

Abstract
The DNA repair proteins poly(ADP-ribose) polymerase-1 (PARP-1), Ku86, and catalytic subunit of DNA-PK (DNA-PKcs) have been involved in telomere metabolism. To genetically dissect the impact of these activities on telomere function, as well as organismal cancer and aging, we have generated mice doubly deficient for both telomerase and any of the mentioned DNA repair proteins, PARP-1, Ku86, or DNA-PKcs. First, we show that abrogation of PARP-1 in the absence of telomerase does not affect the rate of telomere shortening, telomere capping, or organismal viability compared with single telomerase-deficient controls. Thus, PARP-1 does not have a major role in telomere metabolism, not even in the context of telomerase deficiency. In contrast, mice doubly deficient for telomerase and either Ku86 or DNA-PKcs manifest accelerated loss of organismal viability compared with single telomerase-deficient mice. Interestingly, this loss of organismal viability correlates with proliferative defects and age-related pathologies, but not with increased incidence of cancer. These results support the notion that absence of telomerase and short telomeres in combination with DNA repair deficiencies accelerate the aging process without impacting on tumorigenesis.
Introduction
Aging and aging-associated pathologies such as cancer and degenerative diseases are likely to result from irreversible somatic damage accumulated during life (Hasty et al., 2003). In particular, integrity of the ends of chromosomes, or telomeres, is necessary to maintain chromosomal integrity and determines the replicative life span of many cell types (Blasco, 2003). Mammalian telomeres are composed of several kb-long TTAGGG tandem repeats that, through invasion of the 3′ overhang into the duplex part of the telomeric repeat array, have been proposed to form a t-loop structure. The t-loop is stabilized by TRF2 together with other telomere-binding proteins (de Lange, 2002). After repeated cell division cycles, TTAGGG repeats are lost and telomeres eventually become critically short and dysfunctional. Critically short telomeres are thought to be detected by the DNA repair machinery as a DNA double-strand break (DSB) (d'Adda di Fagagna et al., 2003). Thus, chromosomes that have suffered telomere attrition become substrates for unscheduled repair, primarily by nonhomologous end joining (NHEJ) pathways, leading to end-to-end fusions and other chromosomal aberrations (Goytisolo and Blasco, 2002). Therefore, the final outcome of impaired telomere integrity is chromosomal instability and activation of DNA damage signaling pathways, which in turn triggers cell cycle arrest or apoptosis (de Lange, 2002; d'Adda di Fagagna et al., 2003). Such progressive telomere erosion can be prevented when cells express sufficiently high levels of telomerase, an enzyme composed of a telomerase RNA component (Terc) and a catalytic telomerase reverse transcriptase (Tert) subunit with the unique capacity to add TTAGGG repeats onto chromosome ends (Blasco and Hahn, 2003). Ablation of Terc activity in mice leads to a high frequency of end-to-end chromosomal fusions and, at the organismal level, results in loss of fertility, proliferative defects, and symptoms of premature aging (Blasco, 2003).
Apart from telomere shortening, genome integrity is further jeopardized by ruptures of the DNA backbone, which continuously occur as a consequence of housekeeping cellular functions and exposure to genotoxic factors. Among the different types of damage inflicted on DNA, DSBs are of particular biological relevance because they are more difficult to repair and frequently give rise to translocations and loss or amplification of chromosomal material. The accumulation of DSBs compromises viability and can eventually provoke neoplasms, tissue atrophy, and a variety of other aging-related pathologies (Khanna and Jackson, 2001). To cope with DSBs, organisms have evolved two distinct and complementary signaling and repair pathways, homologous recombination and NHEJ. Although homologous recombination provides a high degree of fidelity during repair, NHEJ is considered a rapid template-free and, consequently, error-prone process catalyzing the simple fusion of broken DNA strands without the requirement of any homology and at the expense of creating local microdeletions. In higher eukaryotes, the NHEJ repair pathway predominates, particularly in the case of radiation-induced DSBs. The first step in NHEJ is the recognition of broken DNA termini by the Ku86–Ku70 heterodimer that recruits the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs). At DSBs, the DNA-PK holoenzyme facilitates binding and activation of additional proteins that collaborate in processing, aligning, and joining the broken ends (Khanna and Jackson, 2001).
One additional enzyme, on which we will focus in this paper, is the poly(ADP-ribose) polymerase-1 (PARP-1) that recognizes and binds to DNA strand breaks. Apart from its role in base excision repair, accumulating evidence supports the notion that PARP-1 interacts with the DSB repair machinery. First, PARP-1 was shown to interact with both DNA-PKcs and Ku86 (Ruscetti et al., 1998; Galande and Kohwi-Shigematsu, 1999); second, loss of PARP-1 function impairs DSB repair (Rudat et al., 2001; Veuger et al., 2003); and third, a genetic interaction has been shown for PARP-1 and the DNA-PK complex (Morrison et al., 1997; Henrie et al., 2003).
Interestingly, besides their role in the repair of DNA lesions, Ku86, DNA-PKcs, and PARP-1 have been also implicated in the maintenance of telomeres (Goytisolo and Blasco, 2002; Bailey and Goodwin, 2004). Mounting evidence suggests that mutation of telomere binding proteins in the absence of telomere shortening may result in loss of telomere capping and, consequently, in chromosomal fusions containing TTAGGG repeats at the fusion point (de Lange, 2002; Goytisolo and Blasco, 2002; Bailey and Goodwin, 2004). In fact, increased end-to-end telomere fusions have been reported for mice deficient in Ku86 and DNA-PKcs (Bailey et al., 1999, 2001; Hsu et al., 2000; Samper et al., 2000; d'Adda di Fagagna et al., 2001; Gilley et al., 2001; Goytisolo et al., 2001). In contrast, PARP-1–deficient mice did not show a significant increase in end-to-end fusions involving telomeric sequences, thus indicating that telomere function was intact in these mice (Samper et al., 2001; Bailey and Goodwin, 2004). Finally, there are conflicting data regarding the impact of PARP-1 deficiency at telomeres, and a role for PARP-1 in telomere length regulation remains to be clearly established (d'Adda di Fagagna et al., 1999; Samper et al., 2001). Interestingly, deletion of Ku86 (Vogel et al., 1999) and, to a lesser extent, DNA-PKcs (Espejel et al., 2004) cause an early onset of aging in mice, whereas PARP-1–deficient mice apparently age normally (Wang et al., 1995; de Murcia et al., 1997).
Recently, we have focused on the function of DNA-PKcs and Ku86 on a telomerase-deficient background (i.e., in a setting that recapitulates more closely the situation of human somatic cells, which generally do not express telomerase). Analysis of mice doubly deficient in Ku86 and telomerase showed that Ku86 mediates NHEJ-dependent telomere fusions and male germ line apoptosis triggered by short telomeres (Espejel et al., 2002a). A similar conclusion was obtained for mice doubly deficient in DNA-PKcs and telomerase. However, in contrast to loss of Ku86, ablation of DNA-PKcs further accelerates the rate of telomere shortening in telomerase-deficient cells (Espejel et al., 2002a,b). Genetic interaction between PARP-1 and telomerase in regulating telomere length and function, however, has not been demonstrated to date, although such interaction has been suggested (d'Adda di Fagagna et al., 1999).
Together, mounting evidence points to a complex functional interplay between telomerase at one hand and the DNA repair proteins Ku86, DNA-PKcs, and possibly PARP-1, at the other hand. Nonetheless, the impact of these putative interactions on organismal aging and cancer remains to be investigated. Here, we report life-long follow-up data of mice double deficient for telomerase and any of the DNA repair proteins Ku86 (Terc−/−/Ku86−/−), DNA-PKcs (Terc−/−/DNA-PKcs−/−), or PARP-1 (Terc−/−/PARP-1−/−), and show that abrogation of Ku86 or DNA-PKcs, but not of PARP-1, cooperates with progressive telomere shortening to compromise organismal viability. Finally, by generating mice doubly deficient in PARP-1 and telomerase, we demonstrate here that PARP-1 deficiency does not result in accelerated rate of telomere shortening even in the absence of telomerase activity, thus suggesting that PARP-1 does not have a major role in controlling telomere metabolism.
Results
Absence of PARP-1 does not affect the rate of telomere shortening or chromosomal instability in telomerase-deficient cells
As outlined above, functional interactions between telomerase and the DNA-PK complex (Ku86 and DNA-PKcs) in telomere length maintenance have been already established (Espejel et al., 2002a,b). However, the role of PARP-1 in telomere maintenance remains controversial (d'Adda di Fagagna et al., 1999; Samper et al., 2001; Bailey and Goodwin, 2004). In particular, the question of whether PARP-1 impacts on telomere erosion and chromosomal instability in telomerase-deficient cells in vivo has not been clarified. To this end, we generated successive generations (G1–G4) of mice doubly deficient for PARP-1 and telomerase, Terc−/−/PARP-1−/− mice, and compared telomere length in mouse embryonic fibroblasts (MEFs) derived from these mice to that of the corresponding Terc−/− controls. Comparisons were always made between littermate mice. As shown in Fig. 1, PARP-1 deficiency did not significantly accelerate telomere shortening in increasing Terc−/− generations. Total telomere length reduction along four successive generations of Terc deficiency was 18.2 and 17.9 kb in the presence or absence of PARP-1, respectively. Consistent with this fact, the percentage of critically short telomeres, i.e., chromosomes without detectable TTAGGG repeats, in MEFs from late generation (G4) Terc−/− mice was comparable in the absence (17.8%) or presence (14.7%) of PARP-1 (Fig. 1). Such signal-free ends provoke end-to-end fusions between chromosomes, chromosome breaks, as well as aneuploidies. Specifically, we did not observe significant differences between G4 Terc−/−/PARP-1+/+ and G4 Terc−/−/PARP-1−/− MEFs regarding Robertsonian-like fusions (0.04 vs. 0 per metaphase), dicentric fusions (0.04 vs. 0.04 per metaphase), or telomere associations (0.08 vs. 0 per metaphase), as well as aneuploidies (0.16 vs. 0.24 per metaphases). Thus, these data argue against a role of PARP-1 in controlling telomere length and telomere capping.
Figure 1.

Telomere length in MEFs derived from successive generations of telomerase-deficient mice lacking PARP-1. Telomere length distribution in primary MEFs from littermate mice of the indicated genotypes. One telomere fluorescence unit (TFU) corresponds to 1 kb of TTAGGG repeats (Zijlmans et al., 1997). The respective genotype, average telomere length, and SD are indicated. Note that SD and not SEM is shown. In addition, the total number of telomeres analyzed and the number of signal-free ends, i.e., telomeres that do not contain any detectable TTAGGG signal as determined by the Q-FISH technique (detection limit is 150 bp), are given. The vertical dashed line is shown to facilitate comparisons between genotypes.
Lack of Ku86 and DNA-PKcs, but not of PARP-1, aggravate the detrimental effects of progressive telomere shortening on organismal survival
To analyze the functional interactions between telomerase and the DNA repair proteins Ku86, DNA-PKcs, or PARP-1 in the context of the organism, we generated three independent mouse colonies simultaneously deficient for Terc and either of these DNA repair activities (see Materials and methods).
In the case of the Ku86/Terc mouse colony (Fig. 2 A), mortality at 1 yr of age in single Terc−/− mice increased from <15% in G1 and G2 to 23 and 66% in G3 and G4, respectively (Fig. 2 A, solid lines and open symbols). Similarly, we found that Ku86-deficient mice on a telomerase-positive background showed a 64% mortality at 1 yr of age, concurring with previous reports (Vogel et al., 1999). Importantly, we observed a further increase of mortality rates in mice doubly deficient for Ku86 and Terc (Terc−/−/Ku86−/−) compared with the single controls (Fig. 2 A). In particular, mortality at 1 yr of age was 79% for G1 Terc−/−/Ku86−/− mice and >90% for G2 and G3 (Fig. 2 A; dashed lines, closed symbols). In fact, in G4 Terc−/−/Ku86−/− mice the combined deficiency of telomerase and Ku86 resulted in complete extinction of the mouse colony at 7 mo of age, with a 50% mortality rate below 2 mo (Fig. 2 A). Interestingly, we have previously shown that Ku86 abrogation in the context of telomerase deficiency does not result in accelerated rate of telomere shortening with increasing mouse generations compared with single Terc-deficient controls (Espejel et al., 2002a), indicating that the synergistic effects of simultaneous Terc and Ku86 abrogation on organismal aging are not due to an accelerated rate of telomere loss. Instead, decreased survival of Terc−/−/Ku86−/− mice is likely to result from the combined effects of short telomeres due to Terc deficiency, and of increased DNA DSBs and loss of telomere protection due to Ku86 abrogation (see Discussion).
Figure 2.
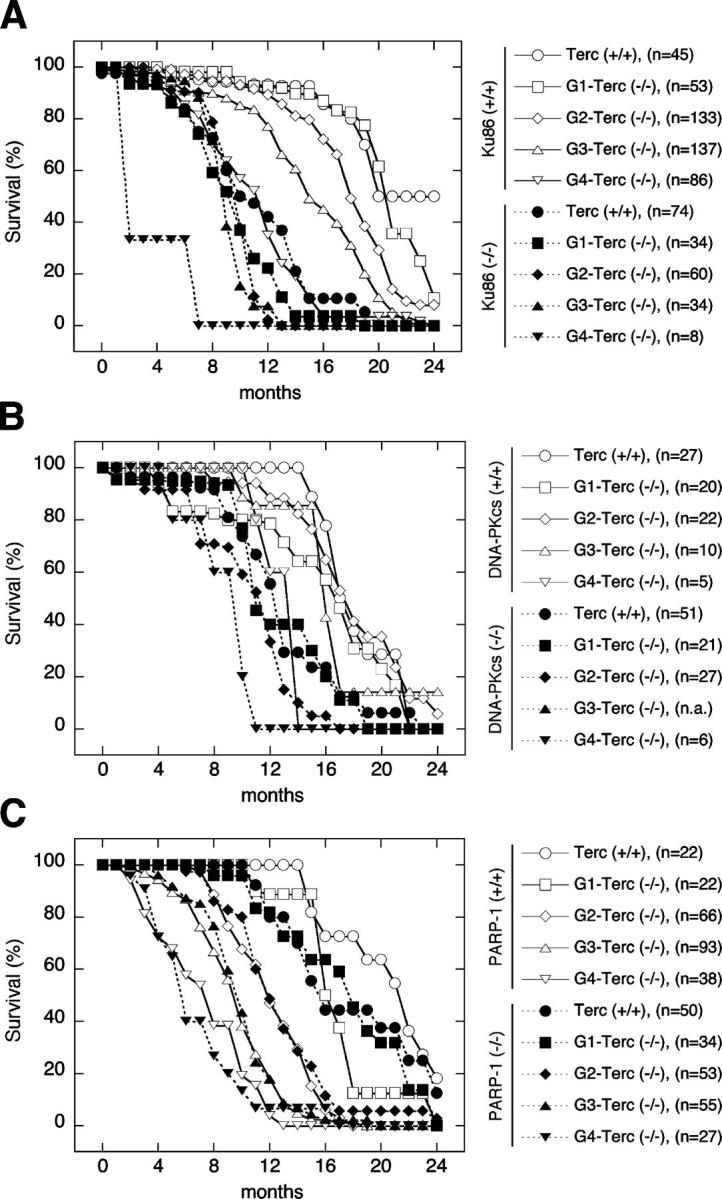
Effect of Ku86, DNA-PKcs, or PARP-1 deficiency on the life span of successive generations of telomerase-deficient mice. Overall survival of mice with intact telomerase (Terc+/+, circles) and four successive generations of telomerase-deficient (Terc−/−) mice, including G1 (squares), G2 (rhombus), G3 (upward triangles), and G4 (downward triangles), which are either wild-type (solid lines, open symbols) or deficient (dashed lines, closed symbols) for Ku86 (A), DNA-PKcs (B), or PARP-1 (C); n.a. = not analyzed.
Interestingly, a similar result was obtained when we deleted DNA-PKcs on a telomerase-deficient background (Fig. 2 B). Mortality in successive generations of Terc−/−/DNA-PKcs+/+ controls at 1 yr of age did not increase to >40% in G4 (Fig. 2 B, solid lines and open symbols). Similarly, mortality in single DNA-PKcs−/− mice at 1 yr of age was 41%. Interestingly, mice doubly deficient for DNA-PKcs and telomerase showed a further increase in mortality at 1 yr of age of 57, 67, and 100% for G1, G2, and G4, respectively (Fig. 2 B, dashed lines and closed symbols). We have previously shown that simultaneous abrogation of telomerase and DNA-PKcs accelerates the rate of telomere shortening by twofold compared with single Terc-deficient controls (Espejel et al., 2002b). Furthermore, DNA-PKcs deficiency also results in loss of telomere protection and increased end-to-end fusions involving telomeric sequences (Bailey et al., 2001; Gilley et al., 2001; Goytisolo et al., 2001). These roles of DNA-PKcs in telomere metabolism, together with the role of DNA-PKcs in NHEJ, are likely to contribute to the detrimental effects on organismal viability of simultaneous Terc and DNA-PKcs deletion (Discussion). In summary, loss of either Ku86 or DNA-PKcs exacerbates the detrimental effects of telomere shortening on the life span of mice.
In the case of the PARP-1/Terc mouse colony (Fig. 2 C), successive generations of single telomerase-deficient cohorts (solid lines and open symbols) displayed a mortality of 18, 50, 80, and 95% at 1 yr of age in G1, G2, G3, and G4, respectively. Of notice, mortality in successive generations of single Terc−/− mice in this particular colony was increased compared with the corresponding Terc−/− cohorts in the Ku86/Terc and DNA-PKcs/Terc colonies (compare Fig. 2 A with Fig. 2 B). This fact might be explained by differences in genetic background impacting on the survival of different mouse colonies (see Materials and methods). Remarkably, Fig. 2 C shows that survival curves of Terc−/−/PARP-1−/− mice (dashed lines and closed symbols) were virtually identical to those obtained from their corresponding single Terc-deficient littermate controls (solid lines and open symbols). Thus, in contrast to what occurred when Ku86 or DNA-PKcs were deleted (see Fig. 2, A and B), we did not observe any aggravation of the loss of viability produced by critically short telomeres in the absence of PARP-1. The lack of genetic interaction between telomerase and PARP-1 is consistent with the apparent lack of interaction between PARP-1 and Terc in the maintenance of telomere length and telomere capping (Fig. 1). However, it is relevant to point out that PARP-1 deficiency alone resulted in a slight decrease in survival of the mouse colony (Fig. 2 C), suggesting a role of PARP-1 on mouse viability during aging, which could be related to the known role of PARP-1 in DNA damage repair (Burkle et al., 2002).
Abrogation of either Ku86 or DNA-PKcs cooperates with telomere attrition to increase proliferative defects and degenerative pathologies
Loss of genome integrity due to impaired DNA repair or telomere dysfunction in somatic cells seriously compromises cell viability. In the context of the aging organism, the two apparently opposed biological endpoints that can result from such accumulation of DNA damage may either be increased neoplasms or, alternatively, tissue atrophy caused by a proliferative defect. To determine how, in the particular setting of telomere attrition, loss of Ku86, DNA-PKcs, and PARP-1 affects the occurrence of age-associated tumors and/or proliferative defects, we killed aged animals that showed signs of poor health, such as reduced activity or dramatic weight loss, and subjected them to exhaustive histopathological analysis (see Materials and methods). In particular, we found that in the presence of telomerase, abrogation of Ku86 did not increase tumor formation compared with wild-type controls (6.7 vs. 10.0%, P = 0.9; Fig. 3 A). Similar to Ku86 deficiency, telomere attrition caused by Terc ablation in increasing generations of Terc−/− mice did not promote tumorigenesis in these mice (Fig. 3 A). The very few preneoplastic lesions and tumors that we could observe in the different wild-type and mutant mouse colonies did not affect a particular tissue, but appeared to be randomly scattered over various organs including the liver (adenoma, hepatoma, sarcoma, and carcinoma), spleen (lymphoma and sarcoma), lung (adenoma), the skin (lipoma and subcutaneous hemangioma), the thymus (lymphoma), ovary (hemangioma), and uterus (hemangioma). Overall, there was no increase of tumor incidence with increasing generations of single Terc−/− mice compared with early generation Terc−/− mice or to wild-type controls (Fig. 3, A, D, and G). In line with our previous observation that short telomeres can act as tumor suppressors (Gonzalez-Suarez et al., 2000), comparisons of G3 Terc−/−/Ku86+/+ mice (1.5% developed tumors) with the corresponding G1 (7.8%, P = 0.05) or wild-type (10.0%, P = 0.04) cohorts revealed a significant reduction of tumors with increasing mouse generations in the absence of telomerase. The combined loss of Ku86 and telomerase in the context of critical short telomeres (G3 Ku86−/−/Terc−/−, 6.7%) did not significantly affect the incidence of tumors compared with late generation (G3) single Terc−/− cohorts (1.5%, P = 0.7; Fig. 3 A). Importantly, tumor incidence of mice doubly deficient in Ku86 and telomerase was never higher than that of the wild-type controls (Fig. 3 A), demonstrating that the combined loss of telomerase and Ku86 does not provoke neoplasms.
Figure 3.

Effect of Ku86, DNA-PKcs, or PARP-1 deficiency on tumorigenesis and tissue atrophies in successive generations of telomerase-deficient mice. Mice with telomerase (Terc+/+) and four successive generations of telomerase-deficient (Terc−/−) mice (G1–G4) that were either wild-type (gray bars) or deficient (black bars) for Ku86 (A–C), DNA-PKcs (D–F), and PARP-1 (G–I) were killed when they showed signs of poor health and were analyzed for the occurrence of tumors (A, D, and G) as well as intestinal (B, E, and H) and testicular atrophy (C, F, and I). The number of animals suffering a given pathology in relation to the total number of animals examined is given above each bar. Significant differences (P < 0.05, Fisher's exact test) between single and double mutant animals are indicated by an asterisk; n.a. = not analyzed.
In contrast with this lack of effect of Ku86 ablation on tumor formation, we found an association between Ku86 deficiency and proliferative defects (Espejel et al., 2002a). As evident from Fig. 3 (B and C), lack of telomerase activity by itself caused atrophy of the large intestine and the testis, which in the first case was further enhanced along successive generations of Terc−/− mice. In particular, the incidence of intestinal atrophy in Ku86-deficient mice at time of death was markedly increased as compared with wild-type littermate controls (13 vs. 0%, P = 0.02; Fig. 3 B), and ablation of Ku86 in Terc-deficient mice increased the incidence of severe intestinal atrophy from 25 to 52% (P < 0.01) in G1, from 35 to 74% (P < 0.0001) in G2, and from 32 to 53% (P = 0.06) in G3. The low incidence of intestinal atrophy in G4 double mutants (20%) has to be interpreted with caution given the low number (n = 5) of viable double mutant animals available in G4.
A similar situation as described above for Ku86-deficient animals was observed when we analyzed DNA-PKcs null cohorts. In the case of tumor susceptibility, there was no significant increase in the incidence of tumors in mice doubly deficient for telomerase and DNA-PKcs compared with the single Terc-deficient mice (e.g., G2-Terc−/− vs. G2-Terc−/−/DNA-PKcs−/−: 10.0 vs. 19.0%, P = 0.3; Fig. 3 D). Similarly, no significant change in tumor incidence was observed along successive generations of telomerase deficiency (e.g., G2-Terc−/− vs. G0-Terc−/−: 10.0 vs. 3.4%, P = 0.3; or G2-Terc−/−/DNA-PKcs−/− vs. G0-Terc−/−/DNA-PKcs−/−: 19.0 vs. 4.9%, P = 0.08; Fig. 3 D). In addition, ablation of DNA-PKcs resulted in severe intestinal atrophy in 18% of mice at time of death, but did not provoke testicular atrophy (Fig. 3, E and F). Nonetheless, similar to Ku86 abrogation (Fig. 3, B and C), DNA-PKcs deficiency appeared to exacerbate intestinal atrophy in the absence of telomerase activity and presence of short telomeres (Fig. 3 E). Although the incidence of this pathology was always higher in the Terc/DNA-PKcs double mutants as compared with their Terc single mutant littermates within the same mouse colony, these differences were not significant (P ≥ 0.1). Only in the particular case of intestinal atrophy in the G2 Terc−/− mouse cohort, a significant increase of this degenerative pathology from 23% in Terc single knock out mice to 52% in Terc/DNA-PKcs double mutants (P = 0.02) was observed.
In the case of single PARP-1 knock out mice, tumor incidence was not significantly altered with increasing generations of PARP-1−/−/Terc−/− mice compared with the single Terc−/− animals. Of note, Terc−/− single knockouts showed a slight reduction in tumor incidence as telomeres become progressively short: wild-type controls (15.4%) versus G1 (0%, P = 0.08), G2 (2.0%, P = 0.05), G3 (2.4%, P = 0.07), and G4 (0%, P = 0.16) Terc-deficient mice (Fig. 3 G). PARP-1 ablation by itself provoked neither intestinal nor testicular atrophy, and we observed only some minor degree of synergism between loss of PARP-1 and telomerase deficiency in the case of intestinal atrophy (Fig. 3 H) in G1 (P = 0.03) and G2 (P = 0.08), but not in late generation (G3 and G4) mice (P > 0.3) (Fig. 3 H). The incidence of testicular atrophy increased along successive generations of Terc−/− mice, but PARP-1 ablation in a telomerase null background did not significantly (P > 0.5) exacerbate this defect (Fig. 3 I).
All together, these data suggest that compromised DNA repair caused by loss of either Ku86 or DNA-PKcs functionally cooperates with progressive telomere shortening in the context of the aging organism to accelerate the age-related development of tissue atrophies without increasing spontaneous tumorigenesis. In contrast, PARP-1 deficiency in the context of short telomeres did not significantly increase age-related tissue atrophies.
To further determine whether the severe degenerative lesions observed in double mutant mice were caused by an increase in apoptosis or, alternatively, by a loss of proliferative potential, we conducted a more detailed analysis of the intestinal epithelium (Fig. 4). Cross sections of large intestine from wild-type mice (Fig. 4 A) show an intact submucosa, muscular layer (ML), and mucosa with well-formed crypts (C) and transverse mucosal folds (F). Single deficiency of Ku86, DNA-PKcs, or PARP-1 occasionally caused mild atrophy of the mucosa. Similarly, ablation of telomerase in G2-Terc−/− mice resulted in detectable, albeit not severe, atrophy of the mucosal layer in the large intestine. Importantly, concomitant abrogation of telomerase and of Ku68, DNA-PKcs, or PARP further exacerbated the phenotype of intestinal atrophy (Fig. 3, B, E, and H). In many cases, signs of degeneration included severe glandular depletion with a loss of mucosal architecture and ulcers of the mucosa with submucosal inflammation and glandular cysts (see Fig. 4 A for a section from a Terc/DNA-PKcs double null mouse). Staining for active caspase-3, a marker for apoptotic cells, suggested the virtual absence of apoptotic cells (<1 cell out of 100 cells in the intestinal epithelium) both in telomerase proficient wild-type and single mutant Ku86−/−, DNA-PKcs−/−, and PARP-1−/− tissues (Fig. 4 B). As shown in Fig. 4 B, positive staining for active caspase-3 was barely detectable in intestinal sections from Terc−/− single mutants or Terc−/−/PARP-1−/− double knockout mice. We have recently reported that deletion of Ku86 and DNA-PKcs abrogates apoptosis in telomerase-deficient male germ cells (Espejel et al., 2002a,b). Therefore, it is tempting to speculate that the combined loss of telomerase and Ku86 or DNA-PKcs will not provoke apoptosis in the intestine. In fact, we could not observe any significant amount of cells staining positive for caspase-3, neither in Terc−/−/Ku86−/− nor in Terc−/−/DNA-PKcs−/− double mutant mice.
Figure 4.
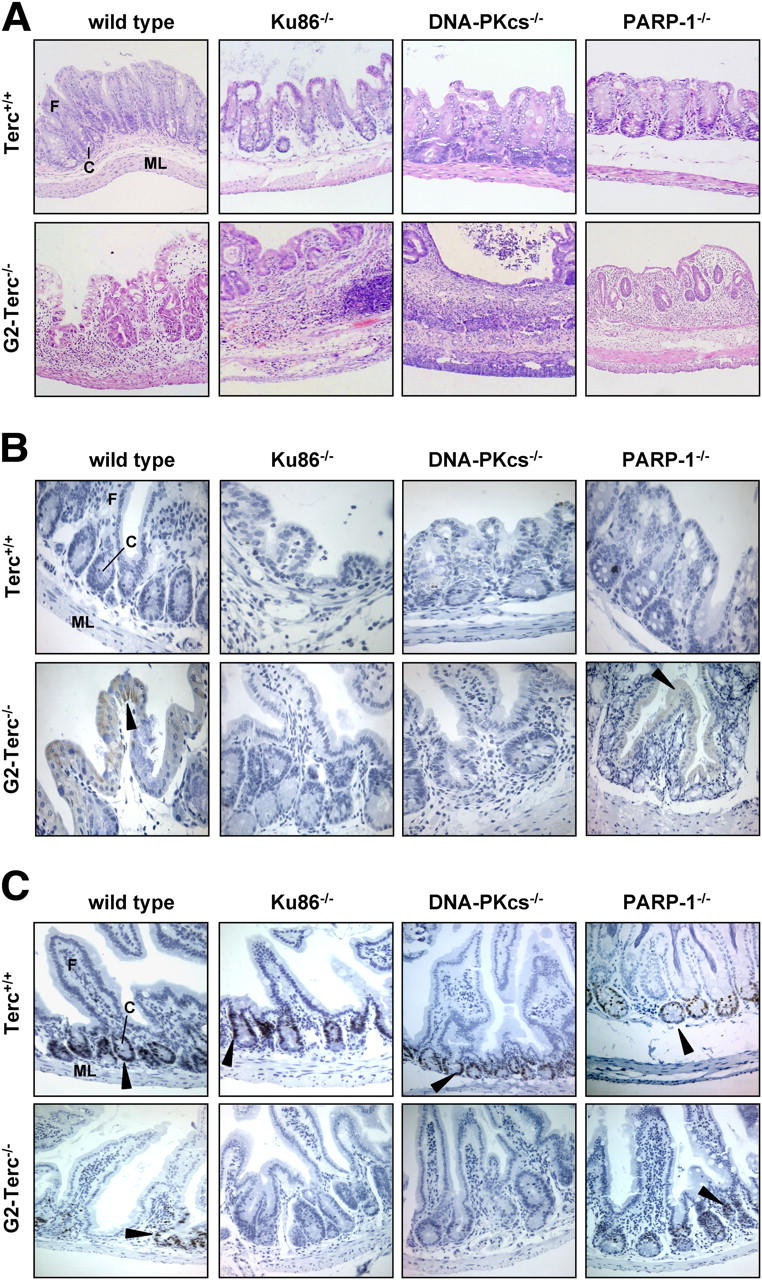
Effect of Ku86, DNA-PKcs, or PARP-1 deficiency on morphology, apoptosis, and proliferative potential of the intestinal epithelium in successive generations of telomerase-deficient mice. Representative photomicrographs of paraffin sections of large intestine from 1-yr-old mice of the indicated genotype stained with Harris hematoxylin and eosin (A), immunostained for active caspase-3 (B), and Ki67 (C). Arrowheads indicate examples of positive immunostaining for active caspase-3 and Ki67. ML, muscular layer; C, crypt; F, fold.
Because these data argue against increased apoptosis as the cause of the intestinal atrophy in double mutant Terc-deficient animals, we next explored the proliferative potential of the intestinal epithelium by Ki67 staining (Fig. 4 C). In accordance with the view that proliferating progenitor cells of the intestinal epithelium reside in the lower half of intestinal crypts (Clatworthy and Subramanian, 2001), intense staining for Ki67 was found to be confined to crypt structures. As compared with wild-type mice (an estimate of 81 ± 7% Ki67-positive crypt cells; mean ± SD from 5 sections with at least 10 crypts each), cross sections from Terc-proficient Ku86−/− (62 ± 16%, n = 5, P = 0.04), DNA-PKcs−/− (41 ± 19%, n = 6, P = 0.004), and PARP−/− (55 ± 18%, n = 6, P = 0.02) mice contained a slightly reduced number of proliferating cells in their crypts. Ablation of Terc in 1-yr-old G2-Terc−/− mice caused a more pronounced reduction of Ki67 expression (38 ± 17%, n = 5, P = 0.001) as compared with wild-type epithelia. Importantly, loss of Terc dramatically reduced the amount of Ki67-positive crypt cells in second generation (G2) Ku86−/−/Terc−/− (3 ± 3%, n = 5, P = 0.01) and DNA-PKcs−/−/Terc−/− (2 ± 4%, n = 6, P = 0.01), but not in PARP−/−/Terc−/− (28 ± 10%, n = 6, P = 0.3) double mutant animals compared with the Terc single mutant. Similarly, a comparison between the Terc-deficient double mutants with their corresponding Terc-proficient controls (Ku86−/− vs. Ku86−/−/Terc−/−: 62 vs. 3%, P = 0.001; DNA-PKcs−/− vs. DNA-PKcs−/−/Terc−/−: 41 vs. 2%, P = 0.002; PARP−/− vs. PARP−/−/Terc−/−: 55 vs. 28%, P = 0.01) suggests that Terc deficiency cooperates with loss of Ku86 and DNA-PKcs, and to a lesser extent, with deletion of PARP-1 in reducing the proliferative potential of epithelial cells in the large intestine. These data further indicate that this proliferative defect might account for the phenotype of intestinal atrophy observed in Terc-deficient animals lacking the DNA repair proteins Ku86, DNA-PKcs, or PARP-1.
Progressive telomere shortening combined with abrogation of Ku86, DNA-PKcs, or PARP-1 results in accelerated organismal aging
The observed relationship between chronological age and mortality (Fig. 2), as well as the frequent occurrence of proliferative defects (Fig. 3) in double mutant mice indicate a genetic interaction between telomerase and DNA repair proteins in longevity. To further study whether telomere shortening cooperates with loss of Ku86, DNA-PKcs, or PARP-1 to accelerate the aging process, we studied the age distribution of age-related pathologies detected in moribund animals (see Materials and methods; Online supplemental material for detailed list of pathologies). With the exception of intestinal and testicular atrophy (Fig. 3), the incidence of any individual pathology was not significantly altered when comparing different genotypes and/or different generations of Terc-deficient animals (see Online supplemental material). Thus, to get an insight into the overall organismal fitness of these animals as a function of their age, we calculated the onset of aging-associated pathologies at <1 yr of age in a given mouse group relative to the total number of pathologies detected in this group; for example, in Terc+/+/Ku86−/− cohorts we detected a total of 21 senile lesions (see Online supplemental material), and 11 of them were detected in animals at < 1 yr of age (Fig. 5 A). In this way, we obtained a more comprehensive measure for the relative incidence of senile lesions in young animals (<1 yr of age) either of the wild type or deficient for Ku86, DNA-PKcs, or PARP-1 along successive generations of telomerase deficiency (Fig. 5). Concurring with our previous observations (Herrera et al., 1999), we found an association of progressive telomere shortening with symptoms of premature aging. Although we did not observe any senile lesion in wild-type animals at <1 yr of age (0%), progressive telomere shortening incremented the incidence of such pathologies to 8% in G1 (P = 0.5), 1% in G2 (P = 0.9), 14% in G3 (P = 0.2), and to 38% in late-generation G4-Terc−/− animals (P = 0.01) (Fig. 5 A, gray bars). In the presence of telomerase, ablation of Ku86 caused a high incidence of senile lesions in young animals (52%), but progressive exhaustion of telomeres further exacerbated this premature aging phenotype with 68, 92, 100, and 100% of senescence-associated pathologies detected in young animals from G1 (P = 0.1), G2 (P < 0.001), and late generation G3 (P < 0.01) and G4 (P = 0.2) Terc−/− cohorts, respectively (Fig. 5 A, black bars). The cooperative impact of telomerase ablation and Ku86 deficiency on the premature occurrence of age-related pathologies was also evident from a comparison of Ku86-deficient cohorts with their respective wild-type littermates in G1 (P < 0.001), G2 (P < 0.001), G3 (P < 0.001), and G4 (P = 0.06) (Fig. 5 A, black bars versus gray bars).
Figure 5.
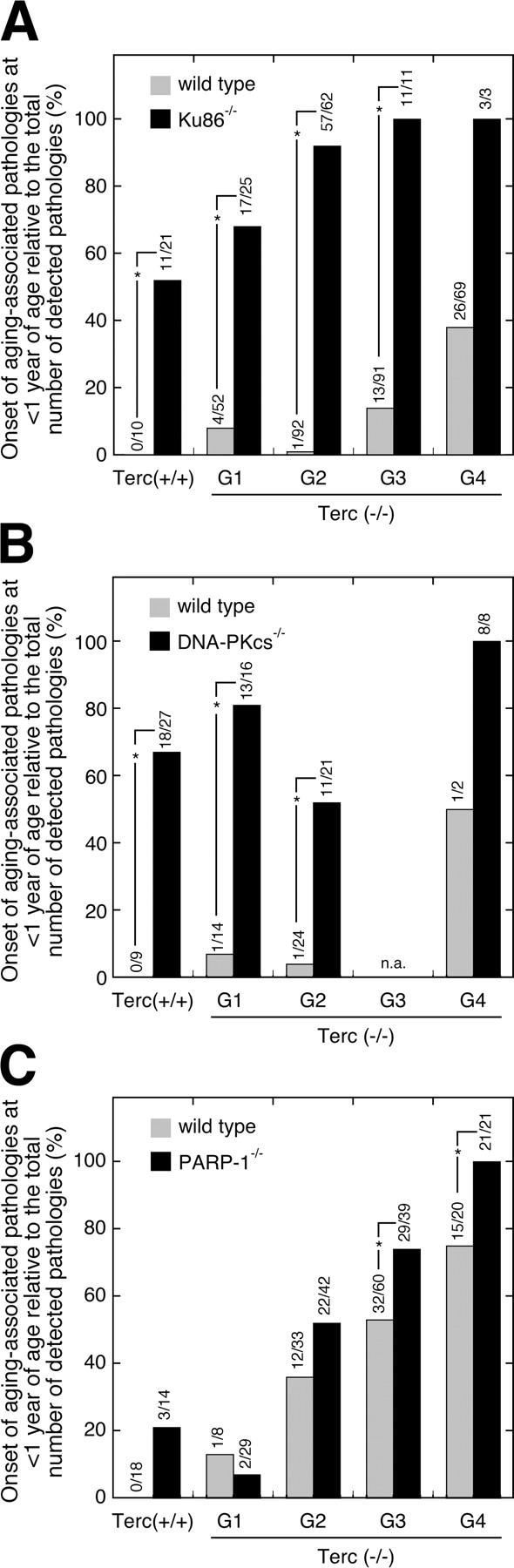
Effect of Ku86, DNA-PKcs, or PARP-1 deficiency on the early onset of age-related pathologies in successive generations of telomerase-deficient mice. Mice with telomerase (Terc+/+) and four successive generations of telomerase-deficient (Terc−/−) mice (G1–G4) that were either wild-type (gray bars) or deficient (black bars) for Ku86 (A), DNA-PKcs (B), and PARP-1 (C) were killed when they showed signs of poor health and analyzed for the occurrence of a variety of age-associated pathologies (see Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200407178/DC1). Here, we show the onset of these aging-associated pathologies in animals younger than 1 yr of age in a given mouse group relative to the total number of detected pathologies in that group. The number of senile lesions detected in young animals (<1 yr) in relation to the total number of lesions detected in the respective mouse group is given above each bar; for example, as shown in A, in the Terc+/+/Ku86−/− mouse cohort we detected a total of 21 senile lesions and 11 of them were detected in mice younger than 1 yr of age. Significant differences (P < 0.05, Fisher's exact test) between single and double mutant animals are indicated by an asterisk; n.a. = not analyzed.
Loss of DNA-PKcs function by itself accelerated aging with 67% of senile lesions affecting young animals (Fig. 5 B). Mice with simultaneous ablation of telomerase and DNA-PKcs exhibited a frequency of senile lesions of 81% in G1 (P = 0.17 vs. G0), 52% in G2 (P = 0.14 vs. G0), and 100% (P = 0.06 vs. G0) in G4 (Fig. 5 B, black bars). The high frequency of senile lesions in late generation G4 double mutants (100%) was not significantly elevated when compared with G1 (P = 0.27), but becomes significant when compared with G2 (P = 0.02). Expectedly, the incidence of premature aging symptoms in the telomerase-deficient controls that were wild-type for DNA-PKcs was much lower and did not exceed 7% in early (G1 and G2) and 50% in late (G4) generation Terc−/− mice, respectively (Fig. 5 B, gray bars). As shown by a comparison of the incidence of these pathologies in DNA-PKcs−/−/Terc+/+, G1 DNA-PKcs−/−/Terc−/−, and G2 DNA-PKcs−/−/Terc−/− cohorts with the corresponding data from their respective DNA-PKcs wild-type littermates, i.e., with DNA-PKcs+/+/Terc+/+, G1 DNA-PKcs+/+/Terc−/−, and G2 DNA-PKcs+/+/Terc−/− cohorts, ablation of DNA-PKcs caused a highly significant (P < 0.01) increase of senile lesions in young mice (Fig. 5 B, black bars versus gray bars). Due to the low number of animals surviving to 1 yr of age, a p-value of only 0.2 was obtained comparing G4 DNA-PKcs−/−/Terc−/− with G4 Ku86+/+/Terc−/− animals. Together, these data indicate that impaired DNA-PK activity, either through ablation of Ku86 or, though less evident, through loss of DNA-PKcs, provoked an early onset of senile pathologies and showed that this premature aging phenotype was further enhanced by progressive telomere attrition.
Although abrogation of PARP-1 did not seem to accelerate the loss of organismal survival associated with increasing mouse generations in the absence of telomerase activity (Fig. 2 C), we observed some deleterious effects of PARP-1 deficiency with respect to aging-related pathologies (Fig. 5 C). In a PARP-1–deficient background, progressive telomere erosion significantly increased the incidence of senile lesions from 21% in telomerase-proficient mice to 52% in G2 (P = 0.03), 74% in G3 (P < 0.001), and 100% in G4 (P < 0.0001) telomerase-deficient cohorts (Fig. 5 C, black bars). Similarly, in G1 Terc−/− mice, deletion of PARP-1 did not increase the overall incidence of senile lesions in mice below 1 yr of age, whereas PARP-1 deficiency exacerbated the early onset of pathologies in later generations of telomerase null mice as seen from a comparison of PARP-1−/−/Terc−/− mice versus their PARP-1+/+/ Terc−/− littermates: 52 vs. 36% (P = 0.1) in G2, 74 vs. 53% (P = 0.05) in G3 and finally, 100 vs. 75% (P = 0.02) in G4 (Fig. 5 C, black bars versus gray bars).
Discussion
Studies in telomerase-deficient mice lend solid experimental support to the concept that telomere shortening limits the replicative potential of organs and tissues in vivo (Blasco, 2003). Mounting evidence further suggests that organismal fitness and longevity also depend on as yet poorly understood functional interactions between telomerase and the activity of a steadily growing number of proteins that have emerged as potential regulators of telomere function and mediators of telomere damage signaling. However, definite conclusions on the implication of such interactions at the telomere on the aging process of the whole organism are largely lacking because only few viable mouse strains with multiple mutations in genome maintenance have been studied over their entire life span with their aging phenotype having been characterized (Wong et al., 2003; Chang et al., 2004). Here, we have generated novel telomerase-deficient mouse models with deletions of three related DNA repair and putative telomere maintenance proteins, namely Ku86, DNA-PKcs, and PARP-1. An important feature of these mouse models is that the mutant Ku86, DNA-PKcs, and PARP-1 alleles were bred through successive generations of Terc−/− mice to generate a series of cohorts with progressive loss of telomere length and function. These double mutant mouse models allow us to study how the aging process is affected by loss of function of these DNA repair proteins in the particular setting of moderate to severe telomere dysfunction.
We found that deficiency of either DNA-PK component, DNA-PKcs and Ku86, reduced the life span of telomerase-deficient mice by increasing their predisposition to succumb from intestinal atrophy as well as to suffer from the early onset of a number of other aging-related pathologies. These observations agree with the fact that Ku86-deficient mice display a severe premature aging phenotype (Vogel et al., 1999). Similarly, we have recently reported that DNA-PKcs deficiency also has an impact on normal aging (Espejel et al., 2004). Interestingly, both Ku86 and DNA-PKcs are important for telomere protection because their deletion results in chromosome end-to-end fusions involving telomeric sequences at the fusion point (Bailey et al., 1999, 2001; Samper et al., 2000; Goytisolo et al., 2001). Furthermore, Ku86 and DNA-PKcs are described to have a differential impact on telomere length maintenance. Although abrogation of Ku86 in telomerase-deficient cells does not result in accelerated rate of telomere loss with increasing mouse generations (Samper et al., 2000; Espejel et al., 2002a), the combined loss of DNA-PKcs and telomerase accelerates the loss of telomeres at a rate that has been estimated to be about twofold higher than that of single telomerase knockouts (Espejel et al., 2002b). Therefore, both DNA-PKcs and Ku86 have important roles in telomere metabolism, which in turn could synergize with telomerase deficiency and short telomeres in accelerating organismal aging as demonstrated in this paper. In addition, it has been recently shown that either short telomeres or uncapped telomeres interfere with the proper repair of DSBs in the genome, thus resulting in loss of organismal viability in response to genotoxic agents (Goytisolo et al., 2000; Latre et al., 2003; Bailey et al., 2004). In this regard, it is likely that unrepaired DSBs in Ku86- and DNA-PKcs–deficient mice, due to a defective NHEJ, could also interfere with short and dysfunctional telomeres in late generation Terc-deficient mice, thus resulting in decreased organismal viability. Finally, we have previously described that lack of either Ku86 or DNA-PKcs could rescue the severe male germ cell apoptosis triggered by short telomeres in telomerase-deficient mice, but not the proliferative arrest (Espejel et al., 2002a,b). Thus, the anticipated implications of either Ku86 or DNA-PKcs mutation in somatic cells that lack telomerase activity would be premature loss of cell viability and proliferative potential at one hand and, due to the lack of apoptosis triggered by dysfunctional telomeres, the accumulation of chromosome fusions at the other hand. In the context of the whole organism, both outcomes are predicted to provoke symptoms of premature aging and/or to lead to an increased risk of cancer. Indeed, here we show a synergism between loss of either Ku86 or DNA-PKcs and telomere dysfunction in reducing overall survival and accelerating aging as evidenced by the early onset of aging-related pathologies in Ku86−/−/Terc−/− and DNA-PKcs−/−/Terc−/− mice. Regarding cancer, simultaneous deficiency in Terc and either Ku86 or DNA-PKcs did not significantly increase tumor incidence compared with the corresponding single Terc−/− controls.
In contrast with a previous report (d'Adda di Fagagna et al., 1999), we and others have reported that PARP-1 deficiency (de Murcia et al., 1997) does not affect telomere length or telomere capping (Samper et al., 2001; Bailey and Goodwin, 2004). This observation fits well with previous reports on the apparent lack of proliferative defects and premature aging in PARP-1−/− mouse strains (Wang et al., 1995; de Murcia et al., 1997). Here, we extend these findings and show that even in the context of telomerase deficiency, PARP-1 is not involved in controlling telomere length and telomere capping. Furthermore, we show that, in marked contrast to Ku86- and DNA-PKcs–deficient animals, a relatively small proportion of the PARP-1 knockout cohort suffered from early onset of aging-related pathologies in a telomerase-deficient background, and this did not have an impact in the overall survival of the strain.
Together, these data indicate that, despite different roles in telomere maintenance and despite different aging phenotypes in single knockouts, loss of Ku86, DNA-PKcs, and to a lesser extent also deletion of PARP-1, share the characteristic to accelerate the aging process in the particular setting of progressive telomere erosion. This finding provides strong support to the notion that short telomeres accelerate aging when in combination with other DNA repair deficiencies, in agreement with recent data obtained with mice doubly deficient for telomerase and either ATM or Werner (Wong et al., 2003; Chang et al., 2004). It is noteworthy that, even though the three double mutant mouse models presented symptoms of an early onset of multiple senile pathologies, we did not encounter any evidence for an increased incidence of tumors in the various cohorts of aged animals. This finding should be seen in the context of the current view that critically short or uncapped telomeres are detected by the cell as DSB-like damaged DNA (d'Adda di Fagagna et al., 2003) and that, consequently, telomere shortening may play a dual role in tumorigenesis, either enhancing the initiation of tumors by induction of chromosomal instability through end-to-end fusions or inhibiting tumor progression by induction of DNA damage responses. Of note, it has been suggested that genetic instability associated with telomere dysfunction does not drive tumorigenesis in any case unless p53 function is abolished (Chin et al., 1999). Our observations on the lack of tumorigenesis in three mouse models with presumably functional intact p53 but severe telomere dysfunction combined with defects in DNA repair pathways would fit with this hypothesis. Importantly, our observations further imply that the endogenous tumor suppressor mechanisms in these double mutant mice are sufficiently robust to cope with combined deficiencies in different key mechanisms of genome maintenance.
Materials and methods
Mice
We independently generated three large mouse colonies: Terc/Ku, Terc/DNA-PKcs, and Terc/PARP-1. Double mutant Terc−/−/Ku86−/− and Terc−/−/ DNA-PKcs−/− mice, as well as the corresponding controls, were obtained as described previously (Espejel et al., 2002a,b). First generation (G1) Terc−/−/PARP-1−/− mice and their corresponding wild-type and single mutant G1 Terc−/− littermates were generated by intercrosses of mice heterozygous for Terc (Blasco et al., 1997) and PARP-1 (de Murcia et al., 1997) following the same breeding scheme as described previously (Espejel et al., 2002a,b). Subsequent generations (G2–G4) were also obtained by intercrossing Terc−/−/PARP-1+/− mice following the scheme described previously (Espejel et al., 2002a). The genetic background of the Terc heterozygous mice used to generate the different mouse colonies was in all cases an ∼95% C57BL6 background (Herrera et al., 1999). Similarly, the genetic backgrounds of the Ku86, DNA-PKcs, and PARP-1 heterozygous mice used to generate the double knockout colonies were described to be largely C57BL6 (de Murcia et al., 1997; Espejel et al., 2002a,b). Nevertheless, because Ku86-, DNA-PKcs–, and PARP-1–deficient mice were obtained from different laboratories, we cannot rule out small differences in the genetic background of the three independent mouse colonies studied here. This may be particularly relevant in the case of telomerase deficiency because the survival of telomerase knockout mice is known to be extremely dependent on the genetic background, due in part to initial differences in telomere length (Herrera et al., 1999). Therefore, to avoid any artifacts due to subtle differences in genetic backgrounds, we strictly followed the breeding protocol described above. Following this breeding protocol, each comparison we make is between littermate mice within a giving mouse colony, which by definition have an identical genetic background. Mice were housed in the barrier area at the National Biotechnology Center (CNB; Madrid, Spain), in which pathogen-free procedures are used in all mouse rooms.
Telomere length analysis
First passage MEFs were prepared and analyzed by Q-FISH as described previously (Samper et al., 2000). Telomere fluorescence was analyzed using the TFL-telo software provided by Peter Lansdorp (British Columbia Cancer Center, Vancouver, Canada).
Scoring of chromosomal abnormalities
Metaphases (50 each) from MEFs prepared from littermate embryos (one embryo per genotype) were scored for chromosomal aberrations by superimposing the telomere image (Q-FISH) on the chromosome image (DAPI) using the TFL-telo software, as described previously (Espejel et al., 2002a,b). We define telomere associations as chromosomes with four distinct telomere signals but aligned less than one-half chromatid apart, and telomere fusions as chromosomes joined at their telomeres and showing at least two overlapping telomeric signals (Goytisolo et al., 2001). Such end-to-end fusions may be chromosomes fused by their p-arms (Robertsonian-like fusions) or by their q-arms (dicentrics).
Histological and immunohistochemical analysis
Mouse tissues were fixed in 10% buffered formalin and embedded in paraffin. Paraffin sections (5 μm) were stained with Harris hematoxylin/eosin according to standard procedures, examined at RT by a microscope (Vanus AHES 3; Olympus) using an S Plan fluorite 10× objective, and photographed with a digital camera (model DP 10; Olympus). Proliferating and apoptotic cells were identified using mAbs against Ki67 (1:1,000 dilution of clone MIB-1 from DAKO Cytomation) and active caspase-3 (1:25 dilution of clone C92-605 from BD Biosciences), respectively. Cells positive for Ki67 and active caspase-3 were visualized using peroxidase-coupled secondary antibodies and 3,3′-DAB tetrahydrochloride as chromogen; nuclei were counterstained with hematoxylin according to standard procedures (Espejel et al., 2002b). Photomicrographs were obtained at RT with a microscope (DM LB; Leica) equipped with N-Plan 20× or 40× objectives (Leica), a digital camera (model DC 100; Leica), and IM1000 imaging software (Leica). The relative amount of immunopositive cells was estimated by visual inspection of at least five different sections from two different tissue preparations and scoring for immunopositive cells in at least 10 crypts per section. Age-related pathologies detected in moribund animals are detailed in the Online supplemental material section.
Statistical analysis
To assess statistical significance, the Fisher exact probability test was performed with the software and according to the guidelines at the VassarStats web site (http://faculty.vassar.edu/lowry/VassarStats.html).
Online supplemental material
Table S1 shows the effects of Ku86, DNA-PKcs, or PARP-1 ablation on the development of senile lesions in successive generations of telomerase-deficient mice. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200407178/DC1.
Acknowledgments
We thank R. Serrano for mouse care, E. Santos and J. Freire for genotyping, and M. Serrano for critical comments on the manuscript.
G. de Murcia's laboratory is supported by the Association pour la Recherche sur le Cancer and La Ligue contre le Cancer. M.A. Blasco's laboratory is funded by the MCyT (SAF2001-1869, GEN2001-4856-C13-08), CAM (08.1/0054/01), European Union (TELOSENS FIGH-CT-2002-00217, INTACT LSHC-CT-2003-506803, ZINCAGE FOOD-CT-2003-506850, RISC-RAD FI6R-CT-2003-508842), and the Josef Steiner Award 2003. S. Espejel is supported by a fellowship from the European Union.
Abbreviations used in this paper: DNA-PK, DNA-dependent protein kinase; DNA-PKcs, catalytic subunit of DNA-PK; DSB, double-strand break; MEF, mouse embryonic fibroblast; NHEJ, nonhomologous end joining; PARP-1, poly(ADP-ribose) polymerase-1; Terc, telomerase RNA component.
References
- Bailey, S.M., and E.H. Goodwin. 2004. DNA and telomeres: beginnings and endings. Cytogenet. Genome Res. 104:109–115. [DOI] [PubMed] [Google Scholar]
- Bailey, S.M., J. Meyne, D.J. Chen, A. Kurimasa, G.C. Li, B.E. Lehnert, and E.H. Goodwin. 1999. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc. Natl. Acad. Sci. USA. 96:14899–14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, S.M., M.N. Comforth, A. Kurimasa, D.J. Chen, and E.H. Goodwin. 2001. Strand-specific postreplicative processing of mammalian telomeres. Science. 293:2462–2465. [DOI] [PubMed] [Google Scholar]
- Bailey, S.M., M.N. Comforth, R.L. Ullrich, and E.H. Goodwin. 2004. Dysfunctional mammalian telomeres join with DNA double-strand breaks. DNA Repair (Amst.). 3:349–357. [DOI] [PubMed] [Google Scholar]
- Blasco, M.A. 2003. Telomeres in cancer and aging: lessons from the mouse. Cancer Lett. 194:183–188. [DOI] [PubMed] [Google Scholar]
- Blasco, M.A., and W.C. Hahn. 2003. Evolving views of telomerase and cancer. Trends Cell Biol. 13:289–294. [DOI] [PubMed] [Google Scholar]
- Blasco, M.A., H.W. Lee, M.P. Hande, E. Samper, P.M. Lansdorp, R.A. DePinho, and C.W. Greider. 1997. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 91:25–34. [DOI] [PubMed] [Google Scholar]
- Burkle, A., S. Beneke, C. Brabeck, A. Leake, R. Meyer, M.L. Muiras, and R. Pfeiffer. 2002. Poly(ADP-ribose) polymerase-1, DNA repair and mammalian longevity. Exp. Gerontol. 37:1203–1205. [DOI] [PubMed] [Google Scholar]
- Chang, S., A.S. Multani, N.G. Cabrera, M.L. Naylor, P. Laud, D. Lombard, S. Pathak, L. Guarente, and R.A. DePinho. 2004. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 36:877–882. [DOI] [PubMed] [Google Scholar]
- Chin, L., S.E. Artandi, Q. Shen, A. Tam, S.L. Lee, G.J. Gottlieb, C.W. Greider, and R.A. DePinho. 1999. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 97:527–538. [DOI] [PubMed] [Google Scholar]
- Clatworthy, J.P., and V. Subramanian. 2001. Stem cells and the regulation of proliferation, differentiation and patterning in the intestinal epithelium: emerging insights from gene expression patterns, transgenic and gene ablation studies. Mech. Dev. 101:3–9. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna, F., M.P. Hande, W.M. Tong, P.M. Lansdorp, Z.Q. Wang, and S.P. Jackson. 1999. Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat Genet. 23:76–80. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna, F., M.P. Hande, W.M. Tong, D. Roth, P.M. Lansdorp, Z.Q. Wang, and S.P. Jackson. 2001. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr Biol. 11:1192–1196. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna, F., P.M. Reaper, L. Clay-Farrace, H. Fiegler, P. Carr, T. Von Zglinicki, G. Saretzki, N.P. Carter, and S.P. Jackson. 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 426:194–198. [DOI] [PubMed] [Google Scholar]
- de Lange, T. 2002. Protection of mammalian telomeres. Oncogene. 21:532–540. [DOI] [PubMed] [Google Scholar]
- de Murcia, J.M., C. Niedergang, C. Trucco, M. Ricoul, B. Dutrillaux, M. Mark, F.J. Oliver, M. Masson, A. Dierich, M. LeMeur, et al. 1997. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA. 94:7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejel, S., S. Franco, S. Rodriguez-Perales, S.D. Bouffler, J.C. Cigudosa, and M.A. Blasco. 2002. a. Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. EMBO J. 21:2207–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejel, S., S. Franco, A. Sgura, D. Gae, S.M. Bailey, G.E. Taccioli, and M.A. Blasco. 2002. b. Functional interaction between DNA-PKcs and telomerase in telomere length maintenance. EMBO J. 21:6275–6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejel, S., M. Martin, P. Klatt, J. Martin-Caballero, J.M. Flores, and M.A. Blasco. 2004. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice. EMBO Rep. 5:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galande, S., and T. Kohwi-Shigematsu. 1999. Poly(ADP-ribose) polymerase and Ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J. Biol. Chem. 274:20521–20528. [DOI] [PubMed] [Google Scholar]
- Gilley, D., H. Tanaka, M.P. Hande, A. Kurimasa, G.C. Li, M. Oshimura, and D.J. Chen. 2001. DNA-PKcs is critical for telomere capping. Proc. Natl. Acad. Sci. USA. 98:15084–15088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Suarez, E., E. Samper, J.M. Flores, and M.A. Blasco. 2000. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat. Genet. 26:114–117. [DOI] [PubMed] [Google Scholar]
- Goytisolo, F.A., and M.A. Blasco. 2002. Many ways to telomere dysfunction: in vivo studies using mouse models. Oncogene. 21:584–591. [DOI] [PubMed] [Google Scholar]
- Goytisolo, F.A., E. Samper, J. Martin-Caballero, P. Finnon, E. Herrera, J.M. Flores, S.D. Bouffler, and M.A. Blasco. 2000. Short telomeres result in organismal hypersensitivity to ionizing radiation in mammals. J. Exp. Med. 192:1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goytisolo, F.A., E. Samper, S. Edmonson, G.E. Taccioli, and M.A. Blasco. 2001. The absence of the dna-dependent protein kinase catalytic subunit in mice results in anaphase bridges and in increased telomeric fusions with normal telomere length and G-strand overhang. Mol. Cell. Biol. 21:3642–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty, P., J. Campisi, J. Hoeijmakers, H. van Steeg, and J. Vijg. 2003. Aging and genome maintenance: lessons from the mouse? Science. 299:1355–1359. [DOI] [PubMed] [Google Scholar]
- Henrie, M.S., A. Kurimasa, S. Burma, J. Menissier-de Murcia, G. de Murcia, G.C. Li, and D.J. Chen. 2003. Lethality in PARP-1/Ku80 double mutant mice reveals physiological synergy during early embryogenesis. DNA Repair (Amst.). 2:151–158. [DOI] [PubMed] [Google Scholar]
- Herrera, E., E. Samper, J. Martin-Caballero, J.M. Flores, H.W. Lee, and M.A. Blasco. 1999. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J. 18:2950–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, H.L., D. Gilley, S.A. Galande, M.P. Hande, B. Allen, S.H. Kim, G.C. Li, J. Campisi, T. Kohwi-Shigematsu, and D.J. Chen. 2000. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 14:2807–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna, K.K., and S.P. Jackson. 2001. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27:247–254. [DOI] [PubMed] [Google Scholar]
- Latre, L., L. Tusell, M. Martin, R. Miro, J. Egozcue, M.A. Blasco, and A. Genesca. 2003. Shortened telomeres join to DNA breaks interfering with their correct repair. Exp. Cell Res. 287:282–288. [DOI] [PubMed] [Google Scholar]
- Morrison, C., G.C. Smith, L. Stingl, S.P. Jackson, E.F. Wagner, and Z.Q. Wang. 1997. Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nat. Genet. 17:479–482. [DOI] [PubMed] [Google Scholar]
- Rudat, V., N. Bachmann, J.H. Kupper, and K.J. Weber. 2001. Overexpression of the DNA-binding domain of poly(ADP-ribose) polymerase inhibits rejoining of ionizing radiation-induced DNA double-strand breaks. Int. J. Radiat. Biol. 77:303–307. [DOI] [PubMed] [Google Scholar]
- Ruscetti, T., B.E. Lehnert, J. Halbrook, H. Le Trong, M.F. Hoekstra, D.J. Chen, and S.R. Peterson. 1998. Stimulation of the DNA-dependent protein kinase by poly(ADP-ribose) polymerase. J. Biol. Chem. 273:14461–14467. [DOI] [PubMed] [Google Scholar]
- Samper, E., F.A. Goytisolo, P. Slijepcevic, P.P. van Buul, and M.A. Blasco. 2000. Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep. 1:244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samper, E., F.A. Goytisolo, J. Menissier-de Murcia, E. Gonzalez-Suarez, J.C. Cigudosa, G. de Murcia, and M.A. Blasco. 2001. Normal telomere length and chromosomal end capping in poly(ADP-ribose) polymerase-deficient mice and primary cells despite increased chromosomal instability. J. Cell Biol. 154:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veuger, S.J., N.J. Curtin, C.J. Richardson, G.C. Smith, and B.W. Durkacz. 2003. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 63:6008–6015. [PubMed] [Google Scholar]
- Vogel, H., D.S. Lim, G. Karsenty, M. Finegold, and P. Hasty. 1999. Deletion of Ku86 causes early onset of senescence in mice. Proc. Natl. Acad. Sci. USA. 96:10770–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z.Q., B. Auer, L. Stingl, H. Berghammer, D. Haidacher, M. Schweiger, and E.F. Wagner. 1995. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 9:509–520. [DOI] [PubMed] [Google Scholar]
- Wong, K.K., R.S. Maser, R.M. Bachoo, J. Menon, D.R. Carrasco, Y. Gu, F.W. Alt, and R.A. DePinho. 2003. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 421:643–648. [DOI] [PubMed] [Google Scholar]
- Zijlmans, J.M., U.M. Martens, S.S. Poon, A.K. Raap, H.J. Tanke, R.K. Ward, and P.M. Lansdorp. 1997. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc. Natl. Acad. Sci. USA. 94:7423–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]