

Abstract
Front–rear asymmetry in motile cells is crucial for efficient directional movement. The uropod in migrating lymphocytes is a posterior protrusion in which several proteins, including CD44 and ezrin/radixin/moesin (ERM), are concentrated. In EL4.G8 T-lymphoma cells, Thr567 phosphorylation in the COOH-terminal domain of ezrin regulates the selective localization of ezrin in the uropod. Overexpression of the phosphorylation-mimetic T567D ezrin enhances uropod size and cell migration. T567D ezrin also induces construction of the CD44-associated polar cap, which covers the posterior cytoplasm in staurosporine-treated, uropod-disrupted EL4.G8 cells or in naturally unpolarized X63.653 myeloma cells in an actin cytoskeleton–dependent manner. Rho-associated coiled coil–containing protein kinase (ROCK) inhibitor Y-27632 disrupts the uropod but not the polar cap, indicating that Rho–ROCK signaling is required for posterior protrusion but not for ERM phosphorylation. Phosphorylated ezrin associates with Dbl through its NH2-terminal domain and causes Rho activation. Moreover, constitutively active Q63L RhoA is selectively localized in the rear part of the cells. Thus, phosphorylated ERM has a potential function in establishing plasma membrane “posteriority” in the induction of the uropod in T lymphocytes.
Introduction
To defend our bodies against invasive pathogens, immune cells such as leukocytes and lymphocytes quickly migrate toward sites of infection or toward appropriate anatomical compartments (Springer, 1994; Kunkel and Butcher, 2002; Hogg et al., 2003). For achieving this migration, immune cells are intrinsically equipped with specialized abilities. One of these is the ability to sense “directional cues” that inform them about the specific sites to be mobilized. Various receptors for chemoattractants and adhesion molecules are responsible for this (Springer, 1994; Kunkel and Butcher, 2002; Hogg et al., 2003). Another contributing ability is the much higher motility of immune cells compared with the cells in the vast majority of tissues. Several factors are considered to cause this high motility, including (1) relatively low adhesiveness, (2) capacity for rapid cytoskeletal reorganization, and (3) definite asymmetry of cellular components along the front–rear axis that is used for effective directional movement. At the front part of the cell (the leading edge), efficient dynamics of actin cytoskeleton organization contribute to pulling the cell body upward in the direction of a chemoattractive gradient (Sánchez-Madrid and del Pozo, 1999; Hogg et al., 2003). Detachment from the substratum and quick retraction at the rear part of the cell (the trailing edge) also facilitate the rapid migration (Sánchez-Madrid and del Pozo, 1999; Hogg et al., 2003). However, the underlying mechanisms by which lymphocytes achieve clear cell polarity remain poorly understood.
Migrating lymphocytes possess a unique cellular structure called the uropod, a spherical membrane protrusion budding from the rear part of the cell body (Sánchez-Madrid and del Pozo, 1999). The roles of the uropod in lymphocyte migration, as well as the molecular mechanisms organizing this structure, are largely unknown. The finding that several molecules are selectively compartmentalized at the uropod provides some clues to clarify these subjects. For instance, some transmembrane adhesion molecules, including CD43, CD44, intercellular adhesion molecules, and PSGL-1, are concentrated at the uropod (Sánchez-Madrid and del Pozo, 1999). These molecules have a motif that can bind ezrin/radixin/moesin (ERM) proteins at the intracellular part; therefore, it is reasonable to suggest that the ERM proteins are also accumulated in the uropod (Mangeat et al., 1999; Sánchez-Madrid and del Pozo, 1999; Tsukita and Yonemura, 1999; Bretscher et al., 2002). ERM proteins act as membrane–cytoskeleton linkers by binding to the membrane proteins at their NH2-terminal (NT) domains and to F-actin at its COOH-terminal (CT) domain (Mangeat et al., 1999; Tsukita and Yonemura, 1999; Gautreau et al., 2000; Bretscher et al., 2002). The linker function is regulated by the phosphorylation of a conserved threonine residue in the CT domain of each ERM protein. This Thr phosphorylation disrupts the intramolecular interaction between the NT and CT domains, allowing them to bind to membrane proteins and F-actin. However, the roles of ERM proteins and Thr phosphorylation in uropod formation or lymphocyte polarity remain to be elucidated.
In this study, by using a T lymphoma cell line, EL4.G8, that constitutively possesses a clear uropod, we show potential roles of the phosphorylated ERM (p-ERM) proteins not only in uropod formation but also in cell polarization in cooperation with Rho–ROCK signaling.
Results
Uropod formation in EL4.G8 cells requires Ser/Thr kinase activity and actin cytoskeleton
In general, the uropod is induced in a chemoattractant- or adhesion-dependent fashion (Sánchez-Madrid and del Pozo, 1999); thus, it seems relatively unstable. We took advantage of the mouse T lymphoma cell line EL4, a well-known cell line that is often used for investigating T cell functions, because it spontaneously constructs a uropod when grown in nonadhesive culture dishes and is therefore suitable for uropod studies. To obtain cells with clearer and more frequent uropod formation, we isolated a subclone, G8, with these features (∼80% of the G8 cells construct a uropod) from the parental line by limiting dilution (Fig. 1 A, arrows). EL4.G8 cells exhibited a hand mirror–shaped morphology, with a narrow neck segregating the cell body from the spherical uropod protrusion. We often observed that these cells attached to each other by their uropods to form aggregates, an observation that indicated that this structure is somewhat stickier than other parts of the cell (Fig. 1 A, right). Confocal microscopic analysis clearly demonstrated that transmembrane proteins such as CD44 and CD43 were selectively localized at the protruding part (Fig. 1, B and C). In addition, ezrin, one of the ERM proteins, also accumulated in this part (Fig. 1 B). A polyclonal antibody that recognizes the phosphorylated CT Thr residues of all three ERM proteins clearly stained the protruding structure (Fig. 1, C and D), suggesting that the ezrin accumulated in this compartment is the Thr567-phosphorylated form. In contrast, a large fraction of F-actin is accumulated at the cell margin opposite to the protruding structure; thus, this part is likely the leading edge, although a faint signal for actin filaments was also detected at the uropod protrusion (Fig. 1 D). A leukocyte integrin, LFA-1 (αLβ2), was also enriched in the leading edge (Fig. 1 E). Therefore, EL4.G8 cells show typical features of lymphocyte polarization (Sánchez-Madrid and del Pozo, 1999).
Figure 1.

EL4.G8 cells show polarized morphology with a clear uropod that is regulated by Ser/Thr kinases and the actin cytoskeleton. (A) Phase-contrast view showing the hand mirror–shaped morphology of EL4.G8 cells with clear uropods (left, arrows). Aggregation of EL4.G8 cells by their uropods (left). (B–E) Selective localization of the uropod markers (CD44 and CD43) and p-ERM in the EL4.G8 cell uropod, whereas F-actin and LFA-1 are enriched in the leading edge. Fixed, permeabilized cells were stained with fluorescence-labeled probes for cell surface CD44, CD43, or LFA-1, and for intracellular ezrin, p-ERM, or F-actin. (F) Staurosporine and cytochalasin D disrupt uropod structure. Cells were treated with DMSO (cont.), staurosporine (stauro.), or cytochalasin D (cyt.D) and stained for p-ERM and CD44. (G) Staurosporine abolishes ERM phosphorylation and the interaction between CD44 and ezrin in EL4.G8 cells. The total amount of ezrin and the p-ERM level were determined by Western blotting using lysates from control or staurosporine-treated cells (top and middle). CD44 ezrin interaction was detected by the immunoprecipitation of CD44 and Western blotting for ezrin (bottom). Bars (A–F), 10 μm.
When EL4.G8 cells were treated with a broad-spectrum Ser/Thr kinase inhibitor, staurosporine, uropod protrusion was completely abolished, as was p-ERM staining, resulting in uniform CD44 distribution on the plasma membrane (Fig. 1 F). In accordance with this observation, staurosporine abolished the phosphorylation of all ERM proteins and the association between ezrin and CD44 (Fig. 1 G). An actin polymerization inhibitor, cytochalasin D, also disrupted the uropod and brought about uniform p-ERM and CD44 distributions (Fig. 1 F). Therefore, Ser/Thr kinase activity and the actin cytoskeleton are both required for maintenance of the uropod in EL4.G8 cells, and ezrin functions appear to be under the control of these cellular processes.
The role of Thr567 phosphorylation in the uropod localization of ezrin
To address the details of ezrin localization in the uropod, we transfected plasmids expressing various GFP-tagged mutant forms of ezrin into EL4.G8 cells and obtained multiple stable lines (Fig. 2, A and D). Transfection with the control vector expressing GFP resulted in cytoplasmic and nuclear distribution of this reporter protein (Fig. 2, B and C). In contrast, wild-type (WT) ezrin-GFP was preferentially localized at the peripheral membrane, especially in the uropod rather than the cell body, although a weak signal was also detected in the cytoplasm. A mutant ezrin in which the CT Thr was replaced by Ala (T567A) was distributed diffusely in the cytoplasm. This mutant cannot be phosphorylated at this residue and, therefore, does not undergo the intramolecular NT and CT domain dissociation and mimics the dormant form of ezrin (Gautreau et al., 2000; Coscoy et al., 2002). In marked contrast, another mutant with replacement of this Thr by Asp, T567D, which does not undergo NT and CT domain association and thus mimics the phosphorylated active form with membrane–cytoskeleton linker activity (Gautreau et al., 2000; Coscoy et al., 2002), exclusively accumulated in the uropod colocalized with CD44 (Fig. 2 B, asterisks). The fluorescent signal of the NT domain fragment tagged with GFP was also detected at the uropod, as well as at the peripheral membrane of the cell body, confirming the requirement of the NT domain for ezrin to localize in the uropod. In contrast, the CT domain fragment of ezrin was accumulated at the leading edge and was well-colocalized with F-actin, but was not abundant in the uropod, indicating that this fragment freely associates with the actin cytoskeleton. Such intracellular localization patterns of ezrin mutants were also observed in another T cell lymphoma line, BW5147, although the cells bearing a uropod in this line are a minor population and the sizes of the uropods are much smaller than in EL4.G8 cells (unpublished data).
Figure 2.
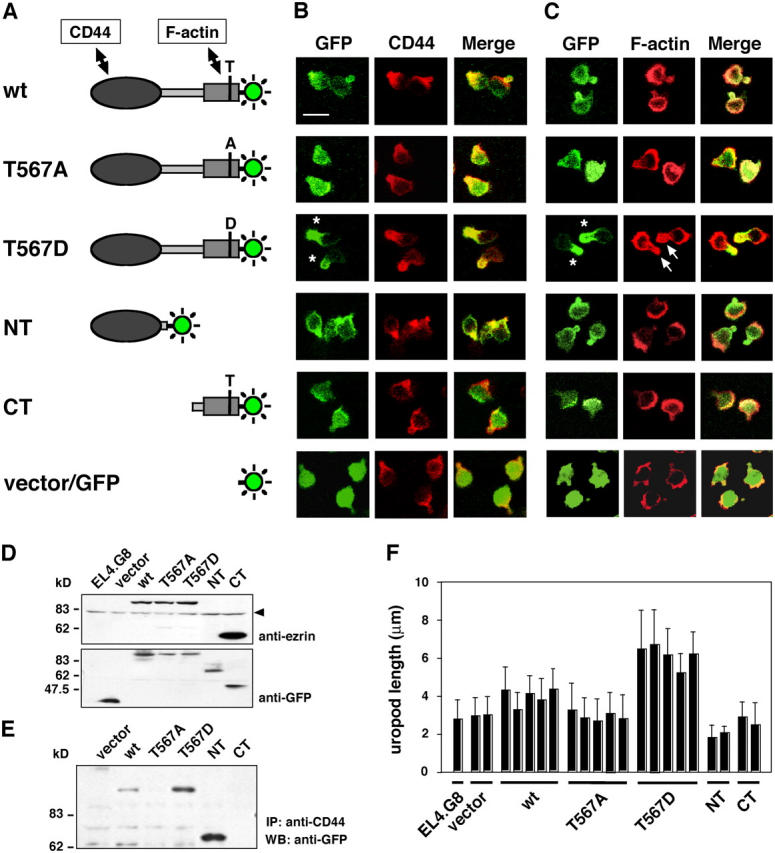
Intracellular localization of ezrin mutants and the enhancement of uropod size in T567D ezrin transfectants. (A) GFP-tagged ezrin constructs used in this study. In point mutant constructs T567A or T567D, a CT phosphorylation site, Thr567 (T) was replaced by Ala (A) or Asp (D), respectively. (B and C) Intracellular localization of ezrin mutants in EL4.G8 cells. Stable transfectants were examined for the localization of GFP as well as CD44 (B) or F-actin (C). Each set of panels corresponds to the construct shown to the left in A. T567D ezrin was exclusively localized at the uropod (asterisks) and colocalized with the enhanced F-actin signal (C, arrows). Bar, 10 μm. (D) GFP-tagged proteins expressed in stable transfectants were detected by Western blotting using antiezrin or anti-GFP antibody. Polyclonal antiezrin antibody recognizes an epitope in the CT domain and, thus, does not detect the NT fragment. The arrowhead indicates endogenous ezrin. (E) Interaction between CD44 and ezrin mutants was detected by the immunoprecipitation of CD44 and Western blotting for GFP. (F) T567D ezrin increases uropod length. The length of the uropod was measured and expressed as mean ± SD (n > 200). Each bar represents an individual stable clone of a different type of ezrin transfectant.
Consistent with the observation of colocalization with CD44 in the uropod, associations between CD44 and WT, T567D, or NT ezrin were readily detected by immunoprecipitation with anti-CD44 antibody followed by Western blotting with anti-GFP antibody, whereas those between CD44 and T567A or CT ezrin were undetectable (Fig. 2 E). These findings suggest that Thr567 phosphorylation, and the consequent conformational changes of ezrin, anchor this protein in the uropod.
T567D ezrin enhances uropod size
The size of the uropod was markedly enhanced by T567D ezrin, whereas T567A ezrin had little effect on it (Fig. 2 F). Forced expression of WT ezrin only slightly augmented the uropod's size, suggesting that the total capacity for ERM Thr phosphorylation is limited in each cell and that a fraction of ezrin-GFP proteins is incorporated into the uropod membrane. Because NT ezrin transfectants showed a slight diminishment of the incidence of cells carrying the uropod (64% for NT, compared with 82% for the vector or WT) and of the uropod's size (Fig. 2 F), this type of deletion mutant might function in a weakly dominant-negative fashion. Interestingly, F-actin was apparently accumulated at the enlarged uropod in T567D ezrin transfectants, demonstrating that the activated ezrin facilitates the organization of the cortical actin cytoskeleton underneath the uropod membrane (Fig. 2 C, arrows). Thus, Thr567 phosphorylation of ezrin can influence the integrity of the uropod in T lymphocytes.
T567D ezrin induces construction of a plasma membrane polar cap in uropod-less situations
As expected, staurosporine treatment disrupted the uropod as well as the polarized distributions of GFP signal and CD44 in WT ezrin transfectants (Fig. 3 A). Surprisingly, in T567D ezrin transfectants, despite the fact that the typical uropod also disappeared upon treatment with staurosporine, the GFP signal and CD44 clearly accumulated at one pole of the cells to form a capped structure (Fig. 3, A [arrows] and B). A similar cap structure was constructed in BW5147 cells transfected with T567D ezrin and treated with staurosporine (unpublished data). These findings strongly suggest that the T567D ezrin can preserve the plasma membrane polarity even in the uropod-abolished cells. Because polar cap formation was not observed in NT ezrin transfectants, this phenomenon requires the phosphorylated CT domain. In addition, cytochalasin D canceled the induction of polar cap formation by T567D ezrin (Fig. 3, A and B), suggesting that F-actin is also required for this event.
Figure 3.

T567D ezrin induces a plasma membrane polar cap in uropod-less cells. (A) T567D ezrin can cause formation of the polar cap in staurosporine-treated (arrows) but not in cytochalasin D–treated EL4.G8 cells. WT, T567D, and NT ezrin transfectants were treated with inhibitors and stained for CD44. Bar, 10 μm. (B) Percentage of cells exhibiting the polar cap in A (n > 130). (C) T567D-ΔAB ezrin lacks the ability to induce polar cap formation in staurosporine-treated EL4.G8 cells. Cells were transiently transfected with constructs for T567D or T567D-ΔAB ezrin, and after 24 h the cells were treated with staurosporine and stained for CD44. In contrast with the results for T567D-ΔAB, clear polar caps are observed in T567D ezrin–transfected cells (arrows). Bar, 10 μm. (D) Percentage of cells exhibiting the polar cap in C (n > 130). (E) T567D ezrin can induce organization of the polar cap in X63.653 myeloma cells (arrows), which are naturally unpolarized with regard to the plasma membrane. X63.653 cells were transiently transfected with constructs for WT, T567D, NT, or T567D-ΔAB ezrin and stained for CD44 24 h after the transfection. Bar, 10 μm. (F) Percentage of cells exhibiting the polar cap in C (n > 120).
To further address the importance of the actin cytoskeleton in polar cap formation, we constructed a variant form of T567D ezrin that has a 6-aa deletion at the CT end and lacks actin-binding ability (T567D-ΔAB; Turunen et al., 1994). Comparing T567D with T567D-ΔAB in transient transfection followed by staurosporine treatment, it is apparent that T567D-ΔAB was unable to construct the polar caps in EL4.G8 cells (Fig. 3, C and D). As the deletion of the CT six amino acids is a minimal alteration except for its effect on actin-binding ability, T567D-ΔAB is thought to preserve most other functions of activated ezrin. Therefore, this finding suggests that polar cap formation is dependent on actin-binding ability as well as on membrane anchorage of phosphorylated ezrin, but independent of other interactive partners.
To confirm the ability of T567D ezrin to induce the polar cap in naturally unpolarized cells, we transfected the construct into the myeloma cell line X63.653, which does not carry a uropod or show specific CD44 (Fig. 3 E) and F-actin localization (not depicted). Strikingly, T567D ezrin induced the construction of the polar cap accompanied by CD44 accumulation in these cells, although the uropod was not induced, whereas WT, NT, or T567D-ΔAB ezrin had no influence on CD44 localization (Fig. 3, E [arrows] and F). These results indicate that X63.653 cells seem to be inactive in the machinery not only for ezrin phosphorylation but also for uropod protrusion, and, hence, T567D ezrin induces only plasma membrane polarization in these cells. Together, these findings suggest that Thr567-phosphorylated ezrin has the ability to organize the plasma membrane polarity in several uropod-less situations, whereas uropod protrusion requires an additional mechanism.
Rho–ROCK signaling is required for uropod protrusion, but not for polar cap formation
Because several Ser/Thr kinases (including the Rho effector ROCK and PKCs) have been shown to phosphorylate ERM proteins (Mangeat et al., 1999; Tsukita and Yonemura, 1999; Bretscher et al., 2002), we next treated EL4.G8 cells with an inhibitor for ROCK (Y-27632) or for PKCs (bisindolylmaleimide I [BIM]). BIM had no remarkable effect on the morphology of the cells (not depicted). In contrast, Y-27632 completely eliminated the uropod, although the CD44/p-ERM polar cap was maintained (Fig. 4, A and B). The preservation of p-ERM was confirmed by Western blotting (Fig. 4 C). The phosphorylation status was also unchanged in Y-27632–treated BW5147 cells, despite the fact that the uropod was completely disrupted (unpublished data). These experiments demonstrate that endogenous p-ERM proteins are also capable of constructing the polar cap in uropod-less cells. Likewise, transfectants of WT as well as T567D ezrin exhibited a polar cap but not a uropod under the ROCK-inhibited condition (Fig. 4, A and B). Thus, ERM phosphorylation is independent of ROCK as well as of PKCs in EL4.G8, but ROCK activity is indispensable for the formation of the mature uropod.
Figure 4.
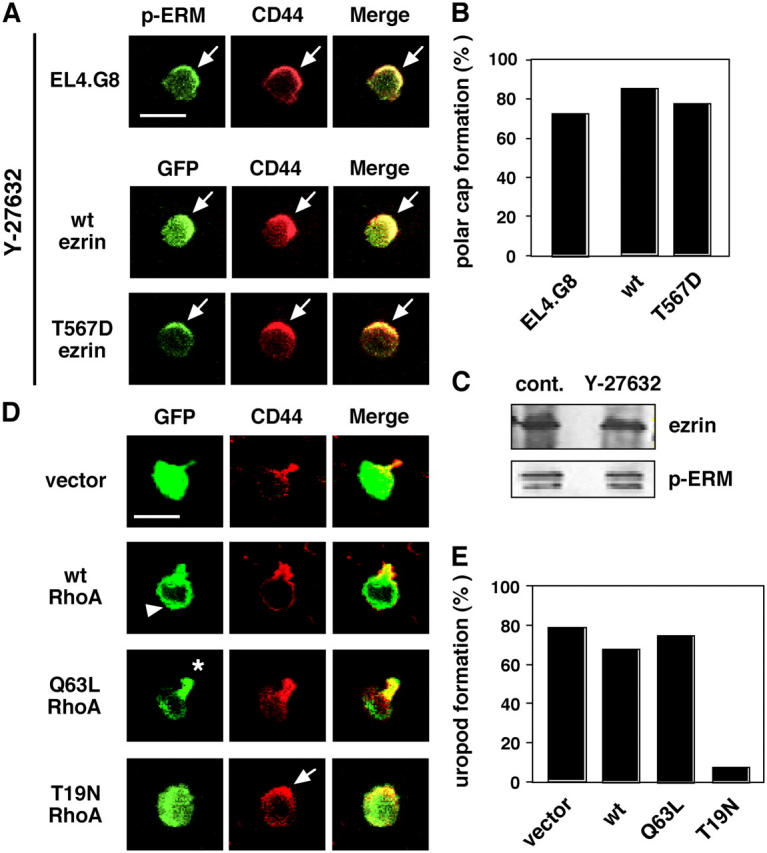
The Rho–ROCK pathway is involved in uropod formation. (A) Y-27632 abolishes uropod but not polar cap formation (arrows). Y-27632–treated parental EL4.G8 cells were stained for p-ERM and CD44 (top panels). GFP-tagged WT and T567D ezrin were also colocalized with CD44 in Y-27632–treated stable transfectants (middle and bottom panels). Bar, 10 μm. (B) Percentage of cells exhibiting the polar cap in A (n > 150). (C) Y-27632 does not alter the phosphorylation status of ERM proteins. The total amount of ezrin and the p-ERM level were determined by Western blotting using lysates from control or Y-27632–treated EL4.G8 cells. (D) WT RhoA shows a diffuse distribution in the cytoplasm, including the leading edge (arrowhead). Constitutively active (Q63L) RhoA is preferentially localized at the rear part of the cells (asterisk), and a dominant-negative form (T19N) abolishes uropod formation but not polar cap formation (arrow). EL4.G8 cells were transiently transfected with WT, Q63L, or T19N RhoA and examined for GFP and CD44 24 h after the transfection. Bar, 10 μm. (E) Percentage of cells exhibiting the uropod in D (n > 75).
Next, we performed transient expression of RhoA mutants tagged with GFP (Subauste et al., 2000) in EL4.G8 cells. WT RhoA showed only a diffuse distribution in the cytoplasm, including the leading edge, without affecting uropod formation (Fig. 4, D and E). In contrast, a constitutively active RhoA (Q63L) was selectively accumulated in the rear part of EL4.G8 cells, including the uropod (Fig. 4, D and E). This suggests that activated Rho and likely the downstream ROCK activation are spatially restricted to this part. On the other hand, forced expression of a dominant-negative mutant of RhoA, T19N, remarkably suppressed uropod formation (Fig. 4, D and E). Among the uropod-less cells, however, the majority (71%) retained the polar cap, further supporting the notion that the formation of the protrusive uropod requires Rho–ROCK activity, although the p-ERM–mediated construction of the plasma membrane polarity is independent of this pathway.
Phosphorylated ezrin interacts with Dbl and induces Rho activation
To address the possibility that p-ERM is involved in Rho–ROCK signaling, we checked the level of Rho-GDP/GTP exchange activity associated with p-ERM in EL4.G8 cells. Using immunoprecipitated p-ERM protein complex from the cell lysate, the exchange activity was assessed by an in vitro nucleotide exchange reaction for recombinant Rho-GDP, followed by RBD (Rho-binding domain)-GST–mediated pull-down detection of Rho-GTP. Strikingly, we detected a substantial nucleotide exchange activity for Rho in p-ERM–containing precipitate, whereas no activity was detected using control antibody (Fig. 5 A). When p-ERM was depleted previously from the cell lysate, the exchange activity associated with ezrin was markedly reduced (Fig. 5 D).
Figure 5.

Rho-GEF activity associated with p-ERM and interaction of Dbl and open-form ezrin in EL4.G8 cells. (A) Protein complex containing p-ERM has Rho-GEF activity. p-ERM was immunoprecipitated from EL4.G8 cell lysate, and the associated Rho-GEF activity was measured compared with that when the immunoprecipitation was performed with control antibody. Rho-GTP in the reaction was pulled down and detected by Western blotting (top). The presence of p-ERM was also confirmed by Western blotting (bottom). (B) Ezrin interacts with Dbl. Dbl coimmunoprecipitated with ezrin was detected by Western blotting. (C) Open-form ezrin specifically binds to Dbl by its NT domain. Ezrin mutants were immunoprecipitated with anti-GFP antibody from lysates of stable transfectants and the interaction with Dbl was analyzed. (D) Most of the Rho-GEF activity associated with ezrin is associated with the phosphorylated form. p-ERM was immunodepleted from EL4.G8 cell lysate, and Rho-GEF activity associated with ezrin was measured compared with that when the immunodepletion was performed with control antibody. (D and E) The relative activity compared with the control level was normalized by the amount of immunoprecipitated ezrin and is shown as a percent of control (histograms). (E) Dbl mediates a major part of the association of Rho-GEF activity with ezrin. Dbl was immunodepleted and Rho-GEF activity associated with ezrin was analyzed. (F) Rho-GEF activity is increased in lysates containing T567D compared with WT ezrin and the activity is associated with the NT but not the CT domain. Rho-GEF activity was analyzed in lysates containing ezrin mutants. The activity in lysates containing T567D relative to that in lysates containing WT ezrin is shown (histogram). (G) Overexpression of Dbl-AID significantly inhibits uropod formation in EL4.G8 cells. EL4.G8 cells were transiently transfected with a construct for GFP-tagged Dbl-AID, and the percentage of cells exhibiting the uropod is shown (n > 180).
The activation of Rho is known to be mediated by various guanine nucleotide exchange factors (GEFs; Schmidt and Hall, 2002), raising the possibility that ezrin could elicit its exchange activity by binding to some GEFs. Thus, we next searched for Rho-GEFs associated with ezrin. One of them, Dbl, was coprecipitated with ezrin from EL4.G8 cell lysate (Fig. 5 B). Similar experiments were also performed using various transfectants for ezrin mutants by immunoprecipitation using anti-GFP antibody. Interestingly, the binding of Dbl to T567D ezrin was markedly augmented compared with its binding to WT ezrin, whereas only a faint Dbl signal was detected in the T567A mutant, suggesting that Dbl is able to associate selectively with activated ezrin (Fig. 5 C). Dbl was coprecipitated with the NT fragment of ezrin but not with the CT fragment (Fig. 5 C), indicating that ezrin binds to Dbl by its NT domain. The level of Rho-GEF activity associated with ezrin mutants was correlated with the Dbl interaction (Fig. 5 F). Moreover, depletion of Dbl from EL4.G8 cell lysate, followed by the precipitation of ezrin, markedly diminished the GEF activity (Fig. 5 E), indicating that a large fraction of the association of GEF activity with ezrin is likely mediated by Dbl.
To determine whether Dbl is involved in uropod formation in vivo, we transfected Dbl-AID, a construct for the GFP-tagged autoinhibitory domain (AID) of Dbl that potentially interferes with the GEF activity mediated by Dbl (Bi et al., 2001), into EL4.G8 cells. Compared with the results of control experiments, the percentage of cells exhibiting the uropod in the Dbl-AID–expressing cells was significantly decreased (Fig. 5 G), suggesting that Dbl plays a substantial role in uropod formation. However, because the inhibitory effect of Dbl-AID is insufficient, it is suggested that the dominant-negative action of this artificial protein fragment might be limited in vivo, or that the other GEFs might associate with p-ERM (Fig. 5, compare the residual GEF activity in E with that in D). Together, these results suggest that phosphorylated open-form ezrin, but not the latent form, potentially functions as an upstream regulator for Rho activation and uropod formation, through its binding to Dbl in EL4.G8 cells.
A polar cap is constructed over the posterior cytoplasm
It is well-established that migrating lymphocytes exhibit unique intracellular polarity, i.e., that the microtubule organizing center (MTOC) and the Golgi apparatus are arranged in the posterior part behind the nucleus, thus revealing a cytoplasmic front–rear axis (Sánchez-Madrid and del Pozo, 1999). MTOC positioning in EL4.G8 cells was rather flexible: 70.5% of cells showed the rear MTOC, whereas in some cells, the MTOC was positioned clearly in the front cytoplasm, near the leading edge (Fig. 6 A). The Golgi apparatus was located in the middle or front part of the cell (in 72.6% of cells; Fig. 6 A), as was revealed by staining with anti-GM130 antibody. These unstable locations of the MTOC or the Golgi apparatus might be caused by the rapid cell cycle in this cell line. Ceramide analogues have been reported to label the Golgi apparatus (Pagano et al., 1991). However, fluorescence-labeled C5-ceramide clearly stained the cytoplasmic components in the rear part of the cell body and the uropod, in addition to the Golgi apparatus, in EL4.G8 cells (Fig. 6 A). In comparison to the MTOC or Golgi positioning, the intracellular arrangement of the front nucleus and the C5-ceramide–labeled rear cytoplasm was obviously stable (93.3% of cells showed this pattern). A similar staining pattern was observed in BW5147 cells bearing a uropod, although the MTOC and the Golgi apparatus in these cells were mostly detected in the rear cytoplasm (>95%; unpublished data). Thus, C5-ceramide was considered to be a useful probe for visualizing the posterior cytoplasm in EL4.G8 cells. Therefore, although they exhibit a slightly different feature of cell polarity from that observed in migrating lymphocytes, EL4.G8 cells show a unique asymmetry along the front–rear axis (Fig. 6 E).
Figure 6.
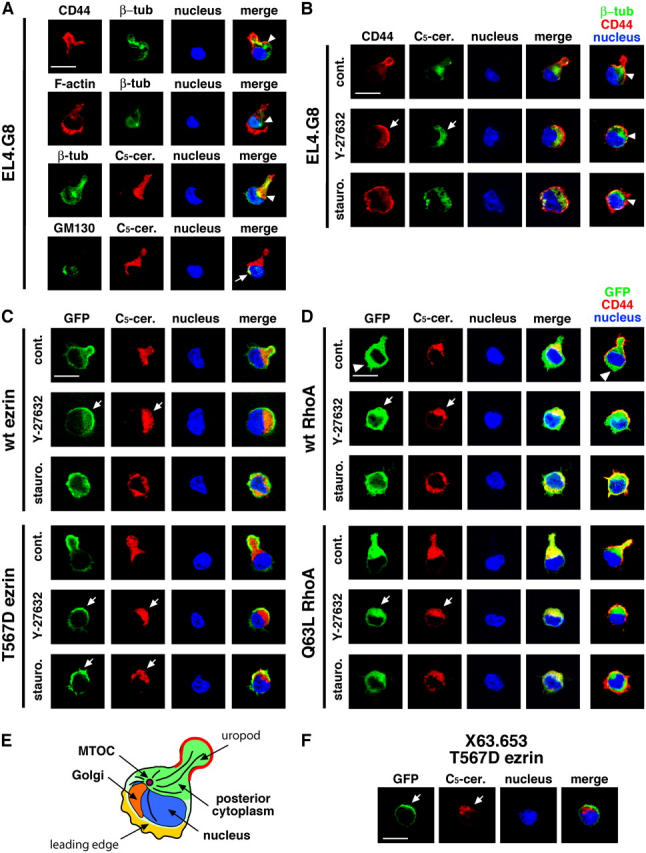
A polar cap covers the posterior part of the cell. (A) EL4.G8 cells exhibit a unique cytoplasmic polarity. EL4.G8 cells were stained for CD44 and β-tubulin, F-actin and β-tubulin, β-tubulin and the posterior cytoplasm (C5-ceramide), or GM130 and the posterior cytoplasm (C5-ceramide). Arrowheads and an arrow indicate the positions of the MTOC and the Golgi apparatus, respectively, in each cell. (B) The polar cap is constructed over the posterior part of the cytoplasm (arrows). Y-27632– or staurosporine-treated EL4.G8 cells were stained for CD44 and the posterior cytoplasm. Composite images for β-tubulin/CD44/nucleus are shown (right) together with the MTOC position (arrowheads). (C) The T567D ezrin–associated polar cap covers the posterior cytoplasm in Y-27632– or staurosporine-treated transfectants (arrows). A similar situation is observed in Y-27632 but not in staurosporine treatment in WT ezrin transfectants (arrows). (D) WT and Q63L RhoA show different intracellular localization in the stable transfectants. WT RhoA is distributed diffusely in the cytoplasm, including the leading edge (arrowheads), whereas Q63L RhoA is localized in the posterior cytoplasm beneath the uropod, or beneath the polar cap when the transfectant is treated with Y-27632 (arrows). Similar but less clear localization of WT RhoA in the posterior cytoplasm is also observed in Y-27632-treated WT RhoA transfectants (arrows). (E) Schematic representation of a typical arrangement of cellular structures in an EL4.G8 cell. (F) The T567D ezrin–associated polar cap covers the posterior cytoplasm in X63.653 cells (arrows). Bars (A–D and F), 10 μm.
Interestingly, in Y-27632–treated cells, the CD44-associated polar cap covered the posterior cytoplasm (Fig. 6 B, arrows), indicating that the plasma membrane polarization induced by p-ERM is aligned with the cytoplasmic polarity axis. A similar pattern was observed in Y-27632–treated WT or T567D ezrin transfectants, and in staurosporine-treated T567D ezrin transfectants (Fig. 6 C). Because the treatment with staurosporine induced a disorganized and fragmented posterior cytoplasm, maintenance of this structure seems to require proper kinase activity. Next, we obtained cells stably transfected with GFP-tagged WT or Q63L RhoA for further analysis. We confirmed that the Q63L RhoA was localized in the rear cytoplasm, whereas WT RhoA was distributed diffusely in the cytoplasm, including the leading edge (Fig. 6 D). As expected, when these cells were treated with Y-27632, the polar cap was apparently correlated with the GFP-Q63L RhoA location. We further obtained stable T567D ezrin transfectants in X63.653 cells, and again observed the polar cap located over the posterior cytoplasm containing the T567D ezrin (Fig. 6 F). Together, these findings suggest that p-ERM has an ability to adapt the plasma membrane polarity, which is established on its own, to the posterior pole of the cytoplasm.
T567D ezrin augments cell migration
In general, the uropod is observed during lymphocyte migration (Sánchez-Madrid and del Pozo, 1999), suggesting that this structure might be important for efficient lymphocyte motility. EL4.G8 cells exhibited clear chemotactic migration toward a chemokine, stromal cell–derived factor-1 (SDF-1), and this chemotaxis was prevented by pertussis toxin, which inactivates Gαi-coupled chemokine receptor signaling, as well as by staurosporine and cytochalasin D (Fig. 7 A). Y-27632 also inhibited the chemotaxis, although withdrawal of the drug restored the activity (Fig. 7 B), indicating that the effects of Y-27632 are reversible. Therefore, ROCK activity is required for the proper chemotactic migration of EL4.G8 cells. Interestingly, among the transfectants of various ezrin mutants, only T567D ezrin transfectants showed higher chemotactic activity toward SDF-1, whereas other transfectants had activity comparable to that of the control cells (Fig. 7 C). We confirmed similar enhancement of chemotaxis in other transfectant clones for T567D ezrin. Together, these findings support the notion that ezrin phosphorylation and Rho–ROCK signaling regulate the efficiency of lymphocyte migration.
Figure 7.

Chemotactic migration of EL4.G8 cells toward SDF-1 and augmentation of the activity in T567D ezrin transfectants. Chemotactic activity was determined by constant-period counting using a flow cytometer and is shown as mean ± SD (A–C). (A) EL4.G8 cells exhibit typical chemotaxis toward SDF-1 (10 ng/ml), and this chemotaxis is markedly inhibited by pretreatment of the cells with pertussis toxin (PTx), staurosporine, or cytochalasin D. (B) EL4.G8 cell chemotaxis is dependent on ROCK activity. Cells were pretreated with Y-27632 and the chemotaxis assay was performed in medium containing the drug (in) or in medium only (wash out [w/o]). (C) T567D ezrin enhances chemotaxis. Transfectants preincubated with or without PTx were examined by the chemotaxis assay. Numbers represent the fold increase of chemotaxis toward SDF-1 (specific activity) compared with that of cells incubated in medium alone. A result typical of at least three experiments is shown.
Discussion
In this study, chiefly using EL4.G8 cells, we showed that the preferential localization of an ERM protein, ezrin, in the uropod requires Thr567 phosphorylation, and that a phosphorylation-mimetic form, T567D ezrin, induces various cellular phenomena, including enhancement of uropod integrity, augmentation of chemotaxis, and polar cap formation. Our findings support the notion that p-ERM proteins are crucial components of lymphocyte morphology and motility. Moreover, we suggest that there is a functional link between the p-ERM–mediated structural framework of the plasma membrane and Rho–ROCK signaling underneath the plasma membrane of the uropod. Uropod protrusion in EL4.G8 cells depends on Rho and ROCK activity. Not only the uropod, but also the p-ERM–associated polar cap, clearly overlaid the rear part of the cytoplasm, in which active Rho is concentrated. Such correlation between specific plasma membrane and cytoplasmic components might stabilize the “posteriority,” and consequently establish a firm front–rear polarity axis, of the cell.
The formation of the polar cap seems to depend on both membrane anchorage and actin binding induced by CT phosphorylation of ERM proteins, but not on other interactive partners. It could be hypothesized that F-actin gathers multiple p-ERM molecules to “cross-link” them at one place underneath the plasma membrane. Under normal conditions, the p-ERM cap structure is likely to be reinforced by further construction of a kind of mature “uropod scaffold” that is mediated by other cellular components but sensitive to staurosporine. This seems reasonable, because even the ezrin mutants lacking actin-binding ability (such as NT and T567D-ΔAB) are able to localize in the uropod, probably by the free NT domain, whereas staurosporine prevents this localization.
ERM proteins have been suggested to participate in Rho activation (Mackay et al., 1997; Sasaki and Takai, 1998; Tsukita and Yonemura, 1999). In vitro interaction of radixin and Dbl was previously reported (Takahashi et al., 1998). In this study, we showed that the protein complex containing phosphorylated ezrin exhibits Rho-GDP/GTP exchange activity, and that only the open-form ezrin can associate with Dbl through its NT domain. These findings suggest that accumulation of p-ERM is likely to augment the local activation of Rho beneath the uropod membrane. The overexpression of Dbl-AID reduced the ability of uropod formation in EL4.G8 cells, indicating that Dbl plays a substantial role in vivo. However, because various pathways can potentially induce Rho activation (Bishop and Hall, 2000; Ridley, 2001), it is also possible that Rho might be activated independently of ERM. The p-ERM–Dbl complex might be important for the maintenance of Rho activity restricted in the uropod membrane. There is a controversy concerning ROCK as a kinase for the CT Thr residues of ERM proteins, and this function has been suggested to depend on the cell type (Matsui et al., 1999; Yonemura et al., 2002). At least in EL4.G8 cells, ROCK is not responsible for ERM phosphorylation. In addition, though we treated EL4.G8 cells with a variety of chemical inhibitors for PKCs (BIM and Gö6976), PKA (KT5720), MEK (PD98059), p38MAPK (SB203580), or MLCK (ML-7), these drugs had no remarkable effect on the morphology of the cells. Thus, what maintains the p-ERM followed by the posterior Rho–ROCK signaling for uropod formation in EL4.G8 cells remains undetermined at this time.
The triggering of lymphocyte migration by various attractants involves complicated signaling (Ward et al., 1998; Hogg et al., 2003), although the overall picture of it remains incomplete. Environmental directional cues are thought to trigger the sequential signaling events upstream of the ERM phosphorylation, as well as the Rho–ROCK pathway, in unpolarized lymphocytes, and to induce the polarized morphology of lymphocytes that are able to migrate efficiently (Fig. 8). Our observations suggest that this process can be dissected into two steps: p-ERM–mediated plasma membrane polarization and Rho–ROCK-mediated uropod protrusion (Fig. 8). In future studies, it will be important to identify any kinases that phosphorylate ERM CT Thr residues, to link the upstream signaling with p-ERM during lymphocyte migration.
Figure 8.

Model of lymphocyte polarization and uropod formation. During the construction of the motile polarized morphology in lymphocytes in response to chemoattraction or adhesion signals (directional cues), two codependent steps are proposed: “posteriorization” of the plasma membrane mediated by ERM phosphorylation, and “uropod protrusion” mediated by Rho–ROCK activation.
In some situations, the uropod of lymphocytes becomes a “sticky” part (Sánchez-Madrid and del Pozo, 1999). This might be because several kinds of adhesion molecules are collected at this part, whereas rapidly regulatable adhesion components, such as integrins, are placed on the leading edge and cell body. In addition, higher membrane–cytoskeleton linking activity by p-ERM proteins likely reduces the local deformability of the plasma membrane. We think that both factors may reduce the rapid motility of the cell. Therefore, uropod formation, by which the relatively sticky and rigid cellular components are restricted to the posterior part, is beneficial for high-speed migration in lymphocytes.
In addition, the uropod may function in effective tail retraction by concentrating the contraction machinery in the rear part of the cell. The Rho–ROCK pathway is known to be involved in a broad spectrum of cell contraction phenomena (Bishop and Hall, 2000; Ridley, 2001). The retraction of the trailing edge has been demonstrated to require this pathway in migrating leukocytes (Alblas et al., 2001; Worthylake et al., 2001; Smith et al., 2003), suggesting that these proteins are crucial for the maintenance of the posterior structure. Recently, it has been shown that the “backness” signal is generated by the activation of Rho by chemoattractant receptors in neutrophils (Meili and Firtel, 2003; Xu et al., 2003). In EL4.G8 cells, we show that Q63L RhoA, which is constitutively active independent of GEFs, is accumulated in the posterior cytoplasm. Because no association between ezrin and Rho could be detected (unpublished data), it can be postulated that there is some mechanism by which Rho-GTP is selectively anchored in the rear part of the cell, which is independent of p-ERM/Dbl function, or actively excluded from the leading edge.
Several reports have shown that Y-27632 induces a long tail that remains behind the cells because of blockage of the posterior retraction (Worthylake et al., 2001; Smith et al., 2003; Xu et al., 2003). However, the requirement of Rho–ROCK signaling for the uropod protrusion in the EL4.G8 model, as revealed by the ability of Y-27632 and dominant-negative Rho to completely abolish the uropod, seems to contradict the idea that the major function of this pathway is only for contraction. The uropod is usually observed as a membrane protrusion lifted off from, rather than attached to, the substratum in lymphocytes (Sánchez-Madrid and del Pozo, 1999). These facts indicate that the uropod, at least in lymphocytes, is not a simple tailing structure but rather is an active machinery for efficient contraction accompanied by somewhat of a protrusive effect, and thus is basically different from the trailing edges observed in adherent cells such as fibroblasts and epithelial cells.
A curious point is that the cytoplasmic polarity (i.e., the intracellular arrangement) of the nucleus, the MTOC, and the Golgi apparatus that is observed in migrating lymphocytes is, in general, in the opposite direction from that observed in many types of adherent cells with migrating morphology. In fibroblasts and astrocytes, the MTOC and the Golgi apparatus are positioned on the leading edge side, relative to the nucleus, during migration (Etienne-Manneville and Hall, 2002; Fukata et al., 2003). This suggests that the construction of the cell polarity in lymphocytes may follow different rules from those followed in adherent cells. Furthermore, although p-ERM is generally localized in an actin-rich compartment of the cell (Mangeat et al., 1999; Tsukita and Yonemura, 1999; Bretscher et al., 2002), it is rather excluded from the actin-rich leading edge of migrating lymphocytes. It is likely that p-ERM–specific anchoring components exist in the posterior compartment in lymphocytes.
ERM proteins are involved in the integrity of the microvilli that constitute the apical compartment in epithelial cells (Mangeat et al., 1999; Tsukita and Yonemura, 1999; Gautreau et al., 2000; Bretscher et al., 2002). Specific localization of ERM proteins has also been reported at the nodes of Ranvier in Schwann cells (Melendez-Vasquez et al., 2001; Gatto et al., 2003). In Drosophila, the sole member of the ERM family, Dmoesin, has been demonstrated to be required for oocyte polarity (Polesello et al., 2002). However, the cellular compartmentalization in these examples is largely dependent on interactions with the neighboring cells or matrix. In highly motile immune cells, in contrast, the dependency of the cell morphology and polarity formation on the environmental structural support seems to be relatively weak. Hence, these cells might possess an intrinsic self-organizing system for plasma membrane polarization, using ERM proteins as the scaffold that connects selective transmembrane proteins to factors involved in cytoplasmic polarity.
Materials and methods
Cells
The mouse T cell lymphoma line, EL4.G8, was obtained from the parental EL4 line by limiting dilution. EL4.G8, BW5147, or X63.653 (P3/X63-Ag8.653) cells were grown in nonadherent culture dishes in RPMI 1640 medium (Invitrogen) supplemented with 10% FCS. For treatment with pharmacological inhibitors, the cells were incubated with 0.5 μM staurosporine, 25 μM BIM, 10 μM Y-27632 (Calbiochem), or 10 μM cytochalasin D (Nacalai Tesque) in the medium for 1 h at 37°C.
Antibodies
The following antibodies or fluorescent probes were used in this study: phycoerythrin-anti–mouse CD43 and CD44 (BD Biosciences), FITC–anti-CD43 (BD Biosciences), Alexafluor 546 phalloidin and Alexafluor 488 goat anti–rabbit IgG (Molecular Probes), anti–β-tubulin (Sigma-Aldrich), FITC–anti-GM130 (BD Biosciences), anti-GFP (CLONTECH Laboratories, Inc.), anti–mouse CD44 (KM201; IQ Products), antiezrin (Upstate Biotechnology), anti–p-ERM (Cell Signaling Technology), normal rabbit IgG anti-Dbl (Santa Cruz Biotechnology, Inc.), anti–LFA-1 (FD441.8 hybridoma culture supernatant), and HRP-anti–rabbit IgG (Jackson ImmunoResearch Laboratories).
Immunofluorescence microscopy
For immunofluorescence staining, cells were fixed with 3% PFA in PBS and permeabilized with 0.2% Triton X-100/PBS or 0.5% saponin/PBS. To detect the intracellular localization of p-ERM, we performed a TCA fixation method, which inactivates phosphatases and maintains the level of p-ERM proteins during sample processing (Hayashi et al., 1999), with a slight modification. In brief, cells were fixed in suspension with 10% TCA solution for 15 min at room temperature, and were rinsed three times with PBS. For detection of the posterior cytoplasm, cells were labeled with BODIPY-FL or -TR C5-ceramide (Molecular Probes) for 30 min at 37°C, before fixation. After being blocked with 5% BSA-PBS, cells were stained with antibodies for 30–60 min. The nucleus was visualized by staining with 0.1 μg/ml DAPI. Stained cells were mounted with Permafluor aqueous mounting medium (Immunotech) and examined with confocal laser scanning microscopes (model MRC-1024 [Bio-Rad Laboratories]; model TSC-SP2 [Leica]). Digital images obtained were processed by Adobe Photoshop software (Katakai et al., 2002).
Counting and measuring uropods and polar caps
We determined the number of cells bearing the uropod by counting cells in the digital images obtained with a phase-contrast microscope (model TND330; Nikon) equipped with a 40× objective and a measuring gauge. Images captured with a 3CCD camera (model C5810; Hamamatsu Photonics) were prepared with Adobe Photoshop software. The length of the uropod was determined by measuring the distance between the imaginary line of the rounded cell body and the tip of the protrusion. Polar caps (indicated by CD44 or GFP localization) formed in drug-treated or transfected cells were counted under a fluorescence microscope.
Plasmids
Expression vectors containing cDNAs encoding EGFP-tagged human WT ezrin, T567A ezrin, and T567D ezrin were provided by Monique Arpin (Institut Curie, Paris, France) (Gautreau et al., 2000; Coscoy et al., 2002). The NT fragment 1–366 and CT fragment 466–586 of ezrin were amplified from full-length ezrin cDNA by PCR, using the following oligonucleotide primers: EzrinNF-EcoRI, 5′-CGTGAATTCCCGAAAATGCCGAAACCAATC-3′; EzrinNIR-SalI, 5′-CGTCGTCGACTTCTCTGTTTCCAGCTGTTG-3′; EzrinCF-EcoRI, 5′-CGTGAATTCGTGATGACAGCA-CCCCCGCCC-3′; and EzrinCR-SalI, 5′-CGTGTCGACGACAGGCCCTCGAACTCGTCG-3′. Dbl-AID was amplified from EL4 cDNA using the following primer pair: N2-XhoI-F, 5′-CCCTCGAGTGGCATCGAATCACCACTATG-3′ and N2-SalI-R, 5′-GGGGTCGACTGATCAGAATTCTTCTCTTGG-3′. The PCR products were subcloned into pEGFP-N1 (CLONTECH Laboratories, Inc.). To construct T567D-ΔAB ezrin, we performed PCR using a plasmid vector for T567D ezrin-EGFP as a template and the following primer pair: SmaI-T567DF, 5′-GAAGGCCCAGGCCCGGGAGGAGAAGC-3′ and AgeI-T567D-ΔABR, 5′-CCGGACCGGTATGCGCTGCTTGGTGTTGCC-3′. The PCR products were subcloned into the T567D ezrin-EGFP plasmid. Expression vectors for EGFP-tagged WT, Q63L, and T19N Rho in a pcDNA3 background were a gift from Klaus Hahn (The Scripps Research Institute, La Jolla, CA) (Subauste et al., 2000).
Transfection
Cells (107 cells per 0.4 ml PBS) were transfected with 30 μg of plasmids in 4-mm-diam cuvettes by electroporation using a Gene Pulser (Bio-Rad Laboratories) at 380 V and 960 μF. After the clump of dead, lysed cells was removed, the cells were diluted 10-fold in medium (containing 10% FCS) and analyzed 24 h after electroporation. For stable transfection, cells electroporated with linearized plasmids were selected with 400 μg/ml G418 in 96-well plates. GFP-positive cells were further enriched by subcloning via limiting dilution, when necessary. Five stable clones for WT ezrin, T567A ezrin, and T567D ezrin, and two for NT ezrin, CT ezrin, and GFP-control vectors, were obtained.
Western blotting
Cells pretreated with or without 10% TCA were lysed in 5× SDS sample buffer. After the samples were boiled, equal amounts of total lysates were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The membranes were soaked in a blocking solution (5% skim milk and 0.2% Tween 20-PBS) for 1 h, and then incubated with primary antibodies for 1 h. After being washed with Tween 20-PBS, membranes were incubated with appropriate HRP-conjugated secondary antibodies for 1 h. Specific bands were visualized by an ECL method (ECL+; Amersham Biosciences).
Immunoprecipitation
Cells were lysed with RIPA buffer (150 mM NaCl, 50 mM Tris, pH 8.0, 0.1% SDS, 0.5% deoxycholate, 1% NP-40, and protease inhibitors [1 mM PMSF, 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 2 μg/ml pepstatin A]) on a rotator for 1 h at 4°C. After insoluble materials were removed by centrifugation, the soluble supernatants were precleared with protein G–Sepharose 4 fast-flow (Amersham Biosciences). Samples were then immunoprecipitated with antibodies and 20 μl of protein G beads. Protein G–bound protein complexes were washed with lysis buffer and eluted by boiling in sample buffer for SDS-PAGE. The immunoprecipitated proteins were detected by Western blotting.
Detection of Rho guanine nucleotide exchange activity
To detect Rho-GEF activity associated with ezrin or p-ERM, we used an assay system combining immunoprecipitation, an in vitro exchange reaction, and pull-down detection of activated Rho. More than 3 × 108 cells were lysed with 2 ml RIPA buffer, and the protein complex was immunoprecipitated using an appropriate antibody. Alternatively, specific antibody (anti–p-ERM or anti-Dbl) was added to the lysate at the preclear step for the depletion of certain protein complexes. The in vitro exchange reaction was performed as described previously (Debant et al., 1996), with a slight modification. 0.5 μg of recombinant His6-RhoA (Cytoskeleton, Inc.) was preloaded with GDP incubating in 100 μl of loading buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5 mM EDTA, 1 mM DTT, and 1 mg/ml BSA) containing 10 μM GDP for 20 min at 25°C. After the incubation, the reaction was quenched with 100 μl of stop exchange buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, and 1 mM DTT), and then diluted with 1.5 ml of exchange buffer (50 mM Tris-HCl, pH 7.5, 200 μM GTPγS, and 2 mM MgCl2). 500 μl of Rho-GDP solution was transferred to a tube containing the immunoprecipitated protein complex, and then incubated with gentle agitation on a rocker at 25°C for 20 min. Then, the reaction was quenched by adding 0.1 volume of stop buffer (50 mM Tris-HCl, pH 7.5, and 60 mM MgCl2). The amount of Rho-GTP in the reaction solution was measured by a pull-down method based on specific binding to Rhotekin-RBD followed by Western blotting using anti-Rho antibody (Rho activation assay biochem kit; BK306; Cyoskeleton, Inc.). The relative amount of active Rho compared with that in the control was calculated by measuring the band density of Rho and normalized to ezrin density.
Chemotaxis assay
The chemotaxis assay was performed using Transwell chambers (6.5-mm diam, 5-mm pore size, Costar). 200,000 cells suspended in 100 μl of medium were placed into the top chamber, and 600 μl of medium containing 10 ng/ml mouse SDF-1 (PeproTech) was added to the bottom well. Alternatively, cells were pretreated with 1 μg/ml pertussis toxin, 0.5 μM staurosporine, 10 μM Y-27632 (Calbiochem), or 10 μM cytochalasin D (Nacalai Tesque) in RPMI 1640 medium for 30 min at 37°C. After 4 h of chemotaxis, cells in the bottom well were collected and the cell number was counted using a FACScalibur flow cytometer (Becton Dickinson) with a constant time period (60 s) (Katakai et al., 2002).
Acknowledgments
We thank Dr. M. Arpin for ezrin cDNAs, Dr. K.M. Hahn and The Scripps Research Institute for RhoA cDNAs, Dr. T. Honjo for allowing us to use a confocal microscope, and Ms. T. Ohfuji for technical assistance.
This work was supported in part by Grants-In-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
The authors have no conflicting financial interests.
J.-H. Lee and T. Katakai contributed equally to this work.
Abbreviations used in this paper: AID, autoinhibitory domain; BIM, bisindolylmaleimide I; CT, COOH-terminal; ERM, ezrin/radixin/moesin; GEF, guanine nucleotide exchange factor; MTOC, microtubule organizing center; NT, NH2-terminal; p-ERM, phosphorylated ERM; ROCK, Rho-associated coiled coil–containing protein kinase; SDF-1, stromal cell–derived factor-1; WT, wild-type.
References
- Alblas, J., L. Ulfman, P. Hordijk, and L. Koenderman. 2001. Activation of RhoA and ROCK are essential for detachment of migrating leukocytes. Mol. Biol. Cell. 12:2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, F., B. Debreceni, K. Zhu, B. Salani, A. Eva, and Y. Zheng. 2001. Autoinhibition mechanism of proto-Dbl. Mol. Cell. Biol. 21:1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, A.L., and A. Hall. 2000. Rho GTPases and their effector proteins. Biochem. J. 348:241–255. [PMC free article] [PubMed] [Google Scholar]
- Bretscher, A., K. Edwards, and R.G. Fehon. 2002. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3:586–599. [DOI] [PubMed] [Google Scholar]
- Coscoy, S., F. Waharte, A. Gautreau, M. Martin, D. Louvard, P. Mangeat, M. Arpin, and F. Amblard. 2002. Molecular analysis of microscopic ezrin dynamics by two-photon FRAP. Proc. Natl. Acad. Sci. USA. 99:12813–12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debant, A., C. Serra-Pages, K. Seipel, S. O'Brien, M. Tang, S.-H. Park, and M. Streuli. 1996. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA. 93:5466–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature. 420:629–635. [DOI] [PubMed] [Google Scholar]
- Fukata, M., M. Nakagawa, and K. Kaibuchi. 2003. Roles of Rho-family GTPases in cell polarization and directional migration. Curr. Opin. Cell Biol. 15:590–597. [DOI] [PubMed] [Google Scholar]
- Gatto, C.L., B.J. Walker, and S. Lambert. 2003. Local ERM activation and dynamic growth cones at Schwann cell tips implicated in efficient formation of nodes of Ranvier. J. Cell Biol. 162:489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautreau, A., D. Louvard, and M. Arpin. 2000. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J. Cell Biol. 150:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, K., S. Yonemura, T. Matsui, S. Tsukita, and S. Tsukita. 1999. Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues. J. Cell Sci. 112:1149–1158. [DOI] [PubMed] [Google Scholar]
- Hogg, N., M. Laschinger, K. Giles, and A. McDowall. 2003. T-cell integrins: more than just sticking points. J. Cell Sci. 116:4695–4705. [DOI] [PubMed] [Google Scholar]
- Katakai, T., T. Hara, M. Sugai, H. Gonda, Y. Nambu, E. Matsuda, Y. Agata, and A. Shimizu. 2002. Chemokine-independent preference for T-helper-1 cells in transendothelial migration. J. Biol. Chem. 277:50948–50958. [DOI] [PubMed] [Google Scholar]
- Kunkel, E.J., and E.C. Butcher. 2002. Chemokines and the tissue-specific migration of lymphocytes. Immunity. 16:1–4. [DOI] [PubMed] [Google Scholar]
- Mackay, D.J.G., F. Esch, H. Furthmayr, and A. Hall. 1997. Rho- and Rac-dependent assembly of focal adhesion complexes and actin filaments in permeabilized fibroblasts: an essential role for ezrin/radixin/moesin proteins. J. Cell Biol. 138:927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat, P., C. Roy, and M. Martin. 1999. ERM proteins in cell adhesion and membrane dynamics. Trends Cell Biol. 9:187–192. [DOI] [PubMed] [Google Scholar]
- Matsui, T., S. Yonemura, S. Tsukita, and S. Tsukita. 1999. Activation of ERM proteins in vivo by Rho involves phosphatidylinositol 4-phosphate 5-kinase and not ROCK kinases. Curr. Biol. 9:1259–1262. [DOI] [PubMed] [Google Scholar]
- Meili, R., and R.A. Firtel. 2003. Two poles and a compass. Cell. 114:153–156. [DOI] [PubMed] [Google Scholar]
- Melendez-Vasquez, C.V., J.C. Rios, G. Zanazzi, S. Lambert, A. Bretscher, and J.L. Salzer. 2001. Nodes of Ranvier form in association with ezrin-radixin-moesin (ERM)-positive Schwann cell processes. Proc. Natl. Acad. Sci. USA. 98:1235–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano, R.E., O.C. Martin, H.C. Kang, and R.P. Haugland. 1991. A novel fluorescent ceramide analogue for studying membrane traffic in animal cells: accumulation at the Golgi apparatus results in altered spectral properties for the sphingolipid precursor. J. Cell Biol. 113:1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesello, C., I. Delon, P. Valenti, P. Ferrer, and F. Payre. 2002. Dmoesin controls actin-based cell shape and polarity during Drosophila melanogaster oogenesis. Nat. Cell Biol. 4:782–789. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J. 2001. Rho GTPases and cell migration. J. Cell Sci. 114:2713–2722. [DOI] [PubMed] [Google Scholar]
- Sánchez-Madrid, F., and M.A. del Pozo. 1999. Leukocyte polarization in cell migration and immune interactions. EMBO J. 18:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, T., and Y. Takai. 1998. The Rho small G protein family-Rho GDI system as a temporal and spatial determinant for cytoskeletal control. Biochem. Biophys. Res. Commun. 245:641–645. [DOI] [PubMed] [Google Scholar]
- Schmidt, A., and A. Hall. 2002. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16:1587–1609. [DOI] [PubMed] [Google Scholar]
- Smith, A., M. Bracke, B. Leitinger, J.C. Porter, and N. Hogg. 2003. LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. J. Cell Sci. 116:3123–3133. [DOI] [PubMed] [Google Scholar]
- Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- Subauste, M.C., M. Von Herrath, V. Benard, C.E. Chamberlain, T.-H. Chuang, K. Chu, G.M. Bokoch, and K.M. Hahn. 2000. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J. Biol. Chem. 275:9725–9733. [DOI] [PubMed] [Google Scholar]
- Takahashi, K., T. Sasaki, A. Mammoto, I. Hotta, K. Takaishi, H. Imamura, K. Nakano, A. Kodama, and Y. Takai. 1998. Interaction of radixin with Rho small G protein GDP/GTP exchange protein Dbl. Oncogene. 16:3279–3284. [DOI] [PubMed] [Google Scholar]
- Tsukita, S., and S. Yonemura. 1999. Cortical actin organization: lessons from ERM (ezrin/radixin/moesin) proteins. J. Biol. Chem. 274:34507–34510. [DOI] [PubMed] [Google Scholar]
- Turunen, O., T. Wahlström, and A. Vaheri. 1994. Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J. Cell Biol. 126:1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, S.G., K. Bacon, and J. Westwick. 1998. Chemokines and T lymphocytes: more than an attraction. Immunity. 9:1–11. [DOI] [PubMed] [Google Scholar]
- Worthylake, R.A., S. Lemoine, J.M. Watson, and K. Burridge. 2001. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 154:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J., F. Wang, A. Van Keymeulen, P. Herzmark, A. Straight, K. Kelly, Y. Takuwa, N. Sugimoto, T. Mitchison, and H.R. Bourne. 2003. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 114:201–214. [DOI] [PubMed] [Google Scholar]
- Yonemura, S., T. Matsui, S. Tsukita, and S. Tsukita. 2002. Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: an essential role for polyphosphoinositides in vivo. J. Cell Sci. 115:2569–2580. [DOI] [PubMed] [Google Scholar]