

Abstract
The apical surface of mouse urothelium is covered by two-dimensional crystals (plaques) of uroplakin (UP) particles. To study uroplakin function, we ablated the mouse UPII gene. A comparison of the phenotypes of UPII- and UPIII-deficient mice yielded new insights into the mechanism of plaque formation and some fundamental features of urothelial differentiation. Although UPIII knockout yielded small plaques, UPII knockout abolished plaque formation, indicating that both uroplakin heterodimers (UPIa/II and UPIb/III or IIIb) are required for plaque assembly. Both knockouts had elevated UPIb gene expression, suggesting that this is a general response to defective plaque assembly. Both knockouts also had small superficial cells, suggesting that continued fusion of uroplakin-delivering vesicles with the apical surface may contribute to umbrella cell enlargement. Both knockouts experienced vesicoureteral reflux, hydronephrosis, renal dysfunction, and, in the offspring of some breeding pairs, renal failure and neonatal death. These results highlight the functional importance of uroplakins and establish uroplakin defects as a possible cause of major urinary tract anomalies and death.
Introduction
The apical surface of mammalian urothelium that is in contact with the urine is highly specialized, featuring two-dimensional (2D) crystals (urothelial plaques) of hexagonally packed 16-nm protein particles (Hicks and Ketterer, 1969; Vergara et al., 1969; Staehelin et al., 1972; Brisson and Wade, 1983; Walz et al., 1995; Kachar et al., 1999; Oostergetel et al., 2001; Min et al., 2003). These plaques contain four major uroplakins (UPs): Ia (27 kD), Ib (28 kD), II (15 kD), and III (47 kD) (Wu et al., 1994; Sun et al., 1999). Uroplakins Ia and Ib have four transmembrane domains, are ∼40% identical in sequence, and belong to the “tetraspanin” family (Yu et al., 1994). The tetraspanin family contains many cell surface molecules, including CD9, CD63, CD81, CD82, and CD151, that play key roles in cellular events involving membrane organization (Maecker et al., 1997; Levy et al., 1998; Berditchevski, 2001; Boucheix and Rubinstein, 2001; Hemler, 2001, 2003; Tarrant et al., 2003; Yunta and Lazo, 2003). Uroplakins II and III both have a single transmembrane domain, and they share a common stretch of ∼12 amino acid residues on the exoplasmic side of the transmembrane domain (Wu and Sun, 1993; Lin et al., 1994; Deng et al., 2002). Within the plaques, these four major uroplakins are organized into two heterodimers consisting of Ia/II and Ib/III, as demonstrated by chemical cross-linking (Wu et al., 1995) and protein isolation (Liang et al., 2001). In addition, transfection studies show that when individual uroplakins are expressed in 293T cells, they are retained in the ER; however, coexpression of uroplakins Ia plus II or Ib plus III permits the heterodimers to exit from the ER; these events suggest that the formation of specific heterodimers is a prerequisite for uroplakins to reach the cell surface (Tu et al., 2002).
To study the functional and disease implications of uroplakins, we previously generated uroplakin III–deficient mice using the gene-targeting approach (Hu et al., 2000). Such mice lack a typical urothelial umbrella cell layer. They have a reduced urothelial plaque size, compromised urothelial permeability barrier function, and retrograde flow of urine from the bladder into the ureters (vesicoureteral reflux [VUR]). In ∼70% of the mice, accumulation of urine in the renal pelvis (hydronephrosis) occurs (Hu et al., 2000, 2002). These results establish that uroplakin III is an integral subunit of urothelial plaques, which contribute to the permeability barrier function of the urothelial apical surface, and suggest that urothelial defects may play a role in VUR (Hu et al., 2000, 2002).
Some of the phenotypes of these UPIII-deficient mice remain hardly understood. For example, the observation that UPIII deficiency leads to the formation of small plaques seems incompatible with our current assumption that all four major uroplakins are required for the formation of the 16-nm particle (Sun et al., 1999). One possible explanation is that UPIIIb, a newly discovered, minor UPIII isoform (Deng et al., 2002), is responsible for the formation of the residual plaques observed in the UPIII-deficient urothelium. If it is, one would predict that knockout of uroplakin II, which does not have a known isoform, should completely abolish plaque formation. Another question has to do with the possible role of uroplakin mutation in VUR. Although knockout of the UPIII gene leads to VUR in mice, screening of human VUR patients revealed that polymorphism of uroplakin genes is only marginally associated with VUR (Giltay et al., 2004; Jiang et al., 2004). Moreover, although polymorphism was found in these VUR patients, no deletion, truncation, or frameshift mutations have been detected so far (Giltay et al., 2004; Jiang et al., 2004). The role of uroplakin defects in VUR and other lower urinary tract anomalies thus remains unclear.
To address these issues, we have genetically ablated the mouse UPII gene to perturb the other uroplakin pair consisting of UPIa/II. This led to the entrapment of UPIa in the ER, but allowed the remaining UPIb/III pair to reach the apical urothelial surface. No 16-nm particle or apical plaques were formed, however, which indicates that both uroplakin heterodimers were required for plaque formation. The small size of the superficial cells in both UPII- and UPIII-deficient urothelia suggested that the incorporation of fully assembled uroplakin plaques into the apical surface through fusion of the fusiform vesicles with the apical membrane played a role in the enlargement of umbrella cells. Because entire litters of UPII-deficient mice from some breeding pairs (originating from the inbred 129/SvEv and outbred Swiss Webster strains) reproducibly died 8–10 d postnatally, as a result of renal failure, it seems that major uroplakin defects such as gene deletion, frameshift, or truncation might cause urinary tract anomalies that, in severe cases, could lead to death. These results highlight the critical functional importance of uroplakins in the formation of a specialized urothelial apical surface, and shed light on the mechanisms of plaque formation, umbrella cell enlargement, and certain urinary tract abnormalities.
Results
Inactivation of the mouse uroplakin II gene
After transfecting embryonic stem (ES) cells (from 129/SvEv mice) with a vector designed to delete the first four exons and a part of the fifth exon of the mouse uroplakin II gene (Fig. 1 A), we screened 320 ES cell colonies and found 4 that were harboring the correct homologous recombination events as determined by long template PCR (Fig. 1 B) and Southern blot (not depicted). 18 chimeric mice from three ES cell lines were germline transmitting and were bred with black Swiss Webster mice to yield homozygous UPII knockout mice. Northern (Fig. 1, C and D) and Western (Fig. 1 E) blotting confirmed the absence of UPII in homozygous uroplakin II–deficient (−/−) mice. The protein levels of uroplakin Ia, UPII's partner, and of uroplakins Ib and III (of the uroplakin Ib/III pair) were reduced 10–20-fold (on a per total cellular protein basis) when compared with those of the normal control mice (+/+; Fig. 1 E). The mRNA levels of UPIa and UPIII were slightly up-regulated (about twofold), whereas that of UPIb was drastically up-regulated by 10–20-fold (Fig. 1 C; see Discussion).
Figure 1.
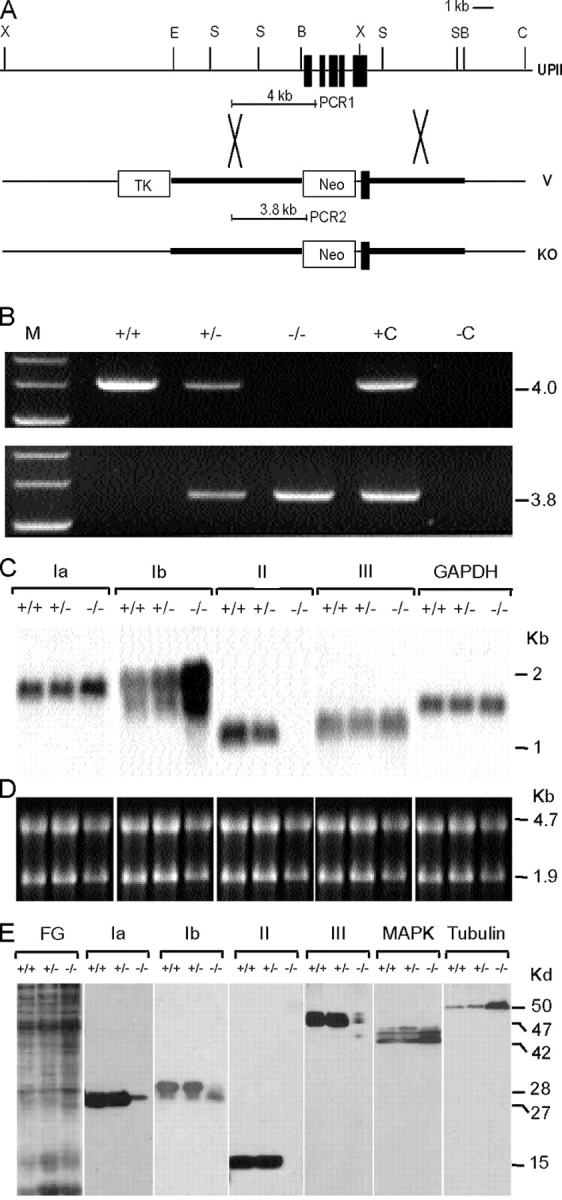
Inactivation of the mouse UPII gene (upk2) perturbed the expression of other uroplakins. (A) Alignment of the mouse UPII locus, the targeting vector (V), and the mutated locus (KO). Black boxes, exons; thick lines, mouse genomic sequences included in the vector; crosses, potential crossover sites; PCR1 and 2, PCR products characteristic of the wild-type and KO alleles, respectively; Neo, neomycin-resistant gene; TK, thymidine kinase of the herpes simplex virus; B, BamHI; C, ClaI; E, EcoRI; S, SacI; X, Xhol. (B) PCR analysis of the genomic DNA using a mixture of two pairs of primers that generated 4.0-kb and 3.8-kb PCR products characteristic of the wild-type (+) and KO (−) alleles, respectively. M, +C, and −C denote size markers, positive controls (+/+ genomic DNA or vector used as templates), and a negative control (no template), respectively. (C) Northern blot analysis. 5 μg of total RNA samples was resolved electrophoretically and probed with cDNA of mouse uroplakins Ia, Ib, II, and III, and glyceraldehyde phosphate dehydrogenase (GAPDH) as a loading control. Note, in the −/− animals, the absence of UPII message and the ∼2× and 10–20× increases in UPIa and UPIb message, respectively. (D) Ethidium bromide–stained rRNA of the samples in C serving as a loading control. (E) Immunoblot analysis of mouse uroplakin proteins Ia (27 kD), Ib (28 kD), II (15 kD), and III (47 kD). FG denotes the fast green–stained total mouse urothelial proteins used for immunoblotting. MAPK and tubulin of the same samples were immunoblotted for comparison. Note in the −/− mice the absence of UPII protein, the significant decrease in the levels of the other three uroplakin proteins, and the absence of the highest mol wt (∼30 kD) UPIb species due to defective glycosylation.
Uroplakin expression and plaque formation
Consistent with the UPIII knockout results, the UPII-deficient urothelium lacked a typical superficial umbrella cell layer (Fig. 2 b). This urothelium was hyperplastic, as indicated by an elevated level of BrdU incorporation (the labeling index increased almost 100-fold from 0.12%, which is normal, to 10.7%; Fig. 2, c and d), and was three- to sixfold thicker than normal (Fig. 2, a and b). Immunohistochemical staining with monospecific rabbit and mouse antibodies to individual uroplakins confirmed that all four major uroplakins were preferentially expressed in superficial umbrella cells in normal mouse urothelium (Fig. 3, a, c, e, g, and i; Hu et al., 2000). The staining of the UPII-deficient urothelium showed that UPIa (the partner of UPII) was diffusely distributed in all upper cells, which is consistent with its entrapment in the ER (Fig. 3 d). Uroplakins Ib and III (of the other uroplakin pair), although expressed at a lower level than normal, were clearly associated with the apical, as well as some basal/lateral, cell surface(s) (Fig. 3, f and j).
Figure 2.

UPII-deficient urothelium lacked a typical umbrella cell layer and became hyperplastic. Paraffin sections of the normal (a) and UPII-deficient (b) bladders of 1-mo-old male mice were stained with hematoxylin and eosin. Those of normal (c) and UPII-deficient (d) bladders of 19-day-old male mice were isolated 2 hr after the final i.p. injection of BrdU and were stained immunohistochemically for BrdU. The inset in c shows an area with some BrdU-incorporating superficial cells. Note the increased thickness and the elevated BrdU incorporation (10.7 ± 1.1% vs. 0.12 ± 0.07%) of the UPII-deficient urothelium. Also note that although most of the labeling occurred in basal cells (arrows), there was occasional labeling of superficial cells (arrowheads). A total of eight UPII-deficient mice and eight normal mice, half of them male, were studied; 1,000–2,000 urothelial cells were examined per section from each animal; P < 0.001; with no significant gender differences. Bar, 50 μm.
Figure 3.

Altered uroplakin expression pattern in the UPII-deficient urothelium. Paraffin sections of wild-type (WT; a, c, e, g, and i) or UPII-deficient (UPII KO; b, d, f, h, and j) mouse urothelium were immunohistochemically stained for keratins (a and b) using a mixture of AE1 and AE3 mouse monoclonal antibodies (Tseng et al., 1982; for review see Cooper et al., 1985), uroplakin Ia (c and d), UPIb (e and f), UPII (g and h), and UPIII (i and j). k shows the silver nitrate–stained (SN) SDS-PAGE patterns of the detergent-insoluble membrane proteins from (lanes 1) bovine urothelium, (2) normal mouse urothelium, (3) UPIII-deficient mouse urothelium, and (4) UPII-deficient mouse urothelium, and the immunoblotting of these proteins using antibodies to UPs Ia, Ib, II, IIIa, and IIIb. Note, in the thickened UPII KO urothelium, the absence of UPII, the diffused cytoplasmic distribution and lack of apical surface association of UPIa, the greatly reduced but still noticeably apical surface association (arrowheads) of UPIb and UPIII, and the reduced amounts of detergent-insoluble uroplakins in the UPIII-deficient urothelium (lanes 3) and their further reduction in the UPII-deficient urothelium (lanes 4). Bar, 50 μm.
Unlike normal urothelium, which was covered by large squamous superficial umbrella cells that could be as large as 100 μm in diameter (Fig. 4 a) and were covered by rigid-looking plaques (Figs. 4 c and 5 a), superficial cells of the UPII-deficient urothelium were uniformly small (20–30 μm; Fig. 4 b) and were completely devoid of the rigid-looking, apical plaques (Fig. 4 d). In addition, the uroplakin-delivering fusiform vesicles (0.7–1 μm in diameter; Fig. 5 a) were totally replaced by numerous small, spherical vesicles (Fig. 5 b). These small vesicles (150–200 nm in diameter) in the UPIII- and UPII-deficient urothelia were moderately and weakly labeled, respectively, by antibodies to uroplakins (Fig. 5, e–h), which indicates that they were involved in delivering the remaining uroplakins to the cell surface. Consistent with these findings, the detergent-insoluble membrane fraction of the UPIII-deficient mouse urothelium showed a significantly reduced number of uroplakins (Fig. 3 k, lanes 3), whereas that of the UPII-deficient urothelium contained almost no detectable uroplakins (Fig. 3 k, lanes 4). Together, these results indicated that UPII ablation completely abolished plaque formation.
Figure 4.

The UPII-deficient urothelium had small superficial cells and a particle-free apical surface. The urothelia from age-matched 3-mo-old wild-type (a and c) and UPII-deficient (b and d) mice were examined by scanning EM (a and b) and quick-freeze deep etch transmission EM (c and d). Note the replacement of large surface umbrella cells (U) by small superficial cells (S) (a and b), and the complete absence of the 16-nm particles and urothelial plaques on the apical surface of the UPII-deficient urothelium (d). cyt, cytoplasm; m, membrane; ups, uroplakin particles. Bars: (a and b) 20 μm; (c and d) 0.2 μm.
Figure 5.

The replacement of the fusiform vesicles by small spherical vesicles that delivered the remaining uroplakin pair in uroplakin-deficient urothelium. The urothelia from age-matched 3-mo-old wild-type (a, c, and d), UPII-deficient (b, g, and h), and UPIII-deficient (e and f) mice were examined by thin section (a and b) and immunolabeling (c–h) transmission EM. Normal uroplakin-delivering fusiform vesicles (a, c, and d) were completely replaced by numerous small, spherical, immature-looking spherical vesicles in UPIII-deficient (e and f) and UPII-deficient (b, g, and h) urothelia. Note the strong labeling of normal fusiform vesicles (*) by antibodies to uroplakins Ia (c) and Ib (d), the moderate staining of the spherical vesicles in the UPIII-deficient urothelium (which still expressed the UPIIIb isoform; e and f), and the weak staining of the small vesicles (arrows) in the UPII-deficient urothelium by anti-UPIII (g) and by a rabbit antiserum to total uroplakins (h). Arrowheads (b) indicate the smooth apical surface of the UPII-deficient urothelium. Fv, fusiform vesicle; P, plaque; Sv, small vesicle. Bars: (a and b) 0.5 μm; (c–h) 1 μm.
VUR, ureteral obstruction, and hydronephrosis
By administering an India ink solution into the bladders of live, anesthetized mice (Fig. 6, a and b), we determined the hydrostatic pressures at which micturition (PM) and VUR (PR) occurred. Normal mice had a PM of 28 cm of H2O pressure (Fig. 6 c), and most of them did not reflux—unless the urethra was ligated, causing outlet obstruction, and the hydrostatic pressure was increased to 40–80 cm of H2O (Fig. 6 d, WT). Although the UPII knockout mice had a normal micturition pressure of ∼24 cm of H2O (Fig. 6 c), >50% (11 out of 20) of the mice refluxed at a pressure lower than this (Fig. 6, d and e). Many of the (−/−) mice developed severe hydronephrosis with a greatly expanded renal pelvis (Fig. 7, b and d) and associated renal morphological changes (Fig. 7, a–f). To determine whether reflux caused hydronephrosis, we calculated the difference between the PM and the PR of each ureter (PK [hydrostatic pressure to the kidney] = PM − PR). For a normal mouse that had a PM of 25 cm of H2O and a PR of 50 cm, an intravesicular pressure of >25 cm of H2O would result in micturition and dissipation of pressure. However, for a UPII knockout mouse that had a PM of 25 cm of H2O but a lower PR of 18 cm of H2O, intravesicular pressures of 18–25 cm of H2O would lead to reflux, thus potentially transmitting up to 7 cm of hydrostatic pressure to the kidney (PK). We wanted to see whether this renal pressure correlated with the grade (G) of hydronephrosis—which we defined as G = D/T, where D was the internal diameter of the renal pelvis and T was the thickness of the remaining renal parenchyma (Fig. 7, c and d). If reflux were a main cause of hydronephrosis, there should have been a positive correlation between PK and G. However, we found that these two parameters were independent (P > 0.2), which suggests that reflux could not be the major etiology of hydronephrosis in this system (Fig. 7 g).
Figure 6.

Inactivation of the UPII gene led to vesicoureteral reflux. An India ink suspension (0.1% in PBS) was instilled into the bladders of 11 normal 3-mo--old mice and 10 UPII knockout mice, and the hydrostatic pressure at which micturition (micturition pressure [PM]) and reflux (reflux pressure [PR]) occurred for individual ureters were recorded. Note in a the presence of India ink in the bladder (B) and ureters (U) and, in b, in the left kidney (K)—but not in the right kidney, suggesting obstruction of the right ureter. Also note that, although the UPII-deficient (KO) mice had a normal micturition pressure (c), 11 out of 20 ureters (55%) had retrograde flow of urine into the ureters (occurring at a pressure lower than the micturition pressure; d, closed circles), compared with 3 out of 22 ureters (13.6%) in wild-type (WT) mice. Open circles denote cases where reflux occurred at a pressure higher than the micturition pressure (and thus would not occur naturally). e shows the mean values of PR based on the data shown in d. Error bars represent 1 SD.
Figure 7.

Inactivation of the UPII gene caused hydronephrosis and renal abnormalities. The kidneys of normal (a, c, and e) and UPII-deficient (b, d, and f) mice were compared in terms of their gross appearance (a and b), whole kidney section (c and d), and histological appearance (e and f). The mice used were 6 mo old for a and b, and 3 mo old for c–g. g shows a plot of renal pressure (PM − PR) versus the grade of hydronephrosis (D/T, where D and T were the diameter of the renal pelvis cavity and the thickness of the renal parenchyma, respectively). More than 100 knockout mice were studied and ∼80% of them showed various degrees of hydronephrosis. Note a lack of positive correlation between the grade of hydronephrosis and renal pressure (Pk) (t test, t = 0.177, P > 0.2), which suggests that reflux could not be a major cause of hydronephrosis. (c, e, and f) C, cortex; G, glomeruli; M, medulla; T, tubules; U, urothelium. Bar, 50 μm.
Another possible cause of hydronephrosis is ureteral obstruction, which impedes the flow of urine from the kidney. To investigate this possibility, we performed i.v. pyelogram (IVP) by injecting 3 μl/g (of body weight) of Omnipaque into the orbital sinus, followed by serial radiography (Fig. 8, a and b). The results indicated that UPII knockout mice had a significantly delayed excretion of Omnipaque, indicating obstruction. Serial sectioning of the urinary tracts of UPII knockout mice revealed areas of the ureter with epithelial polyps or complete epithelial occlusion, which caused structural obstruction (Fig. 8, c–h; see Discussion). These results suggest that both VUR and structural and/or functional obstruction of the ureters may be responsible for the observed hydronephrosis in UPII knockout mice.
Figure 8.
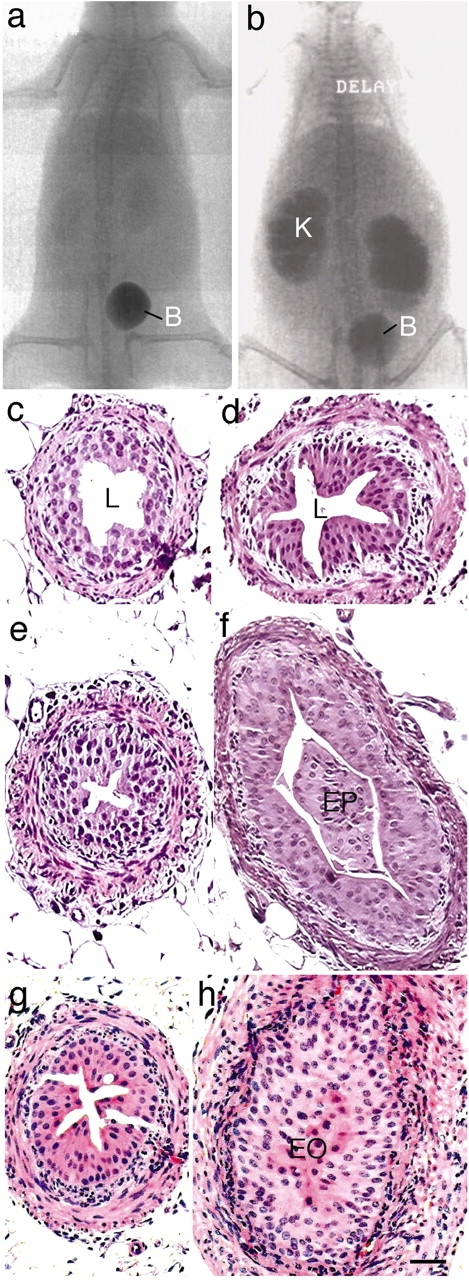
Inactivation of the UPII gene caused ureteral obstruction. I.v. pyelogram of normal (a) and UPII knockout mice (b) established delayed excretion of contrast in the latter. c–h show the histological sections of the ureters of normal (c and e), UPII knockout (d and f), and UPIII knockout (g and h) mice. Samples were taken from proximal (c and d) or distal (e–h) ureter. Note the formation of epithelial outgrowth filling the ureteral lumen (f), sometimes forming a complete occlusion (h). B, bladder; EO, epithelial occlusion; EP, epithelial plug; K, kidney; L, lumen. Bar, 50 μm.
Renal dysfunction and death
Using a filter paper assay, we found that UPII knockout mice had higher volume per micturition (Fig. 9 b), micturition frequency (Fig. 9 c), and total urine output (Fig. 9 d) than normal mice; no statistically significant gender differences were noted (Fig. 9, b–d). The levels of many urinary components, including uric acid, creatinine, potassium, sodium, and chloride ions, were slightly reduced (Fig. 9, f–j), possibly because of defects in mechanisms of urine concentration (Fig. 9 d). The concentration of blood urea nitrogen (BUN) almost doubled (Fig. 9 e), suggesting a compromised renal function. Interestingly, although most of the breeding pairs yielded litters that survived into adulthood, the litters of some breeding pairs (129/SvEv × Swiss Webster) reproducibly died around days 8–10 postnatally (Fig. 10, a and b). These litters were characterized by retarded growth (Fig. 10 c) and a sharp surge in the BUN level (Fig. 10 d), which suggests renal failure as a cause of death, possibly resulting from ureteral obstruction (Fig. 8, c– h) and a defective renal pelvis urothelium that normally expressed uroplakins (Fig. 10 e).
Figure 9.

Inactivation of the UPII gene caused renal malfunction. (a) The standard curve of a filter paper assay for measuring the urine volume. (b) Volume per micturition. (c) Frequency of micturition. (d) Cumulative total micturition volume. (e) Blood urea nitrogen (BUN). Note the twofold increase of BUN in UPII-deficient mice (P = 0.00001) compared with wild-type mice. f–j show the urinary concentrations of uric acid (f), creatinine (g), potassium (h), sodium (i), and chloride (j). Urine and sera from 12 wild-type and 12 knockout 3-mo-old mice were pooled for these analyses. Error bars represent 1 SD.
Figure 10.

Inactivation of the UPII gene can cause neonatal death of the entire litters from some mouse breeding pairs. (a) The pedigree of one pair of founder mice. Note that the entire litter of pups from some breeding pairs, but not from other pairs, died around day 10 postnatally. (b) The fractional survival curves of two litters from different breeding pairs. One underwent neonatal death (2KO-death), whereas the other (2KO-health), like the normal wild-type control (WT), survived into adulthood. (c) The growth (increase in body weight) of the 2KO-death mice was severely retarded. (d) The blood urea nitrogen (BUN) of the 2KO-death mice increased drastically (reaching >10-fold the normal value) before the animals' death. (e) Immunohistochemical staining of normal mouse kidney (embryonic day 16.5), showing that the renal pelvis (RP) urothelium (U) and the simple epithelium covering the papilla (arrowheads) are uroplakin positive. Arrowheads indicate the single-layered papillary epithelium. CD, collecting ducts. Bar, 50 μm.
Discussion
Mammalian urothelium has three unique biological features: its apical membrane is highly specialized, harboring 2D crystals of hexagonally packed 16-nm particles (Hicks and Ketterer, 1969; Vergara et al., 1969; Staehelin et al., 1972; Brisson and Wade, 1983); its superficial umbrella cells are greatly flattened and expanded, with a diameter reaching over 100 μm (Porter and Bonneville, 1963; Hicks, 1965; Koss, 1969; Lewis, 2000; Veranic et al., 2004; for review see Hicks, 1975); and it is one of the slowest-cycling stratified epithelia, with a BrdU-labeling index of <0.1% (Martin, 1972; Farsund, 1975). The ablation of a single gene that encodes uroplakin II perturbed all of these urothelial properties and led to major urinary tract abnormalities.
Mechanism of uroplakin plaque formation
As mentioned earlier, the four major uroplakins can form two heterodimer pairs consisting of uroplakins Ia/II and Ib/III (Wu et al., 1995; Liang et al., 2001; Tu et al., 2002). Transfection studies indicated that the formation of correct heterodimer is required for uroplakins (except UPIb; see below) to exit from the ER and to reach the cell surface (Tu et al., 2002). Because 293T (a human embryonic kidney–derived cell line) cells were used in these transfection studies, the physiological significance of in vitro data obtained using nonurothelial cells might be questioned. Our in vivo data show that although UPIa is distributed throughout the cytoplasm of the UPII-deficient urothelium, suggesting ER entrapment (Fig. 3 d), uroplakins Ib and III (of the Ib/III pair) can reach the urothelial surface (Fig. 3, f and j). These in vivo results validate the transfection data obtained using the 293T cells (Tu et al., 2002), and further support the existence of the Ia/II and Ib/III heterodimers.
Although cultured bovine urothelial cells continue to synthesize large amounts of all the uroplakins, these cells do not form any mature-looking, uroplakin-delivering fusiform vesicles that are characteristic of the in vivo umbrella cells, and their apical cell surface is completely devoid of urothelial plaques (Surya et al., 1990; Chen et al., 2003). Based on what we have learned from cultured keratinocytes (Weiss et al., 1984; Schermer et al., 1989), cultured urothelial cells most likely mimic in vivo regenerating urothelium (Surya et al., 1990; Chen et al., 2003). The inability of regenerating urothelial cells to assemble plaques could be functionally important, because a cell surface laden with the rigid-looking plaques may be incompatible with cell migration and proliferation during wound repair. Therefore, although we can study certain early steps of uroplakin interaction in the currently available, cultured urothelial cells and transfected 293T cells, the final stages of uroplakin assembly and 2D crystal formation need to be studied using the in vivo urothelium. Our present finding that the uroplakin II–deficient urothelium lacks 16-nm particles (Fig. 4, c and d) indicates that, although the UPIb/III pair can be delivered to the apical surface (Fig. 3, f and j) using the small vesicles (∼150–200 nm in diameter) accumulated in the UPII-deficient superficial cells (Fig. 5, b, g, and h), the UPIb/III pair by itself cannot form the 16-nm particle, let alone the urothelial plaques (Fig. 4 d). Severs and Hicks (1979) have shown that, in normal urothelium, 16-nm particles can be detected in early vesicles that have just budded off the Golgi apparatus, which suggests that the two uroplakin heterodimers (Ia/II and Ib/III) interact before they leave the Golgi apparatus to form the 16-nm particle. Together, these data suggest that uroplakins first form, in the ER, heterodimers (Tu et al., 2002), and possibly heterotetramers; the heterotetramers then assemble into the 16-nm particles in the Golgi apparatus or the trans-Golgi network, and these particles, once reaching a sufficient density in the post-Golgi vesicles, can then aggregate to form small and, later, large 2D crystals.
Given our current assumption that both uroplakin pairs are required for the formation of the 16-nm particle, it was baffling that the UPIII-deficient urothelium still made 2D crystals, albeit small ones, from the 16-nm uroplakin particles (Hu et al., 2000). However, we now know that highly purified urothelial plaques contain a minor isoform of UPIII, which we recently characterized (Deng et al., 2002). This 35-kD protein was named uroplakin IIIb because it is urothelium specific, it shares a similar transmembrane topology and significant sequence homologies with UPIII (also known as UPIIIa), and it forms a heterodimer with UPIb, UPIIIa's partner (Deng et al., 2002). This newly found uroplakin IIIb, which is up-regulated in UPIIIa-deficient urothelium (Deng et al., 2002), should allow the formation of small amounts of heterotetramers containing heterodimers UPIb/UPIIIb and UPIa/UPII. The heterodimers can then be delivered by the 150–200-nm vesicles (Fig. 5, e and f) and account for the formation of small surface plaques in the UPIIIa-deficient urothelium. This interpretation is consistent with our finding that the ablation of UPII, which does not have a known isoform, greatly diminishes the amounts of small vesicle-associated uroplakins and completely abolishes the formation of the 16-nm particles (Fig. 4, c and d).
We showed previously that the ablation of UPIII led to abnormal synthesis and processing of UPIb, i.e., the level of UPIb mRNA was greatly increased, whereas the amount of UPIb protein was reduced, became hypoglycosylated, and was mistargeted to the basal/lateral cell surface (Hu et al., 2000). Because UPIII and UPIb were known to interact, we speculated that these UPIb changes were caused by the removal of its partner, UPIII (Hu et al., 2000). Our finding that UPII ablation led to a similar up-regulation of the UPIb mRNA level (Fig. 1 C) and hypoglycosylation of the UPIb protein (Fig. 1 E) indicates, however, that these UPIb changes represent a general response to a perturbed uroplakin assembly. This may explain why, in cultured human urothelial cells, the level of mRNA for UPIb is greatly elevated compared with those for other uroplakins (Varley et al., 2004). Uroplakin Ib is also unique in that it is the only uroplakin that, when expressed alone in 293T cells, can exit from the ER to reach the cell surface (although it is required for its partner, UPIII, to exit from the ER [Tu et al., 2002]); and in that it is the only uroplakin that is expressed in nonurothelial tissues, including corneal and conjunctival epithelia (Adachi et al., 2000) and, possibly, lung epithelium (Kallin et al., 1991; Olsburgh et al., 2003). These data indicate that uroplakin Ib is unique among the uroplakins in its regulation and function.
Uroplakin assembly and umbrella cell enlargement
Normal mouse urothelium is covered by large umbrella cells that have an average diameter of >100 μm (Hicks, 1965; Koss, 1969). The greatly enlarged superficial cells can minimize the intercellular space, thus contributing to the permeability barrier function of the apical urothelial surface (Negrete et al., 1996; Zeidel, 1996; Lewis, 2000; Apodaca, 2004). The mechanism by which these large umbrella cells, which are frequently tetraploid or octoploid (Farsund, 1975), are formed is unclear, although both arrested cytokinesis (Farsund, 1976; Farsund and Dahl, 1978) and cell fusion (Martin and Wong, 1981) have been suggested. Our finding that the superficial cells of both UPII- and UPIII-deficient urothelia failed to enlarge (Fig. 4, a and b; Hu et al., 2000) suggests that urothelial plaque formation may play a role in umbrella cell enlargement. It is possible that the continued insertion of fusiform vesicles into the apical surface not only expands (Porter and Bonneville, 1963; Hicks, 1965; Koss, 1969; Staehelin et al., 1972; Lewis and de Moura, 1982; Truschel et al., 2002; Apodaca, 2004), but also alters/stabilizes, the luminal surface, thus hindering cytokinesis; the fact that some umbrella cells can incorporate BrdU (Fig. 2, c and d; Erman et al., 2004) makes it plausible that arrested cytokinesis could contribute to polyploidy and cell enlargement. A combination of cell surface expansion and arrested cytokinesis may thus account for the formation of large polyploid umbrella cells, without resorting to cell fusion. This “surface alteration hypothesis” is supported by cell kinetic data suggesting arrested cytokinesis (Farsund, 1976; Farsund and Dahl, 1978); however, additional studies are needed to test this hypothesis.
Uroplakins and urothelial growth
A striking phenotype of both uroplakin II– and uroplakin III–deficient urothelia is that they become hyperplastic, with a BrdU-labeling index of >10%, versus a normal value of 0.1% (Fig. 2, c and d) (Hu et al., 2000). It is possible that the permeability barrier function of the uroplakin-deficient urothelia is compromised (Hu et al., 2002) so that the EGF-related mitogens that are known to be present in high concentrations in the mouse urine (Lakshmanan et al., 1990; Parries et al., 1995) can reach urothelial basal cells to stimulate cell proliferation (Rebel et al., 1994). Alternatively, fully assembled urothelial plaques can trigger certain growth-inhibiting signals, which are now absent in the uroplakin-deficient urothelia, leading to hyperplasia. Additional studies are needed to distinguish these possibilities.
Roles of uroplakin defects in urinary tract diseases
Our earlier observation that inactivation of the mouse uroplakin III gene resulted in VUR and hydronephrosis raised the possibility that uroplakin defects may cause VUR in humans (Hu et al., 2000; Mak and Kuo, 2003). By surveying 76 VUR patients, we identified 18 single-nucleotide polymorphisms; 7 of them are missense, with no truncation or frameshift mutations (Jiang et al., 2004). Only two of the missense polymorphisms (an Ala7Val change in the UPIa gene and a Pro154Ala change in the UPIII gene) were found to be marginally associated with VUR (both P = 0.08). These results, as well as those by Giltay et al. (2004), suggest that the uroplakin point mutations that have been detected so far do not play a major role in causing VUR (Jiang et al., 2004). There are two possible explanations for this apparent inconsistency between the human and mouse data. First, the results of mouse studies cannot be directly extrapolated to humans because of minor, but still significant, differences in the development of the lower urinary tracts. Perhaps for this reason, inactivation of angiotensin II receptor (AT2) gene can cause VUR in mice (Nishimura et al., 1999), but polymorphisms in this gene do not seem to be involved in causing primary familial VUR in humans (Hohenfellner et al., 1999; Yoneda et al., 2002). Second, and perhaps more importantly, no truncation or frameshift mutations have been detected in any of the VUR patients that have been studied so far, and some breeding pairs of the UPII and UPIII knockout mice reproducibly die postnatally around days 8–10, as a result of renal failure (Fig. 10; Hu et al., 2000), suggesting that certain uroplakin defects (such as deletion, truncation, or mutations in some structurally crucial amino acid residues), in combination with certain genetic backgrounds (Sanford et al., 2001; Wolfer et al., 2002), may be embryologically or postnatally lethal and are thus undetected in VUR patients. It is interesting to note that a recent survey showed that urinary tract abnormalities account for 20% of all congenital anomalies detected during pregnancies and up to one year after birth (Scott, 2002). Of the 560 cases of prenatal or neonatal deaths resulting from congenital abnormalities, 58% were because of renal anomalies, including a significant number with obstruction (Scott, 2002).
An important finding of this study is that urothelial abnormalities caused by uroplakin defects can lead to renal failure and death (Fig. 10). There are several possible mechanisms by which this could occur. First, defects in the uroplakin-positive renal pelvis urothelium, as well as in the simple epithelium that covers the renal papilla expressing uroplakin prenatally (Fig. 10 e; Jiang et al., 2004), may lead to renal dysfunction and failure. Second, urothelial hyperplasia can cause obstruction of the ureteral lumen (Fig. 8, f and h). Although it has been well-established that mechanical obstruction can induce urothelial hyperplasia (Monson et al., 1992; Curhan et al., 2000), it is unclear whether urothelial hyperplasia, per se, can cause obstruction. In our system, urothelial hyperplasia seems to be the cause, rather than the effect, of obstruction because (a) urothelial hyperplasia precedes obstruction and hydronephrosis; and (b) although only 70–80% of the mice develop hydronephrosis, 100% of the animals show urothelial hyperplasia throughout the urinary tract, thus ruling out obstruction as a cause of urothelial hyperplasia (but see Gamp et al., 2003). Third, defects in the ureteral urothelium may interfere with the development of its underlying smooth muscle (Master et al., 2003), leading to abnormal peristalsis or functional obstruction (Santicioli and Maggi, 1998)—a possibility supported by our recent finding that uroplakin defects lead to abnormal bladder muscle contractility and/or function (Christ, G., personal communication). These possible mechanisms, or a combination thereof, may cause a functional/structural obstruction of the urinary tract, leading to abnormal renal development (Chung and Chevalier, 1996), obstructive nephropathy, hydronephrosis, and, in severe cases, renal failure and death.
Materials and methods
Production of the UPII knockout mice
Genomic clones of the mouse UPII gene were isolated from a 129/Ola mouse P1 genomic library (Genome Systems). The targeting vector was designed to delete the first four exons, and a part of the fifth, of the UPII gene (Fig. 1 A). Three primers were used for genotyping; one forward (5′-gagggagttaagactcaagaatcaatcaagga-3′) and two reverse (5′-cttctatcgccttcttgacgagttcttctgagg-3′ for detecting a 3.8-kb product of the neomycin sequence, and 5′-cagatttctagcagtccactcttgtagaacgg-3′ for a 4.0-kb product of the native UPII gene; Fig. 1, A and B).
Morphological studies
Mouse urothelium was examined by scanning EM (model JSM-840; JEOL) and transmission EM (model 200CX; JEOL) (Hu et al., 2000). Quick-freeze deep etch was performed as described previously (Kachar et al., 1999). Cell proliferation was assessed based on the nuclear incorporation of BrdU (Sigma-Aldrich). 19-d-old mice received five intraperitoneal injections of BrdU (100 mg/kg of body weight, in PBS), 1.5 h apart, and were killed 2 h after the final injection. Various tissues, including esophagus and urinary bladder tissues, were removed, fixed in 10% formalin overnight, and processed for immunostaining with a horseradish peroxidase–conjugated anti-BrdU monoclonal antibody (CHEMICON International). Samples were visualized with a microscope (Axiophot; Carl Zeiss MicroImaging, Inc.) with 10×/0.32 and 20×/0.60 (Plan-Apochromat) or 40×/0.75 (Plan-Neofluar) objective lenses. Images were captured with a digital camera (model DKC-5000; Sony) at room temperature. The images were processed in size and contrast/brightness with Adobe Photoshop 6.0.
Isolation of detergent-insoluble proteins from mouse urothelium
Mouse urothelial cells were homogenized in buffer A (10 mM Hepes, pH 7.5, 1 mM EDTA, 1 mM EGTA, and 1 mM PMSF), loaded onto a 1.6-M sucrose cushion (in buffer A), and centrifuged at 16,000 rpm for 25 min at 4°C (SW41; Beckman Coulter). The crude membranes concentrated at the interface were isolated, washed with buffer A, treated with 2% Sarkosyl in buffer A for 10 min at 25°C, and pelleted. The detergent-insoluble membranes, in which the urothelial plaques were highly enriched, were washed with buffer A before they were solubilized in SDS-PAGE sample buffer and analyzed by SDS-PAGE, followed by silver nitrate staining, or by immunoblotting (Wu et al., 1994; Liang et al., 1999; Zhou et al., 2001).
Determination of reflux pressure and hydronephrosis
The reflux pressure was recorded as the hydrostatic pressure (i.e., cm of H2O) at which an India ink suspension in PBS backflowed from the bladders into the ureters of anesthetized mice (Hu et al., 2000). At the end of the experiment, the kidneys were dissected longitudinally to confirm the presence of the ink in the renal pelvis (Fig. 6 b).
Determination of the micturition pattern and urine/blood chemistry
The micturition pattern of the mice was recorded using special cages, as described previously (Hu et al., 2000). A total of ∼500 μl of mouse urine was collected from each mouse and used for assaying the concentrations of uric acid, creatinine, potassium, sodium, and chloride ions (Tufts Veterinary Diagnostic Laboratory). Mouse sera were used for assaying the BUN level (Anilytics).
Determination of obstruction by IVP and serial sectioning of the mouse ureter
IVP was performed in anesthetized 4-mo-old wild-type (n = 2) and uroplakin II–deficient (n = 4) mice. The contrast Omnipaque (Iohexol; 3 μl/g of body weight) was injected into the orbital sinus, and serial radiographs were made at 0, 5, 15, 30, 45, 60, and 90 min. After lethal anesthesia, the urinary tracts were dissected and fixed in 10% formalin overnight. The ureters were embedded in paraffin and serially sectioned. Sections were numbered from superior to inferior orientation and stained with hematoxylin and eosin.
Acknowledgments
We thank Alexandra Joyner for her help in generating the knockout mice; Songshan Jiang and Jordan Gitlin for experimental assistance; Gloria Gallo and Adrian S. Woolf for reviewing renal histology; the Interstitial Cystitis Association and the NYU Urology and Dermatology Research Programs for support; David Sabatini, Xiang-Peng Kong, Gert Kreibich, Angel Pellicer, and Xue-Ru Wu for helpful discussions; and Herbert Lepor and Irwin M. Freedberg for their encouragement and support.
This work was funded by National Institutes of Health grants DK39753, DK52206, and DK66491.
X.-T. Kong, F.-M. Deng, and P. Hu contributed equally to this paper.
P. Hu's present address is Procter & Gamble Corporate Research Biotechnology, Cincinnati, OH 45252.
Abbreviations used in this paper: 2D, two-dimensional; BUN, blood urea nitrogen; ES, embryonic stem; IVP, i.v. pyelogram; UP, uroplakin; VUR, vesicoureteral reflux.
References
- Adachi, W., K. Okubo, and S. Kinoshita. 2000. Human uroplakin Ib in ocular surface epithelium. Invest. Ophthalmol. Vis. Sci. 41:2900–2905. [PubMed] [Google Scholar]
- Apodaca, G. 2004. The uroepithelium: not just a passive barrier. Traffic. 5:117–128. [DOI] [PubMed] [Google Scholar]
- Berditchevski, F. 2001. Complexes of tetraspanins with integrins: more than meets the eye. J. Cell Sci. 114:4143–4151. [DOI] [PubMed] [Google Scholar]
- Boucheix, C., and E. Rubinstein. 2001. Tetraspanins. Cell. Mol. Life Sci. 58:1189–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisson, A., and R.H. Wade. 1983. Three-dimensional structure of luminal plasma membrane protein from urinary bladder. J. Mol. Biol. 166:21–36. [DOI] [PubMed] [Google Scholar]
- Chen, Y., X. Guo, F.M. Deng, F.X. Liang, W. Sun, M. Ren, T. Izumi, D.D. Sabatini, T.T. Sun, and G. Kreibich. 2003. Rab27b is associated with fusiform vesicles and may be involved in targeting uroplakins to urothelial apical membranes. Proc. Natl. Acad. Sci. USA. 100:14012–14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, K.H., and R.L. Chevalier. 1996. Arrested development of the neonatal kidney following chronic ureteral obstruction. J. Urol. 155:1139–1144. [DOI] [PubMed] [Google Scholar]
- Cooper, D., A. Schermer, and T.-T. Sun. 1985. Classification of human epithelia and their neoplasms using monoclonal antibodies to keratins: strategies, applications, and limitations. Lab. Invest. 52:243–256. [PubMed] [Google Scholar]
- Curhan, G.C., W.S. McDougal, and M.L. Zeidel. 2000. Urinary tract obstruction. The Kidney. G.M. Brenner, editor. W.B. Saunders, Philadelphia. 1820–1843.
- Deng, F.M., F.X. Liang, L. Tu, K.A. Resing, P. Hu, M. Supino, C.C. Hu, G. Zhou, M. Ding, G. Kreibich, and T.T. Sun. 2002. Uroplakin IIIb, a urothelial differentiation marker, dimerizes with uroplakin Ib as an early step of urothelial plaque assembly. J. Cell Biol. 159:685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erman, A., G. Vidmar, and K. Jezernik. 2004. Temporal and spatial dimensions of postnatal growth of the mouse urinary bladder urothelium. Histochem. Cell Biol. 121:63–71. [DOI] [PubMed] [Google Scholar]
- Farsund, T. 1975. Cell kinetics of mouse urinary bladder epithelium. I. Circadian and age variations in cell proliferation and nuclear DNA content. Virchows Arch. B Cell Pathol. 18:35–49. [DOI] [PubMed] [Google Scholar]
- Farsund, T. 1976. Cell kinetics of mouse urinary bladder epithelium. II. Changes in proliferation and nuclear DNA content during necrosis regeneration, and hyperplasia caused by a single dose of cyclophosphamide. Virchows Arch. B Cell Pathol. 21:279–298. [PubMed] [Google Scholar]
- Farsund, T., and E. Dahl. 1978. Cell kinetics of mouse urinary bladder epithelium. III. A histologic and ultrastructural study of bladder epithelium during regeneration after a single dose of cyclophosphamide, with special reference to the mechanism by which polyploid cells are formed. Virchows Arch. B Cell Pathol. 26:215–223. [PubMed] [Google Scholar]
- Gamp, A.C., Y. Tanaka, R. Lullmann-Rauch, D. Wittke, R. D'Hooge, P.P. De Deyn, T. Moser, H. Maier, D. Hartmann, K. Reiss, et al. 2003. LIMP-2/LGP85 deficiency causes ureteric pelvic junction obstruction, deafness and peripheral neuropathy in mice. Hum. Mol. Genet. 12:631–646. [PubMed] [Google Scholar]
- Giltay, J.C., J. van de Meerakker, H.K. van Amstel, and T.P. de Jong. 2004. No pathogenic mutations in the uroplakin III gene of 25 patients with primary vesicoureteral reflux. J. Urol. 171:931–932. [DOI] [PubMed] [Google Scholar]
- Hemler, M.E. 2001. Specific tetraspanin functions. J. Cell Biol. 155:1103–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemler, M.E. 2003. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu. Rev. Cell Dev. Biol. 19:397–422. [DOI] [PubMed] [Google Scholar]
- Hicks, R.M. 1965. The fine structure of the transitional epithelium of rat ureter. J. Cell Biol. 26:25–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks, R.M. 1975. The mammalian urinary bladder: an accommodating organ. Biol. Rev. Camb. Philos. Soc. 50:215–246. [DOI] [PubMed] [Google Scholar]
- Hicks, R.M., and B. Ketterer. 1969. Hexagonal lattice of subunits in the thick luminal membrane of the rat urinary bladder. Nature. 224:1304–1305. [DOI] [PubMed] [Google Scholar]
- Hohenfellner, K., T.E. Hunley, E. Yerkes, P. Habermehl, R. Hohenfellner, and V. Kon. 1999. Angiotensin II, type 2 receptor in the development of vesico-ureteric reflux. BJU Int. 83:318–322. [DOI] [PubMed] [Google Scholar]
- Hu, P., F.M. Deng, F.X. Liang, C.M. Hu, A.B. Auerbach, E. Shapiro, X.R. Wu, B. Kachar, and T.T. Sun. 2000. Ablation of uroplakin III gene results in small urothelial plaques, urothelial leakage, and vesicoureteral reflux. J. Cell Biol. 151:961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, P., S. Meyers, F.X. Liang, F.M. Deng, B. Kachar, M.L. Zeidel, and T.T. Sun. 2002. Role of membrane proteins in permeability barrier function: uroplakin ablation elevates urothelial permeability. Am. J. Physiol. Renal Physiol. 283:F1200–F1207. [DOI] [PubMed] [Google Scholar]
- Jiang, S., J. Gitlin, F.M. Deng, F.X. Liang, A. Lee, A. Atala, S.B. Bauer, G.D. Ehrlich, S.A. Feather, J.D. Goldberg, et al. 2004. Lack of major involvement of human uroplakin genes in vesicoureteral reflux: implications for disease heterogeneity. Kidney Int. 66:10–19. [DOI] [PubMed] [Google Scholar]
- Kachar, B., F. Liang, U. Lins, M. Ding, X.R. Wu, D. Stoffler, U. Aebi, and T.-T. Sun. 1999. Three-dimensional analysis of the 16 nm urothelial plaque particle: luminal surface exposure, preferential head-to-head interaction, and hinge formation. J. Mol. Biol. 285:595–608. [DOI] [PubMed] [Google Scholar]
- Kallin, B., R. de Martin, T. Etzold, V. Sorrentino, and L. Philipson. 1991. Cloning of a growth arrest-specific and transforming growth factor beta-regulated gene, TI 1, from an epithelial cell line. Mol. Cell. Biol. 11:5338–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss, L.G. 1969. The asymmetric unit membranes of the epithelium of the urinary bladder of the rat. An electron microscopic study of a mechanism of epithelial maturation and function. Lab. Invest. 21:154–168. [PubMed] [Google Scholar]
- Lakshmanan, J., E.C. Salido, R. Lam, L. Barajas, and D.A. Fisher. 1990. Identification of pro-epidermal growth factor and high molecular weight epidermal growth factors in adult mouse urine. Biochem. Biophys. Res. Commun. 173:902–911. [DOI] [PubMed] [Google Scholar]
- Levy, S., S.C. Todd, and H.T. Maecker. 1998. CD81 (TAPA-1): a molecule involved in signal transduction and cell adhesion in the immune system. Annu. Rev. Immunol. 16:89–109. [DOI] [PubMed] [Google Scholar]
- Lewis, S.A. 2000. Everything you wanted to know about the bladder epithelium but were afraid to ask. Am. J. Physiol. Renal Physiol. 278:F867–F874. [DOI] [PubMed] [Google Scholar]
- Lewis, S.A., and J.L. de Moura. 1982. Incorporation of cytoplasmic vesicles into apical membrane of mammalian urinary bladder epithelium. Nature. 297:685–688. [DOI] [PubMed] [Google Scholar]
- Liang, F., B. Kachar, M. Ding, Z. Zhai, X.R. Wu, and T.-T. Sun. 1999. Urothelial hinge as a highly specialized membrane: detergent- insolubility, urohingin association, and in vitro formation. Differentiation. 65:59–69. [DOI] [PubMed] [Google Scholar]
- Liang, F.X., I. Riedel, F.M. Deng, G. Zhou, C. Xu, X.R. Wu, X.P. Kong, R. Moll, and T.T. Sun. 2001. Organization of uroplakin subunits: transmembrane topology, pair formation and plaque composition. Biochem. J. 355:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J.H., X.R. Wu, G. Kreibich, and T.-T. Sun. 1994. Precursor sequence, processing, and urothelium-specific expression of a major 15-kDa protein subunit of asymmetric unit membrane. J. Biol. Chem. 269:1775–1784. [PubMed] [Google Scholar]
- Maecker, H.T., S.C. Todd, and S. Levy. 1997. The tetraspanin superfamily: molecular facilitators. FASEB J. 11:428–442. [PubMed] [Google Scholar]
- Mak, R.H., and H.J. Kuo. 2003. Primary ureteral reflux: emerging insights from molecular and genetic studies. Curr. Opin. Pediatr. 15:181–185. [DOI] [PubMed] [Google Scholar]
- Martin, B.F. 1972. Cell replacement and differentiation in transitional epithelium: a histological and autoradiographic study of the guinea-pig bladder and ureter. J. Anat. 112:433–455. [PMC free article] [PubMed] [Google Scholar]
- Martin, B.F., and Y.C. Wong. 1981. Development and maturation of the bladder epithelium of the guinea pig. Acta Anat. (Basel). 110:359–375. [DOI] [PubMed] [Google Scholar]
- Master, V.A., G. Wei, W. Liu, and L.S. Baskin. 2003. Urothlelium facilitates the recruitment and trans-differentiation of fibroblasts into smooth muscle in acellular matrix. J. Urol. 170:1628–1632. [DOI] [PubMed] [Google Scholar]
- Min, G., G. Zhou, M. Schapira, T.T. Sun, and X.P. Kong. 2003. Structural basis of urothelial permeability barrier function as revealed by cryo-EM studies of the 16 nm uroplakin particle. J. Cell Sci. 116:4087–4094. [DOI] [PubMed] [Google Scholar]
- Monson, F.C., B.A. McKenna, A.J. Wein, and R.M. Levin. 1992. Effect of outlet obstruction on 3H-thymidine uptake: a biochemical and radioautographic study. J. Urol. 148:158–162. [DOI] [PubMed] [Google Scholar]
- Negrete, H.O., J.P. Lavelle, J. Berg, S.A. Lewis, and M.L. Zeidel. 1996. Permeability properties of the intact mammalian bladder epithelium. Am. J. Physiol. 271:F886–F894. [DOI] [PubMed] [Google Scholar]
- Nishimura, H., E. Yerkes, K. Hohenfellner, Y. Miyazaki, J. Ma, T.E. Hunley, H. Yoshida, T. Ichiki, D. Threadgill, J.A. Phillips III, et al. 1999. Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol. Cell. 3:1–10. [DOI] [PubMed] [Google Scholar]
- Olsburgh, J., P. Harnden, R. Weeks, B. Smith, A. Joyce, G. Hall, R. Poulsom, P. Selby, and J. Southgate. 2003. Uroplakin gene expression in normal human tissues and locally advanced bladder cancer. J. Pathol. 199:41–49. [DOI] [PubMed] [Google Scholar]
- Oostergetel, G.T., W. Keegstra, and A. Brisson. 2001. Structure of the major membrane protein complex from urinary bladder epithelial cells by cryo-electron crystallography. J. Mol. Biol. 314:245–252. [DOI] [PubMed] [Google Scholar]
- Parries, G., K. Chen, K.S. Misono, and S. Cohen. 1995. The human urinary epidermal growth factor (EGF) precursor. Isolation of a biologically active 160-kilodalton heparin-binding pro-EGF with a truncated carboxyl terminus. J. Biol. Chem. 270:27954–27960. [DOI] [PubMed] [Google Scholar]
- Porter, K.R., and M.A. Bonneville. 1963. An Introduction to the Fine Structure of Cells and Tissues. Lea & Febiger, Philadelphia. 196 pp.
- Rebel, J.M., W.I. De Boer, C.D. Thijssen, M. Vermey, E.C. Zwarthoff, and T.H. Van der Kwast. 1994. An in vitro model of urothelial regeneration: effects of growth factors and extracellular matrix proteins. J. Pathol. 173:283–291. [DOI] [PubMed] [Google Scholar]
- Sanford, L.P., S. Kallapur, I. Ormsby, and T. Doetschman. 2001. Influence of genetic background on knockout mouse phenotypes. Methods Mol. Biol. 158:217–225. [DOI] [PubMed] [Google Scholar]
- Santicioli, P., and C.A. Maggi. 1998. Myogenic and neurogenic factors in the control of pyeloureteral motility and ureteral peristalsis. Pharmacol. Rev. 50:683–722. [PubMed] [Google Scholar]
- Schermer, A., J.V. Jester, C. Hardy, D. Milano, and T.-T. Sun. 1989. Transient synthesis of K6 and K16 keratins in regenerating rabbit corneal epithelium: keratin markers for an alternative pathway of keratinocyte differentiation. Differentiation. 42:103–110. [DOI] [PubMed] [Google Scholar]
- Scott, J.E. 2002. Fetal, perinatal, and infant death with congenital renal anomaly. Arch. Dis. Child. 87:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severs, N.J., and R.M. Hicks. 1979. Analysis of membrane structure in the transitional epithelium of rat urinary bladder. 2. The discoidal vesicles and Golgi apparatus: their role in luminal membrane biogenesis. J. Ultrastruct. Res. 69:279–296. [DOI] [PubMed] [Google Scholar]
- Staehelin, L.A., F.J. Chlapowski, and M.A. Bonneville. 1972. Lumenal plasma membrane of the urinary bladder. I. Three-dimensional reconstruction from freeze-etch images. J. Cell Biol. 53:73–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, T.-T., F.X. Liang, and X.R. Wu. 1999. Uroplakins as markers of urothelial differentiation. Adv. Exp. Med. Biol. 462:7–18. [DOI] [PubMed] [Google Scholar]
- Surya, B., J. Yu, M. Manabe, and T.-T. Sun. 1990. Assessing the differentiation state of cultured bovine urothelial cells: elevated synthesis of stratification-related K5 and K6 keratins and persistent expression of uroplakin I. J. Cell Sci. 97:419–432. [DOI] [PubMed] [Google Scholar]
- Tarrant, J.M., L. Robb, A.B. van Spriel, and M.D. Wright. 2003. Tetraspanins: molecular organisers of the leukocyte surface. Trends Immunol. 24:610–617. [DOI] [PubMed] [Google Scholar]
- Truschel, S.T., E. Wang, W.G. Ruiz, S.M. Leung, R. Rojas, J. Lavelle, M. Zeidel, D. Stoffer, and G. Apodaca. 2002. Stretch-regulated exocytosis/endocytosis in bladder umbrella cells. Mol. Biol. Cell. 13:830–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng, S.C., M.J. Jarvinen, W.G. Nelson, J.W. Huang, M.J. Woodcock, and T.-T. Sun. 1982. Correlation of specific keratins with different types of epithelial differentiation: monoclonal antibody studies. Cell. 30:361–372. [DOI] [PubMed] [Google Scholar]
- Tu, L., T.T. Sun, and G. Kreibich. 2002. Specific heterodimer formation is a prerequisite for uroplakins to exit from the endoplasmic reticulum. Mol. Biol. Cell. 13:4221–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley, C.L., J. Stahlschmidt, W.C. Lee, J. Holder, C. Diggle, P.J. Selby, L.K. Trejdosiewicz, and J. Southgate. 2004. Role of PPARγ and EGFR signalling in the urothelial terminal differentiation programme. J. Cell Sci. 117:2029–2036. [DOI] [PubMed] [Google Scholar]
- Veranic, P., R. Romih, and K. Jezernik. 2004. What determines differentiation of urothelial umbrella cells? Eur. J. Cell Biol. 83:27–34. [DOI] [PubMed] [Google Scholar]
- Vergara, J.A., W. Longley, and J.D. Robertson. 1969. A hexagonal arrangement of subunits in membrane of mouse urinary bladder. J. Mol. Biol. 46:593–596. [DOI] [PubMed] [Google Scholar]
- Walz, T., M. Haner, X.R. Wu, C. Henn, A. Engel, T.-T. Sun, and U. Aebi. 1995. Towards the molecular architecture of the asymmetric unit membrane of the mammalian urinary bladder epithelium: a closed “twisted ribbon” structure. J. Mol. Biol. 248:887–900. [DOI] [PubMed] [Google Scholar]
- Weiss, R.A., R. Eichner, and T.-T. Sun. 1984. Monoclonal antibody analysis of keratin expression in epidermal diseases: a 48- and 56-kdalton keratin as molecular markers for hyperproliferative keratinocytes. J. Cell Biol. 98:1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfer, D.P., W.E. Crusio, and H.P. Lipp. 2002. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci. 25:336–340. [DOI] [PubMed] [Google Scholar]
- Wu, X.R., and T.-T. Sun. 1993. Molecular cloning of a 47 kDa tissue-specific and differentiation-dependent urothelial cell surface glycoprotein. J. Cell Sci. 106:31–43. [DOI] [PubMed] [Google Scholar]
- Wu, X.R., J.H. Lin, T. Walz, M. Haner, J. Yu, U. Aebi, and T.-T. Sun. 1994. Mammalian uroplakins. A group of highly conserved urothelial differentiation-related membrane proteins. J. Biol. Chem. 269:13716–13724. [PubMed] [Google Scholar]
- Wu, X.R., J.J. Medina, and T.-T. Sun. 1995. Selective interactions of UPIa and UPIb, two members of the transmembrane 4 superfamily, with distinct single transmembrane-domained proteins in differentiated urothelial cells. J. Biol. Chem. 270:29752–29759. [DOI] [PubMed] [Google Scholar]
- Yoneda, A., S. Cascio, A. Green, D. Barton, and P. Puri. 2002. Angiotensin II type 2 receptor gene is not responsible for familial vesicoureteral reflux. J. Urol. 168:1138–1141. [DOI] [PubMed] [Google Scholar]
- Yu, J., J.H. Lin, X.R. Wu, and T.-T. Sun. 1994. Uroplakins Ia and Ib, two major differentiation products of bladder epithelium, belong to a family of four transmembrane domain (4TM) proteins. J. Cell Biol. 125:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunta, M., and P.A. Lazo. 2003. Tetraspanin proteins as organisers of membrane microdomains and signalling complexes. Cell. Signal. 15:559–564. [DOI] [PubMed] [Google Scholar]
- Zeidel, M.L. 1996. Low permeabilities of apical membranes of barrier epithelia: what makes watertight membranes watertight? Am. J. Physiol. 271:F243–F245. [DOI] [PubMed] [Google Scholar]
- Zhou, G., W.J. Mo, P. Sebbel, G. Min, T.A. Neubert, R. Glockshuber, X.R. Wu, T.T. Sun, and X.P. Kong. 2001. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: evidence from in vitro FimH binding. J. Cell Sci. 114:4095–4103. [DOI] [PubMed] [Google Scholar]