

Abstract
The mechanism by which the β-cell transcription factor Pax4 influences cell function/mass was studied in rat and human islets of Langerhans. Pax4 transcripts were detected in adult rat islets, and levels were induced by the mitogens activin A and betacellulin. Wortmannin suppressed betacellulin-induced Pax4 expression, implicating the phosphatidylinositol 3-kinase signaling pathway. Adenoviral overexpression of Pax4 caused a 3.5-fold increase in β-cell proliferation with a concomitant 1.9-, 4-, and 5-fold increase in Bcl-xL (antiapoptotic), c-myc, and Id2 mRNA levels, respectively. Accordingly, Pax4 transactivated the Bcl-xL and c-myc promoters, whereas its diabetes-linked mutant was less efficient. Bcl-xL activity resulted in altered mitochondrial calcium levels and ATP production, explaining impaired glucose-induced insulin secretion in transduced islets. Infection of human islets with an inducible adenoviral Pax4 construct caused proliferation and protection against cytokine-evoked apoptosis, whereas the mutant was less effective. We propose that Pax4 is implicated in β-cell plasticity through the activation of c-myc and potentially protected from apoptosis through Bcl-xL gene expression.
Introduction
Diabetes mellitus comprises a heterogeneous group of hyperglycemic disorders resulting from inadequate mass and function of pancreatic islet β-cells. Two studies have associated mutations in the pax4 gene to type 2 diabetes in the Japanese population, while two haplotypes of this gene have been linked to type 1 diabetes in Scandinavian families (Shimajiri et al., 2001, 2003; Kanatsuka et al., 2002; Holm et al., 2004). The functional role of Pax4 in β-cell physiology, and thus its potential implication in diabetes, is still poorly understood. Pax4 is detected in the pancreatic bud at mouse embryonic day 9.5, but expression becomes progressively restricted to the β- and δ-cells of the islet of Langerhans, producing, respectively, insulin and somatostatin (Sosa-Pineda et al., 1997). Several independent studies have detected Pax4 mRNA in adult human, rat, and mouse pancreatic islets (Heremans et al., 2002; Kojima et al., 2003; Zalzman et al., 2003). Targeted disruption of the pax4 gene in mice results in the absence of mature pancreatic β- and δ-cells with a commensurate increase in glucagon-containing α-cells (Sosa-Pineda et al., 1997). However, the earliest insulin-producing precursor cells, detected at embryonic day 8.5–9 (Gittes and Rutter, 1992), are present, indicating that Pax4 expression is not mandatory for the generation of β-cell precursors but rather is critical for the proliferation and/or survival of these cells (Sosa-Pineda et al., 1997). Accordingly, elevated expression levels of Pax4 mRNA are found in human insulinomas (Miyamoto et al., 2001).
To better understand the impact of Pax4 in β-cell function, pharmacological and molecular studies were performed on isolated rat islets. Our work suggests that mitogens such as betacellulin activate Pax4 through the phosphatidylinositol 3-kinase (PI3-kinase) pathway. Furthermore, we found that forced expression of Pax4 stimulates β-cell proliferation and survival through concomitant regulation of the oncogene c-myc and the antiapoptotic gene bcl-xl. In contrast, the diabetes-linked mutant R129W elicited an attenuated response. Consistent with Bcl-xL induction, mitochondrial function such as ATP production and Ca2+ homeostasis was altered, resulting in curtailed glucose-induced insulin secretion. Similarly, human islets transduced with a novel doxycycline-inducible adenoviral construct harboring the mouse Pax4 cDNA exhibited graded proliferation and protection against apoptosis, whereas the diabetes-linked mutant conferred a modest effect. Together, these findings suggest that Pax4 participates in the regulation of β-cell plasticity and that loss-of-function mutations result in the gradual loss of insulin-producing cells, and ultimately diabetes.
Results
Activin A and betacellulin increase Pax4 gene transcription as well as β-cell proliferation in rat islets
Basal mRNA expression levels for Pax4 were established in islets and found to give a relative abundance value of 4.7 when normalized to the housekeeping transcript cyclophilin. In contrast, Pax4 mRNA was barely detectable in rat liver cells. The ubiquitously expressed mitochondrial transcription factor TFAM was found with similar relative abundance of 5 and 6.5 in liver and islets, confirming tissue-specific expression of Pax4 in mature islets (Fig. 1 A). Of note, Pax4 mRNA was 25-fold higher in the insulin-producing INS-1E cell line (unpublished data), which is consistent with elevated expression levels detected in human insulinomas (Miyamoto et al., 2001). The responses of the pax4 gene to activin A (a member of the TGF-β family) and betacellulin (a member of the EGF family) were investigated in rat islets (Demeterco et al., 2000). Treatment of islets for 24 h with a range of concentrations resulted in a dose-dependent increase of Pax4 mRNA levels. Maximal induction was observed with 0.5 nM of activin A or betacellulin that elicited a 4.3- and 4.2-fold increase in Pax4 mRNA, respectively (Fig. 1 B). As in insulinoma cells (Ueda, 2000), the related factor TGF-β1 had no significant effect on Pax4 expression in islets. Of note, insulin mRNA levels were unaffected by both treatments (unpublished data). The main intracellular signaling step of betacellulin via interaction with the EGF receptor is the activation of PI3-kinase. To elucidate whether or not this pathway, which has been shown to promote β-cell replication (Buteau et al., 2003), was also involved in Pax4 activation, islets were incubated with the PI3-kinase inhibitor wortmannin. The inhibitor (100 nM) almost completely abolished betacellulin-induced pax4 gene expression, suggesting that the transcription factor is a downstream target of the PI3-kinase (Fig. 1 C). In parallel, we confirmed the mitogenic effect of activin A and betacellulin by measuring β-cell replication using BrdU incorporation. Both growth factors (at 0.5 nM) increased β-cell proliferation by approximately threefold, whereas TGF-β1–treated islets remained quiescent (Fig. 1 D). Together, these results suggest that stimulation of Pax4 gene expression by activin A and betacellulin coincides with islet proliferation induced by the two mitogens.
Figure 1.

Activin A and betacellulin increase Pax4 mRNA levels as well as β-cell proliferation in rat islets. (A) Pax4 is expressed in adult rat islets but not in the liver. Quantitative RT-PCR using RNA purified from freshly isolated islets and hepatocytes were performed on Pax4 and TFAM. Data are presented as relative mRNA abundance levels normalized to the transcript cyclophilin and represent the mean ± SEM of six independent experiments performed in triplicates. A representative agarose gel depicting the amplified Pax4 transcript is shown on the right. The fragment was subcloned and confirmed to be Pax4 by sequencing. White line indicates that intervening lanes have been spliced out. (B) PAX4 mRNA levels in islets treated with increasing doses of activin A, betacellulin, or TGF-β1 as indicated. (C) Islets were incubated with 0.5 nM of betacellulin in the absence or presence of 50 and 100 nM of the PI3-kinase inhibitor wortmannin. Pax4 transcript abundance levels were estimated by quantitative RT-PCR. (D) β-Cell proliferation was measured by BrdU incorporation in islets treated with the indicated growth factors at 0.5 nM. Data represent the mean ± SEM of four independent experiments, comprising more than 900 cells per condition. Statistical significance was tested by t test. *, P < 0.05; **, P < 0.01.
Adenovirus-mediated Pax4 overexpression in rat pancreatic islets induces β-cell proliferation
To evaluate the importance of Pax4 in β-cell replication, isolated islets were infected with a CMV promoter–driven Pax4/GFP-expressing adenovirus (AdCMVPax4IRESGFP) or control adenovirus (AdCAlacZ). Because the antibody against Pax4 is unable to detect the protein by immunohistochemistry or by Western blotting (unpublished data), we monitored its overexpression via the reporter GFP cotranslated from a bi-cistronic transcript. Approximately 25% and 50% of β-cells expressed GFP 48 h after infection with 1 and 2.4 × 107 pfu/ml of AdCMVPax4IRESGFP, respectively (Fig. 2 A). Pax4 transcript was estimated to reach levels 22 ± fivefold higher (n = 3) than those found in control AdCALacZ-infected islets (unpublished data). Like mitogen-stimulated islets, insulin mRNA levels (Fig. 3 C) were unchanged, indicating that Pax4 overexpression did not alter the phenotypic profile of the β-cell. Production of a functional protein was confirmed by electrophoretic mobility shift assay (EMSA) using a cognate radiolabeled G3 element of the glucagon gene promoter (Ritz-Laser et al., 2002). A single complex previously identified as Pax6 (Ritz-Laser et al., 2002) was observed in nuclear extracts derived from AdCaLacZ-infected islets (Fig. 2 B, lane 4). An additional complex of similar migration pattern to that produced by recombinant Pax4 was generated in islets infected with increasing amounts of AdCMVPax4IRESGFP (Fig. 2 B, lanes 1 and 5–7). This complex was supershifted by the Pax4 antiserum, confirming the binding of Pax4 to this site (Fig. 2 B, lane 2 and 8). The capacity of Pax4 to promote islet proliferation was then evaluated by BrdU incorporation (Fig. 2 C). Quantification revealed a 3.5-fold increase in BrdU labeling of β-cells expressing Pax4 as compared with AdCALacZ-transduced islets. In contrast, proliferation was unaffected by overexpression of Pax6 and neurogenin3, confirming the specificity of Pax4-associated β-cell replication (Fig. 2 D). Thus, forced expression of Pax4 specifically induced DNA synthesis in β-cells, recapitulating the effect observed with both activin A and betacellulin.
Figure 2.
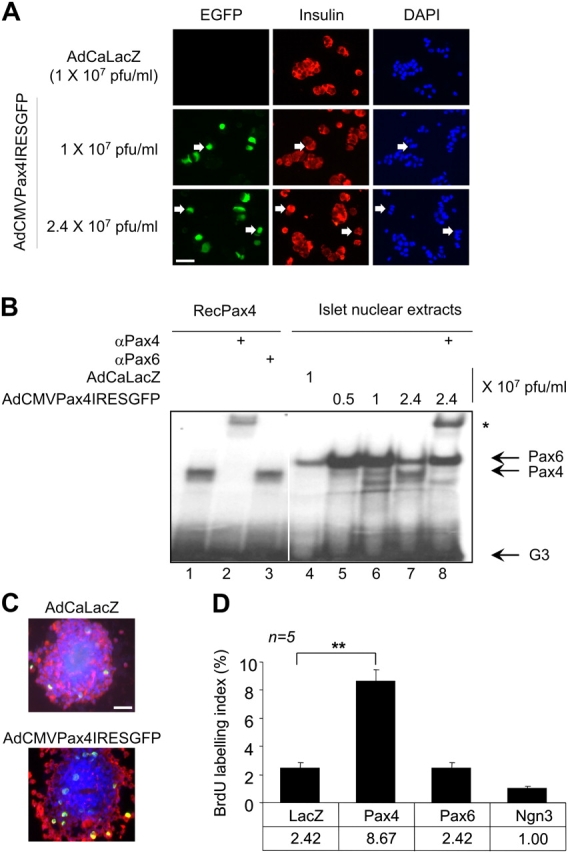
AdCMVPax4IRESGFP-transduced rat islets express Pax4 and exhibit increased β-cell replication. (A) Immunofluorescent detection of EGFP (green) and insulin (red) as well as DAPI nuclei staining (blue) in dispersed islet cells 48 h after infection with the indicated doses of adenovirus. Pax4 is identified via the reporter cotranslated EGFP in insulin-positive cells (arrows). (B) EMSA using a radiolabeled G3 element of the glucagon gene promoter and full-length mouse Pax4, produced by the coupled TNT system (lanes 1–3), as well as 6 μg of nuclear protein extracts from infected rat islets (lanes 4–8). Infection for 48 h with the indicated amounts of the adenovirus increased Pax4 DNA binding activity to the G3 element in a dose-dependent manner (lanes 5–7). The asterisk delineates the formation of a supershift complex in the presence of anti-Pax4 serum (lanes 2 and 8). White line indicates that intervening lanes have been spliced out. (C) β-Cell proliferation was measured by BrdU incorporation in islets infected either with AdCaLacZ or AdCMVPax4IRESGFP (2.4 × 107 pfu/ml). A representative composite image of an islet immunostained for BrdU (green), insulin (red), and DAPI (blue) is shown. (D) Dispersed β-cells immunostained for both insulin and BrdU were counted under a fluorescent microscope and results are depicted as a percentage of BrdU/insulin-positive cells over the total amount of insulin-positive cells. Data show the mean ± SEM of five independent experiments, each representing more than 1,000 cells per condition. **, P < 0.01. Bars, 50 μM.
Figure 3.

Time-dependent gene expression profiling of Pax4-overexpressing islets. (A) EMSA using 6 μg of nuclear protein extracts from AdCMVPax4IRESGFP-transduced rat islets cultured in RPMI 1640 medium over a period of 6 d. Pax4 DNA binding activity to the G3 element is maximal 1 d after infection. The asterisk represents the supershifted complex in the presence of anti-Pax4 serum. (B and C) Quantitative RT-PCR analysis performed on RNA isolated from AdCaLacZ (LacZ; •)- and AdCMVPaxIRESGFP (PAX4; ▪)-infected islets (2.4 × 107 pfu/ml, 50% infectibility). Transcript levels were grouped into four categories: proliferative genes comprising c-myc and Id2; apoptotic genes composed of Bcl-xL, Bcl-2, and caspase-3; the transcription factor Pdx-1; and endocrine hormone genes comprising insulin, glucagon, and somatostatin. Expression patterns were measured over a period of 6 d. Each value represents mean ± SEM of three independent experiments. Statistical significance was tested between LacZ- and PAX4-infected islets by unpaired t test. *, P < 0.05; **, P < 0.01.
Pax4 induces genes implicated in proliferation and survival
The c-myc oncogene was shown to be an important regulator of both cell proliferation and apoptosis in mouse islet β-cells (Pelengaris et al., 2002). Thus, a temporal expression profiling of this factor was performed in rat islets infected for up to 6 d with either AdCMVPax4IRESGFP or AdCaLacZ. EMSA revealed a transient Pax4 DNA binding activity to the G3 element reaching maximal levels 1 d after infection with AdCMVPax4IRESGFP and returning to low levels by day 6 (Fig. 3 A). In parallel, c-myc mRNA levels were induced fourfold in Pax4-expressing islets 24 and 96 h after infection as compared with corresponding time points of cells expressing LacZ (Fig. 3 B). Because c-myc stimulates proliferation through activation of the Id2 cell cycle progression regulator, we explored whether or not this pathway was triggered in Pax4-overexpressing islets (Lasorella et al., 2000). As anticipated, the c-myc target Id2 was increased 5-fold in AdCMVPax4IRESGFP-transduced islets as compared with those detected in control 4 d after infection (Fig. 3 B). Bcl-xL was shown to prevent c-myc–induced β-cell apoptosis and to promote proliferation by suppressing the mitochondrial apoptotic pathway (Pelengaris et al., 2002). A similar phenomenon was reported in a mouse model recapitulating human plasma cell neoplasms (Cheung et al., 2004), indicating an intimate coupling between c-myc and bcl-xl gene expression in promoting proliferation and survival. Consistent with this hypothesis, expression levels of Bcl-xL were found to be 2.2- and 1.9-fold higher in Pax4-expressing cells 24 and 96 h after infection. Caspase-3 mRNA levels remained constant for the duration of the experiment, whereas Bcl-2 mRNA levels were transiently induced (Fig. 3 B). These results suggest that Pax4 may stimulate β-cell proliferation through the activation of the c-myc–Id2 pathway and potentially Bcl-xL gene expression.
Hormone expression profiling of AdCMVPax4IRESGFP-infected islets
Pax4 was reported to inhibit expression of insulin and glucagon in various β and α cell lines (Campbell et al., 1999; Ritz-Laser et al., 2002). We found that insulin, glucagon, and somatostatin mRNA levels were unaltered in AdCMVPax4IRESGFP-infected islets for up to 6 d after transduction (Fig. 3 C). Consistent with these findings, mRNA levels for the transcription factor Pdx1, a major stimulator of insulin and somatostatin gene transcription, also remained stable (Fig. 3 C). Glucagon and insulin protein contents were next determined by radioimmunoassay 48 h after infection. A small but significant increase in insulin protein content was measured in islets transduced with the highest concentration of AdCMVPax4IRESGFP, whereas glucagon protein levels remained constant (Table I). This increase in insulin could be attributed to Pax4-induced increase in cell number as mRNA levels for the hormone remained constant. Thus, Pax4 does not function as a transcriptional repressor of insulin and glucagon in mature islet cells.
Table I. Insulin and glucagon protein contents in Pax4-overexpressing islets.
| Insulin | Glucagon | |
|---|---|---|
| ng/islet | ng/islet | |
| Control | 58.3 ± 2.4 | 1.0 ± 0.3 |
| AdCALacZ | 42.3 ± 0.8 | 1.1 ± 0.1 |
| AdCMVPax4IRESGFP | ||
| 1 × 107 | 59.5 ± 2.2 | 1.0 ± 0.2 |
| 2.4 × 107 | 74.9 ± 3.3 | 1.1 ± 0.2 |
Total insulin and glucagon protein contents were quantified by radioimmunoassay 48 h after infection, and results were expressed as nanograms of protein per islet. Data show the mean ± SEM of three independent experiments. Statistical significance was tested between control and PAX4-infected islets (2.4 × 107 pfu/ml) by unpaired t test and was found to be P < 0.01.
Pax4 transactivates both the c-myc and Bcl-xL gene promoter
To examine whether or not Pax4 is involved in the regulation of c-myc and Bcl-xL transcription, transient transfection assays were performed in BHK-21 cells with luciferase reporter constructs harboring either gene promoter along with increasing amounts of Pax4. The impact of the type 2 diabetes–associated Pax4 mutation R129W, located in the paired DNA binding domain, was also evaluated (Shimajiri et al., 2001). We generated two expression vectors containing either a wild type (wt; Pax4-myc wt) or mutant Pax4 (Pax4-myc R129W) cDNA fused to the myc epitope. We first validated expression and localization of the proteins encoded by the two constructs in rat insulinoma INS-1E cells. Immunofluorescence using a myc antibody revealed nuclear localization of Pax4 (wt and mutant) in transfected cells (Fig. 4 A). Transfection with the vesicular protein synaptotagmin VII/myc tag resulted in cytoplasmic staining, indicating that the epitope did not interfere with proper compartmentalization (Fig. 4 A, bottom). EMSA using equal amounts of in vitro transcribed and translated recombinant proteins (verified by Western blotting, Fig. 4 C) and the G3 element confirmed the binding activity of the myc-fused wt and mutant Pax4 proteins (Fig. 4 B, lanes 1 and 3). The specificity of the complex was demonstrated by supershift assay using the myc antibody (Fig. 4 B, lanes 2 and 4). Interestingly, the G3 binding affinity of the Pax4-myc R129W protein was much weaker than the Pax4-myc wt. Transient transfections revealed that increasing amounts of the Pax4-myc wt expression vector dose dependently stimulated luciferase activity of the c-myc and Bcl-xL gene promoter constructs reaching up to 3.5- and 2.7-fold, respectively (Fig. 4 D). However, Pax4-myc R129W was less efficient in transactivating both constructs, reaching maximal induction levels of only 2.1- and 1.5-fold for the c-myc and Bcl-xL reporter constructs, respectively (Fig. 4 C). Transactivation was promoter specific because Pax4 was unable to induce the telomerase promoter Tert-Luc (Fig. 4 D). These results indicate that Pax4 regulates c-myc and Bcl-xL transcription, whereas the mutant form is less efficient in stimulating the expression of the two genes.
Figure 4.
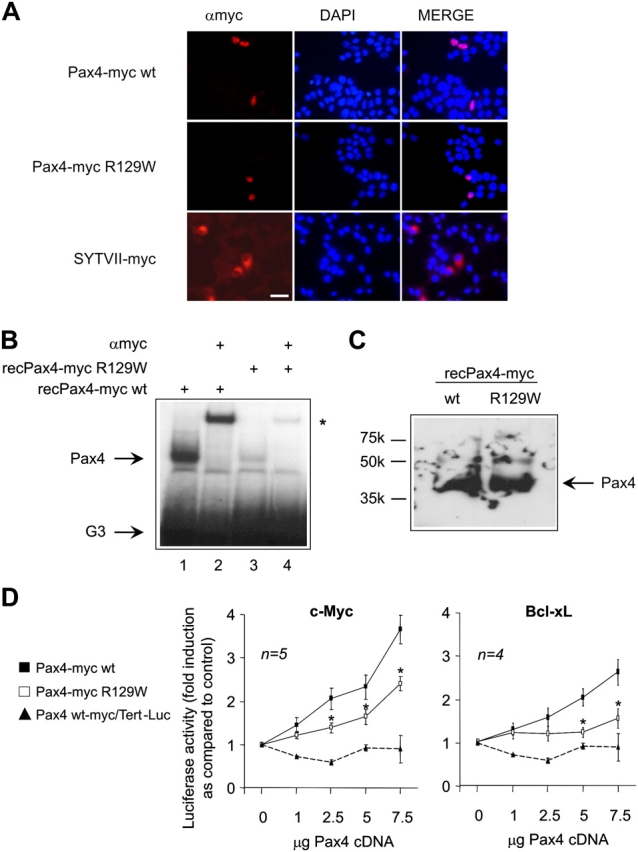
Analysis of the expression and function of Pax4 wt and its mutant R129W. (A) Immunofluorescent detection of the myc-tagged Pax4 or synaptotagmin VII proteins (red) and DAPI nuclei staining (blue) in INS-1E cells 48 h after transfection with the indicated constructs. Pax4 and synaptotagmin VII were detected via the myc epitope in the nuclei and cytoplasm of INS-1E cells, respectively. (B) EMSA using the G3 element and the recombinant proteins Pax4-myc wt (lanes 1 and 2) and Pax4-myc R129W (lanes 3 and 4). An equal amount of protein was applied in each lane (see Fig. 4 C). Pax4 wt bound to the G3 element (lane 1), whereas the binding of the R129W mutant was less efficient (lane 3). The asterisk delineates the formation of a supershift complex due to the addition of anti-myc epitope antibody (lanes 2 and 4). (C) Western blotting of the recombinant proteins Pax4-myc wt and R129W using an anti-myc epitope antibody. (D) Effects of Pax4-myc wt (▪) and its mutant R129W (□) on the human c-myc and murine Bcl-xL promoters. Cotransfection studies using BHK-21 cells were performed with increasing amounts of wt and R129W Pax4. The telomerase promoter construct (▴) was used as a negative control. The pSV-β-galactosidase control vector was used as internal control to normalize for transfection efficiency (∼15%). Data are presented as fold induction of basal luciferase activity and expressed as the mean ± SEM of four to five independent experiments. *, P < 0.05, for comparison between Pax4 wt and R129W for each of the promoter constructs. Bar, 50 μM.
Pax4 overexpression attenuates insulin secretion in islets
Although other antiapoptotic genes may be implicated in the protection of c-myc–induced cell death, we pursued the potential protective function of Bcl-xL in view of its link with c-myc in β-cell survival and proliferation (Pelengaris et al., 2002). Small increases in Bcl-xL, similar to those observed in our work, were shown to protect β-cells against thapsigargin-induced apoptosis in a transgenic mouse model. Increased levels of this mitochondrially targeted protein were also found to impair insulin secretion (Zhou et al., 2000). Consistent with these studies, we found that glucose-stimulated insulin exocytosis was attenuated by 50% in Pax4-overexpressing islets 48 h after infection. β-Galactosidase–expressing islets and noninfected controls exhibited an expected threefold increase in hormone release (Fig. 5 A). However, inhibition was transient as glucose-induced insulin secretion was restored 6 d after infection (unpublished data). Inclusion of 1 μM forskolin/100 μM IBMX, which modulates the effect of glucose on secretion by raising cAMP levels, restored glucose-induced insulin exocytosis, indicating that events downstream of plasma membrane depolarization are functional in Pax4-expressing cells. To evaluate whether or not Pax4-induced Bcl-xL expression curtails the metabolism–secretion coupling cascade, glucose metabolism as well as ATP levels and mitochondrial calcium concentrations ([Ca2+]m) were measured in transduced islets. The rate of glucose oxidation was estimated by measuring the conversion of D-[14C(U)] to 14CO2 and found to be equally efficient in both control and infected islets (Fig. 5 B). However, total cellular ATP levels were fourfold higher in islets expressing Pax4 as compared with control LacZ islets (Fig. 6 A). Cellular ATP levels largely reflect sequestered pools in organelles, in particular in the mitochondria (Detimary et al., 1995). These results prompted us to investigate whether or not glucose was able to raise cytosolic ATP levels in Pax4-overexpressing islets, which are essential in the coupling of metabolism to insulin secretion (Gauthier et al., 2004). Addition of 16.5 mM glucose to control/LacZ islets resulted in a 23% increase of cytosolic ATP, which was sustained until the injection of azide, a compound that dissipates the mitochondrial membrane potential and thus interrupts ATP formation (Fig. 6 B). Cytosolic ATP from islets maintained in 2.5 mM glucose gradually decreased to levels 80% of those at time of glucose injection consistent with low sustained energy consumption. Unexpectedly, basal cytosolic ATP in AdCMVPax4IRESGFP-infected islets was 30% of that measured in control islets, and a small nonsignificant increase in production was detected after exposure to 16.5 mM glucose (Fig. 6 B). Changes in cytosolic calcium are relayed to the mitochondria (Kennedy et al., 1996; Ishihara et al., 2003). Resting [Ca2+]m was elevated in β-cells of Pax4-transduced islets, nearly twofold higher than controls (Fig. 6 C). High concentrations of extracellular potassium trigger calcium influx across the plasma membrane independently of ATP production and KATP channel closure. The potassium-induced rise in [Ca2+]m was normal in transduced islets, as assessed by the total increase in [Ca2+]m (area under peak [AUP]). However, the glucose-induced increase in [Ca2+]m (AUP) was attenuated by 40 ± 5% in Pax4-expressing islets. Together, these results indicate that increased Pax4 expression provokes alterations in both mitochondrial calcium levels and ATP synthesis, which may underlie the blunted glucose-induced insulin secretion (Fig. 6 D).
Figure 5.

Effects of Pax4 overexpression on insulin secretion and glucose oxidation in isolated rat islets. (A) Glucose-induced insulin secretion was inhibited by AdCMVPax4IRESGFP. 2 d after infection, islet hormone secretion was assayed as described in Materials and methods. Data are expressed as the mean ± SEM of four independent experiments. **, P < 0.01. (B) 2 d after infection with 2.4 × 107 pfu/ml of indicated adenoviruses, islet CO2 generation was measured in the presence of 2.5 or 16.7 mM glucose to assess glucose oxidation rate as described in the experimental procedures. Data represent the mean ± SEM of five independent experiments.
Figure 6.

Total cellular ATP and mitochondrial calcium levels are increased in AdCMVPax4IRESGFP-infected islets. (A) Total cellular ATP levels were measured in islets overexpressing either β-galactosidase or PAX4 (2.4 × 107 pfu/ml, 50% of cell infected) and maintained in 1 mM glucose for 10 min. Results represent the means ± SEM. **, P < 0.01. (B) Cytosolic ATP production in response to 2.5 or 16.5 mM glucose was determined over a period of 20 min using the ATP-sensitive bioluminescence probe luciferase (3.6 × 107 pfu/ml). Glucose and azide were added at indicated times (arrows). Results are the mean ± SEM of at least five experiments performed in duplicates (*, P < 0.05). (C) Mitochondrial calcium was monitored in β-galactosidase or PAX4 overexpressing islets using β-cell–specific/mitochondrial-targeted aequorin as described in Materials and methods. After the establishment of baseline luminescence (30 min; LacZ = 210 ± 49 nM and Pax4 = 387 ± 46 nM, left), islets were superfused for 5 min in basal conditions (0 glucose) before stimulation with glucose (16.7 mM), and then KCl (60 mM) for 5 min intervals each, as shown (middle). The induced increases in [Ca2+]m were evaluated on the basis of the AUP and a presented on the right. Each value represents the mean ± SEM of a minimum of six separate experiments. *, P < 0.05. (D) Proposed model of Pax4-induced β-cell proliferation. Mitogens activate Pax4, which will stimulate c-myc and Bcl-xL gene transcription. c-Myc will promote Id2 gene expression and activate the cell cycle replication program. Bcl-xL increased expression will promote survival by preventing mitochondria from initiating the apoptotic program. However, cells become refractory to glucose-evoked insulin secretion due to altered ATP production and calcium handling.
Induction of Pax4 stimulates human islet β-cell proliferation and protects against apoptosis
Next, we assessed the impact of Pax4 and its mutant variant R129W on human islet proliferation using novel doxycycline inducible recombinant adenoviruses engineered to express these proteins tagged to the myc epitope (Ad-mPax4-myc wt or Ad-mPax4-myc R129W). In the absence of doxycycline, the immunoreactive myc epitope was not detected in transduced islet cells (Fig. 7 A). Addition of 0.5 μg/ml doxycycline resulted in the induction of mPax4-myc wt and R129W in the nuclei of ∼70% of islet cells (Fig. 7 A). Quantitative RT-PCR revealed a 10- and 20-fold increase in Pax4 transcript in islets treated with 0.5 and 1 μg/ml doxycycline, respectively (unpublished data). Similarly, a dose-dependent increase in Pax4 protein levels was detected by Western blotting using the myc epitope antibody (Fig. 7 B). To achieve physiological levels of Pax4, similar to those induced by mitogens, proliferation experiments were performed using the lower concentration of doxycycline (0.5 μg/ml). No visible proliferation was detected in the absence of doxycycline (<1%). Concomitant with induction of mPax4 wt expression, ∼10% of cells incorporated BrdU, whereas only 2% of BrdU-positive cells were detected in islets expressing Pax4 R129W (Fig. 8 A). Interestingly, 7% of cells infected with Ad-mPax4-myc wt were BrdU and insulin positive (as shown in a representative experiment in Fig. 8 B, bottom right arrow), consistent with values obtained in rat islets (Fig. 2 D). However, an additional 3% were only BrdU positive, indicating that other cell types may also be prone to proliferation in the presence of Pax4 (Fig. 8 B, top left arrows). To appraise the potential protective role conferred by Pax4-induced Bcl-xL expression observed in rat islets, transduced human islets were exposed to a mixture of cytokines (2 ng/ml IFN-γ, IL-1β, and TNF-α) to induce apoptosis. Adenoviral-infected islets in the absence of both doxycycline and cytokines exhibited a 1.3-fold increase in apoptosis as compared with control noninfected islets (Fig. 9, A and B). Treatment with cytokines provoked a further 4.4-fold increase in cell death. In contrast, Pax4 wt expression (induced by the two concentrations of doxycycline) conferred complete protection against apoptosis (Fig. 9 A). At the low dose of doxycycline, the R129W mutation showed an attenuate protection against cytokine-induced cell death (8.8% TUNEL-positive cells compared with 4% in Pax4 wt; ANOVA: P < 0.00245; Fig. 9 B). Together, our results suggest that Pax4 promotes human islet cell replication as well as conferring survival potentially through the activation of Bcl-xL. The diabetes-linked mutant form of Pax4 is incapable of reproducing these effects.
Figure 7.
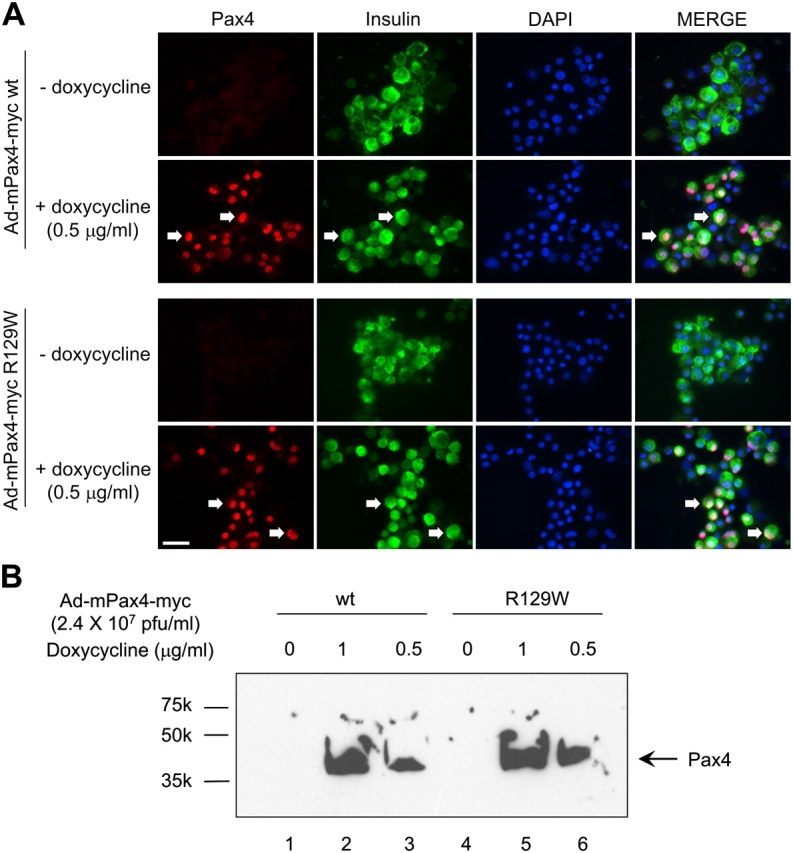
Pax4 and its diabetes-linked mutant are induced by doxycycline in a dose-dependent manner in human islets. (A) Islets were coinfected with either Ad-mPax4-myc wt or R129W as described in Materials and methods. Doxycycline-dependent activation of PAX4 wt and mutant was assessed 48 h later by immunohistochemistry; myc epitope (red), insulin (green), and DAPI (blue). Arrows depict Pax4-expressing β-cells. Pax4 was detected in the nuclei of ∼70% of human islet cells cultured in the presence of doxycycline, whereas no basal induction of Pax4 was observed in the absence of doxycycline. Bar, 50 μM. (B) Western blotting of nuclear extracts derived from infected islet cells cultured in the presence of 0 (lanes 1 and 4), 0.5 (lanes 3 and 6), and 1 μg/ml (lanes 2 and 5) of doxycycline. The same myc anti-serum was used for Western blotting and immunofluorescence.
Figure 8.
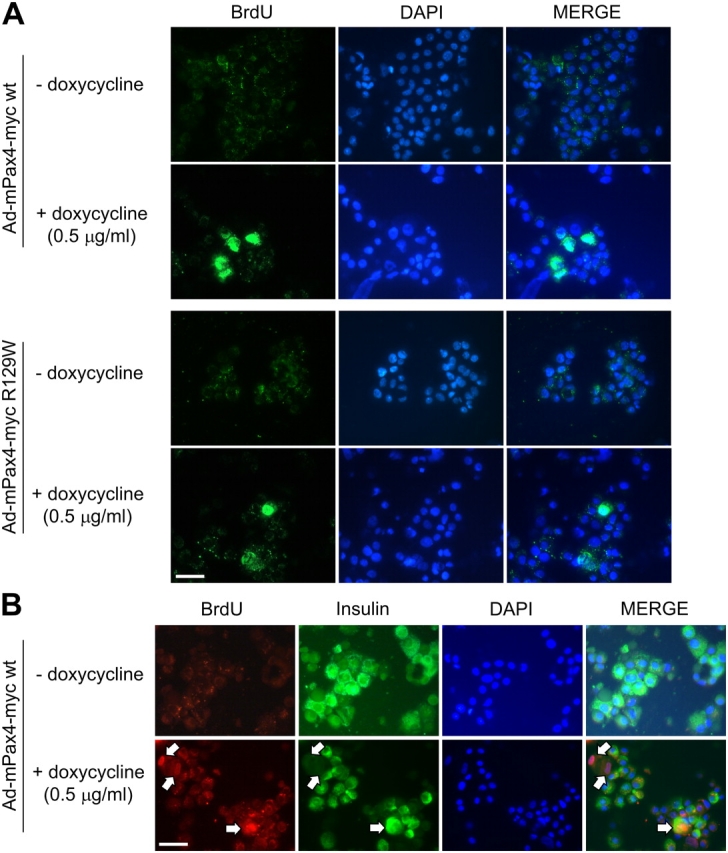
Doxycycline-induced Pax4 stimulates β-cell proliferation in human islets. (A) Proliferation was measured by BrdU incorporation in dispersed islets expressing either Pax4 wt or R129W. Islets were cultured with or without 0.5 μg/ml of doxycycline in the presence of 10 μM BrdU. Immunocytochemical detection of BrdU incorporation (green) and nuclei staining (DAPI in blue) was performed 48 h later. (B) Forced expression of Pax4-induced proliferation in human islet β-cell replication as assessed by costaining of BrdU and insulin (arrows). Bars, 50 μM.
Figure 9.

Doxycycline-stimulated Pax4 expression protects human islets from cytokine-induced apoptosis. Islets were infected with either Ad-mPax4-myc wt (A) or R129W (B) as described in Materials and methods and cultured for 24 h with the indicated concentrations of doxycycline. Islets were subsequently treated for 24 h with IFN-γ, IL-1β, and TNF-α (2 ng/ml each) to promote apoptosis. Cell death was measured by the TUNEL assay. More than 700 cells were counted for each condition. *, P < 0.05; **, P < 0.01. An ANOVA with Bonferroni/Dunn post hoc analysis between Pax4 wt and R129W revealed a statistical significance of P < 0.00245 at 0.25 μg/ml of doxycycline.
Discussion
The endocrine pancreas is considered a dynamic tissue that undergoes perpetual cell renewal (neogenesis and replication) as well as apoptosis throughout a lifetime (Bonner-Weir, 2000; Finegood et al., 2001; Dor et al., 2004). Although growth factors regulating both proliferation and apoptosis in islets are being progressively identified (Garcia-Ocana et al., 2001; Nielsen et al., 2001), underlying molecular mechanisms and target genes remain largely unknown. The present work provides evidence that Pax4 functions as mediator of mitogen-induced β-cell replication by orchestrating the activation of key factors such as c-myc and Bcl-xL.
Consistent with several reports, we detected Pax4 transcript in adult islets but not in the liver. Furthermore, a recent paper has demonstrated that EGFP+ β-cells could be FACS purified from a transgenic mouse model harboring the EGFP cDNA under the control of the Pax4 gene promoter (Theis et al., 2004). These results clearly demonstrate that the promoter is active in mature β-cells and corroborates the detection of the Pax4 transcript.
The importance of Pax4 in β-cell proliferation was highlighted in this work using pharmacological and molecular approaches. A concomitant increase between Pax4 mRNA levels and cell proliferation was found in islets treated with activin A and betacellulin. These mitogens were shown to influence islet proliferation and differentiation (Demeterco et al., 2000). Prentki and coworkers have demonstrated that the proliferative effects of betacellulin and of glucagon-like peptide 1 in β-cells are mediated by PI3-kinase and two of its downstream targets, p38 MAPK and PKCζ (Buteau et al., 2001, 2003). In our work, wortmannin suppressed betacellulin-induced Pax4 expression, implicating the PI3-kinase in this signaling pathway leading to Pax4 activation and subsequent proliferation.
In agreement with published studies, ∼2.5% of β-cells were proliferating in islets cultured in the presence of 10% FCS and 11.5 mM glucose (Scharfmann et al., 1990). Adenoviral-mediated overexpression of Pax4 in rat islets resulted in a more than threefold increase in replication, whereas overexpression of Pax6 and neurogenin3 had no effect. Consistent with the putative proliferative role of Pax4, other members of the Pax family were shown to stimulate cell replication. For instance, increased levels of Pax3 were observed in human tumors of neural crest origin, whereas Pax2 expression was shown to be indispensable for survival of ovarian and bladder cancer cell lines (Muratovska et al., 2003; Parker et al., 2004). Interestingly, Pax5 was identified as a key factor for the maintenance of the tumorigenic phenotype of neuroblastoma, whereas its repression resulted in extensive self-renewal of B cell clones in mice (Schaniel et al., 2002; Baumann Kubetzko et al., 2004). These studies emphasize the critical role of Pax members in cell growth with actions depending on the cellular context.
An increase in c-myc mRNA levels as well as its downstream target Id2 accompanied pax4-induced β-cell proliferation. This member of the Id family is a dominant-negative protein that sequesters the retinoblastoma protein pRb, thus preventing the antiproliferative effect of the tumor suppressor protein (Lasorella et al., 2000). The importance of this pathway was recently demonstrated by showing that suppression of E2F, a target of pRB, results in impaired pancreatic growth and β-cell mass (Fajas et al., 2004). Paradoxically, activation of c-myc in mature β-cells was shown to induce β-cell proliferation and simultaneously promote apoptosis that rapidly erodes β-cell mass (Laybutt et al., 2002; Pelengaris et al., 2002). Concurrent overexpression of Bcl-xL in these β-cells converted c-myc from an apoptotic gene to a growth inducer (Pelengaris et al., 2002). Consistent with these studies, we found a sustained increase in Bcl-xL gene expression, which may thus protect β-cells from apoptosis. Although increases in Bcl-xL transcripts were relatively modest, similar levels were shown to protect islet β-cells from thapsigargin-induced apoptosis (Zhou et al., 2000). We confirmed by transient transfection assays that Pax4 stimulates c-myc and Bcl-xL gene promoter activities. More importantly, we demonstrate that the mutation in the paired DNA binding domain of Pax4, which has been linked to type 2 diabetes, was less efficient in transactivating both genes. A precedent for Bcl-xL transcriptional regulation by Pax family members exists in that Pax3 binds and transactivates the promoter of this gene (Margue et al., 2000).
Normal nutrient-stimulated insulin release is initiated by mitochondrial ATP production. This causes the closure of ATP-dependent K+ channels, plasma membrane depolarization promoting an increase in cytosolic Ca2+, which is the main trigger for exocytosis (Maechler and Wollheim, 2001; Wollheim and Maechler, 2002). Rises in cytosolic Ca2+ are relayed to the mitochondria and reflect β-cell activation (Kennedy et al., 1996; Ishihara et al., 2003). We found that total ATP levels and resting [Ca2+]m were markedly higher in Pax4-transduced islets. Similar alterations in total ATP levels were reported in a mouse model overexpressing Bcl-xL in β-cells as well as in cardiomyocytes treated with IGF-1 (Zhou et al., 2000; Yamamura et al., 2001). Furthermore, Bcl-xL has recently been shown to induce ion channel activity in mitochondria (Jonas et al., 2003) providing an explanation for the elevated [Ca2+]m. Thus, increased Bcl-xL may render β-cells refractory to further stimulation by nutrients. Indeed, glucose-evoked increases in both cytosolic ATP generation and [Ca2+]m were attenuated in Pax4-overexpressing islets, indicating that perhaps Bcl-xL rather than Pax4 is directly responsible for blunted glucose-induced insulin secretion. Despite the elevated total ATP content, basal cytosolic ATP levels were drastically reduced in Pax4-expressing islets indicating defective ATP transport across the mitochondrial membrane. However, mRNA levels for the predominant transporter of ATP, the adenine nucleotide translocase (ANT1), were unaltered (unpublished data), suggesting other consequences of Bcl-xL up-regulation. Therefore, Pax4-stimulated Bcl-xL expression may confer protection against cell death prone to c-myc expression while concomitantly impeding insulin secretion by altering mitochondrial signaling. Incidentally, the raised mitochondrial ATP concentration will inhibit pyruvate dehydrogenase activity and force pyruvate carbons toward pyruvate carboxilase and the anaplerosis pathway. Such a shift was shown to allow normal or even increased CO2 production from glucose despite attenuated PDH activity, providing an explanation for normal steady-state levels of glucose oxidation in Pax4-overexpressing islets (Liu et al., 2004).
A major finding of this work was the capacity of Pax4 to also promote β-cell replication and survival in human islets. Doxycycline-inducible adenoviral vectors allowed us to convincingly show that the wt Pax4 upon drug stimulation promoted proliferation and protected islet cells from cytokine-induced apoptosis, whereas the mutant was less efficacious. Of note, it was recently demonstrated that estrogen-stimulated Bcl-xL expression in neurons protects against cytokine-induced apoptosis reinforcing the potential involvement of Bcl-xL in islet cell survival (Koski et al., 2004). Furthermore, Pax4 levels were maintained close to physiological ranges providing for a specific effect of the transcription factor on proliferation and cell survival. Thus, by modulating apoptosis through Bcl-xL expression and proliferation via c-myc levels, Pax4 may regulate the total population of β-cells and ultimately islet mass.
A recent paper has shown that pancreatic β-cells are replenished exclusively from preexisting mature islet β-cells rather than from precursor cells without providing a molecular mechanism (Dor et al., 2004). We propose in the current study that Pax4 operates as a key regulator of adult β-cell mass by orchestrating the replicating effect of several signal transduction pathways toward the c-myc/Id2 cascade. We further suggest that Pax4 induces Bcl-xL in parallel, thus preventing c-myc–induced apoptosis to the detriment of insulin secretion (see proposed model, Fig. 6 D). Down-regulation of Bcl-xL by RNA interference should confirm this specific protective function. However, we cannot exclude the involvement of other potential anti- or proapoptotic genes in Pax4-induced β-cell survival, a quest that we are currently investigating. The involvement of Pax4 mutations in the development of type 2 diabetes (Shimajiri et al., 2001, 2003; Kanatsuka et al., 2002) and haplotype association with type 1 diabetes (Holm et al., 2004) could be linked to the failure of islets to compensate for the loss of β-cells aggravated by additional genetic and environmental factors.
Materials and methods
Islet isolation and culture
Pancreatic islets were isolated from Wistar rats as described previously (Gauthier et al., 2004). In several instances, islets were exposed to 0.1, 0.5, and 2 nM of activin A, betacellulin, and TGF-β1 as well as 50 or 100 nM of wortmannin (Sigma-Aldrich) for 24 h. Freshly isolated human islets, obtained from D. Bosco (The Cell Isolation and Transplantation Laboratory, Geneva, Switzerland), were maintained in CMRL-1066 supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 μg/ml gentamycin for 2–4 d before experiments.
Plasmid and adenovirus constructions
The full-length mouse Pax4 cDNA was amplified by PCR and the product was cloned into the expression vector pcDNA3.1/myc-His (Invitrogen). The Pax4-myc wt was subjected to mutagenesis to generate mutant Pax4-myc R129W (arginine at codon 129 to tryptophan). The mouse mutant R129W of pax4 gene was shown to correspond to the human mutation R121W (Shimajiri et al., 2001). Subsequently, wt and mutant Pax4-myc cDNAs were subcloned into the pTRE-Shuttle2 vector (CLONTECH Laboratories, Inc.). The inducible cassettes were transferred into the Adeno-X viral DNA to generate recombinant adenoviruses (Ad-mPax4-myc wt and Ad-mPax4-myc R129W).
Adenoviral infection of islets
The recombinant adenoviruses AdCMVPax4IRESGFP, AdCMVNgn3IRESGFP, and AdCMVPax6 were provided by L. St-Onge (DeveloGen AG, Göttingen, Germany). AdCAlacZ, containing the bacterial β-galactosidase cDNA, was used as control (Ishihara et al., 1999). Rat islets were infected with various amounts of recombinant adenoviruses (as indicated in figures) for 90 min, washed, and replenished with fresh medium. Human islets were coinfected with either Ad-mPax4-myc wt or Ad-mPax4-myc R129W along with the adenoviral construct harboring the tetracycline transcriptional activator (Ad-X Tet-On) at a ratio of 2:1 (3.6 × 107 pfu/ml of total viral particles). Cells were rinsed 90 min after infection and cultured in fresh media supplemented with the indicated concentration of doxycycline.
Quantitative RT-PCR
Total RNA from 50 islets was extracted using the Trizol reagent (Invitrogen) and 2 μg were converted into cDNA as previously described (Gauthier et al., 1999b). Primers for cyclophilin, somatostatin, glucagon, insulin, Pdx1, c-myc, Id2, Bcl-xL, Bcl-2, Pax4, and caspase 3 were designed using the Primer Express Software (Applera Europe). Quantitative RT-PCR was performed described as previously (Gauthier et al., 2004).
Transient transfection assays
The c-myc (pDEL-1-Luc), Bcl-xL (Bcl-xL/515), and telomerase (pTERT-luc) gene promoter luciferase reporter constructs were provided by B. Vogelstein (The Johns Hopkins Oncology Center, Baltimore, MD), B. Schaefer (University of Zurich, Zurich, Switzerland), and R. Dalla-Favera (Columbia University, New York, NY), respectively. The BHK-21 cell line was transiently transfected using the calcium phosphate precipitation technique as described previously (Gauthier et al., 1999a). The pSV-β-galactosidase control vector (Promega) was used as internal control to normalize for transfection efficiency (∼15%) in all experiments. Values correspond to the mean and standard error of at least four to five individual transfections performed in duplicates. Results are presented as fold induction of the control sample obtained from cells transfected with empty expression vector.
Nuclear extract preparation and EMSA
Nuclear protein extracts and DNA binding assays were performed as described previously (Gauthier et al., 2002). Recombinant Pax4 as well as Pax6 were prepared using an in vitro transcription and translation system as described by the manufacturer (Promega). Antibodies generated against Pax4 and Pax6 were provided by M.S. German (University of California, San Francisco, San Francisco, CA) and S. Saule (Institut Curie, Orsay Cedex, France), respectively.
Hormone radioimmunoassays
Insulin secretion from 15 matched-size islets per condition was measured over a period of 30 min in Krebs-Ringer bicarbonate Hepes buffer containing the indicated stimulators. Insulin radioimmunoassays were performed as outlined previously (Gauthier et al., 2004). Secreted insulin was expressed as a percentage of total cellular insulin content. Glucagon radioimmunoassays were adapted from a protocol derived from Salehi et al. (1999).
Glucose oxidation and ATP production
Carbon dioxide production derived from glucose oxidation was measured using the multiwell 14CO2-capture assay developed by Collins et al. (1998). ATP measurements were performed as previously outlined (Gauthier et al., 2004).
Mitochondrial calcium measurements
Islets were infected with rAdRIP-maequorin (4.8 × 107 pfu/ml) and either AdCaLacZ or AdCMVPax4IRESGFP (2.4 × 107 pfu/ml) for 90 min. Islets were washed and seeded onto A431 extracellular matrix-coated coverslips (Ishihara et al., 2003). Coverslips were placed in a sealed, thermostatted (37°C) chamber, 5 mm from a photonmultiplier, which was used to detect emitted luminescence, as previously described (Kennedy et al., 1996). Islets were superfused (1.0 ml min−1) with Krebs-Ringer bicarbonate Hepes buffer supplemented with either 16.7 mM glucose or 60 mM KCl where indicated. Luminescence output was recorded every second using a photon-counting board (model C660; Thorn EMI) after a 30-min equilibration period to establish the baseline. Recorded counts were converted to [Ca2+]m as published elsewhere (Challet et al., 2001).
Immunohistochemistry
Islets or single cell suspensions were cultured on polyornithine-treated glass coverslips for 2 d, washed with PBS, and fixed in 4% PFA in PBS for 20 min at RT. Recombinant Pax4 wt or R129W myc-tagged proteins as well as doxycycline-dependent activation of PAX4 were visualized by immunohistochemistry using an antibody against the myc epitope (dilution 1:200; Invitrogen). Immunochemical detection of β-cells was performed as described previously (Ishihara et al., 2003). Nuclei were stained with 10 μg/ml DAPI (Sigma-Aldrich). Coverslips were mounted using fluorescent mounting medium (DakoCytomation) and visualized using an Axiophot I (Carl Zeiss MicroImaging, Inc.).
Cell proliferation and TUNEL assays
For proliferation, 24 h before fixation, infected or mitogen-treated cells were labeled with 10 μM BrdU. Proliferation was estimated using an immunohistochemical assay kit as described by the manufacturer (BrdU labeling and detection kit; Roche). Cells were also costained for insulin as described, and results are expressed as a percentage of BrdU/insulin-positive cells over the total amount of insulin-positive cells. Alternatively, human islets were cultured with or without doxycycline for 24 h after infection and incubated for 24 h in the presence of IFN-γ, IL-1β, and TNF-α (2 ng/ml each) to promote apoptosis. Cell death was measured by the TUNEL assay (In Situ Cell Death Detection Kit; Roche). Results are expressed as a percentage of fluorescein-labeled nuclei (TUNEL-positive cells) over the total amount of islet cells (nuclei staining by DAPI).
Statistical analysis
Results are expressed as mean ± SEM. Where indicated, the statistical significance of the differences between groups was estimated by unpaired t test. * and ** indicate statistical significance with P < 0.05 and P < 0.01, respectively. In some instances, ANOVA with Bonferroni/Dunn post hoc analysis was performed.
Acknowledgments
We are grateful to Dominique Duhamel, Eve-Julie Sarret, Tania Nguyen, Nicole Aebischer, Olivier Dupont, and Aslan Gjinovci for their expert technical assistance.
This work was supported by the European Foundation for the Study of Diabetes and a Johnson and Johnson Research grant (C.B. Wollheim), the Swiss National Science Foundation (grant 32-66907.01 to C.B. Wollheim and B.R. Gauthier), the European Network grant (GrowBeta) through the Swiss Office for Education and Science (grant 01.0260 to C.B. Wollheim), and by a seeding grant from the National Institutes of Health Beta Cell Biology Consortium. We are also indebted to the Bonizzi-Theler Foundation for their financial contribution.
L. St-Onge's present address is NeuroNova AG, 80804 Munich, Germany.
Abbreviations used in this paper: AUP, area under peak; EMSA, electrophoretic mobility shift assay; PI3-kinase, phosphatidylinositol 3-kinase; wt, wild type.
References
- Baumann Kubetzko, F.B., C. Di Paolo, C. Maag, R. Meier, B.W. Schafer, D.R. Betts, R.A. Stahel, and A. Himmelmann. 2004. The PAX5 oncogene is expressed in N-type neuroblastoma cells and increases tumorigenicity of a S-type cell line. Carcinogenesis. 25:1839–1846. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir, S. 2000. Life and death of the pancreatic beta cells. Trends Endocrinol. Metab. 11:375–378. [DOI] [PubMed] [Google Scholar]
- Buteau, J., S. Foisy, C.J. Rhodes, L. Carpenter, T.J. Biden, and M. Prentki. 2001. Protein kinase Czeta activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes. 50:2237–2243. [DOI] [PubMed] [Google Scholar]
- Buteau, J., S. Foisy, E. Joly, and M. Prentki. 2003. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. 52:124–132. [DOI] [PubMed] [Google Scholar]
- Campbell, S.C., H. Cragg, L.J. Elrick, W.M. Macfarlane, K.I. Shennan, and K. Docherty. 1999. Inhibitory effect of Pax4 on the human insulin and islet amyloid polypeptide (IAPP) promoters. FEBS Lett. 463:53–57. [DOI] [PubMed] [Google Scholar]
- Challet, C., P. Maechler, C.B. Wollheim, and U.T. Ruegg. 2001. Mitochondrial calcium oscillations in C2C12 myotubes. J. Biol. Chem. 276:3791–3797. [DOI] [PubMed] [Google Scholar]
- Cheung, W.C., J.S. Kim, M. Linden, L. Peng, B. Van Ness, R.D. Polakiewicz, and S. Janz. 2004. Novel targeted deregulation of c-Myc cooperates with Bcl-X(L) to cause plasma cell neoplasms in mice. J. Clin. Invest. 113:1763–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, C.L., B.P. Bode, W.W. Souba, and S.F. Abcouwer. 1998. Multiwell 14CO2-capture assay for evaluation of substrate oxidation rates of cells in culture. Biotechniques. 24:803–808. [DOI] [PubMed] [Google Scholar]
- Demeterco, C., G.M. Beattie, S.A. Dib, A.D. Lopez, and A. Hayek. 2000. A role for activin A and betacellulin in human fetal pancreatic cell differentiation and growth. J. Clin. Endocrinol. Metab. 85:3892–3896. [DOI] [PubMed] [Google Scholar]
- Detimary, P., J.C. Jonas, and J.C. Henquin. 1995. Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release. Unmasking by decreasing a stable pool of adenine nucleotides in mouse islets. J. Clin. Invest. 96:1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor, Y., J. Brown, O.I. Martinez, and D.A. Melton. 2004. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 429:41–46. [DOI] [PubMed] [Google Scholar]
- Fajas, L., J.S. Annicotte, S. Miard, D. Sarruf, M. Watanabe, and J. Auwerx. 2004. Impaired pancreatic growth, beta cell mass, and beta cell function in E2F1−/− mice. J. Clin. Invest. 113:1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegood, D.T., M.D. McArthur, D. Kojwang, M.J. Thomas, B.G. Topp, T. Leonard, and R.E. Buckingham. 2001. Beta-cell mass dynamics in Zucker diabetic fatty rats. Rosiglitazone prevents the rise in net cell death. Diabetes. 50:1021–1029. [DOI] [PubMed] [Google Scholar]
- Garcia-Ocana, A., R.C. Vasavada, K.K. Takane, A. Cebrian, J.C. Lopez-Talavera, and A.F. Stewart. 2001. Using beta-cell growth factors to enhance human pancreatic Islet transplantation. J. Clin. Endocrinol. Metab. 86:984–988. [DOI] [PubMed] [Google Scholar]
- Gauthier, B., M. Robb, F. Gaudet, G.S. Ginsburg, and R. McPherson. 1999. a. Characterization of a cholesterol response element (CRE) in the promoter of the cholesteryl ester transfer protein gene: functional role of the transcription factors SREBP-1a, -2, and YY1. J. Lipid Res. 40:1284–1293. [PubMed] [Google Scholar]
- Gauthier, B., M. Robb, and R. McPherson. 1999. b. Cholesteryl ester transfer protein gene expression during differentiation of human preadipocytes to adipocytes in primary culture. Atherosclerosis. 142:301–307. [DOI] [PubMed] [Google Scholar]
- Gauthier, B.R., V.M. Schwitzgebel, M. Zaiko, A. Mamin, B. Ritz-Laser, and J. Philippe. 2002. Hepatic nuclear factor-3 (HNF-3 or Foxa2) regulates glucagon gene transcription by binding to the G1 and G2 promoter elements. Mol. Endocrinol. 16:170–183. [DOI] [PubMed] [Google Scholar]
- Gauthier, B.R., T. Brun, E.J. Sarret, H. Ishihara, O. Schaad, P. Descombes, and C.B. Wollheim. 2004. Oligonucleotide microarray analysis reveals PDX1 as an essential regulator of mitochondrial metabolism in rat islets. J. Biol. Chem. 279:31121–31130. [DOI] [PubMed] [Google Scholar]
- Gittes, G.K., and W.J. Rutter. 1992. Onset of cell-specific gene expression in the developing mouse pancreas. Proc. Natl. Acad. Sci. USA. 89:1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heremans, Y., M. Van De Casteele, P. in't Veld, G. Gradwohl, P. Serup, O. Madsen, D. Pipeleers, and H. Heimberg. 2002. Recapitulation of embryonic neuroendocrine differentiation in adult human pancreatic duct cells expressing neurogenin 3. J. Cell Biol. 159:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, P., B. Rydlander, H. Luthman, and I. Kockum. 2004. Interaction and association analysis of a type 1 diabetes susceptibility locus on chromosome 5q11-q13 and the 7q32 chromosomal region in Scandinavian families. Diabetes. 53:1584–1591. [DOI] [PubMed] [Google Scholar]
- Ishihara, H., H. Wang, L.R. Drewes, and C.B. Wollheim. 1999. Overexpression of monocarboxylate transporter and lactate dehydrogenase alters insulin secretory responses to pyruvate and lactate in beta cells. J. Clin. Invest. 104:1621–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara, H., P. Maechler, A. Gjinovci, P.L. Herrera, and C.B. Wollheim. 2003. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat. Cell Biol. 5:330–335. [DOI] [PubMed] [Google Scholar]
- Jonas, E.A., D. Hoit, J.A. Hickman, T.A. Brandt, B.M. Polster, Y. Fannjiang, E. McCarthy, M.K. Montanez, J.M. Hardwick, and L.K. Kaczmarek. 2003. Modulation of synaptic transmission by the BCL-2 family protein BCL-xL. J. Neurosci. 23:8423–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsuka, A., Y. Tokuyama, O. Nozaki, K. Matsui, and T. Egashira. 2002. Beta-cell dysfunction in late-onset diabetic subjects carrying homozygous mutation in transcription factors NeuroD1 and Pax4. Metabolism. 51:1161–1165. [DOI] [PubMed] [Google Scholar]
- Kennedy, E.D., R. Rizzuto, J.M. Theler, W.F. Pralong, C. Bastianutto, T. Pozzan, and C.B. Wollheim. 1996. Glucose-stimulated insulin secretion correlates with changes in mitochondrial and cytosolic Ca2+ in aequorin-expressing INS-1 cells. J. Clin. Invest. 98:2524–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima, H., M. Fujimiya, K. Matsumura, P. Younan, H. Imaeda, M. Maeda, and L. Chan. 2003. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat. Med. 9:596–603. [DOI] [PubMed] [Google Scholar]
- Koski, C.L., S. Hila, and G.E. Hoffman. 2004. Regulation of cytokine-induced neuron death by ovarian hormones: involvement of antiapoptotic protein expression and c-JUN N-terminal kinase-mediated proapoptotic signaling. Endocrinology. 145:95–103. [DOI] [PubMed] [Google Scholar]
- Lasorella, A., M. Noseda, M. Beyna, Y. Yokota, and A. Iavarone. 2000. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature. 407:592–598. [DOI] [PubMed] [Google Scholar]
- Laybutt, D.R., H. Kaneto, W. Hasenkamp, S. Grey, J.C. Jonas, D.C. Sgroi, A. Groff, C. Ferran, S. Bonner-Weir, A. Sharma, and G.C. Weir. 2002. Increased expression of antioxidant and antiapoptotic genes in islets that may contribute to beta-cell survival during chronic hyperglycemia. Diabetes. 51:413–423. [DOI] [PubMed] [Google Scholar]
- Liu, Y.Q., J.A. Moibi, and J.L. Leahy. 2004. Chronic high glucose lowers pyruvate dehydrogenase activity in islets through enhanced production of long chain acyl-CoA: prevention of impaired glucose oxidation by enhanced pyruvate recycling through the malate-pyruvate shuttle. J. Biol. Chem. 279:7470–7475. [DOI] [PubMed] [Google Scholar]
- Maechler, P., and C.B. Wollheim. 2001. Mitochondrial function in normal and diabetic beta-cells. Nature. 414:807–812. [DOI] [PubMed] [Google Scholar]
- Margue, C.M., M. Bernasconi, F.G. Barr, and B.W. Schafer. 2000. Transcriptional modulation of the anti-apoptotic protein BCL-XL by the paired box transcription factors PAX3 and PAX3/FKHR. Oncogene. 19:2921–2929. [DOI] [PubMed] [Google Scholar]
- Miyamoto, T., T. Kakizawa, K. Ichikawa, S. Nishio, S. Kajikawa, and K. Hashizume. 2001. Expression of dominant negative form of PAX4 in human insulinoma. Biochem. Biophys. Res. Commun. 282:34–40. [DOI] [PubMed] [Google Scholar]
- Muratovska, A., C. Zhou, S. He, P. Goodyer, and M.R. Eccles. 2003. Paired-Box genes are frequently expressed in cancer and often required for cancer cell survival. Oncogene. 22:7989–7997. [DOI] [PubMed] [Google Scholar]
- Nielsen, J.H., E.D. Galsgaard, A. Moldrup, B.N. Friedrichsen, N. Billestrup, J.A. Hansen, Y.C. Lee, and C. Carlsson. 2001. Regulation of beta-cell mass by hormones and growth factors. Diabetes. 50:S25–S29. [DOI] [PubMed] [Google Scholar]
- Parker, C.J., S.G. Shawcross, H. Li, Q.Y. Wang, C.S. Herrington, S. Kumar, R.M. MacKie, W. Prime, I.G. Rennie, K. Sisley, and P. Kumar. 2004. Expression of PAX 3 alternatively spliced transcripts and identification of two new isoforms in human tumors of neural crest origin. Int. J. Cancer. 108:314–320. [DOI] [PubMed] [Google Scholar]
- Pelengaris, S., M. Khan, and G. Evan. 2002. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 109:321–334. [DOI] [PubMed] [Google Scholar]
- Ritz-Laser, B., A. Estreicher, B. Gauthier, A. Mamin, H. Edlund, and J. Philippe. 2002. The pancreatic b-cell-specific transcription factor Pax-4 inhibits glucagon gene expression through Pax-6. Diabetologia. 45:97–107. [DOI] [PubMed] [Google Scholar]
- Salehi, A., D. Chen, R. Hakanson, G. Nordin, and I. Lundquist. 1999. Gastrectomy induces impaired insulin and glucagon secretion: evidence for a gastro-insular axis in mice. J. Physiol. 514:579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaniel, C., M. Gottar, E. Roosnek, F. Melchers, and A.G. Rolink. 2002. Extensive in vivo self-renewal, long-term reconstitution capacity, and hematopoietic multipotency of Pax5-deficient precursor B-cell clones. Blood. 99:2760–2766. [DOI] [PubMed] [Google Scholar]
- Scharfmann, R., A. Basmaciogullari, and P. Czernichow. 1990. Effect of growth hormone and glucose on rat islet cells replication using 5-bromo-2-deoxyuridine incorporation. Diabetes Res. 15:137–141. [PubMed] [Google Scholar]
- Shimajiri, Y., T. Sanke, H. Furuta, T. Hanabusa, T. Nakagawa, Y. Fujitani, Y. Kajimoto, N. Takasu, and K. Nanjo. 2001. A missense mutation of Pax4 gene (R121W) is associated with type 2 diabetes in Japanese. Diabetes. 50:2864–2869. [DOI] [PubMed] [Google Scholar]
- Shimajiri, Y., M. Shimabukuro, T. Tomoyose, H. Yogi, I. Komiya, and N. Takasu. 2003. PAX4 mutation (R121W) as a prodiabetic variant in Okinawans. Biochem. Biophys. Res. Commun. 302:342–344. [DOI] [PubMed] [Google Scholar]
- Sosa-Pineda, B., K. Chowdhury, M. Torres, G. Oliver, and P. Gruss. 1997. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 386:399–402. [DOI] [PubMed] [Google Scholar]
- Theis, M., C. Mas, B. Doring, J. Degen, C. Brink, D. Caille, A. Charollais, O. Kruger, A. Plum, V. Nepote, et al. 2004. Replacement by a lacZ reporter gene assigns mouse connexin36, 45 and 43 to distinct cell types in pancreatic islets. Exp. Cell Res. 294:18–29. [DOI] [PubMed] [Google Scholar]
- Ueda, Y. 2000. Activin A increases Pax4 gene expression in pancreatic beta cell lines. FEBS Lett. 480:101–105. [DOI] [PubMed] [Google Scholar]
- Wollheim, C.B., and P. Maechler. 2002. Beta-cell mitochondria and insulin secretion: messenger role of nucleotides and metabolites. Diabetes. 51 Suppl 1:S37–S42. [DOI] [PubMed] [Google Scholar]
- Yamamura, T., H. Otani, Y. Nakao, R. Hattori, M. Osako, and H. Imamura. 2001. IGF-I differentially regulates Bcl-xL and Bax and confers myocardial protection in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 280:H1191–H1200. [DOI] [PubMed] [Google Scholar]
- Zalzman, M., S. Gupta, R.K. Giri, I. Berkovich, B.S. Sappal, O. Karnieli, M.A. Zern, N. Fleischer, and S. Efrat. 2003. Reversal of hyperglycemia in mice by using human expandable insulin-producing cells differentiated from fetal liver progenitor cells. Proc. Natl. Acad. Sci. USA. 100:2426–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y.P., J.C. Pena, M.W. Roe, A. Mittal, M. Levisetti, A.C. Baldwin, W. Pugh, D. Ostrega, N. Ahmed, V.P. Bindokas, et al. 2000. Overexpression of Bcl-x(L) in beta-cells prevents cell death but impairs mitochondrial signal for insulin secretion. Am. J. Physiol. Endocrinol. Metab. 278:E340–E351. [DOI] [PubMed] [Google Scholar]