI] [PubMed] [Google Scholar]
- Coleman, C.S., H. Huang, and A.E. Pegg. 1996. Structure and critical residues at the active site of spermidine/spermine-N1-acetyltransferase. Biochem. J. 316:697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir, S.M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 411:494–498. [DOI] [PubMed] [Google Scholar]
- Goldfinger, L.E., J. Han, W.B. Kiosses, A.K. Howe, and M.H. Ginsberg. 2003. Spatial restriction of α4 integrin phosphorylation regulates lamellipodial stability and α4β1-dependent cell migration. J. Cell Biol. 162:731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J., S. Liu, D.M. Rose, D.D. Schlaepfer, H. McDonald, and M.H. Ginsberg. 2001. Phosphorylation of the integrin α4 cytoplasmic domain regulates paxillin binding. J. Biol. Chem. 276:40903–40909. [DOI] [PubMed] [Google Scholar]
- Holtta, E. 1977. Oxidation of spermidine and spermine in rat liver: purification and properties of polyamine oxidase. Biochemistry. 16:91–100. [DOI] [PubMed] [Google Scholar]
- Huang, X.Z., J.F. Wu, R. Ferrando, J.H. Lee, Y.L. Wang, R.V. Farese Jr., and D. Sheppard. 2000. Fatal bilateral chylothorax in mice lacking the integrin α9β1. Mol. Cell. Biol. 20:5208–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, D.A., C. Fields, A. Fallon, M.E. Fitzgerald, M.J. Viar, and L.R. Johnson. 2002. Polyamine-dependent migration of retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 43:1228–1233. [PubMed] [Google Scholar]
- Kassner, P.D., and M.E. Hemler. 1993. Interchangeable α chain cytoplasmic domains play a positive role in control of cell adhesion mediated by VLA-4, a β1 integrin. J. Exp. Med. 178:649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner, P.D., R. Alon, T.A. Springer, and M.E. Hemler. 1995. Specialized functional properties of the integrin α4 cytoplasmic domain. Mol. Biol. Cell. 6:661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger, D.A., and A.F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell. 84:359–369. [DOI] [PubMed] [Google Scholar]
- Liao, Y.F., P.J. Gotwals, V.E. Koteliansky, D. Sheppard, and L. Van De Water. 2002. The EIIIA segment of fibronectin is a ligand for integrins α9β1 and α4β1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J. Biol. Chem. 277:14467–14474. [DOI] [PubMed] [Google Scholar]
- Liu, S., S.M. Thomas, D.G. Woodside, D.M. Rose, W.B. Kiosses, M. Pfaff, and M.H. Ginsberg. 1999. Binding of paxillin to α4 integrins modifies integrin-dependent biological responses. Nature. 402:676–681. [DOI] [PubMed] [Google Scholar]
- McCloskey, D.E., C.S. Coleman, and A.E. Pegg. 1999. Properties and regulation of human spermidine/spermine N1-acetyltransferase stably expressed in Chinese hamster ovary cells. J. Biol. Chem. 274:6175–6182. [DOI] [PubMed] [Google Scholar]
- Palmer, E.L., C. Ruegg, R. Ferrando, R. Pytela, and D. Sheppard. 1993. Sequence and tissue distribution of the integrin α9 subunit, a novel partner of β1 that is widely distributed in epithelia and muscle. J. Cell Biol. 123:1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto, A.L., G.M. Edelman, and K.L. Crossin. 1993. Multiple integrins mediate cell attachment to cytotactin/tenascin. Proc. Natl. Acad. Sci. USA. 90:10154–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, R.M., S.A. McCormack, C. Covington, M.J. Viar, Y. Zheng, and L.R. Johnson. 2003. The requirement for polyamines for intestinal epithelial cell migration is mediated through Rac1. J. Biol. Chem. 278:13039–13046. [DOI] [PubMed] [Google Scholar]
- Smith, L.L., H.-K. Cheung, L.E. Ling, J. Chen, D. Sheppard, R. Pytela, and C.M. Giachelli. 1996. Osteopontin N-terminal domain contains a cryptic adhesive sequence recognized by α9β1 integrin. J. Biol. Chem. 271:28485–28491. [PubMed] [Google Scholar]
- Takahashi, H., T. Isobe, S. Horibe, J. Takagi, Y. Yokosaki, D. Sheppard, and Y. Saito. 2000. Tissue transglutaminase, coagulation factor XIII, and the pro-polypeptide of von Willebrand factor are all ligands for the integrins α9β1 and α4β1. J. Biol. Chem. 275:23589–23595. [DOI] [PubMed] [Google Scholar]
- Taooka, Y., J. Chen, T. Yednock, and D. Sheppard. 1999. The integrin α9β1 mediates adhesion to activated endothelial cells and transendothelial neutrophil migration through interaction with vascular cell adhesion molecule-1. J. Cell Biol. 145:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, A., Y. Yokosaki, R. Ferrando, J. Balmes, and D. Sheppard. 1996. Differential regulation of airway epithelial integrins by growth factors. Am. J. Respir. Cell Mol. Biol. 15:664–672. [DOI] [PubMed] [Google Scholar]
- Yokosaki, Y., E.L. Palmer, A.L. Prieto, K.L. Crossin, M.A. Bourdon, R. Pytela, and D. Sheppard. 1994. The integrin α9β1 mediates cell attachment to a non-RGD site in the third fibronectin type III repeat of tenascin. J. Biol. Chem. 269:26691–26696. [PubMed] [Google Scholar]
- Yokosaki, Y., N. Matsuura, S. Higashiyama, I. Murakami, M. Obara, M. Yamakido, N. Shigeto, J. Chen, and D. Sheppard. 1998. Identification of the ligand binding site for the integrin alpha9 beta1 in the third fibronectin type III repeat of tenascin-C. J. Biol. Chem. 273:11423–11428. [DOI] [PubMed] [Google Scholar]
- Yokosaki, Y., N. Matsuura, T. Sasaki, I. Murakami, H. Schneider, S. Higashiyama, Y. Saitoh, M. Yamakido, Y. Taooka, and D. Sheppard. 1999. The integrin α9β1 binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J. Biol. Chem. 274:36328–36334. [DOI] [PubMed] [Google Scholar]
- Young, B.A., Y. Taooka, S. Liu, K.J. Askins, Y. Yokosaki, S.M. Thomas, and D. Sheppard. 2001. The cytoplasmic domain of the integrin α9 subunit requires the adaptor protein paxillin to inhibit cell spreading but promotes cell migration in a paxillin-independent manner. Mol. Biol. Cell. 12:3214–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]