

Abstract
Integrin αv is required for melanoma cell survival and tumor growth in various models. To elucidate integrin αv-mediated melanoma cell survival mechanisms, we used a three-dimensional (3D) collagen gel model mimicking the pathophysiological microenvironment of malignant melanoma in the dermis. We found that integrin αv inactivated p53 and that suppression of p53 activity by dominant negative p53 or p53-small interfering RNA obviated the need for integrin αv for melanoma cell survival in 3D-collagen and for tumor growth in vivo. This indicates that integrin αv-mediated inactivation of p53 functionally controls melanoma cell survival. Furthermore, we found that melanoma cell integrin αv was required for MAPK kinase (MEK) 1 and extracellular signal-regulated kinase (ERK)1/2 activity in 3D-collagen, whereas inhibition of MEK1 activity induced apoptosis. Surprisingly, MEK1 and ERK1/2 activities were restored in integrin αv-negative melanoma cells by suppression of p53, whereas concomitant block of MEK1 induced apoptosis. This suggests that integrin αv controls melanoma cell survival in 3D-collagen through a pathway involving p53 regulation of MEK1 signaling.
Introduction
Integrins play critical roles for the regulation of tumor growth and invasion (Hood and Cheresh, 2002). For example, expression of integrin αvβ3 has been linked to malignant melanoma progression, in which the vertical growth phase of dermal malignant melanoma displays high expression levels of integrin αvβ3 as compared with horizontally growing melanoma in the epidermis (Albelda et al., 1990; Van Belle et al., 1999). Moreover, in vivo gene delivery of integrin β3 promoted invasive melanoma growth from the epidermis into the dermis in three-dimensional (3D) skin reconstructs (Hsu et al., 1998). Consistently, integrin αv controls melanoma tumorigenicity (Felding-Habermann et al., 1992), by promoting melanoma cell survival as shown in a 3D collagen gel model in vitro, and in full thickness human skin in vivo (Montgomery et al., 1994; Petitclerc et al., 1999). Importantly, block of integrin αvβ3 by an antagonistic anti-integrin αvβ3 mAb induced melanoma cell apoptosis and thereby prevented melanoma tumor growth in mice, whereas reconstitution of the integrin αv subunit into αv-negative melanoma cells rescued cell survival in 3D-collagen as well as in human dermis and thereby restored melanoma tumor growth in vivo (Montgomery et al., 1994; Petitclerc et al., 1999). However, it is unclear how integrin αv may promote melanoma cell survival within 3D microenvironments.
Integrin-mediated cell–matrix interactions trigger a variety of signaling pathways (Giancotti and Ruoslahti, 1999). Signal transduction in cells within 3D-matrices appears to be markedly different from signaling events in cells attached onto two-dimensional (2D) substrates (Cukierman et al., 2002). For example, tyrosine phosphorylation of FAK and EGF-receptor signaling was different in response to cell adhesion within 3D-matrices as compared with attachment to 2D substrates coated onto tissue culture plates (Wang et al., 1998; Cukierman et al., 2001). The integrin-induced MAPK kinase (MEK)-extracellular signal-regulated kinase (ERK) MAPK cascades are key signaling pathways involved in the regulation of adhesion-dependent cell growth and survival (Howe et al., 2002). In melanoma cells, MEK and ERK1/2 can be activated by active mutations of BRAF in 2D cultures (Satyamoorthy et al., 2003). Given that BRAF is mutated in most melanomas, BRAF-dependent MEK activation might be associated with oncogenic behavior of melanoma (Smalley, 2003). However, the role of the Raf–MEK1–ERK1/2 pathway in the regulation of melanoma growth and cell survival is not well characterized. Furthermore, although cell anchorage is needed for activating ERK1/2 in melanocytes (Conner et al., 2003), it is unclear if integrin αv may regulate melanoma cell MEK1–ERK1/2 activity within 3D environments and if this may play a role for the control of melanoma cell survival.
p53-induced apoptotic cell death plays a central role for suppression of tumor growth (Schmitt et al., 2002). Upon activation by various types of stress stimuli, p53 transcriptionally regulates target genes, including PUMA, Apaf 1, Bax, and Bcl-2, which critically regulate mitochondrial apoptotic cascades (Vousden and Lu, 2002). p53 may also induce apoptosis by directly affecting mitochondria (Mihara et al., 2003). In addition, p53 has been associated with death receptors and activation of caspase-8 (Ashkenazi and Dixit, 1998). In angiogenesis, ligation of integrin αvβ3 inactivated vascular cell p53, whereas p53 null mice were refractory to an integrin αv-antagonist that blocked angiogenesis in wild-type (wt) mice (Strömblad et al., 1996, 2002). Interestingly, the p53 gene is rarely mutated in melanoma, although p53 is mutated in most human cancers (Geara and Ang, 1996; Jenrette, 1996). Therefore, melanoma cells typically express wt p53 protein and would because of this be expected to be sensitive to DNA-damaging agents. However, most melanoma cells are extremely radio resistant and irradiation of melanoma cells expressing wt p53 leads to accumulation of p53 but not to apoptosis (Satyamoorthy et al., 2000). Similarly, overexpression of wt p53 by adenovirus in melanoma cells did not induce apoptosis (Satyamoorthy et al., 2000). However, it is unclear why melanoma cells harboring wt p53 can still form tumors and survive. Based on our previous finding that integrin αvβ3 inhibits endothelial cell p53 activity, and that dermal malignant melanoma express high levels of integrin αvβ3, we hypothesized that melanoma cell integrin αvβ3 might suppress p53 activity and thereby obviate the need for p53 mutations.
To elucidate molecular mechanisms for how integrin αvβ3 may promote melanoma cell survival, we used a 3D dermal collagen type I gel model because 3D cell culture systems are capable to provide distinct advantages as compared with 2D systems for studying cellular behavior in a 3D tissuelike context (Zahir and Weaver, 2004). For example, a 3D laminin-rich basement membrane model has been effectively used to examine mammary epithelial cell morphogenesis, polarity, differentiation, proliferation, and survival (Bissell et al., 2002; Zahir and Weaver, 2004). The model with a dermal 3D collagen type I gel that we used mimics the physiological dermal microenvironment where ∼90% of the protein is collagen type I (Montgomery et al., 1994). We found that M21 and M0-αv melanoma cells depended on integrin αv for survival in 3D-collagen. Furthermore, integrin αv functionally promoted melanoma cell survival by suppression of p53 activity and by activation of a MEK1 signaling pathway. Importantly, our results indicate that p53 functionally acts upstream of MEK1 in an integrin αv-dependent pathway that regulates melanoma cell survival in 3D environments, but that melanoma cells are independent of this pathway under 2D culture conditions. This also emphasizes the importance of studying functional intracellular pathways within a 3D environment that mimics the tissue composition.
Results
Integrin αv promotes melanoma cell survival in 3D-collagen
Previous studies indicated a role for integrin αvβ3 in melanoma cell survival both in 3D collagen in vitro and in human skin in vivo (Montgomery et al., 1994; Petitclerc et al., 1999). To elucidate potential molecular mechanisms for integrin αvβ3-mediated melanoma cell survival, we used M21 and M0 melanoma cell lines with or without integrin αv. We characterized and compared the cell surface expression of integrin αvβ3 and of the integrin β3 subunit in these cells by flow cytometry (Fig. 1 A). These melanoma cells were then applied in a 3D dermal collagen gel model enriched in collagen type I. Integrin α2β1 is a major receptor for collagen type I, mediating initial M21 cell adhesion and spreading onto collagen type I, but when collagen is degraded, the exposure of cryptic RGD sites renders collagen type I to also allow binding of integrin αvβ3 (Montgomery et al., 1994). To control the potential effect of integrin αv expression on integrin α2β1, the cell surface expression of integrin α2β1 was analyzed by flow cytometry. However, we detected no differences in integrin α2β1 levels between the different integrin αv-positive and αv-negative M21 subpopulations (unpublished data), indicating that alterations of integrin αv did not affect integrin α2β1 and thereby did not affect the capability to attach to collagen type I. M21 (integrin αv+) and M21L (integrin αv−) cells were then incubated in 3D-collagen and analyzed for apoptosis using Annexin-V staining. We found that M21L (αv−) cells underwent apoptosis to a much higher degree as compared with M21 (αv+) cells after 3–7 d of exposure in 3D-collagen (Fig. 1, B and C), confirming previous findings that integrin αv is critical for M21 melanoma cell survival in 3D-collagen (Montgomery et al., 1994). It should be noted that only low levels of apoptosis were observed in M21 (αv+) and M21L (αv−) cells under 2D culture conditions (d 0). Among different experiments, the number of apoptotic M21L (αv−) cells ranged between 30 and 70% and the onset of apoptosis in these cells varied somewhat in time and was typically clearly visible after 5 d in 3D-collagen (unpublished data). In addition, similar results were obtained by TUNEL staining (unpublished data). The role of integrin αv in melanoma cell survival was also addressed in M0 cell variants, an independent set of melanoma cells either lacking integrin αv (M0) or expressing high levels of wt integrin αv (M0-αv) (Chen et al., 1995). M0 (αv−) and M0-αv (αv+) cells displayed low levels of apoptosis under 2D culture conditions (d 0) (Fig. 1 D). However, M0 (αv−) cells became apoptotic to a clearly higher degree as compared with M0-αv (αv+) after culture in 3D-collagen for 3–5 d, indicating a role for integrin αv also in the survival of these melanoma cells in 3D-collagen. Altogether, these results suggest an essential role for integrin αv in control of melanoma cell survival.
Figure 1.

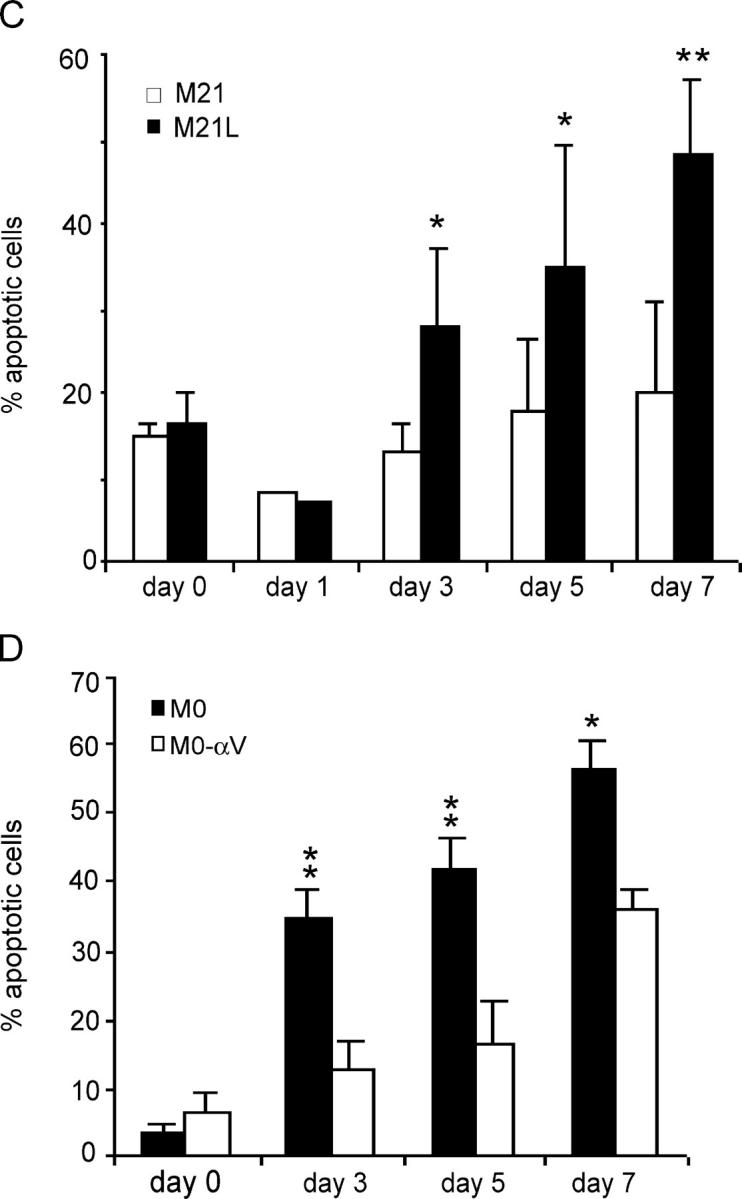
Integrin αv is required for melanoma cell survival in 3D-collagen. (A) Integrin αvβ3 (left) and β3 subunit (right) expression characterized by flow cytometry in the melanoma cells used in this work. The M1 gate marks positive integrin staining as compared with the negative control (not depicted). (B) Apoptosis was detected with Annexin-V staining in integrin αv-positive M21 and αv-negative M21L cells cultured in 3D-collagen for the indicated times of 3–7 d. The displayed staining profiles are representative among three independent experiments. M1 marks Annexin-V positive cells as compared with the control (left row) and the given numbers represent the quantification of the fraction of Annexin-V positive cells in the displayed representative experiment. (C and D) Bar graphs display apoptosis in M21 (αv+) and M21L (αv−) cells (C) and in M0 (αv−) and M0-αv (αv+) (D) cells cultured under 2D conditions (d 0) and within 3D-collagen for the indicated times. Each graph in C and D represents the mean ± SD of Annexin-V positive cells among three independent experiments (* −P < 0.05; ** −P < 0.01; as compared with M21 (C) or M0-αv (D) cells using unpaired two tailed t test).
Melanoma cell integrin αv regulates caspase-9 but not caspase-8
Unligated integrin αvβ3 may induce epithelial cell apoptosis in 3D-collagen by recruitment and activation of caspase-8 (Stupack et al., 2001). To examine potential caspase activation of integrin αv-positive and αv-negative melanoma cells cultured in 3D-collagen, we analyzed the presence of cleaved (active) caspases 8 and 9 in M21 (αv+), M21L (αv−), M0 (αv−), and M0-αv (αv+) cells (Fig. 2, A and B). We found no cleaved caspase-8 in any of these four melanoma cell types when cultured in 3D-collagen, whereas caspase-8 cleavage was clearly visible in cycloheximide-treated Jurkat cells serving as positive control. Interestingly, caspase-9 were cleaved to a higher degree in integrin αv-negative M21L and M0 cells as compared with integrin αv-positive M21 and M0-αv cells, respectively, in 3D-collagen, indicating that loss of integrin αv leads to activation of caspase-9. Given that caspase-9 is active in the mitochondrial apoptotic pathway (Green and Kroemer, 1998), this suggests that the observed apoptosis in M21L (αv−) and M0 (αv−) cells might be mediated via mitochondria.
Figure 2.

Regulation of melanoma cell caspase cleavage by integrin αv in 3D-collagen. Cleaved caspase-8 and caspase-9 were detected by immunoblotting in the integrin αv-positive and αv-negative melanoma M21 (αv+) and M21L (αv−) cells (A) as well as M0 (αv−) and M0-αv (αv+) cells (B) cultured under 2D conditions (d 0) and within 3D-collagen for the indicated times. Active caspase-8 from cycloheximide-treated Jurkat cells served as a positive control for caspase-8 cleavage. Detection of actin served as loading control. The displayed blots are representative among three independent experiments.
Melanoma cell integrin αv inhibits p53 activity in 3D-collagen
Active p53 can induce the mitochondrial apoptotic pathway (Vousden and Lu, 2002) and activation of p53 has also been associated with block of integrin αvβ3 during angiogenesis (Strömblad et al., 1996, 2002). Therefore, we investigated whether integrin αv may also regulate p53 activity in melanoma cells cultured in 3D-collagen. Using electrophoretic mobility shift analysis (EMSA), we examined p53 DNA-binding activity by detecting specific p53 supershift bands with an anti-p53 mAb. First, we verified the specificity by specific and nonspecific competition (Fig. 3 A). Then, p53 activities were examined in M21 (αv+) and M21L (αv−) cells after exposure in 3D-collagen. We found no difference in p53 activity between M21 (αv+) cells and M21L (αv−) cells before exposure in 3D-collagen (d 0), indicating that p53 activity is not regulated by melanoma cell integrin αv under 2D culture conditions. However, the p53 activity was clearly higher in M21L (αv−) cells as compared with M21 (αv+) cells after 5–7 d in 3D-collagen (Fig. 3 B), whereas p53 protein levels displayed no difference (Fig. 3 C). Consistently, we found that the p53 activity was higher in M0 (αv−) cells than in M0-αv (αv+) cells after exposure in 3D-collagen (Fig. 3 D), but without any change in p53 protein levels (Fig. 3 E). Together, our results demonstrate that melanoma cell integrin αv inhibits p53 DNA-binding activity within a 3D dermal collagen environment.
Figure 3.

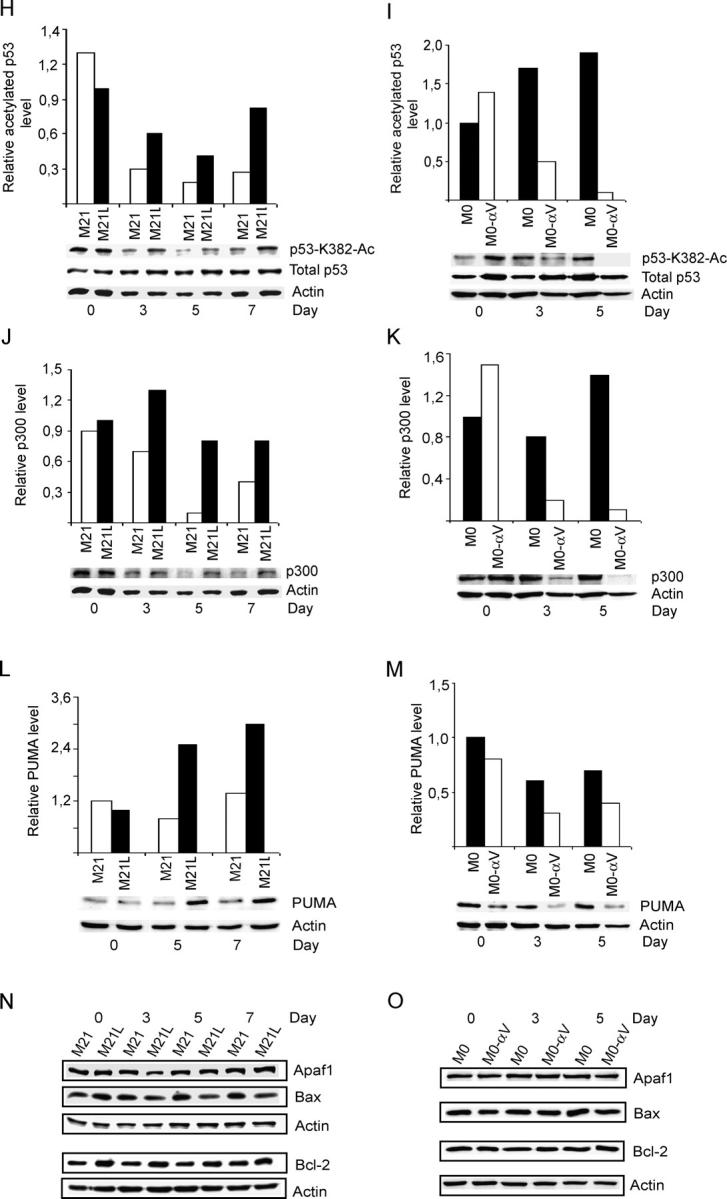
Integrin αv inhibits p53 activity in 3D-collagen. (A) Nuclear extracts from integrin αv-negative M21L cells cultured in 3D-collagen for 5 d were used for detecting p53 DNA-binding activity by EMSA. Specific p53 supershift was obtained by anti-p53 mAb Pab421. Specific or nonspecific competition was generated by incubating the nuclear extracts with the 32P-labeled probe in the presence or absence of 50-fold excess of unlabeled specific or nonspecific oligonucleotides. (B and D) p53-DNA binding activity was detected by EMSA in M21 (αv+) and M21L (αv−) cells (B) and in M0 (αv−) and M0-αv (αv+) cells (D) under 2D conditions (d 0) or within 3D-collagen for the indicated times. Large bar graphs show quantifications of p53 DNA-binding activity EMSA supershift bands displayed immediately below. Boxed bar graphs show the mean ± SD of the ratio of p53 DNA-binding activity bands between M21L (αv−) and M21 (αv+) (B) or between M0 (αv−) and M0-αv (αv+) (D) at each time point based on three to four independent experiments as described in Materials and methods. (* −P < 0.05; ** −P < 0.01, as compared with the corresponding p53 DNA-binding activity ratios (αv−/αv+) at d 0 using unpaired two-tailed t test). (C and E) p53 protein levels were determined by Western blotting in M21 (αv+) and M21L (αv−) cells (C) and in M0 (αv−) and M0-αv (αv+) cells (E). Bar graphs show mean ± SD of quantifications of p53 protein levels relative to actin loading control from three independent experiments. (F and G) Phosphorylated p53 at Ser 15 and Ser 20 were detected by Western blotting in M21 (αv+) and M21L (αv−) cells (F) and in M0 (αv−) and M0-αv (αv+) cells (G). The protein levels of p53 and actin served as controls. (H–O) The levels of acetylated p53-K382, p300, PUMA, Apaf1, Bax, and Bcl-2 proteins were detected by Western blotting in M21 (αv+) and M21L (αv−) cells (H, J, L, and N) and in M0 (αv−) and M0-αv (αv+) cells (I, K, M, and O) cultured under 2D conditions (d 0) or in 3D-collagen for the indicated times. Bar graphs (H and I) show quantifications of p53-K382-Ac bands relative to total p53 levels of the blots shown below, whereas bar graphs in J–M show quantifications of the respective targeted protein bands relative to the corresponding actin levels in blots displayed below the respective graph. Statistical analyses were performed using t-test by analyzing the ratios between the M21L and M21 or M0 and M0-αv values at each time point compared with the corresponding ratios at d 0 as described in Materials and methods based on three independent experiments. p53-K382-Ac (H and I): P < 0.001 for M21L/M21 at d 7 and M0/M0-αv at d 3 and d 5; P < 0.05 for M21L/M21 at d 3. p300 (J and K): P < 0.001 for M0/M0-αv at d5; P < 0.05 for M21L/M21 at d 5 and d 7 and for M0/M0-αv at d 3. PUMA (L and M): P < 0.001 for M21L/M21 at d 5; P < 0.05 for M21L/M21 at d 7 and M0/M0-αv at d 5. Displayed blots and gel shifts are representative among at least three independent experiments.
Posttranslational modifications, including phosphorylations and acetylations, are very important mechanisms for regulation of p53 activity (Vousden, 2002; Brooks and Gu, 2003). To elucidate whether posttranslational modifications of p53 were regulated by integrin αv, we examined potential phosphorylations of p53 at Ser15 and Ser20 and potential acetylation at Lys382 in integrin αv-positive and αv-negative melanoma cells cultured in 3D-collagen. We found that phosphorylation of p53 Ser20 was not altered by integrin αv (Fig. 3, F and G). Although phosphorylation of p53 Ser15 showed a lower level in M0-αv (αv+) as compared with M0 (αv−) cells, this difference was not found when comparing M21 (αv+) and M21L (αv−) cells in 3D-collagen (Fig. 3 F), indicating that differences in phosphorylation of p53 Ser15 is not responsible for the observed regulation of p53 activity. Importantly, melanoma cell integrin αv inhibited acetylation of p53-K382 in 3D-collagen, an acetylation known to promote p53 activity, whereas the p53 protein levels remained constant (Fig. 3, H and I). Given that p53-K382 is an acetylation site for the cofactor p300, p300 protein levels were also quantified. Similar to acetylation of p53-K382, the levels of p300 were reduced in integrin αv-positive M21 and M0-αv cells as compared with the αv-negative M21L and M0 cells, respectively, within 3D-collagen (Fig. 3, J and K). This suggests that integrin αv controls p300 levels in melanoma cells and that this regulation of p300 might be responsible for the differences in p53-K382 acetylation. Given that p53 functions as a transcriptional activator, with transcriptional targets playing critical roles in p53-induced apoptosis (Vousden and Lu, 2002), we also examined the potential transcriptional p53 downstream targets PUMA, Apaf1, Bax, and Bcl-2, all known to be involved in apoptosis regulation. We found that PUMA protein levels were significantly increased in integrin αv-negative M21L and M0 cells as compared with those in αv-positive M21 and M0-αv cells, respectively, (Fig. 3, L and M), thus correlating with the p53-DNA binding activities (Fig. 3, B and C). However, the protein levels of Apaf1, Bax, or Bcl-2 were not regulated by integrin αv in these melanoma cells within 3–7 d in 3D-collagen (Fig. 3, N and O).
Inactivation of p53 compensates for lack of integrin αv in melanoma cell survival and tumor growth
We then examined whether the regulation of p53 activity by integrin αv could functionally mediate the role of integrin αv in melanoma cell survival in 3D-collagen and tumor growth in vivo. To this end, we blocked integrin αv-negative M21L cell p53 activity by stably transfecting a dominant negative (dn) p53-His175 cDNA. A number of p53-His175 expressing clones were identified using Western blotting (unpublished data). The M21L-p53-His175 clones, M21L (αv−) and M21 (αv+) cells were cultured in 3D-collagen and analyzed for apoptosis. In contrast to the parental M21L (αv−) cells, all four M21L-p53His175 clones survived to a similar degree as the integrin αv-positive M21 cells (Fig. 4 A, top). To control for the inhibitory effect of p53-His175 in these clones within 3D-collagen, p53 DNA-binding activities were analyzed after 5 d in 3D-collagen using EMSA (Fig. 4 A, bottom). These results indicate that block of p53 function by p53-His175 compensates for the lack of integrin αv in melanoma cell survival. Furthermore, M21 (αv+) and M21L (αv−) cells as well as two of the M21L-p53-His175 clones were injected subcutaneously (s.c.) into the back of C57/BL nude mice, to test if block of p53 function could compensate for lack of integrin αv in melanoma tumor growth in vivo. Consistent with previous studies (Felding-Habermann et al., 1992), M21 (αv+) cells formed large tumors, whereas M21L (αv−) only formed small or no tumors at all (Fig. 4, B–D). Importantly, M21L (αv−) cells overexpressing dn p53-His175 formed tumors to a similar extent and with a similar growth rate as the integrin αv-positive M21 cells (Fig. 4, B–D). This indicates that inhibition of p53 is essential for melanoma tumor growth.
Figure 4.


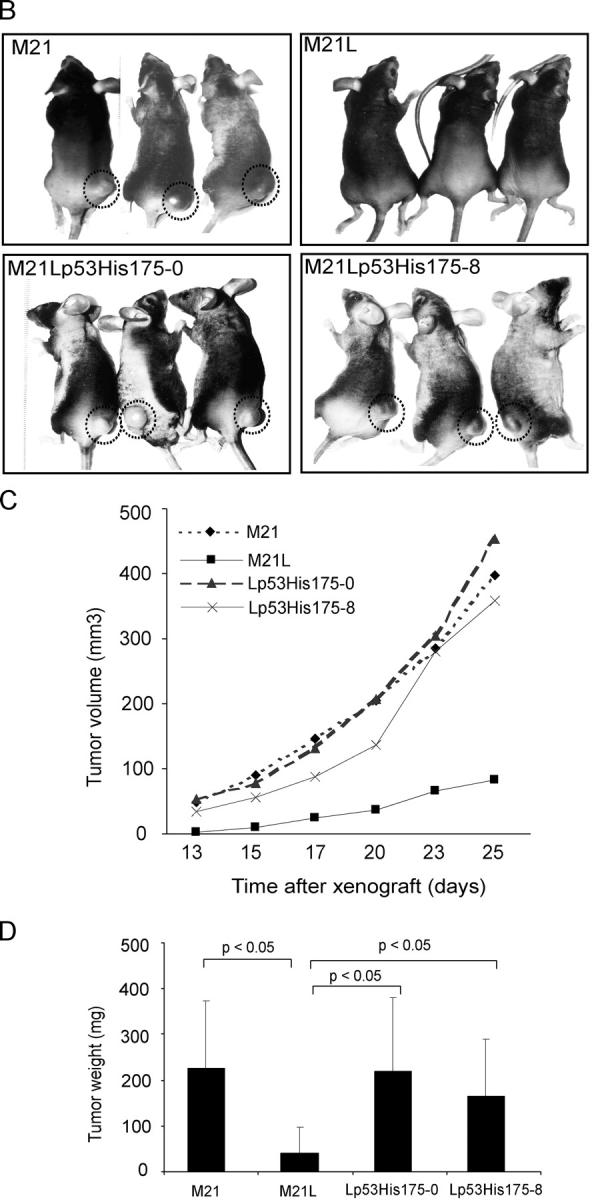
Suppression of p53 compensates for the lack of integrin αv for melanoma cell survival and tumor growth. (A) Apoptosis was analyzed by Annexin-V staining in M21L-p53His175 clones (Lp53His175), M21L (αv−) and M21 (αv+) cells, cultured under 2D conditions (day 0) and within 3D-collagen for the indicated times. The bar graph (top) shows the quantifications of Annexin-V staining of one representative among three independent experiments. p53 activities were detected by EMSA in the indicated cells cultured in 3D-collagen for 5 d (bottom) to control for suppression of p53 activity by p53-His175 within the 3D collagen model used. (B–D) Melanoma cells were injected s.c. into the back of C57/BL mice as described in Materials and methods. (B) Photographs show representative mice from each group 25 d after tumor cell injections. (C) The graph shows the mean tumor volumes for each group of mice over time (n = 7). (D) The wet weights were measured in dissected tumors after 25 d. The graph shows mean weight ± S.D. for each group (n = 7; P < 0.05, unpaired two-tailed t test). (E) M21 (αv+), M21L (αv−), and five individual M21L-p53-siRNA clones (Lp53siRNA) were cultured under 2D conditions (d 0) and within 3D-collagen for the indicated times. Apoptosis was detected by Annexin-V staining. The Western blots for p53 and actin protein levels are shown in the vertical display for the cells indicated immediately to the left of each lane. The displayed results are representative among three independent experiments. (F) The protein levels of PUMA, Bax, and Apaf1 were detected by Western blotting in M21L-p53-siRNA clones, M21L (αv−) and M21 (αv+) cells, cultured in 3D-collagen for the indicated times, controlled by p53 and actin levels.
However, it can be argued that dn constructs are not entirely specific. Therefore, in separate experiments, p53 protein levels were knocked down by stable overexpression of p53-small interfering RNA (siRNA). A number of M21L (αv−) clones with stable expression of p53-siRNA were identified by detection of p53 protein levels, showing that p53 protein levels were almost wiped out (Fig. 4 E, vertical blot). However, suppression of p53 did not impact on integrin αvβ3 or integrin α2β1 expression levels as detected by flow cytometry (unpublished data). The M21L-p53siRNA clones, M21L (αv−) and M21 (αv+) cells were then cultured in 3D-collagen and analyzed for apoptosis. In agreement with the results using dn p53, apoptosis was significantly decreased in the M21L-p53siRNA clones in 3D-collagen as compared with the parental M21L (αv−) cells and were instead similar to that of M21 (αv+) cells (Fig. 4 E), suggesting that inhibition of p53 by p53-siRNA compensated for loss of integrin αv in promotion of melanoma cell survival. Thereby, our results indicate that regulation of p53 activity mediates the function of integrin αv in melanoma cell survival and tumor growth. Furthermore, to investigate whether the p53 transcriptional targets PUMA, Bax, and Apaf1 might be regulated by p53 within 3D-collagen, their protein levels were determined in the M21L-p53siRNA clones and compared with M21L (αv−) and M21 (αv+) cells (Fig. 4 F). The PUMA levels were reduced in M21L-p53siRNA clones as compared with the parental M21L (αv−) cells in 3D-collagen, whereas the levels of Bax and Apaf1 were not affected. This indicates that PUMA might be a direct p53 downstream target in melanoma cells in 3D-collagen.
Integrin αv-dependent MEK1 activity is required for melanoma cell survival in 3D-collagen
Integrin αvβ3 ligation promotes sustained activation of the MEK1–ERK1/2 signaling pathway in vascular cells during angiogenesis (Eliceiri et al., 1998). Because of an active mutation of BRAF, MEK1 and ERK1/2 are constitutively activated in most melanoma cell lines under conventional 2D culture conditions (Satyamoorthy et al., 2003). However, it is unclear if integrin αvβ3 may play a role in regulation of MEK1 and ERK1/2 activities in melanoma cells within a 3D environment. To address this question, we examined MEK1 and ERK1/2 activities in integrin αv-positive M21 and integrin αv-negative M21L cells cultured in 3D-collagen. We detected no differences in MEK1 or ERK1/2 activities between M21 (αv+) and M21L (αv–) cells under 2D culture conditions (Fig. 5 A, d 0). However, both MEK1 and ERK1/2 activities were markedly reduced in αv-negative M21L cells in contrast to αv-positive M21 cells after 3–7 d of exposure in 3D-collagen (Fig. 5 A). This indicates that whereas MEK1 and ERK1/2 is active in melanoma cells under 2D culture conditions, integrin αv appears to be needed for activation of MEK1 and ERK1/2 within a 3D environment. To examine the potential role of MEK1 signaling in integrin αv-mediated melanoma cell survival, we blocked MEK1 activity by treatment of integrin αv-positive M21 cells with two specific MEK1 inhibitors, PD98059 and U0126. Both inhibitors strongly induced apoptosis in M21 (αv+) cells within 3D-collagen (Fig. 5, B and C), but with no changes in integrin αvβ3 cell surface expression levels (not depicted). This shows that integrin αv-dependent MEK1 signaling is required for melanoma cell survival within a 3D environment.
Figure 5.

Integrin αv-dependent MEK1 activity is required for melanoma cell survival in 3D-collagen. (A) M21 (αv+) cells and M21L (αv−) cells were cultured under 2D conditions (d 0) and within 3D-collagen for the indicated times. The levels of active MEK1 and ERK1/2 as well as of total MEK1 and ERK1/2 were detected by Western blotting, with actin as control and the displayed blots are representative among five experiments. (B and C) M21 (αv+) cells within 3D-collagen were treated with the MEK1 inhibitors PD98059 (B) and U0126 (C) or DMSO as a vehicle control. Annexin-V staining detected apoptosis and the bar graphs show the mean ± SD of apoptotic cells among three independent experiments (** −P < 0.01, as compared with vehicle control using unpaired two-tailed t test). Phosphorylated ERK1/2 was detected by Western blotting to control for the suppressive effect of the MEK1 inhibitors. (D) p53 DNA-binding activities were detected by EMSA and p53 protein levels determined by Western blotting in M21 (αv+) cells treated with PD98059 within 3D-collagen for 3–7 d. Phosphorylated ERK1/2 was detected to monitor the inhibitory effect of PD98059, and total ERK1/2 and actin levels were analyzed as controls. Note that the exposure time in this EMSA was longer than for EMSAs displayed in other figures (Fig. 2, B and D).
To examine if block of MEK1 might induce p53 activity within 3D-collagen, p53 DNA-binding activity and p53 protein levels were examined in PD98059-treated integrin αv-positive M21 cells (Fig. 5 D). However, both p53 activity and protein levels were not induced by PD98059, but instead appeared to be inhibited, demonstrating that the suppression of p53 activity by integrin αv is not mediated by MEK1 signaling. In addition, overexpression of a constitutively active (ca) mutant MEK1 (S218D/S222D) did not affect p53 activity in integrin αv-negative M21L cells (unpublished data), likewise indicating that MEK1 does not mediate integrin αv-dependent p53 inactivation.
Suppression of p53 rescues MEK1 activity in integrin αv-negative melanoma cells
Given that MEK1 signaling did not act as a mediator of integrin αv-dependent regulation of p53, we instead tested if suppression of p53 might affect MEK1–ERK1/2 activity within 3D-collagen. We used the M21L (αv−) clones stably expressing dn p53-His175 or p53-siRNA. Under 2D culture conditions (d 0), we observed no influence on MEK1 by expression of p53-His175 or p53-siRNA (unpublished data). However, surprisingly, suppression of p53 by both p53-His175 and p53-siRNA expression rescued MEK1 and ERK1/2 activities in integrin αv-negative M21L cells to similar levels as in integrin αv-positive M21 cells within 3D-collagen (Fig. 6, A and B). However, no changes were observed at MEK1 or ERK1/2 protein levels (Fig. 6, A and B), indicating that the suppression of p53 in cells lacking integrin αv instead only influenced MEK1 and ERK1/2 activities. Furthermore, we examined whether MEK1 activity was still required for melanoma cell survival after knockdown of p53 by siRNA and thus whether MEK1 may functionally act downstream of p53 in regulation of melanoma cell survival. As shown in Fig. 6 C, M21L-p53siRNA (αv−) clones that normally survived to the same extent as M21 (αv+) cells underwent apoptosis to a high degree when treated with the MEK1 inhibitor PD98059 within 3D-collagen, indicating that these cells were still dependent on MEK1 for survival. This indicates that MEK1 is critical and functionally acts downstream of p53 in integrin αv-mediated melanoma cell survival. However, overexpression of ca MEK1 (S218D/S222D) did not rescue M21L (αv−) cell survival in 3D-collagen (unpublished data). Given that block of p53 rescued the same M21L (αv−) cells, this suggests that p53 may act not only through MEK1 regulation, but also through additional downstream pathways to induce melanoma cell apoptosis.
Figure 6.

p53 controls a MEK1-dependent melanoma cell survival pathway. (A and B) The levels of activated and total MEK1 as well as activated and total ERK1/2 were detected by Western blotting in M21 (αv+), M21L (αv−), and in M21L-p53His175 clones (Lp53His175) (A) and M21L-p53-siRNA clones (Lp53siRNA) (B) after 5 d of culture within 3D-collagen. The p53 protein levels were measured as control for the effect of p53-siRNA and actin levels as control. (C) M21L (αv−) clones stably expressing p53-siRNA (Lp53siRNA) were treated with the MEK1 inhibitor PD98059 in 3D-collagen for 5 d and compared with untreated M21L (αv−) and M21 (αv+) cells. Apoptosis was detected by Annexin-V staining. The displayed results are representative among three independent experiments.
Discussion
Several reports have demonstrated that integrin-mediated cell–matrix interactions trigger different signaling and cellular behavior in 3D environments as compared with regular 2D culture conditions (for reviews see Cukierman et al., 2002; Jacks and Weinberg, 2002). Our results reveal another example of how important a 3D environment can be for the cellular response to integrins. We investigated regulatory mechanisms for melanoma cell survival in a 3D dermal collagen model mimicking the pathophysiological environment of malignant melanoma in the dermis (Montgomery et al., 1994). Our results showed that p53 activity as well as MEK1–ERK1/2 signaling was regulated by integrin αv only within 3D-collagen. Moreover, integrin αv-mediated suppression of p53 activity functionally controlled a MEK1-dependent melanoma cell survival pathway. However, none of these components appeared to be critical for regulation of cell survival in 2D cultures. Importantly, our results from the 3D-collagen gel model are similar to our results on melanoma tumor growth in vivo, where suppression of p53 activity rescued tumor growth of αv-integrin negative melanoma cells that otherwise did not grow. Thus, integrin αv-mediated inhibition of p53 was essential for the control of melanoma cell survival within 3D-collagen in vitro as well as for tumor growth in vivo. In fact, a similar pathway has been implicated in angiogenesis, where integrin αvβ3 antagonist-induced vascular cell apoptosis corresponded to induction of p53 activity (Strömblad et al., 1996, 2002). Our results might explain why melanoma that overexpresses integrin αvβ3 appears to have an advantage for growth, survival, invasion, and metastasis (Hsu et al., 1998; Petitclerc et al., 1999; Seftor et al., 1999). However, the molecular mechanisms by which melanoma cell integrin αvβ3 regulates p53 activity remain unclear.
Acetylations are known to be important modulators of p53 activity (Brooks and Gu, 2003). We found that p53 activation caused by lack of integrin αv correlated to p53-K382 acetylation as well as to regulation of p300. Acetylation of p53-K382 by p300 is known to be an important regulatory mechanism for p53 activity (Sakaguchi et al., 1998) and p300 levels are mainly regulated by the rate of proteasomal degradation (Poizat et al., 2000). Given our recent finding that ligation of integrins induces specific proteasomal proteolysis (Bao et al., 2002), it is possible that melanoma cell integrin αvβ3 may specifically regulate the proteasomal turnover of p300 and thereby modulate acetylation of p53-K382 and p53 activity. However, it remains to be elucidated if p300-mediated p53-acetylation is functional in integrin αv-mediated regulation of p53 activity.
A notable fact is that most malignant melanoma harbor wt p53, but display strong resistance to chemotherapy and radiation, suggesting that p53 may be dysfunctional (Satyamoorthy et al., 2001). A number of ways have been suggested to result in the dysfunction of wt p53 in melanoma, including defective downstream effectors. For example, Apaf1, an essential downstream component in p53-induced apoptosis, has been suggested to be lost in malignant melanoma leading to melanoma cells escaping from apoptosis and thereby exhibiting strong chemotherapy resistance (Soengas et al., 2001). However, our results displayed no loss of Apaf1 protein in M21 melanoma cells. Moreover, caspase-9 cleavage was still regulated by integrin αv, suggesting no defect in Apaf1 activity because Apaf1 is needed for caspase-9 cleavage (Zou et al., 1999). However, in preliminary experiments, we tested the integrin αvβ3 cell surface expression in 13 different malignant melanoma cell lines carrying wt p53 and found that eleven of these cell lines expressed high levels of integrin αvβ3, whereas four out of five melanoma cell lines tested with mutant p53 had low or no integrin αvβ3 expression (unpublished data). This indicates a correlation between the p53 mutational status and integrin αvβ3 expression in malignant melanoma cells. Moreover, integrin αvβ3 is known to be highly expressed in most dermal malignant melanoma in patients (Albelda et al., 1990; Seftor et al., 1999; Van Belle et al., 1999). Together, with the role for integrin αv in suppressing wt p53 activity presented here, integrin αv-mediated inactivation of wt p53 might help to explain the lack of need for malignant melanoma p53 mutations in vivo.
Concomitant with enhanced p53 activity, PUMA levels were increased upon lack of integrin αv, but were reduced when also p53 was suppressed, implicating that melanoma cell PUMA might be a downstream target of p53 (Jeffers et al., 2003). Although the levels of Bax protein were not regulated in response to loss of integrin αv, a transcription-independent regulation of bax by p53 might induce apoptosis (Chipuk et al., 2004). Considering that caspase-9 is activated by loss of integrin αv, and also that caspase-9 is known to be essential for p53-induced apoptosis (Soengas et al., 1999), loss of integrin αv might trigger a mitochondrial apoptotic pathway, which is commonly associated with increased PUMA and active caspase-9 (Wang, 2001). However, unlike the suggested caspase-8–dependent apoptotic pathway that may be caused by unligated integrin αvβ3 in epithelial cells (Stupack et al., 2001), our results rule out activation of caspase-8 in the induction of melanoma cell apoptosis caused by lack of integrin αv.
Integrin-activated Raf–MEK1–ERK1/2 signaling regulates various cellular functions, including cell survival (Howe et al., 2002) and melanocytes need anchorage to activate ERK1/2 signaling (Conner et al., 2003). However, although most melanoma cells cultured under 2D conditions display active ERK1/2 induced by a ca BRAF V599E mutation (Satyamoorthy et al., 2003), we found that melanoma cell MEK1–ERK1/2 signaling depended on integrin αv within 3D-collagen. Importantly, this integrin αv-mediated signaling pathway was necessary for melanoma cell survival in 3D-collagen because block of MEK1 activity induced melanoma cell apoptosis, whereas a previous study found no such effect by blocking MEK1 under 2D culture conditions (Smalley and Eisen, 2002). This is another example of how our results display differences in signal transduction and cell survival triggered by cell to 3D matrix interactions as compared with 2D culture conditions. Furthermore, our results are also consistent with an in vivo need for integrin αvβ3 for sustained vascular cell ERK1/2 activation during angiogenesis (Eliceiri et al., 1998). Recently, integrin αvβ3 was found to trigger FAK as well as PAK1-mediated c-Raf-S338 phosphorylation in angiogenic endothelial cells leading to activation of MEK and ERK (Hood et al., 2003). However, we found neither FAK-Y397 or c-Raf-S338 phosphorylation, nor PAK1 kinase activity to be regulated by integrin αv in melanoma cells in 3D-collagen (unpublished data). Because we did not observe any integrin αv-dependent regulation of FAK activity within 3D-collagen, our results do not favor the possibility that integrin-activated FAK may suppress p53 in the regulation of apoptosis (Ilic et al., 1998), although we used a different cell type and a different model compared with previous studies. However, surprisingly, we found that inactivation of p53 functions upstream of MEK1 in regulating integrin αv-mediated melanoma cell survival. p53 can transcriptionally activate MAPK phosphatases, including PAC1, which may play a role in p53-dependent apoptosis by dephosphorylating and inactivating ERK (Yin et al., 2003). However, we found no regulation of PAC1 levels by p53 within our system (unpublished data).
Several ECM proteins can be recognized by αv-integrins, including vitronectin, denatured collagen type I, and osteopontin (Strömblad and Cheresh, 1996). Although integrin α2β1 is a major receptor for collagen type I, M21 melanoma cells are capable of degrading native collagen type I to expose cryptic RGD sites for integrin αvβ3 ligation, suggesting that degraded collagen type I could be a functional ligand for melanoma cell integrin αv-mediated cell survival (Montgomery et al., 1994). Alternatively, given that osteopontin may be involved in melanoma progression from the radial growth phase to the vertical growth phase (Sturm et al., 2002), and that overexpression of osteopontin may promote melanocyte survival in 3D-collagen (Geissinger et al., 2002), endogenously produced osteopontin is another potential ligand for integrin αv within 3D-collagen. It should be noted that although the dermis contains ∼90% collagen type I, it also contains other types of collagen as well as fibronectin and additional ECM components. Furthermore, other cell types are present in the dermis in vivo and the collagen density and assembly in vivo might also differ from the used in vitro 3D-collagen model. Therefore, the used in vitro model is not an exact replica of the dermis environment. However, importantly, our results obtained in the 3D collagen model were consistent with our results obtained in vivo in mouse skin, indicating that the in vitro 3D-collagen model is valid for studies of molecular pathways involved in integrin αv regulation of melanoma cell survival.
In conclusion, we have identified an integrin αv-dependent melanoma cell survival pathway within 3D dermal collagen. We found that integrin αv caused inactivation of wt p53, which functionally promoted melanoma cell survival and tumor growth in vivo. Simultaneously, integrin αv-mediated activation of MEK1 signaling was also required for melanoma cell survival. Surprisingly, we found that whereas p53 acts downstream of integrin αv, it also regulates MEK1 activity in the same functional melanoma cell survival pathway. Although the melanoma cells depended on this integrin αv-mediated pathway for survival in 3D environments, they were independent of this pathway under 2D culture conditions, stressing the importance of studying the role of cell to matrix interactions and their functional signaling pathways within 3D contexts.
Materials and methods
Cell culture and 3D collagen gel model
Human melanoma M21 cells expressing integrin αv and a subpopulation of these cells, M21L cells lacking integrin αv were obtained by negative selection using FACS with an anti-αv mAb (Felding-Habermann et al., 1992). Human melanoma M0 cells lacking the integrin αv subunit and M0-αv cells expressing wt integrin αv were gifts from M. Ginsberg (University of California, San Diego, San Diego, CA; Chen et al., 1995). Cells were cultured in RPMI 1640 medium containing 5% FCS, 2 mM l-glutamine and 50 μg/ml gentamycin. Before culturing cells in a 3D-collagen, cells were adapted into RPMI1640 medium with 1% Nutridoma-SP (Boehringer-Mannheim) for 3 wk as described previously (Montgomery et al., 1994). Dermal collagen type I gels (Cohesion) were also prepared as described previously (Montgomery et al., 1994) and in certain experiments, 50 μM PD98059 (Calbiochem) or 5 μM U0126 (Calbiochem) was mixed into the collagen before polymerization. For the extraction of cells from the collagen gel, 0.25% clostridial collagenase (Worthington) in PBS was used to digest the collagen gel for 10–15 min at 37°C. After washes with cold PBS, cells were used for further analysis.
Flow cytometric determination of integrin expression and apoptosis
1 × 106 cells were incubated with 1 μg/μl of anti-αvβ3 mAb LM609 (CHEMICON International, Inc.), 5 μg/μl of anti-β3 mAb AP3 (GTI) or 10 μg/μl anti-α2β1 mAb 12F1 (CHEMICON International, Inc.) for 1 h at RT. After washing, cells were incubated with FITC-conjugated goat anti–mouse IgG (1:50; Jackson ImmunoResearch Laboratories) for 30 min and analyzed by flow cytometry with CellQuest Software (Becton Dickinson). The fraction of apoptotic cells was detected by flow cytometry after staining with Annexin-V FITC as described by the manufacturer (Biosource International), or by TUNEL using an in situ Cell Death Detection kit (Boehringer-Mannheim).
Stable transfections
Expression vectors with human p53 dn mutant p53-His175 (pCMV-Neo-BAM-p53-His175) and p53-siRNA (pSUPER-p53) were gifts from K. Wiman (Karolinska Institutet) and R. Agami (The Netherlands Cancer Institute, Amsterdam, Netherlands), respectively. M21L (αv−) cells were transfected with 10 μg of p53-His175 cDNA or cotransfected with 10 μg of pSUPER-p53 and 1 μg of pCI-neo plasmid using Lipofectamine 2000 (Invitrogen). 72 h after transfections, cells were treated with 600 μg/ml of G-418 (Invitrogen). After 14 d, G418-resistant clones were picked, expanded and eventually selected based on p53 protein levels and p53 DNA binding activities. ca MEK1 S218D/S222D cDNA, a gift from P. Gerwins (Uppsala University, Uppsala, Sweden), was stably introduced into M21L (αv−) cells by the same method. Positive clones were selected according to induced levels of phosphorylated ERK1/2.
EMSA
Nuclear extracts were prepared and subjected to EMSA as described previously (Selivanova et al., 1996). The protein concentrations of the crude nuclear extracts were determined using a BCA protein quantification kit (Pierce Chemical Co.) using BSA as a standard. 32P-end–labeled double stranded oligonucleotides containing the following human p53 recognition sequence was used: (5′-CAGGCATGTCTGCAGGCAAAGGCATGTCTG-3′). For specific or nonspecific competition, the corresponding unlabeled oligonucleotides or random oligonucleotides were added together in reactions. For supershift, anti-p53 mAb Pab421 (Oncogene) was used and performed as described previously (Selivanova et al., 1996). The analysis was finalized by autoradiography or by a phosphor-imager (Packard).
Western blotting
Cell lysates, protein determination, and Western blotting were performed as described previously (Bao and Strömblad, 2002). The following primary antibodies were used: anti-p53 mAb (DO1), anti-bax pab (N-20), anti-MEK1 pab (C-18), and anti-PAC1 pab (N-19) were purchased from Santa Cruz Biotechnology, Inc.; anti-p53 mAb (Pab421), anti-p53 pab (Ab-7), anti-acetylated p53 Lys382 pab (Ab-1), anti-p300 pab (N-15), anti-PUMA pab (Ab-1), and anti–caspase-8 mAb (clone 1–3) were purchased from Oncogene Research Products; anti–phospho-MEK1/2 pab (Ser 217/221), anti–phospho-ERK1–ERK2 pab (Thr202/Thr204), anti–phospho-p53 pab (Ser 15), anti–phospho-p53 pab (Ser 20), and anti–caspase-9 pab were purchased from New England Biolabs, Inc.; anti-Apaf1 mAb (2E12) was purchased from Alexis; anti-actin mAb (JLA 20) was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa.
In vivo tumor growth
Animal experiments were performed according to a permit from the Stockholm South ethical committee in Sweden. C57BL6 black nude female mice (6–8 w) were obtained from M&B. Melanoma cells (1 × 106) were injected s.c. at the back of the mice. The tumor size was measured every other day with calipers and tumor volumes were calculated by the formula 0.52 × length × width2. The wet weight of dissected tumors was determined at the end of experiments.
Statistical analyses
The following statistical analyses were performed for the results represented in Fig. 3. Although the integrin αv-negative cells constantly displayed higher p53 DNA-binding activity as compared with the integrin αv-positive cells after exposure in 3D-collagen, the absolute values (magnitude) and the time of onset of p53 activity varied between experiments. Therefore, we calculated the ratio of p53 activity between αv-negative cells and the corresponding αv-positive cells at each time point (Fig. 3, B and D, closed bars) and performed a statistical analysis of these ratios compared with the corresponding ratios at the start of the experiments (d 0) by use of unpaired two-tailed t test. Likewise, the ratios between results obtained from integrin αv-negative and integrin αv-positive cells were used for the statistical analyses of Western blot results represented in Fig. 3 (H–M) in the same manner. Statistical evaluations of experiments shown in other figures were performed as described in the figure legends.
Acknowledgments
We are grateful to Dr. Ming Chen for excellent technical assistance during animal experiments. We thank Dr. Mark Ginsberg for providing M0 and M0-αv cells; Drs. Klas Wiman, Reuven Agami, and Pär Gerwins for providing various cDNA vectors; and the Developmental Studies Hybridoma Bank at the University of Iowa for providing mab JLA20. We also thank Drs. Anthony Montgomery, Galina Selivanova, Minna Thullberg, Klas Wiman, and Hongquan Zhang for critical reading of the manuscript. The Clinical Research Center, Huddinge, is acknowledged for the use of their animal facility.
This work was supported by grants to SS from the Swedish Cancer Society, the Swedish Research Council, the Swedish Medical Association and the Cancer Society of Stockholm and to WB from the Robert Lundberg Memorial Fund. SS holds a Senior Scientist position from the Swedish Research Council.
Abbreviations used in this paper: 2D, two-dimensional; 3D, three-dimensional; ca, constitutively active; dn, dominant negative; EMSA, electrophoretic mobility shift analysis; ERK, extracellular signal-regulated kinase; MEK, MAPK kinase; s.c., subcutaneously; siRNA, small interfering RNA.
References
- Albelda, S.M., S.A. Mette, D.E. Elder, R. Stewart, L. Damjanovich, M. Herlyn, and C.A. Buck. 1990. Integrin distribution in malignant melanoma: association of the beta 3 subunit with tumor progression. Cancer Res. 50:6757–6764. [PubMed] [Google Scholar]
- Ashkenazi, A., and V.M. Dixit. 1998. Death receptors: signaling and modulation. Science. 281:1305–1308. [DOI] [PubMed] [Google Scholar]
- Bao, W., and S. Strömblad. 2002. Use of an immobilized monoclonal antibody to examine integrin alpha5beta1 signaling independent of cell spreading. Biol. Proced. Online. 4:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, W., M. Thullberg, H. Zhang, A. Onischenko, and S. Strömblad. 2002. Cell attachment to the extracellular matrix induces proteasomal degradation of p21(CIP1) via Cdc42/Rac1 signaling. Mol. Cell. Biol. 22:4587–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell, M.J., D.C. Radisky, A. Rizki, V.M. Weaver, and O.W. Petersen. 2002. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 70:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, C.L., and W. Gu. 2003. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr. Opin. Cell Biol. 15:164–171. [DOI] [PubMed] [Google Scholar]
- Chen, Y.P., T.E. O'Toole, L. Leong, B.Q. Liu, F. Diaz-Gonzalez, and M.H. Ginsberg. 1995. Beta 3 integrin-mediated fibrin clot retraction by nucleated cells: differing behavior of alpha IIb beta 3 and alpha v beta 3. Blood. 86:2606–2615. [PubMed] [Google Scholar]
- Chipuk, J.E., T. Kuwana, L. Bouchier-Hayes, N.M. Droin, D.D. Newmeyer, M. Schuler, and D.R. Green. 2004. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 303:1010–1014. [DOI] [PubMed] [Google Scholar]
- Conner, S.R., G. Scott, and A.E. Aplin. 2003. Adhesion-dependent activation of the ERK1/2 cascade is by-passed in melanoma cells. J. Biol. Chem. 278:34548–34554. [DOI] [PubMed] [Google Scholar]
- Cukierman, E., R. Pankov, D.R. Stevens, and K.M. Yamada. 2001. Taking cell-matrix adhesions to the third dimension. Science. 294:1708–1712. [DOI] [PubMed] [Google Scholar]
- Cukierman, E., R. Pankov, and K.M. Yamada. 2002. Cell interactions with three-dimensional matrices. Curr. Opin. Cell Biol. 14:633–639. [DOI] [PubMed] [Google Scholar]
- Eliceiri, B.P., R. Klemke, S. Strömblad, and D.A. Cheresh. 1998. Integrin αvβ3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 140:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felding-Habermann, B., B.M. Mueller, C.A. Romerdahl, and D.A. Cheresh. 1992. Involvement of integrin alpha V gene expression in human melanoma tumorigenicity. J. Clin. Invest. 89:2018–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geara, F.B., and K.K. Ang. 1996. Radiation therapy for malignant melanoma. Surg. Clin. North Am. 76:1383–1398. [DOI] [PubMed] [Google Scholar]
- Geissinger, E., C. Weisser, P. Fischer, M. Schartl, and C. Wellbrock. 2002. Autocrine stimulation by osteopontin contributes to antiapoptotic signalling of melanocytes in dermal collagen. Cancer Res. 62:4820–4828. [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Green, D., and G. Kroemer. 1998. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 8:267–271. [DOI] [PubMed] [Google Scholar]
- Hood, J.D., and D.A. Cheresh. 2002. Role of integrins in cell invasion and migration. Nat. Rev. Cancer. 2:91–100. [DOI] [PubMed] [Google Scholar]
- Hood, J.D., R. Frausto, W.B. Kiosses, M.A. Schwartz, and D.A. Cheresh. 2003. Differential αv integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J. Cell Biol. 162:933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, A.K., A.E. Aplin, and R.L. Juliano. 2002. Anchorage-dependent ERK signaling–mechanisms and consequences. Curr. Opin. Genet. Dev. 12:30–35. [DOI] [PubMed] [Google Scholar]
- Hsu, M.Y., D.T. Shih, F.E. Meier, P. Van Belle, J.Y. Hsu, D.E. Elder, C.A. Buck, and M. Herlyn. 1998. Adenoviral gene transfer of beta3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am. J. Pathol. 153:1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic, D., E.A. Almeida, D.D. Schlaepfer, P. Dazin, S. Aizawa, and C.H. Damsky. 1998. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 143:547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks, T., and R.A. Weinberg. 2002. Taking the study of cancer cell survival to a new dimension. Cell. 111:923–925. [DOI] [PubMed] [Google Scholar]
- Jeffers, J.R., E. Parganas, Y. Lee, C. Yang, J. Wang, J. Brennan, K.H. MacLean, J. Han, T. Chittenden, J.N. Ihle, et al. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 4:321–328. [DOI] [PubMed] [Google Scholar]
- Jenrette, J.M. 1996. Malignant melanoma: the role of radiation therapy revisited. Semin. Oncol. 23:759–762. [PubMed] [Google Scholar]
- Mihara, M., S. Erster, A. Zaika, O. Petrenko, T. Chittenden, P. Pancoska, and U.M. Moll. 2003. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 11:577–590. [DOI] [PubMed] [Google Scholar]
- Montgomery, A.M., R.A. Reisfeld, and D.A. Cheresh. 1994. Integrin alpha v beta 3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen. Proc. Natl. Acad. Sci. USA. 91:8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitclerc, E., S. Strömblad, T.L. von Schalscha, F. Mitjans, J. Piulats, A.M. Montgomery, D.A. Cheresh, and P.C. Brooks. 1999. Integrin alpha(v)beta3 promotes M21 melanoma growth in human skin by regulating tumor cell survival. Cancer Res. 59:2724–2730. [PubMed] [Google Scholar]
- Poizat, C., V. Sartorelli, G. Chung, R.A. Kloner, and L. Kedes. 2000. Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Mol. Cell. Biol. 20:8643–8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi, K., J.E. Herrera, S. Saito, T. Miki, M. Bustin, A. Vassilev, C.W. Anderson, and E. Appella. 1998. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 12:2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyamoorthy, K., N.H. Chehab, M.J. Waterman, M.C. Lien, W.S. El-Deiry, M. Herlyn, and T.D. Halazonetis. 2000. Aberrant regulation and function of wild-type p53 in radioresistant melanoma cells. Cell Growth Differ. 11:467–474. [PubMed] [Google Scholar]
- Satyamoorthy, K., T. Bogenrieder, and M. Herlyn. 2001. No longer a molecular black box–new clues to apoptosis and drug resistance in melanoma. Trends Mol. Med. 7:191–194. [DOI] [PubMed] [Google Scholar]
- Satyamoorthy, K., G. Li, M.R. Gerrero, M.S. Brose, P. Volpe, B.L. Weber, P. Van Belle, D.E. Elder, and M. Herlyn. 2003. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 63:756–759. [PubMed] [Google Scholar]
- Schmitt, C.A., J.S. Fridman, M. Yang, E. Baranov, R.M. Hoffman, and S.W. Lowe. 2002. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 1:289–298. [DOI] [PubMed] [Google Scholar]
- Seftor, R.E., E.A. Seftor, and M.J. Hendrix. 1999. Molecular role(s) for integrins in human melanoma invasion. Cancer Metastasis Rev. 18:359–375. [DOI] [PubMed] [Google Scholar]
- Selivanova, G., V. Iotsova, E. Kiseleva, M. Strom, G. Bakalkin, R.C. Grafstrom, and K.G. Wiman. 1996. The single-stranded DNA end binding site of p53 coincides with the C-terminal regulatory region. Nucleic Acids Res. 24:3560–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley, K.S. 2003. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int. J. Cancer. 104:527–532. [DOI] [PubMed] [Google Scholar]
- Smalley, K.S., and T.G. Eisen. 2002. Farnesyl thiosalicylic acid inhibits the growth of melanoma cells through a combination of cytostatic and pro-apoptotic effects. Int. J. Cancer. 98:514–522. [DOI] [PubMed] [Google Scholar]
- Soengas, M.S., R.M. Alarcon, H. Yoshida, A.J. Giaccia, R. Hakem, T.W. Mak, and S.W. Lowe. 1999. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 284:156–159. [DOI] [PubMed] [Google Scholar]
- Soengas, M.S., P. Capodieci, D. Polsky, J. Mora, M. Esteller, X. Opitz-Araya, R. McCombie, J.G. Herman, W.L. Gerald, Y.A. Lazebnik, et al. 2001. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 409:207–211. [DOI] [PubMed] [Google Scholar]
- Strömblad, S., and D.A. Cheresh. 1996. Integrins, angiogenesis and vascular cell survival. Chem. Biol. 3:881–885. [DOI] [PubMed] [Google Scholar]
- Strömblad, S., J.C. Becker, M. Yebra, P.C. Brooks, and D.A. Cheresh. 1996. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin alphaVbeta3 during angiogenesis. J. Clin. Invest. 98:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strömblad, S., A. Fotedar, H. Brickner, C. Theesfeld, E. Aguilar de Diaz, M. Friedlander, and D.A. Cheresh. 2002. Loss of p53 compensates for alpha v-integrin function in retinal neovascularization. J. Biol. Chem. 277:13371–13374. [DOI] [PubMed] [Google Scholar]
- Stupack, D.G., X.S. Puente, S. Boutsaboualoy, C.M. Storgard, and D.A. Cheresh. 2001. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J. Cell Biol. 155:459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm, R.A., K. Satyamoorthy, F. Meier, B.B. Gardiner, D.J. Smit, B. Vaidya, and M. Herlyn. 2002. Osteonectin/SPARC induction by ectopic beta(3) integrin in human radial growth phase primary melanoma cells. Cancer Res. 62:226–232. [PubMed] [Google Scholar]
- Van Belle, P.A., R. Elenitsas, K. Satyamoorthy, J.T. Wolfe, D. Guerry IV, L. Schuchter, T.J. Van Belle, S. Albelda, P. Tahin, M. Herlyn, and D.E. Elder. 1999. Progression-related expression of beta3 integrin in melanomas and nevi. Hum. Pathol. 30:562–567. [DOI] [PubMed] [Google Scholar]
- Wang, F., V.M. Weaver, O.W. Petersen, C.A. Larabell, S. Dedhar, P. Briand, R. Lupu, and M.J. Bissell. 1998. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc. Natl. Acad. Sci. USA. 95:14821–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922–2933. [PubMed] [Google Scholar]
- Vousden, K.H. 2002. Activation of the p53 tumor suppressor protein. Biochim. Biophys. Acta. 1602:47–59. [DOI] [PubMed] [Google Scholar]
- Vousden, K.H., and X. Lu. 2002. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2:594–604. [DOI] [PubMed] [Google Scholar]
- Yin, Y., Y.X. Liu, Y.J. Jin, E.J. Hall, and J.C. Barrett. 2003. PAC1 phosphatase is a transcription target of p53 in signalling apoptosis and growth suppression. Nature. 422:527–531. [DOI] [PubMed] [Google Scholar]
- Zahir, N., and V.M. Weaver. 2004. Death in the third dimension: apoptosis regulation and tissue architecture. Curr. Opin. Genet. Dev. 14:71–80. [DOI] [PubMed] [Google Scholar]
- Zou, H., Y. Li, X. Liu, and X. Wang. 1999. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 274:11549–11556. [DOI] [PubMed] [Google Scholar]