

Abstract
In Xenopus oocytes, the spindle assembly checkpoint (SAC) kinase Bub1 is required for cytostatic factor (CSF)-induced metaphase arrest in meiosis II. To investigate whether matured mouse oocytes are kept in metaphase by a SAC-mediated inhibition of the anaphase-promoting complex/cyclosome (APC/C) complex, we injected a dominant-negative Bub1 mutant (Bub1dn) into mouse oocytes undergoing meiosis in vitro. Passage through meiosis I was accelerated, but even though the SAC was disrupted, injected oocytes still arrested at metaphase II. Bub1dn-injected oocytes released from CSF and treated with nocodazole to disrupt the second meiotic spindle proceeded into interphase, whereas noninjected control oocytes remained arrested at metaphase. Similar results were obtained using dominant-negative forms of Mad2 and BubR1, as well as checkpoint resistant dominant APC/C activating forms of Cdc20. Thus, SAC proteins are required for checkpoint functions in meiosis I and II, but, in contrast to frog eggs, the SAC is not required for establishing or maintaining the CSF arrest in mouse oocytes.
Introduction
After mitotic expansion of diploid germ cells, oocytes enter meiotic differentiation, which is a discontinuous two-step process. After initiation of meiosis, oocytes become arrested in prophase of meiosis I, where oocytes continue to grow until progesterone triggers the completion of meiosis I and entry into meiosis II. Matured oocytes are arrested at metaphase of meiosis II and await fertilization, which completes the meiotic process.
Investigating the nature of this arrest in meiosis II, Masui and Markert (1971) discovered that microinjection of a cytoplasmic extract from metaphase II arrested frog eggs into one blastomere of a two cell stage embryo caused a cleavage arrest in the injected blastomere, whereas the uninjected blastomere continued to divide normally. This activity was called cytostatic factor (CSF). The same extracts, when injected into immature oocytes, which are arrested in the prophase of first meiosis, could induce the resumption and completion of the meiotic maturation process. This second activity was called maturation promoting factor (MPF). Later, the Mos–MAPK pathway was identified as CSF (Sagata et al., 1989), and a complex of Cdk1–cyclin B as MPF (Dunphy et al., 1988; Gautier et al., 1988; Lohka et al., 1988). Both, Mos–MAPK and Cdk1–cyclin B activities are low in prophase/germinal vesicle (GV) stage oocytes, are stimulated by the resumption of the meiotic process and are high at GV breakdown (GVBD), which marks the entry into meiotic maturation. MPF activity drops between the first and the second meiosis, whereas the Mos–MAPK pathway is active throughout meiosis. Upon fertilization, both activities drop and meiosis II is completed with the extrusion of the second polar body (PB; Tunquist and Maller, 2003).
The role of the Mos–MAPK pathway in meiosis has been extensively studied in Xenopus eggs. In this system, the MAPKKK Mos performs several roles, i.e., initiation of GVBD (Sagata et al., 1988; Yew et al., 1992), suppression of S-phase between the first and the second M-phase (Furuno et al., 1994), as well as the CSF arrest (Sagata et al., 1989). In mouse oocytes, Mos is synthesized around GVBD, and, as in the frog, MAPK is active throughout meiosis (Verlhac et al., 1996). To check, whether Mos is essential for meiosis in mouse oocytes, knockout mice were generated (Colledge et al., 1994; Hashimoto et al., 1994). Mos-deficient mice were viable, but females were subfertile due to defects in oocyte maturation. Their oocytes became frequently parthenogenetically activated and extruded the second PB, indicating an essential role for Mos in the meiosis II arrest of mouse oocytes. In contrast to frog oocytes, however, Mos-deficient oocytes were still able to suppress DNA replication between meiosis I and II, as well as after meiosis II. Careful analysis of the Mos-deficient oocytes revealed that, after the extrusion of the second PB, the parthenogenetically activated eggs did not exit M-phase, but stayed in a metaphase-like state. This third metaphase is thought to be mediated by the spindle assembly checkpoint (SAC), induced by the haploid genome (Verlhac et al., 1996).
How DNA replication is suppressed between meiosis I and II, and how Mos arrests cells at metaphase of meiosis II are important, yet still unresolved, issues. Studies using Xenopus egg extracts identified several factors required for CSF activity, including Cdk2/cyclin E (Tunquist et al., 2002), RINGO (Ferby et al., 1999), Emi1 (Reimann and Jackson, 2002), and Bub1 (Tunquist et al., 2002). Among those factors, a direct connection to the MAPK pathway has only been postulated for the Bub1 kinase, which is a SAC component that was shown to be phosphorylated in vitro by p90Rsk, a downstream target of MAPK (Schwab et al., 2001). Bub1 might thus be a link between CSF and the SAC, suggesting that components of the SAC might be recruited to generate the arrest of mature oocytes at metaphase of meiosis II in Xenopus eggs (Tunquist et al., 2002, 2003).
During mitosis, the SAC senses the proper attachment of chromosomes to the spindle apparatus, delaying sister chromatid separation and exit from mitosis until all kinetochores are properly attached in a bipolar manner (Musacchio and Hardwick, 2002; Yu, 2002). The molecular components of the SAC were first discovered in yeast genetic screens, which identified Mad1, 2, and 3 (BubR1), as well as Bub1-3 and Mps1 as essential components (Hoyt et al., 1991; Li and Murray, 1991; Weiss and Winey, 1996). With the exception of Bub2, these proteins are conserved in eukaryotes and generally required for the SAC. In contrast to budding yeast, however, the mitotic checkpoint is essential for proper progression through mitosis in multicellular organisms (Dobles et al., 2000).
Mad1, Mad2, BubR1, Bub1, and Bub3 localize to the kinetochores of unaligned chromosomes, generating the inhibitory signal that delays the onset of anaphase (Basu et al., 1998; Chen et al., 1996; Li and Benezra, 1996; Taylor and McKeon, 1997; Taylor et al., 1998; Chan et al., 1999). Bub1 and BubR1 are protein kinases, which both interact with Bub3 (Taylor et al., 1998). BubR1 interacts with CENP-E (Chan et al., 1998), which senses spindle tension at the kinetochores, and Cdc20, thereby inhibiting the anaphase-promoting complex/cyclosome (APC/C; Tang et al., 2001). Bub1 is required for the recruitment of other proteins to the kinetochores and was proposed to work as a scaffolding protein (Sharp-Baker and Chen, 2001; Musacchio and Hardwick, 2002). Once the checkpoint is activated, Bub1 also interacts with Mad1, which is required for the kinetochore localization of Mad2 and the formation of the Mad2-Cdc20 complex (Luo et al., 2002). The localization of these proteins to the kinetochore is thus interdependent and disruption of one protein is often enough to cause checkpoint defects in cells. However, it is not yet clear whether the SAC components act in a single or in multiple pathways, responding to microtubule attachment at kinetochores, or interkinetochore tension caused by bipolar attachment to the mitotic spindle, respectively (Musacchio and Hardwick, 2002; Yu, 2002; Zhou et al., 2002).
Little is known about a potential involvement of the SAC in the CSF arrest of mouse oocytes. Mad2 and Bub1 localize to kinetochores in metaphase II arrested oocytes, supporting a potential role of the SAC in CSF (Kallio et al., 2000; Brunet et al., 2003). Moreover, a Mad2-dependent checkpoint is functional during meiosis I (Wassmann et al., 2003). SAC components are thus present and can be activated during the meiotic maturation of mouse oocytes. Here, we tested the involvement of several SAC proteins in CSF by injecting dominant-negative interfering mutants of checkpoint proteins (Bub1, Mad2, and BubR1), or gain of function mutants of the APC/C activator Cdc20, into mouse oocytes. We present evidence that, although these proteins disrupt the SAC in meiosis I and II, they do not abrogate the CSF-induced metaphase II arrest.
Results
The effect of dominant-negative Bub1 (Bub1dn) on meiosis I and II
To check whether the SAC is involved in the CSF-mediated metaphase II arrest in mouse oocytes, we chose to microinject interfering mutants of SAC components into oocytes. GV stage (prophase I) oocytes were injected with in vitro transcribed and capped mRNAs encoding the mutant protein, kept in prophase for 1 h by medium containing dbcAMP, and subsequently released for completion of meiosis by exchanging the medium. Resumption of meiotic maturation can be monitored because the oocytes dissolve their nuclear membrane (GVBD) 60–90 min after the release. The first meiotic division, which results in the extrusion of a PB, takes place 9–10 h after GVBD. Oocytes then enter meiosis II without an intermittent S phase, and become arrested at metaphase of meiosis II, by the activity of CSF. This metaphase II arrest is stable for more than 15 h in our in vitro culture system (see Fig. 1 for a schematical representation of our experimental set up).
Figure 1.

Overview of the oocyte injection experiments. MI, AI, and TI indicate metaphase, anaphase, and telophase of meiosis I, respectively. IK, interkinasis; MII, metaphase of meosis II. Time after GVBD is indicated in hours.
A YFP-fusion protein of a dominant-negative version of Bub1 (Bub1dn, amino acids 1–331) was used to test whether Bub1 is required for the metaphase II arrest in mouse oocytes. This mutant lacks the kinase domain, and disrupts the SAC in somatic cells by competing with the endogenous kinase for kinetochore localization. When microinjected into mouse oocytes, the fusion protein localized to kinetochores during meiosis (Fig. 2).
Figure 2.

Localization of Bub1dn-YFP on kinetochores. Bub1dn-YFP and histone H2B-RFP mRNAs were coinjected into GV-stage oocytes, kept in dbcAMP for 1 h, released, and fixed 5–6 h after GVBD (1st meiosis) or overnight culture (2nd meiosis). (Left) The whole oocyte is shown in an overview, with chromosomes in red (histone H2B-RFP) and Bub1dn-YFP in green. The position of the PB is marked. (Right) Chromosomes are enlarged, and the signals from histone H2B-RFP and Bub1dn-YFP individually, as well as overlaid (merge). Note the dotted appearance of the Bub1dn-YFP signal around the chromosomes suggesting a kinetochore localization. Bar, 10 μm.
Because the localization of Bub1dn-YFP to kinetochores might enable it to interfere with Bub1 function during meiosis, we microinjected the mRNA encoding Bub1dn into prophase oocytes, and monitored progression through meiosis. Time-lapse video microscopy (Fig. 3 A) revealed that most of the Bub1dn mRNA injected oocytes extruded their first PB 3 h earlier (i.e., 6–7 h after GVBD) than non- or control-injected oocytes (Table I). Thus, the Bub1dn mutant accelerated progression through meiosis I, probably by interfering with the SAC.
Figure 3.

The effect of Bub1dn injection on meiosis I and II in mouse oocytes. (A) Time-lapse video microscopy. Uninjected, and Bub1dn-injected oocytes were kept 1 h in dbcAMP before they were released from the prophase block. 3 h after GVBD, the oocytes were placed under the microscope in a 37°C heating box. Pictures were taken every 45 min for 12 h. Red arrows indicate the extruded PB. Time points (h) after GVBD are indicated. Bar, 50 μm. (B) Bub1dn disrupts the SAC in meiosis II. Uninjected, and Bub1dn-injected oocytes were cultured for 14–15 h after GVBD and transferred into individual drops of medium containing nocodazole, or strontium, or both, and then cultured for an additional 3–4 h. Oocytes were fixed and stained with PI (red) and an anti-tubulin antibody, followed by an FITC-labeled secondary antibody (green; a and d), or with DAPI alone (b and e). The position of the PB is marked, and the oocyte chromosomes are boxed. (c and f) The DAPI stained chromosomes from b and e were enlarged by 2.6 times. Bar, 10 μm. (C) Single cell H1/MBP kinase assay. Total H1 and MBP kinase activities were assayed in whole cell lysates prepared from uninjected, GV-stage, and Bub1dn-injected oocytes. The autoradiograph is shown.
Table I. Timing of PB extrusion of oocytes injected with constructs encoding mutant checkpoint proteins (time-lapse video).
| Uninjected | Bub1dn | BubR1d | Mad2ΔC | Cdc20-GR | Cdc20-4AV | Cdc20- 4AVGR |
|
|---|---|---|---|---|---|---|---|
| 5a | 0 | 3 | 0 | 0 | 3 | 0 | 3 |
| 6 | 0 | 13 | 1 | 1 | 11 | 0 | 8 |
| 7 | 0 | 16 | 10 | 11 | 10 | 8 | 6 |
| 8 | 0 | 0 | 8 | 9 | 11 | 15 | 3 |
| 9 | 0 | 2 | 2 | 1 | 1 | 1 | 0 |
| 10 | 20 | 0 | 0 | 0 | 0 | 0 | 0 |
| 11 | 9 | 0 | 0 | 0 | 0 | 0 | 0 |
| 12 | 4 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 33/47b | 34/35 | 21/22 | 22/28 | 36/53 | 24/27 | 20/32 |
Hours after GVBD.
No. of oocytes with PB extruded/total oocytes.
To check the effects of Bub1dn on CSF and meiosis II, injected as well as noninjected control oocytes were cultured overnight, and stained for DNA (propidium iodide [PI] or DAPI) and, in some cases, tubulin to visualize the meiotic spindle apparatus. Both, untreated as well as Bub1dn injected oocytes arrested at metaphase II, with condensed chromosomes and well organized meiotic spindles (Fig. 3 B, a–c and d–f, 1st column; Table II).
Table II. MI/MII arrest of oocytes injected with constructs encoding mutant checkpoint proteins.
| Uninjected | Bub1dn | BubR1d | Mad2ΔC | Cdc20- GR |
Cdc20- 4AV |
Cdc20- 4AVGR |
|
|---|---|---|---|---|---|---|---|
|
|
n = 32 | n = 125 | n = 100 | n = 128 | n = 53 | n = 60 | n = 160 |
| % | % | % | % | % | % | % | |
| No PB (MI) | 3 | 25 | 17 | 15 | 17 | 18 | 49 |
| PB (MII) | 97 | 75 | 83 | 85 | 83 | 82 | 51 |
To examine, whether Bub1dn interferes with the SAC in meiosis II, oocytes were treated with strontium to inactivate CSF in the presence or absence of nocodazole. Strontium treatment caused Bub1dn as well as control-injected oocytes to exit from the CSF arrest, as indicated by the decondensation of the chromatin and the formation of anaphase spindles (Fig. 3 B, 2nd column). Nocodazole treatment alone produced highly condensed chromosomes with no visible spindles in both injected and noninjected oocytes (Fig. 3 B, 3rd column). In contrast to nocodazole-treated control oocytes, which remained arrested with highly condensed chromosomes in the presence of strontium, Bub1dn-injected oocytes were released from the metaphase arrest (Fig. 3 B, compare 4th column a–c with d–f; Table III). Thus, Bub1dn is capable of abrogating the SAC in meiosis II.
Table III. SAC analysis in meiosis II.
| Uninjected | Bub1dn | BubR1d | Mad2ΔC | Cdc20-4AVGR | |
|---|---|---|---|---|---|
| Untreated | 0/4a (0%) | 0/3 (0%) | 0/2 (0%) | 0/2 (0%) | See Table IV |
| Strontium | 23/28 (82%) | 18/19 (95%) | 8/8 (100%) | 11/13 (85%) | 21/22 (95%) |
| Nocodazole | 0/14 (0%) | 0/22 (0%) | 0/18 (0%) | 0/24 (0%) | 0/4 (0%) |
| Strontium + nocodazole | 0/20 (0%) | 24/28 (85%) | 21/28 (75%) | 19/21 (90%) | 10/14 (71%) |
No. of oocytes released from CSF/total oocytes.
To make sure that Bub1dn-treated oocytes were really arrested at metaphase II, we measured the kinase activities of MPF and MAPK, as a downstream target of CSF, from individual oocytes 22 h after GVBD. There was no difference in kinase activity between uninjected and Bub1dn-injected mature oocytes (Fig. 3 C), confirming that Bub1dn did not interfere with the CSF arrest.
These results suggested that the SAC induced by nocodazole is able to prevent oocytes from exiting meiosis even if CSF is inactivated. Disruption of the SAC by using dominant-negative Bub1, on the other hand, was not sufficient to allow exit from the CSF arrest.
Mad2ΔC and BubR1dn disrupt the SAC but not CSF
Recent studies indicated that the SAC might consist of individual branches, with Bub1 playing an important role in only one of them (Musacchio and Hardwick, 2002; Yu, 2002). Therefore, it is possible that CSF uses the other pathways that include Mad2 or/and BubR1. To test whether Mad2 or BubR1 are involved in CSF signaling in mouse oocytes, we made use of two previously described dominant-negative mutants of these two proteins, Mad2ΔC and BubR1d. Mad2ΔC lacks 10 aa at the COOH terminus of the protein and cannot interact with Cdc20 (Luo et al., 2002). BubR1d (BubR1 351-700) can bind Bub3 and Cdc20, but cannot inhibit APC/C in vitro, and overexpressed BubR1d was shown to act as a dominant negative in human cells (Tang et al., 2001).
In time course experiments, overexpression of either Mad2ΔC or BubR1d accelerated passage of the oocytes through meiosis I, when compared with uninjected controls (Fig. 4 A; Table I). But, as with the Bub1dn mutant, neither construct was able to overcome CSF and induce exit from the metaphase II arrest. The spindle apparatus as well as the condensed metaphase chromosomes were similar to the control CSF-arrested oocytes (Fig. 4 B). Again, as with Bub1dn, the injected proteins were functional in abolishing the SAC, because they allowed metaphase II-arrested oocytes to proceed to interphase when released from the CSF in the presence of nocodazole (Fig. 4 C; Table III). Together, these results strongly argue against an essential role of the SAC in the CSF-mediated metaphase II arrest in mouse oocytes.
Figure 4.
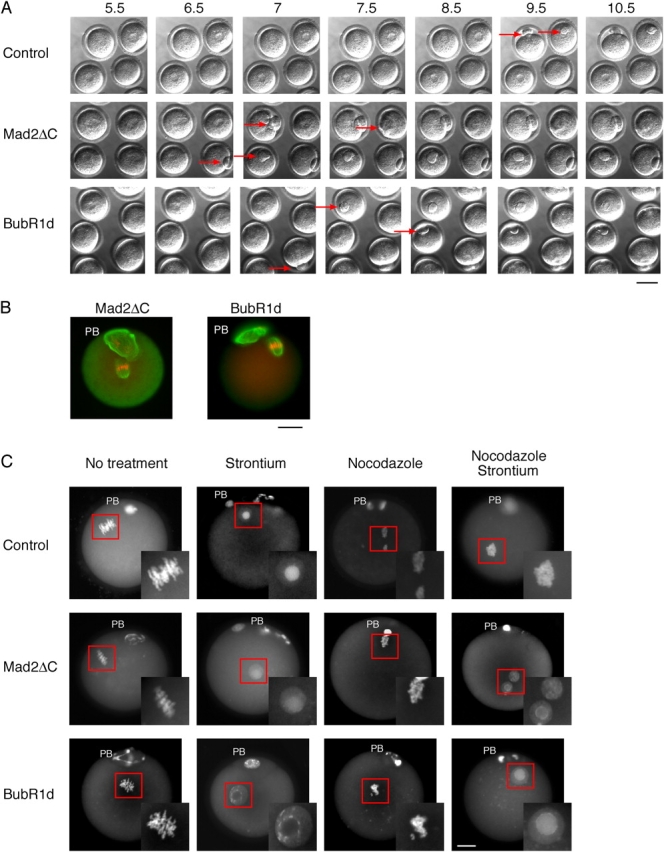
The effect of Mad2ΔC and BubR1d injection on meiosis I and II in mouse oocytes. (A) Time-lapse video microscopy. Mad2ΔC or BubR1d injected oocytes were treated as in Fig. 3 A. Pictures were taken every 30 min for 12 h. Red arrows indicate the extruded PB. Time points (h) after GVBD are indicated. Bar, 50 μm. (B) Immunofluorescence staining of Mad2ΔC (left) or BubR1d-injected (right) oocytes. Mad2ΔC or BubR1d-injected oocytes were kept 1 h in dbcAMP before they were released from the prophase block. 20 h after GVBD, the oocytes were fixed, chromosomes stained with PI (red), and the meiotic spindles visualized using an anti-tubulin antibody, followed by an FITC-labeled secondary antibody (green). Note the extruded PB, and the chromosomes aligned on a metaphase plate with an intact spindle apparatus, indicating a CSF-arrest. Bar, 10 μm. (C) Activation of Mad2ΔC and BubR1d-injected oocytes. Uninjected, and Mad2ΔC- or BubR1d-injected oocytes were cultured for 14–15 h after GVBD and transferred into individual drops of medium containing nocodazole, or strontium, or both, and then cultured for an additional 3–4 h. Chromosomes were stained with DAPI. The position of the PB is marked. The oocyte nuclei are boxed, and shown enlarged in the insets. Bar, 10 μm.
Cdc20 overexpression cannot override CSF
The above results argue against a role of SAC proteins in the CSF-mediated meiosis II arrest in mouse oocytes. However, they rely on dominant-negative mutants that may only abrogate the SAC, but not a potential CSF function of the endogenous proteins. Also, the CSF might still inhibit the APC/C by directly inactivating Cdc20. To test whether Cdc20 might be rate limiting for APC/C activation in CSF-arrested mouse oocytes, we checked whether overexpression of mouse Cdc20 would override the CSF arrest as has been shown before in the Xenopus egg system (Reimann and Jackson, 2002).
First, we expressed YFP-tagged Cdc20 to monitor its subcellular localization in mouse oocytes. Cdc20-YFP localized strongly to kinetochores, and, more pronounced in meiosis II than in meiosis I, to spindles (Fig. 5). Other than in mitotic cells, Cdc20-YFP did not localize to the spindle poles.
Figure 5.

Localization of Cdc20-YFP. Cdc20-YFP and histone H2B-RFP were coinjected into GV-stage oocytes, kept in dbcAMP for 1 h, released and fixed 6 h after GVBD (1st meiosis), 9–10 h after GVBD (1st anaphase and 1st telophase), or after overnight culture (2nd meiosis). (Left) The whole oocyte is shown in an overview, with chromosomes in red (histone H2B-RFP) and Cdc20-YFP in green. The position of the PB is marked in the metaphase II-arrested oocytes. (Right) The chromosomes are enlarged, and the signals from histone H2B-RFP and Cdc20-YFP are shown individually, as well as overlaid (merge). Bars, 10 μm.
Despite correct localization, overexpression of Cdc20-YFP did not overcome the CSF-mediated metaphase II arrest in mouse oocytes. To exclude the possibility that the COOH-terminal YFP tag might have interfered with Cdc20 function, we also expressed untagged wild-type Cdc20 in mouse oocytes (unpublished data). In contrast to frog eggs, however, overexpression of Cdc20 did not interfere with the CSF arrest in the injected mouse oocytes.
Dominant checkpoint-resistant forms of Cdc20 do not override the CSF arrest
Although consistent with the results from the experiments using dominant-negative checkpoint proteins, the failure of Cdc20 overexpression to overcome the CSF arrest might be due to insufficient expression levels, or efficient inactivation of Cdc20. To overcome these limitations, we made use of a gain of function mutant of Cdc20, which harbors mutations in one of its WD40 repeats (Cdc20-50 or Cdc20-GR; Schott and Hoyt, 1998). We introduced the GR mutation into mouse Cdc20, to test whether this potentially checkpoint-resistant Cdc20 mutant might be capable of overcoming the CSF arrest in mouse oocytes.
As shown in Fig. 6, the Cdc20-GR mutant caused early anaphase in the first meiosis (Fig. 6 A; Table I), but could not force exit from metaphase II (Fig. 6 B; Table II). This mutant, thus, behaved similarly to the above tested dominant-negative checkpoint proteins, and could, despite its checkpoint resistance, not activate the APC/C to induce exit from meiosis II.
Figure 6.
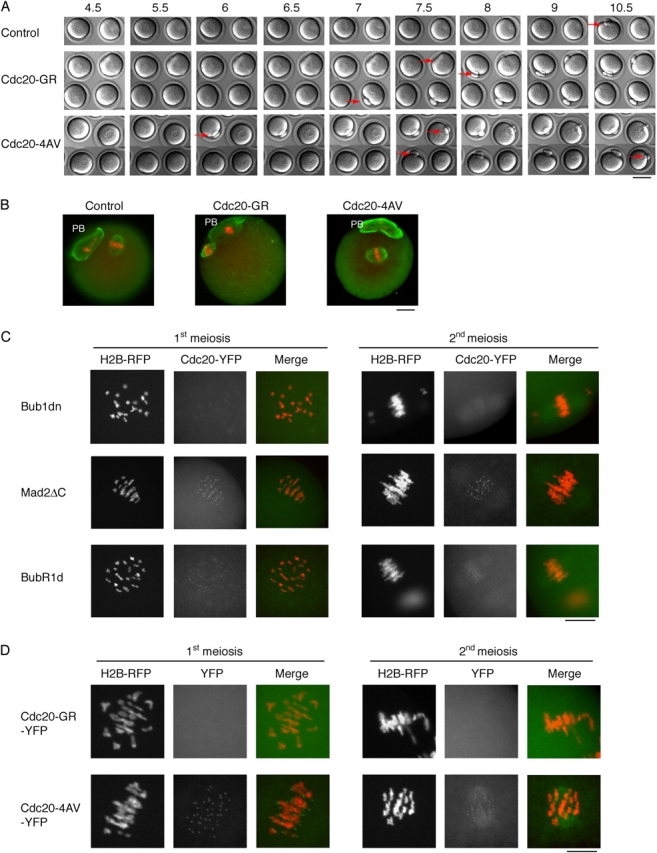
The effect of Cdc20-GR and -4AV injection into mouse oocytes. (A) Time-lapse video microscopy. Uninjected, or oocytes injected with Cdc20-GR or -4AV were treated as in Fig. 3 A. Red arrows indicate the extruded PB. Time points (h) after GVBD are indicated. Bar, 50 μm. (B) Immunofluorescence staining of control (left), or Cdc20-GR injected (middle), or Cdc20-4AV injected (right) oocytes. Oocytes were treated as in Fig. 4 B. Note the extruded PB, and the chromosomes aligned on a metaphase plate with an intact spindle apparatus. (C) The localization of Cdc20 is affected by checkpoint protein mutants. Cdc20-YFP and histone H2B-RFP mRNA and Bub1dn- or Mad2ΔC- or BubR1d-injected oocytes were treated as above, and fixed 5–6 h (1st meiosis) after GVBD, or after overnight culture (2nd meiosis). The histone H2B-RFP and Cdc20-YFP signals are shown individually, as well as overlaid (merge). Note the dotted appearance of the Cdc20-YFP signal around the chromosomes, suggesting a kinetochore localization, in the Mad2ΔC coinjected oocytes, which is weaker in the BubR1d coinjected, and seems absent from the Bub1dn coinjected oocytes. (D) The localization of the Cdc20-GR or -4AV YFP-fusion proteins. Cdc20-GR-YFP– or Cdc20-4AV-YFP– and H2B-RFP–injected oocytes were treated as above, and fixed 6 h (1st meiosis) after GVBD, or after overnight culture (2nd meiosis). Note the dotted appearance of the Cdc20-4AV-YFP signal around the chromosomes, suggesting a kinetochore localization, which is not detectable in Cdc20-GR-YFP–injected oocytes. Bars: (B–D) 10 μm.
Chung and Chen (2003) found that the mutation of 4 putative CDK or MAPK phosphorylation sites in Cdc20 to nonphosphorylatable amino acids (Cdc20-4AV) generated a dominant Cdc20 that was capable of overriding the SAC, as well as CSF, in the Xenopus egg extract. Thus, CSF might inhibit Cdc20 by phosphorylation. To test this hypothesis, we produced a mouse version of Cdc20-4AV.
Injection of this mutant into mouse oocytes resulted, again, in an accelerated exit from the first meiosis, at 6–7 h after GVBD (Fig. 6 A; Table I), but had no effect on the metaphase II arrest (Fig. 6 B; Table II). These results confirmed the previous results with the dominant-negative proteins, suggesting that the CSF did not, at least not exclusively, depend on either inhibitory Cdc20 phosphorylation nor a SAC-mediated down-regulation of Cdc20, to prevent APC/C activation in mouse oocytes.
The mutant checkpoint proteins Bub1dn, BubR1d and Mad2ΔC might exert their effect by interfering with Cdc20 localization. To test this hypothesis, we coinjected Cdc20-YFP with Bub1dn, Mad2ΔC, or BubR1d. Bub1dn/Cdc20-YFP doubly injected oocytes showed weak spindle localization of Cdc20-YFP, but only a very weak signal from the kinetochores throughout maturation, suggesting that Bub1 may be required for kinetochore localization of Cdc20. A similar result was found when BubR1d was coinjected, although the Cdc20-YFP signal at the kinetochore was slightly stronger, especially during first meiosis. On the other hand, Mad2ΔC did not affect the localization of Cdc20 (Fig. 6 C). In addition, whereas the Cdc20-4AV-YFP mutant localized to kinetochores like the wild-type protein, Cdc20-GR-YFP was not detectable at kinetochores (Fig. 6 D).
Cdc20-4AVGR disrupts meiosis
Because the above results suggested that the two Cdc20 mutants acted differently, we combined them to generate a double mutant, Cdc20-4AVGR. This indeed had a synergistic effect on the dominant behavior of Cdc20. When mRNA encoding a Cdc20-4AVGR double mutant was injected into mouse oocytes, the onset of anaphase of meiosis I was even earlier than with any of our other mutant proteins, starting at 5–6 h after GVBD (Fig. 7 A; Table I). However, despite the accelerated entry into anaphase, the exit from meiosis I generally lasted more than an hour (Fig. 7 A) and often could not be completed properly. Some oocytes failed to extrude their PB, despite multiple attempts (Fig. 7 A, green box; the place where the PB should be extruded is indicated by a yellow arrow), or produced two (Fig. 7 A, blue box; Table IV). When Cdc20-4AVGR–injected oocytes were fixed after overnight culture, and stained with PI and a tubulin-specific antibody, a variety of phenotypes was revealed.
Figure 7.

The effect of Cdc20-4AVGR injection into mouse oocytes. (A) Time-lapse video microscopy. Uninjected, or oocytes injected with the Cdc20-4AVGR mutant were treated as in Fig. 3 A. Red arrows indicate the extruded PB. The oocyte marked by the green box failed, despite several attempts, to extrude its PB. The pink arrow indicates the spot where the PB should have been extruded. The oocyte marked by the blue box, however, divided abnormally and extruded two PBs, indicated by the light blue arrow. Time points (h) after GVBD are indicated. Bar, 50 μm. (B) Immunofluorescence staining of uninjected, and Cdc20-4AVGR–injected oocytes. Oocytes were treated as in Fig. 4 B. (Left) The whole oocyte is shown in an overview. (Right) The area of the spindle is enlarged, and the DNA and tubulin staining are shown individually, as well as overlaid (merge). The uninjected oocyte extruded the PB, and the chromosomes are aligned on a metaphase plate with an intact spindle apparatus. Examples of the various phenotypes of Cdc20-4AVGR–injected oocytes are shown below. In b–d, the oocytes did not extrude their first PB. The abnormal spindles in c and d are marked with red arrows. In e, an oocyte with an interphase nucleus in the absence of any extruded PB is shown. In f, a typical example of an oocyte with one PB extruded is shown. In g, an oocyte is shown with two extruded PBs. Bars, 10 μm. (C) Activation of Cdc20-4AVGR–injected oocytes. The oocytes were treated as in Fig. 4 C. The position of the PB is marked. The oocyte nuclei are boxed, and shown enlarged in the insets. (D) Single cell H1/MBP kinase assay. Total H1 and MBP kinase activities were assayed in whole cell lysates prepared from uninjected, GV-stage, and Cdc20-, Cdc20-4AV–, and Cdc20-4AVGR–injected oocytes. The autoradiograph is shown.
Table IV.
Phenotypes observed with Cdc20-4AVGR–injected oocytes ( n = 159)
| No PB extruded | One PB extruded | Two PBs extruded | ||
|---|---|---|---|---|
| cond.a | decond.b | cond. | decond. | chrom. cond. |
| % | % | % | % | % |
| 37 | 12 | 45 | 0 | 6 |
Chromosomes condensed.
Chromosomes decondensed.
About 50% of the injected oocytes did not extrude their first PB and the chromosomes were widely distributed on elongated spindles (Fig. 7 B, b–d; Table IV). In some cases, two spindles were connected to each other (Fig. 7 B, c) or two different types of spindles were observed, one monopolar, the other a late telophase-like spindle (Fig. 7 B, d). Occasionally, an interphase nucleus was observed (Fig. 7 B, e).
Oocytes that succeeded to extrude the first PB often exhibited a disrupted meiotic spindle that only weakly stained with antitubulin antibodies. Chromosomes were not properly aligned at the metaphase plate (Fig. 7 B, f). About 5–10% of the injected oocytes had two PBs extruded, and were arrested (Fig. 7 B, g) with monopolar spindles. A quantification of these results is shown in Table IV.
The Cdc20-4AVGR injected oocytes with anaphase-like spindles and lagging chromosomes, or with two PBs extruded (Fig. 7 B, f and g), seemed to have overcome the CSF arrest, although their chromosomes were still condensed. To test, whether the CSF was still active in Cdc20-4AVGR injected oocytes, we treated them with strontium. This treatment induced decondensation of the chromosomes, indicating an exit to interphase from a metaphase II arrest (Fig. 7 C; Table III), thus arguing against the hypothesis that the Cdc20-4AVGR mutant overcame the CSF arrest.
As an additional test, whether the above oocytes were still arrested at metaphase II, MPF and MAPK activities were assayed from individual uninjected, or Cdc20 wild-type, Cdc20-4AV, or Cdc20-4AVGR mutant–injected oocytes. The H1 kinase activity and MAPK activity from oocytes injected with the double mutant, although slightly reduced in H1 kinase activity when compared with the control oocytes, was readily detectable (Fig. 7 D). Thus, the Cdc20-4AVGR–injected oocytes were, despite their anaphase-like phenotype, still arrested with high levels of Cdk activity. Together, with the fact that strontium induced the decondensation of the chromosomes, these results suggested that Cdc20-4AVGR–expressing cells were still arrested by CSF.
The Cdc20-4AVGR mutant may induce sister chromatid separation in meiosis I
The anaphase-like spindles observed in the Cdc20-4AVGR–injected cells might be the consequence of a premature separation of sister chromatids. To check for split sister chromatids, chromosome spreads were prepared from uninjected, and Cdc20-4AVGR–injected oocytes 16–18 h after GVBD.
The numbers of chromosomes from Cdc20-4AVGR–injected oocytes clearly differed from those of their uninjected counterparts (compare Fig. 8 A, b with c, d; a quantification is shown in Fig. 8 B). In uninjected metaphase II oocytes, we counted 18–24 chromosomes with 20 being the expected number (Fig. 8 A b; Fig. 8 B). In Cdc20-4AVGR–injected oocytes that had extruded a PB, chromosome counts were between 20 and 50 (Fig. 8 A, c and d; Fig. 8 B). The increase in the number of chromosomes indicated that the sister chromatids were at least partially separated in the Cdc20-4AVGR–injected oocytes. The wide variation of counts, and the fact that some were above the expected maximum of 40 chromatids, suggested that chromosome mis-segregation had occurred during meiosis I. Chromosomes prepared from Cdc20-4AVGR–injected oocytes that had failed to extrude their PB (Fig. 8 A, e and f) were not bivalent like those from control oocytes in the first meiosis (Fig. 8 A, a). Chromosome counts from these oocytes were higher than 40, ranging up to the expected maximum of 80 (Fig. 8 B).
Figure 8.

Cdc20-4AVGR mutant induces sister chromatid separation. (A) Chromosome spreads were prepared from noninjected, or Cdc20-4AVGR–injected oocytes after overnight culture. The chromosomes were stained using a 2% Giemsa solution. In the insets, chromosomes are shown that were found outside the main field. a, uninjected in 1st meiosis (6 h after GVBD); b, uninjected in 2nd meiosis; c and d, Cdc20-4AVGR–injected oocytes with the PB extruded; e and f, Cdc20-4AVGR–injected oocytes with no PB extruded. Bar, 10 μm. (B) Quantification of A. On the x axis are the numbers of chromosomes counted per oocyte, on the y axis the number of oocytes. a, control; b, Cdc20-4AVGR–injected oocytes with PB extruded; c, Cdc20-4AVGR–injected oocytes with no PB extruded. (C) Time-lapse video microscopy. The Cdc20-4AVGR construct was coinjected into oocytes either with (a) cyclin B1N170-RFP (a 170–amino acid NH2-terminal fragment of cyclin B1 fused to RFP) or with (b) Pds1-RFP. The oocytes were kept in dbcAMP for 1 h, released 4 h after GVBD, and observed by differential interference contrast, and fluorescence time-lapse video microscopy. Pictures were taken every hour (BN170-RFP) or every 45 min (Pds1-RFP). The complete series is shown. Time points (h) after GVBD are indicated. Bars, 50 μm.
The observation that Cdc20-4AVGR–injected oocytes could still be released from their anaphase-like arrest by strontium, suggested that the activated Cdc20 mutant could either not effectively turn on the APC/C in the presence of CSF, or that cyclin B1 was selectively protected from proteolysis by CSF. To investigate whether Cdc20-4AVGR was able to activate the APC/C, we measured the level of APC/C-dependent proteolysis after the first meiosis in the presence of the activated mutant. As a proxy for destruction box- and APC/C-dependent proteolysis, we monitored the fluorescence of a cyclin B1N170-RFP fusion construct, which comprises the cyclin B1 NH2-terminal 170 aa including the destruction box, but lacking the Cdk1 activation domain (King et al., 1995; Cohen-Fix et al., 1996). The cyclin B1N170-RFP signal behaved as a full-length cyclin B1-YFP fusion protein in mouse oocytes (Ledan et al., 2001), accumulated until 7–8 h after GVBD and disappeared before extrusion of the first PB. B1N170-RFP rapidly reaccumulated during meiosis II and was readily detectable in CSF-arrested control oocytes (Fig. 8 C, a, control).
In oocytes coinjected with Cdc20-4AVGR, B1N170-RFP accumulated to lower levels and was degraded earlier after GVBD, consistent with the more rapid progression through meiosis I caused by the dominant Cdc20 mutant. Surprisingly, however, B1N170-RFP reaccumulated during meiosis II to levels in meiosis I, suggesting that CSF-mediated inhibition of the APC/C could not be overcome by coexpression of Cdc20-4AVGR (Fig. 8 C, a, Cdc20-4AVGR).
Similar results were obtained using a Pds1/securin-RFP fusion protein, suggesting that the APC/C is refractory to Cdc20 in the presence of CSF (Fig. 8 C, b). We concluded from these experiments that expression of Cdc20-4AVGR caused premature sister chromatid separation during meiosis I, as well as an abnormal PB extrusion (cytokinesis), but failed to initiate interphase between meiosis I and II and did not overcome the CSF arrest in oocytes.
In summary, our data suggest that chromosome segregation during meiosis I is controlled by the SAC and that precocious activation of the APC/C can cause premature sister chromatid separation during meiosis I. As oocytes progress into meiosis II, CSF activation is dominant over the effects of constitutively active Cdc20 mutants or dominant-negative interfering mutants of checkpoint proteins, suggesting that, in contrast to frog oocytes, the SAC does not play an essential role in establishing or maintaining the CSF-dependent metaphase arrest in meiosis II.
Discussion
Despite the discovery that the Mos–MAPK–p90Rsk pathway is essential and sufficient for CSF activity in Xenopus oocytes, little is known about the downstream targets of this signal transduction pathway. CSF activity acts by inhibiting the APC/C, preventing sister chromatid separation and cyclin degradation.
The similarities between SAC-arrested cells and CSF-arrested extracts suggested that the CSF might act via activation of the SAC to inhibit the APC/C. Consistent with this hypothesis, the SAC kinase Bub1 has been identified as a direct target of the Mos–MAPK–p90Rsk pathway in Xenopus, highlighting a potential role of the SAC in CSF. Further evidence that CSF uses components of the SAC to inhibit the APC/C include the notion that Mad2, as an inhibitor of Cdc20, is required for the establishment of CSF (Tunquist et al., 2003), and that overexpression of Cdc20 could release Xenopus oocytes from the meiosis II arrest (Reimann and Jackson, 2002).
In mouse oocytes, Mos is required to arrest cells at metaphase of meiosis II. In contrast to Xenopus, however, Mos deficiency does not allow the oocytes to proceed to interphase between meiosis I or II. Rather, MPF activity remains high in the majority of Mos-deficient oocytes, suggesting that cyclin levels are controlled in a Mos-independent manner. Moreover, if the integrity of the meiotic spindle is disrupted, mouse oocytes are able to mount a potent SAC during meiosis I as well as meiosis II, whereas additional nuclei have to be added to frog egg extracts for a checkpoint arrest to be induced (Minshull et al., 1994). Thus, the checkpoint proteins involved in the CSF arrest in frog egg extracts work in the absence of kinetochores. In the mouse oocyte, however, the SAC component Bub1 localizes to kinetochores during metaphase of meiosis I and II (Brunet et al., 2003).
To investigate the function of Bub1 during meiosis of mouse oocytes, we overexpressed a dominant-negative mutant. However, in contrast to the Xenopus system, we did not observe an exit from the CSF-mediated arrest at metaphase II. Upon careful inspection, however, we discovered that chromosomes were not properly aligned at the metaphase plate in otherwise apparently normal metaphase II arrested oocytes, suggesting that the injected Bub1dn may have interfered with some process in meiosis (unpublished data). Time-lapse video microscopy of oocytes undergoing meiosis was used to measure the timing of meiotic events, which revealed that Bub1dn caused an accelerated progression through meiosis I as indicated by the earlier extrusion of the first PB.
Progression through meiosis I is controlled by the SAC in mouse oocytes, as has been shown in experiments aimed to investigate the role of Mad2 during meiosis I (Wassmann et al., 2003). Therefore, it is possible that the overexpression of Bub1dn interfered with SAC function during meiosis I, causing a precocious onset of anaphase, as has been observed in experiments using human tissue culture cells (Taylor and McKeon, 1997; Geley et al., 2001).
In addition, Bub1dn was also able to disrupt the SAC during meiosis II, which can be activated by nocodazole and revealed by inactivating CSF using strontium. These data strongly suggested that mouse oocytes are able to mount a strong and Bub1 dependent SAC response during meiosis I and II. Because the chromosomes are perfectly aligned during metaphase of meiosis II, the conventional signal for SAC activation, i.e., chromosome misalignment, might be missing. Nevertheless, Bub1 localizes to kinetochores (Fig. 2), and is phosphorylated in CSF-arrested mouse oocytes (Brunet et al., 2003) as in checkpoint-activated mitotic cells. But even if the SAC was constitutively activated in meiosis II, it could be overridden by Bub1dn without causing exit from meiosis, indicating that the CSF-induced metaphase arrest is independent from the SAC. This is in contrast to the frog system, in which depletion of Bub1 interfered with the establishment of the Mos-induced CSF arrest. However, because in this study, the NH2 terminus of Bub1 (Bub1dn), believed to act as a dominant negative by competing with the kinetochore localization of the endogenous protein, was used, rather than depletion of the endogenous protein, it is possible that Bub1dn did not interfere with a kinetochore-independent function required for the establishment or maintenance of CSF. Due to the lack of suitable reagents, we have not investigated this possibility, but have pursued the question of whether other SAC components might be activated by the CSF during meiosis II to keep the APC/C in an inactive state and prevent unwanted exit from meiosis II.
Mad2 and BubR1 are known to physically bind and thereby inhibit the APC/C. Mad2 has been shown to localize at kinetochores of the CSF-arrested mouse oocytes, suggesting that the SAC might be active in meiosis II, or that the CSF might induce a SAC-like state (Kallio et al., 2000). However, none of the mutants tested by us overcame the CSF arrest, although they efficiently interfered with the SAC in meiosis I and II. We conclude from these experiments that the SAC can be activated during meiosis I and II, and is essential for progression through meiosis I, but in meiosis II the oocytes become arrested by CSF in a SAC-independent manner.
These results differ from observations using Xenopus egg extracts. Xenopus egg extracts depleted of either of these checkpoint proteins, could not be arrested by the addition of Mos, indicating that these checkpoint proteins were necessary to establish the CSF arrest. However, in the same system, depletion of most of the checkpoint proteins, except of Mad1, did not allow the extract to exit from CSF. Even BubR1 and Mad2, as physical inhibitors of Cdc20, seemed dispensable once CSF was established (Tunquist and Maller, 2003). However, in mouse oocytes, a CSF arrest can be established and maintained even if the function of the SAC components is disturbed by the overexpression of dominant negatives. Because it is currently not clear how Mos regulates these checkpoint proteins to inhibit the APC/C in CSF, and because our analysis was based on the use of dominant-negative Bub1, Mad2, and BubR1, we could still not exclude the possibility that the mutants only interfered with the SAC function, but not with their putative function in CSF.
To go around this problem, we tested the effect of overexpressing wild type, and activated forms of the APC/C activator Cdc20, the downstream target of the SAC, on CSF. In Xenopus egg extracts, the addition of exogenous Cdc20-activated cyclin B degradation and induced exit from mitosis in the absence of calcium (Reimann and Jackson, 2002). Moreover, the expression of a Cdc20-4AV mutant at levels comparable to endogenous Cdc20 was able to mediate exit from CSF in Cdc20-depleted frog egg extracts (Chung and Chen, 2003). These data suggest that Cdc20 is the main target of CSF in frog meiosis and that it may be inactivated by similar mechanisms in meiosis as during SAC responses in mitosis. Neither wild-type, nor any of the activated Cdc20 mutants we tested overcame the CSF arrest, although all of them interfered with the SAC in meiosis I and II.
The effect of the double mutant Cdc20-4AVGR, constructed to overcome currently known inhibitory pathways, is difficult to explain. However, the apparently synergistic effect of the two mutations suggested that, indeed, Cdc20 is controlled by two independent pathways operative during meiosis. Only Bub1dn, when expressed at very high levels, produced a similar phenotype (unpublished data). Oocytes injected with either mutant were arrested with anaphase-like spindles, some of them in the absence of an extruded first PB. Furthermore, in the case of Cdc20-4AVGR–injected oocytes, higher chromosome counts were observed than in their uninjected counterparts, indicating that interfering with APC/C regulation during meiosis I might trigger premature sister chromatid separation. However, because neither Bub1dn nor Cdc20-4AVGR caused the formation of interphase nuclei, it appears that APC/C control was only relaxed for some, but not all targets, i.e., caused the activation of separase (via Pds1/securin degradation), but to a lesser extent the degradation of cyclin B1. This may be similar to the situation in Xenopus CSF-arrested egg extracts, in which cyclin B1 is stable, but cyclin A is barely detectable, and when added as a recombinant protein, is rapidly degraded in a destruction-box–dependent manner (Geley et al., 2001).
Recently, sgo1 has been identified in yeast, and shown to be essential for preventing cleavage of Rec8, the meiotic cohesion, at centromeric regions during meiosis. Sgo1 is a substrate of the APC/C, which becomes unstable only after meiosis I, allowing equational chromosome segregation in meiosis I (Kitajima et al., 2004). The presence of homologous candidate genes for Sgo1 in mammals suggests that a similar mechanism may operate to control sister chromatid separation during meiosis of mouse oocytes. The hyperactive Cdc20 might cause premature degradation of Sgo1, resulting in the cleavage of Rec8, which holds the sister kinetochores together.
However, even under these extreme conditions, which induced sister chromatid separation during meiosis I, and this is important for our main conclusion, CSF activity was induced during further progression through meiosis, APC/C was shut down, and the oocytes arrested at metaphase of meiosis II. Thus, in mouse oocytes, the inhibition of the APC/C during the second metaphase does not depend on the SAC.
Materials and methods
Preparation and culture of oocytes
Immature oocytes arrested at prophase of meiosis I were obtained from ovarian follicles of ovaries removed from 8–12-wk-old FVB mice. They were collected in M2 medium (Sigma-Aldrich) with 50 μg/ml dbcAMP (Sigma-Aldrich) to keep them at GV stage (the first prophase) under mineral oil at 37°C until injection. After injection, oocytes were washed with and cultured in fresh M2 media to allow them to resume meiosis. Nocodazole was used at a final concentration of 10 μM. Oocytes were released from CSF using 10 mM strontium (Sigma-Aldrich) in Ca2+/Mg2+-free KSOM medium.
Plasmid construction
The Bub1dn cDNA was subcloned from pEF dnBub1. All other clones were amplified by PCR from IMAGE clones as templates (BubR1: IMAGp998F0810912Q2, Mad2: IMAGp998A206741Q3, Cdc20: IMAGp998K2411151Q3, histone H2B: IMAGp998E0711102Q2), and sequenced. To detect the expressed proteins, the cDNAs were then either NH2-terminally double myc-tagged using the pBS KS 2myc vector, or fused to either YFP (from pEFplmmB1-YFP) or RFP (from pCS2mRFP). For in vitro transcription reactions, cDNAs were cloned into either pRN3 (a gift from M.-H. Verlhac, CNRS, Université Paris, Paris, France), which has a globin 3′ UTR plus a short (30 A) poly A tail, or pBS-KC NBS 3′ UTR, which has the cyclin B1 3′ UTR plus a 30 poly A tail.
RNA synthesis
The plasmids were linearized by SfiI (for pRN3) or SalI (for pBS-KC NBS 3′ UTR) and purified using a gel extraction kit (QIAGEN). T3 message machine (Ambion) was used for producing capped mRNA, which was purified using the RNeasy kit (QIAGEN) and, if necessary, concentrated using Microcon microconcentrators (Millipore).
Immunofluorescence and chromosome spreads
Immunofluorescence was performed as described previously (Polanski et al., 1998), except that 4% PFA was used to fix the oocytes. The α-tubulin specific YL1/2 rat mAb (CHEMICON International, Inc.) was used at a dilution of 1/500 and developed using an FITC-conjugated α-rat IgG (Santa Cruz Biotechnology, Inc.). Chromosome spreads were done according to a previously described method (Clouston et al., 2002). Pictures were taken using an AxioCam HRc (Carl Zeiss MicroImaging, Inc.) mounted on an Axioplan microscope (Carl Zeiss MicroImaging, Inc.) with 40×/0.75 objective lenses.
Histone H1 kinase assay
Oocytes were collected in 1 μl of a 5 mg/ml BSA solution, and frozen in liquid N2. For the assay, 5 μl of 2× H1 kinase buffer (40 mM Tris-HCl, pH 7.5, 160 mM β-glycerolphosphate, 40 mM EGTA, 30 mM MgCl2, 2 mM DTT, 30 μg/ml Leupeptin and pepstatin, 2 mM PMSF) and 1.5 μl of water were added to one oocyte. To start the reaction, 1.5 μl of the reaction mix (2 mg/ml histone H1 and MBP, 1 mM DTT, 15 mM MgCl2, 0.3 mM ATP, 5% (vol/vol) γ-[32P]ATP (10218; Amersham Biosciences) was added and incubated at 37°C for 20 min. The reaction was stopped with 2 μl 4× SDS sample buffer, and the products were resolved using SDS-PAGE.
Microinjection and time-lapse video microscopy
GV stage oocytes were microinjected using an Eppendorf pressure microinjector and a 3D Hydraulic Coarse/Fine Micromanipulator mounted on a microscope (model DM IRBE; Leica). For microinjections, 0.75 mg/ml (or 2 mg/ml for Mad2ΔC and Bub1dn high concentration) mRNA solutions were used. 0.5 μl mRNA was loaded into a borosilicate glass capillary GC100TF-15 (Harvard Apparatus Ltd.) pulled out using a micropipette puller. For time-lapse video microscopy, oocytes were cultured on a glass bottom dish (WillCo) in an incubator for 3 h in a small drop of media under mineral oil. They were then moved to an Axiovert 200M microscope (Carl Zeiss MicroImaging, Inc.) with 20×/0.40 korr objective lenses equipped with a 37°C heating box. AxioCam MRm or HRc cameras were handled by the Axiovision 3.1 or 4 (Carl Zeiss MicroImaging, Inc.) software package. Time-lapse series were generated by taking images at the intervals indicated, and were subsequently converted to 8-bit images and assembled in Adobe Photoshop.
Acknowledgments
The authors would like to thank Frieda Chen and Nicoletta Bobola for discussions and critically reading the manuscript.
This work was in part supported by Austrian Science Fund (Fonds zur Förderung der wissenschaftlichen Forschung) grant P15000 to S. Geley.
Abbreviations used in this paper: APC/C, anaphase-promoting complex/cyclosome; CSF, cytostatic factor; GV, germinal vesicle; GVBD, GV breakdown; MPF, maturation promoting factor; PB, polar body; PI, propidium iodide; SAC, spindle assembly checkpoint.
References
- Basu, J., E. Logarinho, S. Herrmann, H. Bousbaa, Z. Li, G.K. Chan, T.J. Yen, C.E. Sunkel, and M.L. Goldberg. 1998. Localization of the Drosophila checkpoint control protein Bub3 to the kinetochore requires Bub1 but not Zw10 or Rod. Chromosoma. 107:376–385. [DOI] [PubMed] [Google Scholar]
- Brunet, S., G. Pahlavan, S. Taylor, and B. Maro. 2003. Functionality of the spindle checkpoint during the first meiotic division of mammalian oocytes. Reproduction. 126:443–450. [DOI] [PubMed] [Google Scholar]
- Chan, G.K., B.T. Schaar, and T.J. Yen. 1998. Characterization of the kinetochore binding domain of CENP-E reveals interactions with the kinetochore proteins CENP-F and hBUBR1. J. Cell Biol. 143:49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, G.K., S.A. Jablonski, V. Sudakin, J.C. Hittle, and T.J. Yen. 1999. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 146:941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R.H., J.C. Waters, E.D. Salmon, and A.W. Murray. 1996. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 274:242–246. [DOI] [PubMed] [Google Scholar]
- Chung, E., and R.H. Chen. 2003. Phosphorylation of Cdc20 is required for its inhibition by the spindle checkpoint. Nat. Cell Biol. 5:748–753. [DOI] [PubMed] [Google Scholar]
- Clouston, H.J., M. Herbert, J. Fenwick, A.P. Murdoch, and J. Wolstenholme. 2002. Cytogenetic analysis of human blastocysts. Prenat. Diagn. 22:1143–1152. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix, O., J.M. Peters, M.W. Kirschner, and D. Koshland. 1996. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 10:3081–3093. [DOI] [PubMed] [Google Scholar]
- Colledge, W.H., M.B. Carlton, G.B. Udy, and M.J. Evans. 1994. Disruption of c-mos causes parthenogenetic development of unfertilized mouse eggs. Nature. 370:65–68. [DOI] [PubMed] [Google Scholar]
- Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635–645. [DOI] [PubMed] [Google Scholar]
- Dunphy, W.G., L. Brizuela, D. Beach, and J. Newport. 1988. The Xenopus cdc2 protein is a component of MPF, a cytoplasmic regulator of mitosis. Cell. 54:423–431. [DOI] [PubMed] [Google Scholar]
- Ferby, I., M. Blazquez, A. Palmer, R. Eritja, and A.R. Nebreda. 1999. A novel p34(cdc2)-binding and activating protein that is necessary and sufficient to trigger G(2)/M progression in Xenopus oocytes. Genes Dev. 13:2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuno, N., M. Nishizawa, K. Okazaki, H. Tanaka, J. Iwashita, N. Nakajo, Y. Ogawa, and N. Sagata. 1994. Suppression of DNA replication via Mos function during meiotic divisions in Xenopus oocytes. EMBO J. 13:2399–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier, J., C. Norbury, M. Lohka, P. Nurse, and J. Maller. 1988. Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene cdc2+. Cell. 54:433–439. [DOI] [PubMed] [Google Scholar]
- Geley, S., E. Kramer, C. Gieffers, J. Gannon, J.M. Peters, and T. Hunt. 2001. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 153:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, N., N. Watanabe, Y. Furuta, H. Tamemoto, N. Sagata, M. Yokoyama, K. Okazaki, M. Nagayoshi, N. Takeda, Y. Ikawa, et al. 1994. Parthenogenetic activation of oocytes in c-mos-deficient mice. Nature. 370:68–71. [DOI] [PubMed] [Google Scholar]
- Hoyt, M.A., L. Totis, and B.T. Roberts. 1991. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 66:507–517. [DOI] [PubMed] [Google Scholar]
- Kallio, M., J.E. Eriksson, and G.J. Gorbsky. 2000. Differences in spindle association of the mitotic checkpoint protein Mad2 in mammalian spermatogenesis and oogenesis. Dev. Biol. 225:112–123. [DOI] [PubMed] [Google Scholar]
- King, R.W., J.M. Peters, S. Tugendreich, M. Rolfe, P. Hieter, and M.W. Kirschner. 1995. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 81:279–288. [DOI] [PubMed] [Google Scholar]
- Kitajima, T.S., S.A. Kawashima, and Y. Watanabe. 2004. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature. 427:510–517 [DOI] [PubMed] [Google Scholar]
- Ledan, E., Z. Polanski, M.E. Terret, and B. Maro. 2001. Meiotic maturation of the mouse oocyte requires an equilibrium between cyclin B synthesis and degradation. Dev. Biol. 232:400–413. [DOI] [PubMed] [Google Scholar]
- Li, R., and A.W. Murray. 1991. Feedback control of mitosis in budding yeast. Cell. 66:519–531. [DOI] [PubMed] [Google Scholar]
- Li, Y., and R. Benezra. 1996. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 274:246–248. [DOI] [PubMed] [Google Scholar]
- Lohka, M.J., M.K. Hayes, and J.L. Maller. 1988. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc. Natl. Acad. Sci. USA. 85:3009–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, X., Z. Tang, J. Rizo, and H. Yu. 2002. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell. 9:59–71. [DOI] [PubMed] [Google Scholar]
- Masui, Y., and C.L. Markert. 1971. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J. Exp. Zool. 177:129–145. [DOI] [PubMed] [Google Scholar]
- Minshull, J., H. Sun, N.K. Tonks, and A.W. Murray. 1994. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell. 79:475–486. [DOI] [PubMed] [Google Scholar]
- Musacchio, A., and K.G. Hardwick. 2002. The spindle checkpoint: structural insights into dynamic signalling. Nat. Rev. Mol. Cell Biol. 3:731–741. [DOI] [PubMed] [Google Scholar]
- Polanski, Z., E. Ledan, S. Brunet, S. Louvet, M.H. Verlhac, J.Z. Kubiak, and B. Maro. 1998. Cyclin synthesis controls the progression of meiotic maturation in mouse oocytes. Development. 125:4989–4997. [DOI] [PubMed] [Google Scholar]
- Reimann, J.D., and P.K. Jackson. 2002. Emi1 is required for cytostatic factor arrest in vertebrate eggs. Nature. 416:850–854. [DOI] [PubMed] [Google Scholar]
- Sagata, N., M. Oskarsson, T. Copeland, J. Brumbaugh, and G.F. Vande Woude. 1988. Function of c-mos proto-oncogene product in meiotic maturation in Xenopus oocytes. Nature. 335:519–525. [DOI] [PubMed] [Google Scholar]
- Sagata, N., N. Watanabe, G.F. Vande Woude, and Y. Ikawa. 1989. The c-mos proto-oncogene product is a cytostatic factor responsible for meiotic arrest in vertebrate eggs. Nature. 342:512–518. [DOI] [PubMed] [Google Scholar]
- Schott, E.J., and M.A. Hoyt. 1998. Dominant alleles of Saccharomyces cerevisiae CDC20 reveal its role in promoting anaphase. Genetics. 148:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, M.S., B.T. Roberts, S.D. Gross, B.J. Tunquist, F.E. Taieb, A.L. Lewellyn, and J.L. Maller. 2001. Bub1 is activated by the protein kinase p90(Rsk) during Xenopus oocyte maturation. Curr. Biol. 11:141–150. [DOI] [PubMed] [Google Scholar]
- Sharp-Baker, H., and R.H. Chen. 2001. Spindle checkpoint protein Bub1 is required for kinetochore localization of Mad1, Mad2, Bub3, and CENP-E, independently of its kinase activity. J. Cell Biol. 153:1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z., R. Bharadwaj, B. Li, and H. Yu. 2001. Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell. 1:227–237. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., and F. McKeon. 1997. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 89:727–735. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., E. Ha, and F. McKeon. 1998. The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J. Cell Biol. 142:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist, B.J., P.A. Eyers, L.G. Chen, A.L. Lewellyn, and J.L. Maller. 2003. Spindle checkpoint proteins Mad1 and Mad2 are required for cytostatic factor-mediated metaphase arrest. J. Cell Biol. 163:1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist, B.J., and J.L. Maller. 2003. Under arrest: cytostatic factor (CSF)-mediated metaphase arrest in vertebrate eggs. Genes Dev. 17:683–710. [DOI] [PubMed] [Google Scholar]
- Tunquist, B.J., M.S. Schwab, L.G. Chen, and J.L. Maller. 2002. The spindle checkpoint kinase bub1 and cyclin e/cdk2 both contribute to the establishment of meiotic metaphase arrest by cytostatic factor. Curr. Biol. 12:1027–1033. [DOI] [PubMed] [Google Scholar]
- Verlhac, M.H., J.Z. Kubiak, M. Weber, G. Geraud, W.H. Colledge, M.J. Evans, and B. Maro. 1996. Mos is required for MAP kinase activation and is involved in microtubule organization during meiotic maturation in the mouse. Development. 122:815–822. [DOI] [PubMed] [Google Scholar]
- Wassmann, K., T. Niault, and B. Maro. 2003. Metaphase I arrest upon activation of the Mad2-dependent spindle checkpoint in mouse oocytes. Curr. Biol. 13:1596–1608. [DOI] [PubMed] [Google Scholar]
- Weiss, E., and M. Winey. 1996. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J. Cell Biol. 132:111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew, N., M.L. Mellini, and G.F. Vande Woude. 1992. Meiotic initiation by the mos protein in Xenopus. Nature. 355:649–652. [DOI] [PubMed] [Google Scholar]
- Yu, H. 2002. Regulation of APC-Cdc20 by the spindle checkpoint. Curr. Opin. Cell Biol. 14:706–714. [DOI] [PubMed] [Google Scholar]
- Zhou, J., J. Yao, and H.C. Joshi. 2002. Attachment and tension in the spindle assembly checkpoint. J. Cell Sci. 115:3547–3555. [DOI] [PubMed] [Google Scholar]