

Abstract
VEGF is unique among angiogenic growth factors because it disrupts endothelial barrier function. Therefore, we considered whether this property of VEGF might contribute to tumor cell extravasation and metastasis. To test this, mice lacking the Src family kinases Src or Yes, which maintain endothelial barrier function in the presence of VEGF, were injected intravenously with VEGF-expressing tumor cells. We found a dramatic reduction in tumor cell extravasation in lungs or livers of mice lacking Src or Yes. At the molecular level, VEGF compromises the endothelial barrier by disrupting a VE-cadherin–β-catenin complex in lung endothelium from wild-type, but not Yes-deficient, mice. Disrupting the endothelial barrier directly with anti–VE-cadherin both amplifies metastasis in normal mice and overcomes the genetic resistance in Yes-deficient mice. Pharmacological blockade of VEGF, VEGFR-2, or Src stabilizes endothelial barrier function and suppresses tumor cell extravasation in vivo. Therefore, disrupting Src signaling preserves host endothelial barrier function providing a novel host-targeted approach to control metastatic disease.
Introduction
Hematogenous metastasis is a complex cascade involving tumor cell invasion from the primary tumor, intravasation into the circulation, as well as extravasation at distant sites. The molecular mechanisms regulating tumor cell extravasation in vivo are poorly understood, but clearly depend on the invasive capacity of tumor cells and their ability to breach the endothelial cell barrier. Because VEGF has been implicated in the breakdown of endothelial cell barrier function through disruption of adherens junctions (Esser et al., 1998), the production and release of VEGF by tumor cells (Senger et al., 1993) may contribute to their extravasation and metastasis.
Recently, we demonstrated that VEGF leads to vascular permeability in a manner that depends on Src family kinases (Eliceiri et al., 1999). Although endothelial cells express three primary Src family members (Src, Fyn, and Yes), only mice deficient in Src or Yes are protected from VEGF-induced permeability, yet show normal angiogenesis (Eliceiri et al., 1999). This selective resistance to VEGF-induced permeability protects Src- or Yes-deficient mice from cardiac (Weis et al., 2004) or cerebral (Paul et al., 2001) edema after ischemic injury. The fact that these mice lack a VEGF-mediated permeability response allows us to determine whether VEGF facilitates tumor cell extravasation during the spread of metastatic tumor cells. Here, we use various models of experimental metastasis in which intravenously injected VEGF-expressing tumor cells colonize the lung or liver. These models allow for investigation of the extravasation of circulating tumor cells regardless of their ability to proliferate and migrate at the primary tumor site. Here, we demonstrate that VEGF-mediated Src kinase activity leads to a breakdown in endothelial barrier function which, in turn, hastens tumor cell extravasation and metastasis. In fact, both genetic and pharmacological evidence is presented indicating that blockade of endothelial cell Src activity increases host resistance to metastatic disease.
Results and discussion
Ultrastructural analysis of tumor cell extravasation
To study the extravasation phase of metastasis, we injected VEGF-expressing CT26 murine colon carcinoma cells i.v. into mice and evaluated the lungs from these animals at the ultrastructural level after hours to days. In normal mice 1–3 h after inoculation, CT26 cells can be observed extending processes between gaps in the endothelium (Fig. 1, A and B), suggesting that a breach in endothelial barrier function may facilitate extravasation. In fact, single cells extravasate within hours of entering the circulation (Fig. 1 C), and form multicellular lesions after several days (Fig. 1 D). These studies suggest the possibility that VEGF, which induces such endothelial gaps (Weis et al., 2004), may facilitate tumor cell extravasation.
Figure 1.
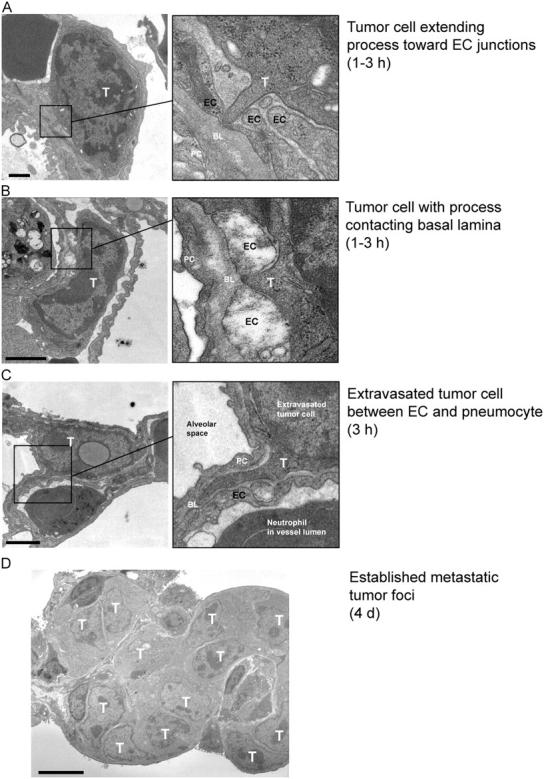
Tumor cell accumulation hours-to-days after tumor cell injection. To determine a time line for CT26 tumor cell extravasation after i.v. inoculation, we prepared lungs for analysis by transmission EM. By 1–3 h, tumor cells (T) had extended protrusions toward endothelial cells (EC) (A) or extended processes between EC junctions to contact the underlying basal lamina (BL) (B). (C) At 3 h, we observed individual extravasated tumor cells immediately outside the blood vessels where they were typically lodged in the extracellular space between an EC and a pneumocyte (PC). (D) This ultimately gave way to metastatic foci containing numerous tumor cells by day 4. Bars: (A–C) 1 μm; (D) 5 μm.
VEGF expression enhances lung colonization of ovarian tumor cells
To evaluate the possibility that endothelial barrier breakdown by VEGF contributes to tumor cell extravasation, we tested ID8 murine ovarian carcinoma cells retrovirally infected to stably express GFP either alone or together with VEGF (Zhang et al., 2002). ID8 cells overexpressing VEGF were previously found to have a metastatic advantage compared with ID8 cells lacking VEGF (Zhang et al., 2002), but it was unclear whether tumor cell extravasation was enhanced. Accordingly, ID8 cells were injected i.v. into the tail vein and tumor metastasis was monitored 9 d later. The VEGF-expressing ID8 cells formed considerably more metastatic lesions in the lung than the cells expressing GFP alone (Fig. 2), suggesting that VEGF contributes to tumor cell extravasation and/or cell proliferation at the metastatic site. Together, these studies suggest that tumor cell extravasation is linked to the ability of VEGF to promote a breakdown in endothelial barrier function.
Figure 2.
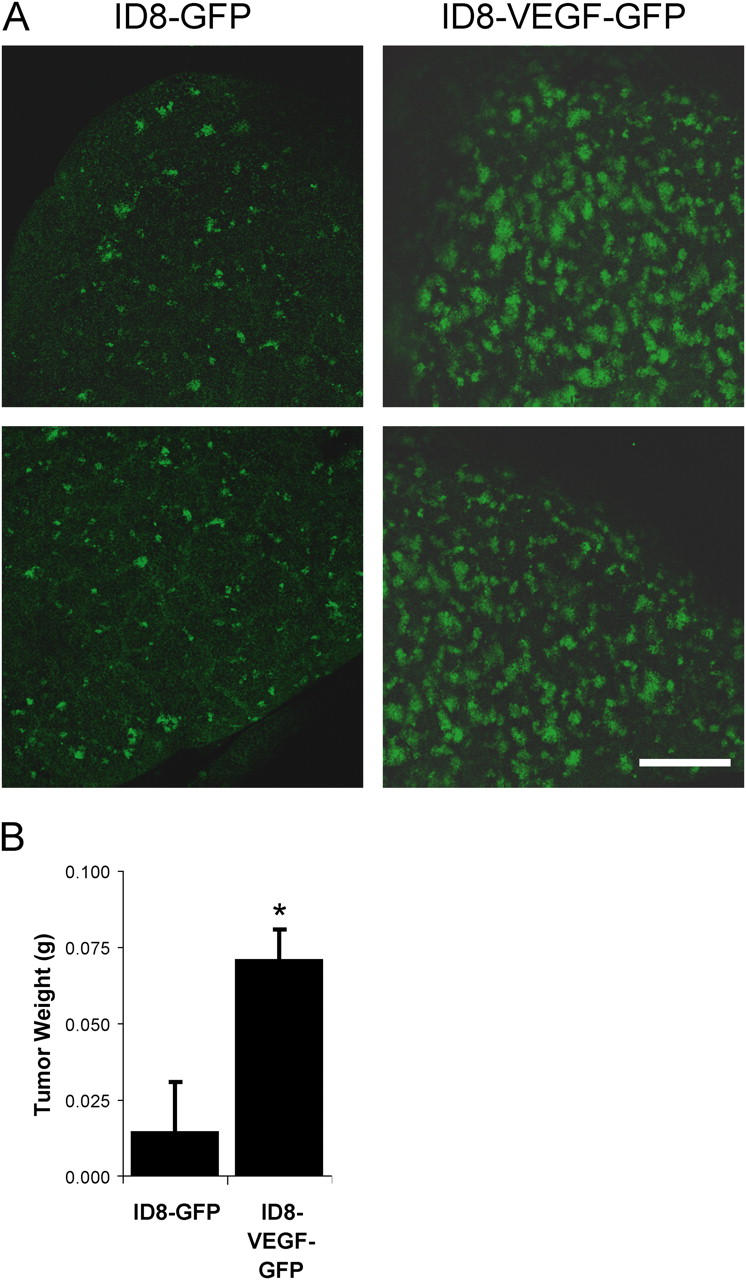
VEGF-expressing tumor cells show enhanced metastasis. ID8 murine ovarian carcinoma cells, which stably express GFP, were injected i.v. to form pulmonary metastatic lesions. (A) Fresh lung tissue was examined using laser scanning confocal microscopy to detect GFP-positive metastatic tumor cells. After 9 d, considerably more metastatic lesions were visible in lungs of mice injected with cells expressing VEGF (ID8-VEGF-GFP) compared with cells expressing GFP alone (ID8-GFP). (B) Lung weight due to tumor was significantly increased. * indicates P < 0.05; n = 8 each bar. Bar, 1 mm.
Mice deficient in Src or Yes are protected from tumor cell extravasation
We demonstrated previously that Src- or Yes-deficient mice or normal mice treated with pharmacological Src inhibitors fail to undergo VEGF-dependent vascular permeability yet show normal VEGF-mediated angiogenesis (Eliceiri et al., 1999). Thus, these mice allow us to evaluate the role that VEGF-mediated vascular permeability plays in tumor cell extravasation in vivo. Src- or Yes-deficient mice showed significantly diminished CT26 or D121 Lewis Lung carcinoma metastatic lesions in the lung compared with wild-type littermates (Fig. 3, A–C). In a second metastatic model, CT26 or D121 cells were injected beneath the spleen capsule followed by splenectomy, which ultimately results in hepatic metastatic lesions (Xiang et al., 1997). Consistent with the lung model, Yes-deficient mice showed sharply reduced hepatic metastases compared with control animals for either cell line (Fig. 3 D).
Figure 3.
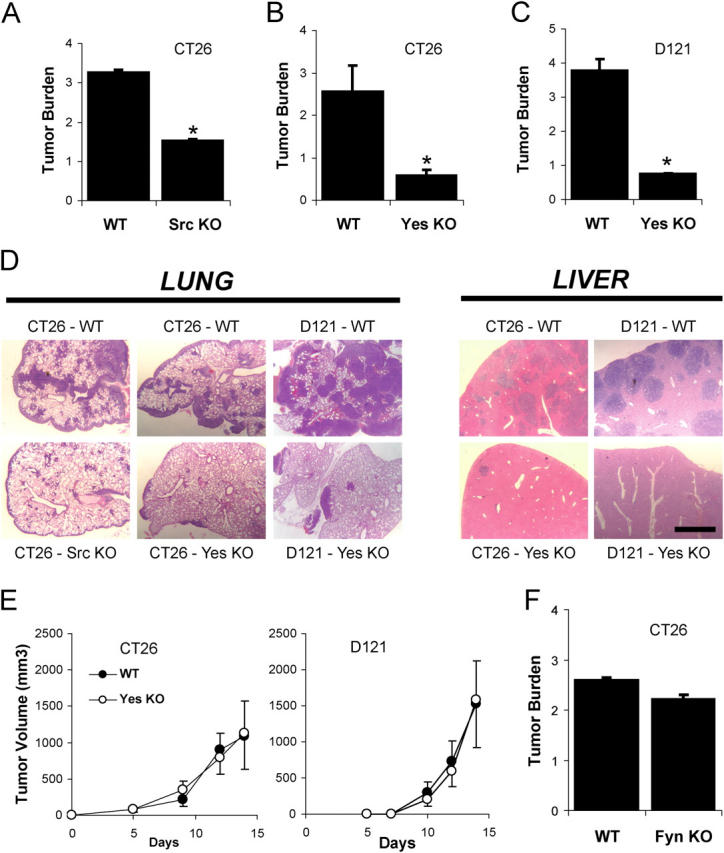
Gene-targeted deletion of individual Src kinases protects against tumor cell extravasation. (A–C) Tumor burden, computed as the increase in lung/heart weight ratio over that for normal mice, was measured 12 d after i.v. inoculation with metastatic tumor cells. Mice lacking Src (A) or Yes (B and C) are protected from experimental pulmonary metastasis of CT26 (A and B) or D121 (C) cells. (D) This phenomenon is not specific to the lung, because Yes-deficient mice are also protected from experimental hepatic metastasis of CT26 or D121 cells. (E) Growth of primary tumors on the flank is not different between genotypes for CT26 or D121 cells. (F) Mice lacking Fyn, which show a normal vascular permeability response to VEGF, are not protected from metastasis. * indicates P < 0.05; n = 8 each bar. Bar, 1 mm.
Importantly, primary tumor growth of each cell line injected subcutaneously on the flank was identical for wild-type and Yes-deficient mice (Fig. 3 E) indicating that these animals have no intrinsic defect in angiogenesis or tumor growth per se. Furthermore, mice lacking the Src family kinase Fyn, which show normal VEGF-induced vascular permeability (Eliceiri et al., 1999), are not resistant to pulmonary metastasis (Fig. 3 F). These findings demonstrate Src- or Yes-, but not Fyn-deficient mice, which show no VEGF-induced permeability are genetically resistant to the extravasation phase of metastasis. These findings confirm that there is specificity among the ubiquitously expressed Src family kinase members (Src, Fyn, and Yes), and reinforces the causal link between vascular permeability and tumor cell extravasation.
Src kinase regulation of VE-cadherin–β-catenin association facilitates tumor cell extravasation
Recently, we showed that during ischemic injury, VEGF-mediated vascular permeability depends on Src kinase-induced uncoupling of VE-cadherin–β-catenin containing junctions in vivo (Weis et al., 2004). Therefore, we evaluated the possibility that VEGF-expressing tumor cells circulating in the lung may compromise the vascular barrier by disrupting the VE-cadherin–β-catenin complex within the lung vasculature. Mice were injected i.v. with VEGF and lung lysates were subjected to immunoprecipitation and immunoblotting to detect the VE-cadherin–β-catenin complex. 5 min after VEGF stimulation, we observed a transient but consistent decrease in the lung endothelial cell VE-cadherin–β-catenin complex (Fig. 4 A). In contrast, this VE-cadherin–β-catenin complex did not dissociate after VEGF treatment in Yes-deficient mice (Fig. 4 A), which are resistant to the permeability-promoting effects of VEGF (Eliceiri et al., 1999) and to tumor cell extravasation (Fig. 3 A). These data not only confirm a role for Src kinases downstream of VEGF in the regulation of cadherin-mediated endothelial junctional barrier function, but help to explain the resistance these animals display toward tumor cell extravasation.
Figure 4.

Disruption of cadherin-mediated adhesion enhances tumor cell extravasation. (A) Direct i.v. injection of VEGF induces rapid and transient dissociation of β-catenin from VE-cadherin in lung from wild-type, but not Yes-deficient mice, determined by immunoprecipitation and immunoblotting of mouse lung homogenates. Representative data from three experiments are shown. (B) Treating mice before tumor cell inoculation with VE-cadherin–disrupting antibody BV13 induces vascular permeability, facilitates CT26 tumor cell extravasation, and increases metastases. VE-cadherin antibody E4G10 which does not produce permeability has no impact on metastasis. Tumor burden represents increase in lung/heart weight ratio over control. * indicates P < 0.05; n = 8 each bar (A) and n = 4 each bar (B).
Perturbation of VE-cadherin–mediated intercellular adhesion enhances tumor cell extravasation
To determine whether disruption of VE-cadherin was sufficient to promote tumor cell extravasation and metastasis, we evaluated the metastatic capacity of circulating CT26 cells in animals whose endothelial barrier function was compromised by a function blocking antibody to VE-cadherin (BV13). Treatment of these animals with BV13, which is known to disrupt endothelial barrier function of normal vessels (Liao et al., 2002), dramatically increased pulmonary metastasis of CT26 tumor cells by four- to sevenfold (Fig. 4 B) compared with mice treated with either saline or the isotype matched anti–VE-cadherin mAb E4G10. E4G10 serves as a strong control, because like BV13, it recognizes an epitope on the extracellular domain of VE-cadherin, but unlike BV13 does not disrupt endothelial barrier function (Liao et al., 2002). The ratio of wet/dry lung weight was not different between mice treated with BV13 or vehicle for 24 h (4.64 ± 0.04 and 4.69 ± 0.00, respectively), demonstrating the increase in lung weight is due to tumor, not edema. Although both BV13 and E4G10 block angiogenesis and suppress primary tumor growth (Liao et al., 2002), our findings demonstrate that disruption of VE-cadherin–mediated intercellular junctions with BV13 is sufficient to promote tumor cell extravasation, suggesting that VE-cadherin adhesion and its regulation of endothelial barrier function plays a role as a “gatekeeper” in the metastatic cascade.
To establish whether the suppression of metastasis in Yes-deficient mice is related to VE-cadherin–dependent barrier function, these animals were treated with the VE-cadherin function blocking antibody BV13. This resulted in a fourfold increase (P = 0.006) in the number of metastatic lesions within the lungs of these animals, and a 27 ± 4% increase (P = 0.0001) in lung weight due to tumor compared with untreated Yes-deficient mice (n = 8 each group). Therefore, disruption of VE-cadherin can overcome the genetic resistance to tumor cell extravasation observed in Yes-deficient mice, supporting the notion that Src kinase regulation of VE-cadherin within the endothelium impacts tumor cell extravasation in vivo.
VEGF, Flk, or Src inhibitors suppress tumor cell extravasation
If VEGF signaling potentiates endothelial barrier breakdown, then pharmacological blockade of VEGF or its downstream effectors might reduce tumor cell extravasation. Thus, we tested if blocking VEGF signaling during the first 2 d after i.v. administration of tumor cells would impact extravasation. We found that treating mice with the VEGF inhibitor Cyclo-VEGI reduced pulmonary metastasis by 47% (Fig. 5 A, P = 0.01), suggesting that an early step in tumor cell extravasation is sensitive to pharmacological inhibition of VEGF.
Figure 5.
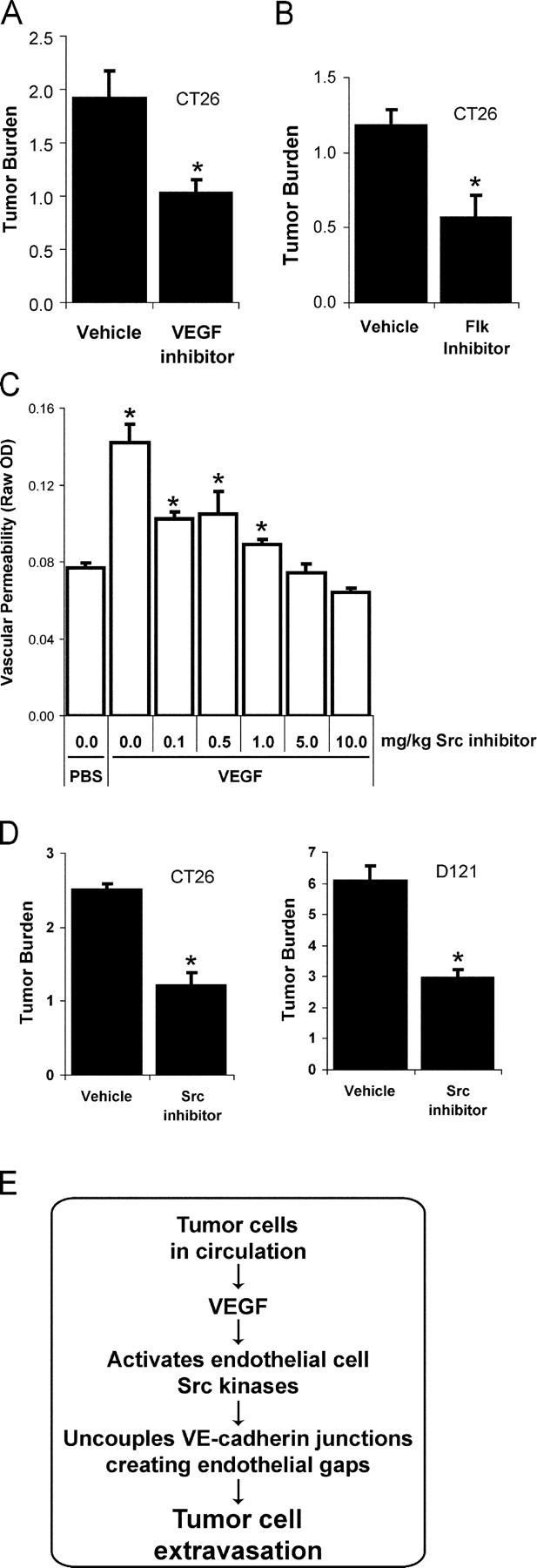
Pharmacological Src, Flk, or VEGF blockade suppresses tumor cell extravasation. (A) Pretreatment with VEGF inhibitor Cyclo-VEGI provides a similar extent of protection from pulmonary metastases as Src or Flk inhibition, further suggesting the permeability-inducing effects of VEGF contribute to tumor cell extravasation. (B) VEGF receptor 2 signaling is required for metastasis of CT26 cells because a single pretreatment with Flk inhibitor SU1498 reduces lung tumor burden after 12 d. (C) Pretreatment with Src inhibitor provides a dose-dependent blockade of VEGF-induced vascular leak in the skin evaluated using the Miles assay. (D) Src inhibitor treatment administered twice daily during days 0–3 after i.v. introduction of CT26 or D121 cells significantly reduces lung tumor burden after 12 d. (E) Preventing VEGF-induced endothelial barrier breakdown via VEGF, Flk, or Src blockade preserves cadherin-mediated endothelial cell adhesion and limits tumor cell extravasation. Tumor burden represents mean ± SEM for increase in lung/heart weight ratio over control. * indicates P < 0.05; n = 8 each bar (A, B, and D); n = 4 each bar (C).
Although VEGF signaling occurs through binding to its three cell surface receptors VEGFR-1 (Flt), VEGFR-2 (Flk/KDR), and/or neuropilin1/2, the permeability-inducing effects of VEGF have been attributed to signaling downstream of Flk exclusively (Ferrara et al., 2003). Therefore, we considered whether blockade of Flk might influence tumor cell extravasation and the number or size of metastatic lesions in the CT26 experimental pulmonary metastasis model. Mice were pretreated with a single injection of a Flk inhibitor (SU1498) 1 h before i.v. inoculation with CT26 cells. This treatment reduced lung metastasis by ∼50% (Fig. 5 B, P < 0.001), suggesting that Flk-dependent signaling during the dissemination phase of metastasis contributes to disease progression. Although this particular Flk inhibitor has recently been shown to affect Erk signaling downstream of other growth factors (Boguslawski et al., 2004), the efficacy of a single dose of this agent delivered before tumor cell inoculation is consistent with the blockade of VEGF-mediated tumor cell extravasation during the hours after injection.
We previously demonstrated that Src activity is required downstream of VEGF for subsequent VE-cadherin and β-catenin phosphorylation, leading to breakdown of endothelial barrier function (Weis et al., 2004). Thus, we considered whether a pharmacological Src inhibitor might suppress tumor cell extravasation and metastasis in genetically normal mice. Administration of the Src inhibitor SKI-606 at the minimal dose required to block vascular leak (Fig. 5 C, 10 mg/kg) during the first 3 d after either CT26 or D121 tumor cell inoculation reduced the formation of metastatic lesions in the lung by nearly 60% (Fig. 5 D), yet did not influence proliferation of these cells in culture (not depicted). SKI-606 like other inhibitors is likely to target a number of structurally related kinases and may directly impact tumor cell activities associated with invasion (Golas et al., 2003; Recchia et al., 2003). Nevertheless, considering the genetic data, it appears that Src kinases may be important targets to regulate host endothelial barrier function during metastatic disease.
Together, these data suggest that disrupting VEGF signaling leading to vascular leak by pretreatment with pharmacological inhibitors of VEGF, Flk, or Src can alleviate lung metastasis (Fig. 5 E). The beneficial effect of these inhibitors was attained by treatment within the first few days after tumor cell inoculation when single tumor cells are beginning to extravasate and proliferate, but before tumor growth and angiogenesis is established.
Conclusions
Previous studies have implicated VEGF (Rowe et al., 2000), Flk (Bruns et al., 2002), or Src (Golas et al., 2003) in tumor growth and metastatic disease. In particular, blockade of tumor cell-associated Src activity has been associated with reduced metastasis (Nam et al., 2002), but this has been attributed to the requirement of Src activity for tumor cell migration and intravasation into the circulation from the primary tumor site. We demonstrate for the first time that regardless of the invasive capacity of individual tumor cells, metastatic disease critically depends on the ability of circulating tumor cells to breach the endothelial barrier. We propose that metastatic tumor cells which express and release high levels of VEGF have the capacity to dysregulate at least one critical endothelial cell–cell junctional complex facilitating their extravasation. These findings suggest an unexpected therapeutic role for pharmacological inhibitors of VEGF or Src kinases in blocking a host response important for metastatic disease.
Materials and methods
Pharmacological agents
The SKI-606 Src inhibitor (10 mg/kg i.p., 236 nM; Boschelli et al., 2001; Golas et al., 2003) was injected twice daily for days 0–3 after tumor cell injection. The SU1498 Flk inhibitor (single injection 1 h before tumor cell injection, 20 mg/kg i.p., 385 nM) and Cyclo-VEGI (CBO-P11) VEGF inhibitor (twice daily days 0–2 after tumor cell injection, 2 mg/kg i.p., 13 nM) were purchased from Calbiochem. VE-cadherin murine mAbs BV13 and E4G10 (Liao et al., 2000, 2002), gifts from D. Hicklin (ImClone Systems, Inc.), were injected i.p. at 50 μg per adult mouse 1 h before tumor cell injection.
Cell culture
ID8 murine ovarian carcinoma cells retrovirally infected to stably express GFP or GFP+VEGF were provided by G. Coukos (University of Pennsylvania, Philadelphia, PA) and have been described previously (Zhang et al., 2002). Murine CT26 colon carcinoma and D121 Lewis Lung carcinoma cells were provided by R. Reisfeld at our institution. Cells were maintained at subconfluency, washed, harvested, counted, and resuspended in sterile PBS for tumor cell injection as described previously (Hood et al., 2002; Zhang et al., 2002).
Mice
Mice with gene-targeted deletion of Src, Yes, or Fyn are available from Jackson Laboratory. Src- and Yes-deficient mice were the gifts of P. Soriano and J. Cooper (Fred Hutchinson Cancer Research Center, Seattle, WA), and P. Stein (University of Pennsylvania, Philadelphia, PA). BalbC and C57By/J mice were generated at The Scripps Research Institute. Appropriate age-matched littermate wild-type controls were used for each gene-targeted strain.
Experimental metastasis
We injected 5 × 105 CT26, D121, or ID8 carcinoma cells either i.v. (for experimental pulmonary metastasis) or beneath the spleen capsule followed by its removal within 2 min (for experimental hepatic metastasis), typically resulting in the formation of lung or liver metastases, respectively, within 4 d (Xiang et al., 1997). Metastases were established for 12 d to ensure that all control animals contained actively growing lung or liver tumors of a consistent size. Fresh tissue was photographed using an research stereoscope (model SZH10; Olympus) with a SPOT camera (model 2.2.1; Diagnostic Instruments) or a laser scanning confocal microscope (emission filter 522 nm for GFP, 2.5×/0.075, MRC 1024; Bio-Rad Laboratories). Tissue was weighed and images were acquired using an BX60 light microscope (Olympus) with UPlanFI (10×/0.30) or UPlanApo (20×/0.70) objectives and a SPOT camera (model 2.2.1; Diagnostic Instruments). To account for differences between mice of different age, gender, or genotype, lung tumor burden was computed as the increase in the ratio of lung/heart weight over control mice (1.43 ± 0.06 g for 31 mice). In some groups, tissue was fixed, sectioned, and stained with hematoxylin and eosin to count the number of lesions per section of equivalent area. Data in each figure are representative of three or more separate experiments, each performed with appropriate littermate controls.
Primary tumor growth
CT26 (5 × 105) or D121 (105) carcinoma cells were injected subcutaneously on the mouse flank, and growth of the primary tumor was measured by microcaliper for 14 d. Tumor volume (V) was computed as V = 1/2 (L × W)2, where L and W represent tumor width (shortest dimension) and length (longest dimension), respectively.
Ultrastructural analysis by electron microscopy
Tissue was prepared from mouse lung 1 h to 4 d after CT26 tumor cell injection. Tissue was prepared as described previously (Weis et al., 2004) and ultra thin sections were viewed using a CM-100 transmission electron microscope (Philips). Negatives were scanned on an Epson Expression 1680 scanner using Adobe Photoshop 7.0 software.
In vivo vascular permeability
The Miles assay was adapted to measure local VEGF-induced leak of dye in the skin as described previously (Eliceiri et al., 1999).
In vivo vascular signaling
Adult mice were injected i.v. with 100 μl of VEGF (0.2 mg/kg in PBS; PeproTech). At specified times, the lungs were rapidly excised, homogenized, and prepared for immunoprecipitation and immunoblotting as described previously (Weis et al., 2004). The following antibodies were purchased from Santa Cruz Biotechnology: VE-cadherin (SC-6458) and β-catenin (SC-7963). Representative data from at least three separate experiments are shown.
Statistical analysis
Data are presented as mean ± SEM, with statistical significance determined from t test (P < 0.05).
Acknowledgments
This is manuscript 16572-IMM from The Scripps Research Institute. We thank Ralph Reisfeld and Rong Xiang at our institution for assistance in establishing the tumor models in our laboratory, and Malcolm Wood at the Core Microscopy Facility for performing transmission electron microscopy.
National Institutes of Health provided grant support to D.A. Cheresh (CA50286, CA45726, CA95262, EY14174, CA78045, HL57900) and S.M. Weis (1F32HL69701).
References
- Boguslawski, G., P.W. McGlynn, K.A. Harvey, and A.T. Kovala. 2004. SU1498, an inhibitor of vascular endothelial growth factor receptor 2, causes accumulation of phosphorylated ERK kinases and inhibits their activity in vivo and in vitro. J. Biol. Chem. 279:5716–5724. [DOI] [PubMed] [Google Scholar]
- Boschelli, D.H., F. Ye, Y.D. Wang, M. Dutia, S.L. Johnson, B. Wu, K. Miller, D.W. Powell, D. Yaczko, M. Young, et al. 2001. Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J. Med. Chem. 44:3965–3977. [DOI] [PubMed] [Google Scholar]
- Bruns, C.J., M. Shrader, M.T. Harbison, C. Portera, C.C. Solorzano, K.W. Jauch, D.J. Hicklin, R. Radinsky, and L.M. Ellis. 2002. Effect of the vascular endothelial growth factor receptor-2 antibody DC101 plus gemcitabine on growth, metastasis and angiogenesis of human pancreatic cancer growing orthotopically in nude mice. Int. J. Cancer. 102:101–108. [DOI] [PubMed] [Google Scholar]
- Eliceiri, B.P., R. Paul, P.L. Schwartzberg, J.D. Hood, J. Leng, and D.A. Cheresh. 1999. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell. 4:915–924. [DOI] [PubMed] [Google Scholar]
- Esser, S., M.G. Lampugnani, M. Corada, E. Dejana, and W. Risau. 1998. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 111:1853–1865. [DOI] [PubMed] [Google Scholar]
- Ferrara, N., H.P. Gerber, and J. LeCouter. 2003. The biology of VEGF and its receptors. Nat. Med. 9:669–676. [DOI] [PubMed] [Google Scholar]
- Golas, J.M., K. Arndt, C. Etienne, J. Lucas, D. Nardin, J. Gibbons, P. Frost, F. Ye, D.H. Boschelli, and F. Boschelli. 2003. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 63:375–381. [PubMed] [Google Scholar]
- Hood, J.D., M. Bednarski, R. Frausto, S. Guccione, R.A. Reisfeld, R. Xiang, and D.A. Cheresh. 2002. Tumor regression by targeted gene delivery to the neovasculature. Science. 296:2404–2407. [DOI] [PubMed] [Google Scholar]
- Liao, F., Y. Li, W. O'Connor, L. Zanetta, R. Bassi, A. Santiago, J. Overholser, A. Hooper, P. Mignatti, E. Dejana, et al. 2000. Monoclonal antibody to vascular endothelial-cadherin is a potent inhibitor of angiogenesis, tumor growth, and metastasis. Cancer Res. 60:6805–6810. [PubMed] [Google Scholar]
- Liao, F., J.F. Doody, J. Overholser, B. Finnerty, R. Bassi, Y. Wu, E. Dejana, P. Kussie, P. Bohlen, and D.J. Hicklin. 2002. Selective targeting of angiogenic tumor vasculature by vascular endothelial-cadherin antibody inhibits tumor growth without affecting vascular permeability. Cancer Res. 62:2567–2575. [PubMed] [Google Scholar]
- Nam, J.S., Y. Ino, M. Sakamoto, and S. Hirohashi. 2002. Src family kinase inhibitor pp2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin. Cancer Res. 8:2430–2436. [PubMed] [Google Scholar]
- Paul, R., Z.G. Zhang, B.P. Eliceiri, Q. Jiang, A.D. Boccia, R.L. Zhang, M. Chopp, and D.A. Cheresh. 2001. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 7:222–227. [DOI] [PubMed] [Google Scholar]
- Recchia, I., N. Rucci, C. Festuccia, M. Bologna, A.R. MacKay, S. Migliaccio, M. Longo, M. Susa, D. Fabbro, and A. Teti. 2003. Pyrrolopyrimidine c-Src inhibitors reduce growth, adhesion, motility and invasion of prostate cancer cells in vitro. Eur. J. Cancer. 39:1927–1935. [DOI] [PubMed] [Google Scholar]
- Rowe, D.H., J. Huang, M.L. Kayton, R. Thompson, A. Troxel, K.M. O'Toole, D. Yamashiro, C.J. Stolar, and J.J. Kandel. 2000. Anti-VEGF antibody suppresses primary tumor growth and metastasis in an experimental model of Wilms' tumor. J Pediatr Surg. 35:30-32; discussion 32-33. [DOI] [PubMed] [Google Scholar]
- Senger, D.R., L. Van de Water, L.F. Brown, J.A. Nagy, K.T. Yeo, T.K. Yeo, B. Berse, R.W. Jackman, A.M. Dvorak, and H.F. Dvorak. 1993. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev. 12:303–324. [DOI] [PubMed] [Google Scholar]
- Weis, S., S. Shintani, A. Weber, R. Kirchmair, M. Wood, A. Cravens, H. McSharry, A. Iwakura, Y.S. Yoon, N. Himes, et al. 2004. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Invest. 113:885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, R., H.N. Lode, C.S. Dolman, T. Dreier, N.M. Varki, X. Qian, K.M. Lo, Y. Lan, M. Super, S.D. Gillies, and R.A. Reisfeld. 1997. Elimination of established murine colon carcinoma metastases by antibody-interleukin 2 fusion protein therapy. Cancer Res. 57:4948–4955. [PubMed] [Google Scholar]
- Zhang, L., N. Yang, J.R. Garcia, A. Mohamed, F. Benencia, S.C. Rubin, D. Allman, and G. Coukos. 2002. Generation of a syngeneic mouse model to study the effects of vascular endothelial growth factor in ovarian carcinoma. Am. J. Pathol. 161:2295–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]