

Abstract
Invasive tumor dissemination in vitro and in vivo involves the proteolytic degradation of ECM barriers. This process, however, is only incompletely attenuated by protease inhibitor–based treatment, suggesting the existence of migratory compensation strategies. In three-dimensional collagen matrices, spindle-shaped proteolytically potent HT-1080 fibrosarcoma and MDA-MB-231 carcinoma cells exhibited a constitutive mesenchymal-type movement including the coclustering of β1 integrins and MT1–matrix metalloproteinase (MMP) at fiber bindings sites and the generation of tube-like proteolytic degradation tracks. Near-total inhibition of MMPs, serine proteases, cathepsins, and other proteases, however, induced a conversion toward spherical morphology at near undiminished migration rates. Sustained protease-independent migration resulted from a flexible amoeba-like shape change, i.e., propulsive squeezing through preexisting matrix gaps and formation of constriction rings in the absence of matrix degradation, concomitant loss of clustered β1 integrins and MT1-MMP from fiber binding sites, and a diffuse cortical distribution of the actin cytoskeleton. Acquisition of protease-independent amoeboid dissemination was confirmed for HT-1080 cells injected into the mouse dermis monitored by intravital multiphoton microscopy. In conclusion, the transition from proteolytic mesenchymal toward nonproteolytic amoeboid movement highlights a supramolecular plasticity mechanism in cell migration and further represents a putative escape mechanism in tumor cell dissemination after abrogation of pericellular proteolysis.
Keywords: cell migration; invasion; fibrosarcoma cells; carcinoma cells; matrix proteases
Introduction
The invasion and migration of tumor cells involves coordinated adhesion as well as proteolytic interaction with the ECM substrate, resulting in the degradation and remodeling of interstitial tissue barriers (Stetler-Stevenson et al., 1993). Upon tumor progression, multiple classes of ECM-degrading enzymes are up-regulated and activated, including matrix metalloproteinases (MMPs),* serine proteases, and cathepsins (Birkedal-Hansen, 1995). Fibrillar collagen, the main constituent of the connective tissue matrix is degraded by several proteases, including MMP-1, -2, -8, and -13, membrane-anchored MT1-MMP, MT3-MMP, and cathepsin B, K, and L (Montcourrier et al., 1990; Aimes and Quigley, 1995; Ohuchi et al., 1997; Sassi et al., 2000). Accordingly, tumor cells expressing MMPs and other proteases generate proteolytic substrate degradation along their migration tracks on gelatin, matrigel, or fibronectin (Nakahara et al., 1997; d'Ortho et al., 1998). Blocking of MMPs and other proteases by pharmacological compounds impairs invasive tumor cell behavior in different migration and invasion models in vitro (Mignatti et al., 1986; Kurschat et al., 1999; Ntayi et al., 2001) as well as experimental metastasis in vivo (Coussens et al., 2002; and references therein). Together, these findings support a concept on proteolytic path generation favoring tumor cell dissemination within the tissues (Murphy and Gavrilovic, 1999; Friedl and Bröcker, 2000).
Conversely, accumulating evidence suggests that, despite their established proinvasive function, ECM-degrading enzymes could be dispensable for tumor cell motility and dissemination. After blocking of MMPs or serine proteases, significant residual migration of individual cells is observed in different migration models (Deryugina et al., 1997; Hiraoka et al., 1998; Kurschat et al., 1999; Ntayi et al., 2001). In vivo, protease inhibitor–based targeting of MMPs and serine proteases has prompted an unexpectedly weak benefit in some animal tumor models as well as clinical trials in humans, suggesting that a principal dissemination capacity remained intact (Della et al., 1999; Zucker et al., 2000; Kruger et al., 2001; Coussens et al., 2002). These studies, however, leave unresolved by which mechanisms cells may maintain migratory dissemination in the absence of ECM-degrading capacity. As one possibility, proteolytic compensation could be provided by enzymes not inhibited in these studies; alternatively, cells could sustain motility via unknown protease-independent compensation strategies.
To understand how proteases contribute to cell migration, the concept of pericellular proteolysis has to be integrated into the current paradigm on cell migration as a supramolecular process (Lauffenburger and Horwitz, 1996; Friedl and Bröcker, 2000). Cell migration results from a three-step cycle of polarized cell extension and substrate binding through leading pseudopod(s), followed by actin-based contraction of the cell body and release of adhesive bonds at the trailing edge (Palecek et al., 1997). Adhesive interaction and traction toward the ECM substrate are generated by adhesion receptors predominantly of the β1 and β3 integrin families (Brooks et al., 1996; Palecek et al., 1997; Sheetz et al., 1998). After binding to extracellular scaffolds, integrins cluster and couple to the actin cytoskeleton to further strengthen cell–ECM linkages, which generates bipolar traction between anterior and posterior cell edges to elongate the cell body toward a spindle-shaped morphology (Sheetz et al., 1998; Maaser et al., 1999). For focalized degradation of ECM substrata, integrins cooperate with cell surface proteases. In invasive tumor cells, αvβ3 integrin colocalizes with MMP-2 (Brooks et al., 1996) and β1 integrins cocluster with MT1-MMP (Belkin et al., 2001), as well as the receptor for urokinase-type plasminogen activator (uPAR) (Wei et al., 1996). Accordingly, overexpression of uPAR and MT-MMPs favor integrin-mediated motility and invasion in two-dimensional (2D) and three-dimensional (3D) ECM models, respectively (Wei et al., 1996; Hiraoka et al., 1998). Consistent with migration-associated proteolysis, integrin-dependent migration of tumor cells results in the formation of tube-like matrix defects that represent paths of least resistance for the advancing cell body (Friedl et al., 1997). However, although proteolytic ECM degradation accompanies and augments tumor invasion and motility, it remains to be clarified whether pericellular proteolysis is indeed indispensable for the penetration of interstitial matrix barriers.
In the present study, using proteolytic tumor cells migrating through 3D fibrillar collagen-rich matrix in vitro and in vivo, we have investigated to what extent abrogation of ECM-degrading capacity, including the blocking of MMPs, serine proteases, and cathepsins, impairs the migratory action. By means of time-lapse videomicroscopy and four-dimensional (4D) confocal and multiphoton microscopy, we have particularly focused on the mechanisms of residual tumor cell movement after protease function was antagonized.
Results
HT-1080 fibrosarcoma cells overexpressing the membrane-localized MT1-MMP (Deryugina et al., 1997, 1998) and MDA-MB-231 mammary carcinoma cells (Ishibashi et al., 1999; Bachmeier et al., 2001) were used as metastatic tumor cells capable of efficiently degrading and invading the ECM.
A mesenchymal type of tumor cell migration
In 3D collagen matrices, the migration of HT-1080/MT1 cells was characterized by polarized binding of the leading edge to collagen fibers, generating traction and spindle-shaped elongation of the cell body (Fig. 1 A; Video 1 A, available online at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1). Both polarization and migration were abrogated by adhesion-perturbing anti–β1 integrin mAb 4B4 (Fig. 1 B; Video 1 B), confirming β1 integrin–mediated migratory force generation. MT1-MMP and β1 integrins were coclustered at interaction sites to collagen fibers (Fig. 1 C), which represented the location of initial fiber binding, traction, and bundling toward the leading edge (Fig. 1, D and E, white arrowheads; Video 2, available online at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1). Upon forward movement of the cell body, the detachment of the trailing edge generated a circumscribed matrix defect that was bordered by aligned multifibrillar collagen bundles (Fig. 1 E, asterisks; Video 2). Cell detachment was further accompanied by the shedding of surface determinants and cell fragments along the migration track (Fig. 1 D, black arrowhead). This mesenchymal migration type of spindle-shaped cells that produce matrix defects is consistent with the integrin-dependent haptokinetic migration reported for fibroblasts (Doane and Birk, 1991; Palecek et al., 1997) and further supports the concept of focalized pericellular proteolysis for the generation of migration tracks (Murphy and Gavrilovic, 1999).
Figure 1.

Proteolytic migration of HT-1080/MT1 fibrosarcoma cells in 3D collagen lattice. (A) Morphology of HT-1080/MT1 cell migrating through 3D collagen lattice monitored by video microscopy. (B) Reduction of migration speed (black bars) and induction of detached, nonmobile spherical morphology (inset) by adhesion-perturbing anti–β1 integrin antibody 4B4. (C) Confocal backscatter (matrix fibers) and fluorescence of MT1-MMP (red), β1 integrins (green), and colocalization (yellow; arrowheads) at fiber traction zone of the leading edge. (D) 3D reconstruction of a calcein-stained migrating cell by time-lapse confocal microscopy and (E) backscatter signal of the same cell from the central section (time, 60 min). Fiber bundling (white arrowheads), deposition of cell fragments (black arrowhead), and newly formed matrix defect (asterisk). Black arrows, direction of migration. Bars, 20 μm.
Inhibition of collagenolysis by protease inhibitor cocktail
Pericellular degradation of native or denatured collagens can be directly or indirectly provided by proteases from different classes, including MMPs, serine proteases (e.g., plasmin and urokinase-type plasminogen activator [uPA]), as well as cysteine and aspartic proteases (e.g., cathepsins) (Montcourrier et al., 1990; Aimes and Quigley, 1995; Ohuchi et al., 1997; Sassi et al., 2000). In addition to MT1-MMP, many of these proteases, including up to five other collagenases toward native type collagen (Fig. 2 A, asterisks), were expressed by HT-1080/MT1 cells, as detected by RT-PCR. Because collagenolytic redundancy was anticipated, we used a cocktail of broad-spectrum protease inhibitors to simultaneously target a wide spectrum of endoproteolytic activity (Table I). To maintain sufficient inhibitory activity within the matrix, the employed concentrations of protease inhibitors were orders of magnitudes higher than the known maximum inhibitory values, unless cell viability was dose limiting (Table I). Because most of the proteases detectable by RT-PCR escape visualization by zymography (Deryugina et al., 1997, 1998; Ntayi et al., 2001), inhibition of cell-dependent collagenolysis in situ was measured using the fibrillar collagen migration substrate as read-out. On a qualitative basis, structural breakdown of the matrix fibers caused by HT-1080/MT1 cells (Fig. 2 B, 1 and 3) was fully abrogated in the presence of protease inhibitor cocktail (Fig. 2 B, 2). This strong inhibitory effect was confirmed by a quantitative 3D fluorometric fluorescein (FITC) release assay, detecting the degradation of FITC-labeled collagen fibers (Fig. 2 C). Upon migration within FITC–collagen, HT-1080/MT1 cells released 20% of the total FITC content (Fig. 2 D), confirming the observed macroscopic (Fig. 2 B, 1) and microscopic collagen fiber degradation (Fig. 1 E; Fig. 2 B, 3). Protease inhibitor cocktail inhibited 95% of the cell-mediated FITC release generated by HT-1080/MT1 cells (Fig. 2 D) and to the same residual level in vector-transfected HT-1080/neo cells (unpublished data). Broad-range MMP inhibitor BB-2516 (marimastat) alone abrogated >80% of the FITC release in HT-1080/MT1 cells, suggesting a major contribution of MMPs to the degradation of collagen (Fig. 2 D).
Figure 2.

Protease expression in HT-1080/MT1 cells and inhibition of collagenolysis. (A) mRNA expression of MMPs, ADAMs, cathepsins, and serine proteases detected by RT-PCR. Asterisks, proteases cleaving native type I collagen. (B) Degradation of 3D fibrillar collagen by HT1080/MT1 cells (500,000 cells) layered on top of a 1-mm-thick collagen matrix in the absence (1) or presence (2) of protease inhibitor cocktail. (B, 3) Confocal backscatter of lysis zone bordered by clumped collagen, as indicated by the square in (1). (B, 4) Negative control matrix overlaid with cell-free medium only. (C) Cell contact to FITC-labeled collagen fibers; confocal reflection (gray) and FITC fluorescence (green). Bar, 20 μm. (D) Migration-associated collagenolysis caused by HT1080/MT1 cells within 3D FITC–collagen lattices was quantified from the FITC release after 40 h of migration in the absence or presence of inhibitors. ***, P < 0.001; two-tailed t test for independent means, difference to control cells. As negative control, T cells did not release FITC above background levels (not depicted).
Table I. Protease inhibitor cocktail.
| Inhibitor | Target protease | IC50 values | Concentration | Reference |
|---|---|---|---|---|
| μM | μM | |||
| BB-2516 | MMPsa | 0.002–0.2 | 100 | |
| E-64 | Cysteine proteases (cathepsin B, H, L, and K) | 0.001–20 | 250 | Bedi and Williams, 1994 |
| Pepstatin A | Aspartatic proteases (including cathepsin D) | 0.0001–5 | 100 | Laurent and Salzet, 1995; Nisbet and Billingsley, 1999 |
| Leupeptin | Cathepsin D | 0.1–10 | 2b | Bedi and Williams, 1994 |
| Aprotinin | Serine proteases (uPA and PA) | 0.15–3.5 | 2.2b | Callas et al., 1994 |
Guidelines for the clinical use of marimastat were from British Biotech Inc. (1998).
Highest nontoxic concentration (for details see Materials and methods).
Unexpectedly, although inhibition of collagenolysis by protease inhibitor cocktail was near complete, the migration efficiency of HT-1080/MT1 cells within 3D collagen lattices was barely reduced (Fig. 3). Persistent migration was monitored by sensitive real-time analysis of time-dependent population speed (Fig. 3 A) and the median speed derived from single cell analysis (Fig. 3 B). Similar high migration rates were obtained by BB-2516 alone as well as from HT-1080/neo and wild-type cells in the presence of inhibitor cocktail (unpublished data). Because the capacity to move, i.e., to generate traction and overcome physical matrix constraints appeared intact after blocking of collagenolysis, we hypothesized the existence of compensation strategies to counterbalance the loss of pericellular proteolysis.
Figure 3.

Sustained migration in the presence of protease inhibitor cocktail. (A) Cell tracking analysis of the steady-state population speed ± SD and (B) mean speed for each individual cell (time, 20 h; n = 3 experiments, 120 cells).
Induced amoeboid migration in vitro
As it became apparent from the video recordings (Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1), the presence of protease inhibitor cocktail changed several aspects of migratory behavior in HT-1080/MT1 cells (Fig. 4). Whereas spontaneously moving cells maintained a spindle-shaped, elongated morphology (Fig. 4 A, left), this mesenchymal migration type was converted to less polarized, more spherical morphodynamics in the presence of protease inhibitor cocktail (Fig. 4 A, right; Video 3). Spherical-shaped cells sustained migration (Fig. 4 B) and exhibited a significant reduction in median axis length compared with control cells (Fig. 4 C). In the presence of protease inhibitors, spherical morphodynamics included flexible shape change and forward propulsion guided by multiple anterior and outward ruffling filopodia, as detected by high resolution video microscopy (Video 4, available online at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1). Very similar observations were obtained for invasive MDA-MB-231 mammary carcinoma cells, expressing soluble MMPs, MT-MMPs, serine proteases, and cathepsins (Ishibashi et al., 1999; Bachmeier et al., 2001). Inhibition of proteolysis in MDA-MB-231 cells by protease inhibitor cocktail (Fig. 4 D), albeit slightly less efficient than in HT-1080 cells, resulted in a similar transition from constitutive spindle shape to less polarized, elliptoid morphodynamics (Fig. 4 E; Video 5, available online at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1), including a significantly shortened median axis length (Fig. 4 F).
Figure 4.

Transition of spindle-shaped (mesenchymal) to more spherical (amoeboid) migration in HT1080/MT1 and MDA-MB-231 cells in the presence of protease inhibitor cocktail. (A) Conversion of elongated (left) toward spherical shape (right) in HT1080/MT1 cells, and (B) higher magnification of an amoeboid migrating cell in the presence of inhibitor cocktail. Time in B, 117 min. (C) Median elongation (calculated from length divided by width) in the absence and presence of protease inhibitor cocktail (n = 3; 170 cells; ***, P < 0.0001). (D) Inhibition of collagen degradation by MDA-MB-231 cells by protease inhibitor cocktail (n = 3; P < 0.05, unpaired two-tailed t test). (E) Conversion from spindle shaped (left) to more spherical morphology (right), and (F) reduced median elongation in the presence of protease inhibitors in MDA-MB-231 cells (n = 3; 200 cells; ***, P < 0.0001). (G) Frequency of mesenchymal and amoeboid shape in actually migrating cells in the absence (▪) and presence (□) of protease inhibitor cocktail (HT-1080/MT1 cells, n = 3, 100 cells; **, P < 0.001 for difference to untreated control; two-tailed t test for independent means). Cells of indeterminate morphology (15–40%; for details see the Materials and methods) were excluded from analysis. Bars: (A and E) 100 μm; (B) 20 μm.
This novel nonproteolytic phenotype in HT-1080/MT1 and MDA-MB-231 cells was reminiscent of Dictyostelium amoeba migrating across 2D surfaces (Yumura et al., 1984; Killich et al., 1993) as well as leukocytes crawling through 3D ECM substrata (Gunzer et al., 2000; Friedl et al., 2001; for comparison, see migrating T lymphocytes in Gunzer et al. [2000] supplemental material). Morphometric analysis showed a reduced number in mesenchymally moving cells in the presence of protease inhibitor cocktail, whereas the percentage of the more spherical “amoeboid” phenotype was at least tripled (Fig. 4 G) or doubled by BB-2516 alone (unpublished data).
Mechanisms of induced nonproteolytic migration
Consistent with impaired collagenolysis, amoeboid moving HT-1080/MT1 cells did not cause structural remodeling of collagen fibers (Video 6, available online at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1). Induced protease-independent migration resulted from adaptation and alignment of the cell body along preformed fiber strands (Fig. 5 A, white arrowheads) and consecutive migratory guidance along fibrillar scaffolds (Fig. 5 B, black arrowheads). Upon cell detachment, no remodeling, bundling, or destruction of the collagen network was generated, leaving behind the intact reticular texture of individual fibrils at their original position (Fig. 5 A, black arrowheads; Video 6). To overcome regions of narrow space by changing shape, an initial pseudopod elongation through a preformed matrix gap (4 μm pore diameter from the central section, 18 μm cell diameter; Fig. 5 C, black arrowhead) was followed by propulsion of the cell body and the development of a narrow region confined by matrix fibers (constriction ring; Lewis, 1934; Fig. 5 C, black arrowhead). Constriction rings persisted until the cell body had squeezed or pulled forward, while no matrix defect was apparent after cell detachment (Fig. 5 C; Video 7, available at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1).
Figure 5.

Cellular mechanism of nonproteolytic movement within 3D collagen matrix and related changes in β1 integrin, MT1-MMP, and F-actin distribution in HT-1080/MT1 cells. (A) Induced amoeboid migration lacking fiber degradation. Alignment of cell body along a fiber strand (white arrowheads) and intact individual collagen fiber at its original position after cell detachment (black arrowhead). (B) Migratory alignment of the cell depicted in A along the preexisting fiber scaffold. The outline of the cell edge at 2.5-min time intervals (blue lines) was superimposed onto the 3D reconstruction of the transmigrated matrix structure. Bright pixels indicate colocalization of cell boundary and fibers (arrowheads). (C) Migration through a narrow gap bordered by fibers (black arrowhead) resulting in morphological adaptation and the formation of a constriction ring. (D) Reduced F-actin and β1 integrin focalization at fiber binding sites in an amoeboid HT1080/MT1 cell, compared with F and Fig. 1 C. Because of constriction caused by a perpendicular collagen fiber, this cell contains a lobulated main body. Black arrowhead, uropod. (E) Loss of clustered MT1-MMP and β1 integrins from interactions with fibers in induced amoeboid migration. Arrowheads indicate two simultaneous constriction rings bordered by perpendicular collagen fibers. (F) F-actin and β1 integrins in an untreated control cell of elongated shape. Time: (A) 35 min; (B) 65 min; (C) 20 min. Bars, 20 μm. Black arrows, direction of migration.
Because the structure and location of adhesive cell–matrix contacts control cell shape and migration dynamics (Palecek et al., 1997; Sheetz et al., 1998), we investigated the cytoskeletal structure and integrin distribution in amoeboid HT-1080/MT1 cells. Induced amoeboid morphology was characterized by diffuse cortical actin rims and small actin-rich spots at interactions with collagen fibers (Fig. 5 D). In amoeboid HT1080/MT1 cells, β1 integrins showed nonclustered, linear surface distribution at contacts to collagen fibers (Fig. 5, D and E) deviating from clustered β1 integrins and prominent actin nucleation zones in spindle-shaped control cells (Fig. 1 C; Fig. 5 F). MT1-MMP was consistently dissociated from fiber binding sites but accumulated intracellularly (Fig. 5 E).
Mesenchymal–amoeboid transition in vivo
Although collagen lattices provide a complex 3D ECM scaffold and a barrier for moving cells, a putatively different spacing and molecular composition of life connective tissue may impose additional physical and molecular constraints, putatively yielding in distinct migration mechanisms. Therefore, the in vivo migration of HT-1080/MT1 cells within the mouse dermis was investigated by multiphoton microscopy. 3 h after the injection of HT-1080/MT1 cells into the loose connective tissue of the mouse dermis (Fig. 6 A), both nontreated control cells (green) and cells pretreated with protease inhibitor cocktail (red) were detected at and passively scattered around the injection site (Fig. 6 A, asterisk), or located within multicellular cords (Fig. 6 A, black arrowheads). In wash-out experiments in vitro, preincubation of cells with protease inhibitor cocktail for 6 h resulted in stable amoeboid movement for at least 10 h before reversion toward mesenchymal migration occurred (unpublished data), indicating both relatively slow turnover of the target proteases after inhibition and full reversibility of inhibitor-induced phenotypic change. In the dermis, ortotopic 3D reconstruction of the injection site 3 and 8 h after injection identified considerable position changes for both control cells (Fig. 6 B, top) and inhibitor-pretreated HT-1080/MT1 cells (Fig. 6 B, bottom). This position change corresponded to a translocation of 4–10 μm/hr (Fig. 6 B, white arrowheads), in part approximating migration velocities present in collagen matrices in vitro (6–36 μm/h; compare Fig. 3 B). At high-resolution reconstruction, cells pretreated with protease inhibitors developed a significantly reduced elongation, reminiscent of amoeboid morphology (Fig. 6 C, red cells), and a near-round median shape (Fig. 6 D), whereas control cells retained their constitutive spindle-like elongation (Fig. 6 C, green cells; Video 8, available at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1). In summary, similar to 3D collagen matrices, abrogation of matrix protease function in HT-1080/MT1 cells was followed by cell movement and concomitant induction of amoeboid morphology in vivo, confirming migratory plasticity for dermal connective tissue.
Figure 6.
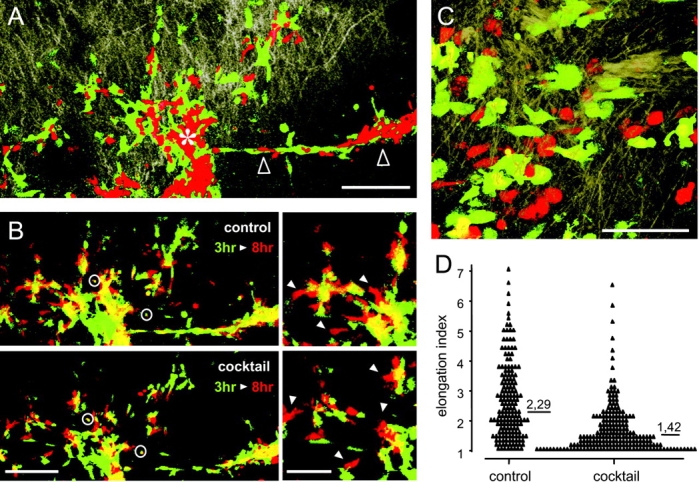
In vivo translocation and morphology of HT-1080/MT1 cells in the mouse dermis. Intravital microscopy of ECM structure (light brown) and labeled cells 3 h (A–D) and 8 h (B) after injection. (A) 3D reconstruction of injection site (asterisk), more distant cells passively scattered around the injection site, and matrix structure at low magnification (10× objective). Control cells (green) and cells pretreated with protease inhibitor cocktail (red). Asterisk, center of injection site. Black arrows, multicellular cord. (B) Position change of control cells (top) and cells pretreated with protease inhibitor cocktail (bottom) from the injection site depicted in A. False-color reconstruction was obtained for each fluorescence channel 3 h (green) and 8 h (red) after injection. Orthotopic superimposition was controlled by the position of coinjected fluorescent beads (circles) as well as the multicellular cord. White arrowheads, regions of cell translocation (right). (C) High resolution imaging of cell morphology and (D) elongation of control cells (green) and cells pretreated with protease inhibitor cocktail (red) at least 2 h after injection (40× objective). Elongation was calculated from 170 cells, six independent experiments; ***, P < 0.0001. Bars: (A and B [left]) 200 μm; (B [right] and C) 100 μm.
Discussion
The transition from proteolytic and “mesenchymal” to protease independent and “amoeboid” movement reveals a novel cellular and molecular adaptation pathway in sustaining cell migration: the switch from a path-generating to a path-finding migration mode that we have termed mesenchymal–amoeboid transition (MAT). Although proteases were used for constitutive migration of proteolytic cells of mesenchymal and epithelial origin, abrogation of pericellular proteolysis was followed by nonproteolytic movement. MAT thus represents a compensatory migration mode in vitro and in vivo to bypass physical matrix resistance by amoeboid shape change.
Constitutive proteolytic migration of mesenchymal cells
Important concepts on cell migration are derived from the movement of fibroblasts and myoblasts (Lauffenburger and Horwitz, 1996). In 3D in vitro and in vivo tissues, fibroblasts develop elongated, spindle-shaped morphology mediated by cortical F-actin and occasional stress fibers (Welch et al., 1990). Fibroblast migration is dependent on β1 integrin function (Doane and Birk, 1991) and is further coupled to the cleavage and remodeling of ECM components using MMPs and other proteases (Langholz et al., 1995). A similar type of mesenchymal behavior is developed by HT-1080 fibrosarcoma cells and MDA-MB-231 mammary carcinoma cells in 3D collagen matrices. These cells move as individual spindle-shaped cells that utilize β1 integrin–mediated binding to collagen fibers for elongation and translocation. Similarly to fibroblasts, a range of ECM-degrading enzymes is expressed, coclustered with integrins at interactions to substrate, and used for the generation of tube-like matrix defects that correspond to known clearance tracks in other models (Nakahara et al., 1997; d'Ortho et al., 1998). Because the cell diameter of mesenchymal-like cells exceeds the mean pore size within the fiber network (Friedl et al., 1997), contact-dependent focal degradation of collagen barriers may facilitate migration by locally lowering the physical resistance of the ECM barrier (Murphy and Gavrilovic, 1999).
Constitutive nonproteolytic amoeboid migration strategies
In some cell types, migration results from more dynamic, transient and less defined cell–substrate contacts associated with amoeboid shape (Friedl and Bröcker, 2000; Friedl et al., 2001; and references therein). The concept of amoeboid movement is most clearly established from the single cell state of the lower eukaryote Dictyostelium discoideum. Dictyostelium is an elliptoid cell that translocates via rapidly alternating cycles of morphological expansion and contraction, relatively low-affinity integrin-independent interactions with the substrate, and extraordinary deformability (Yumura et al., 1984; Devreotes and Zigmond, 1988; Killich et al., 1993; Friedl et al., 2001). In higher eukaryotic cells, such as leukocytes and certain types of tumor cells, hallmarks of amoeboid motion are retained, including rapid low affinity gliding coupled to shape change, squeezing through preformed matrix gaps, and propulsive elongation along paths of least resistance (Lewis, 1934; Schor et al., 1983; Friedl et al., 1998, 2001). In collagen matrices, T cell migration is independent of structural matrix remodeling (Schor et al., 1983) and it is not sensitive to protease inhibitor treatment (unpublished data) or the inhibition of β1 integrin function, the latter both in vitro and in vivo (Friedl et al., 1998; Brakebusch et al., 2002). Thus, amoeboid migration appears to comprise morphodynamic supramolecular mechanisms to bypass tissue barriers independent of ECM degradation.
Mesenchymal–amoeboid transition: induced amoeboid migration
In HT-1080 fibrosarcoma and MDA-MB-231 mammary carcinoma cells, the mechanism of sustained migration after abrogation of pericellular proteolysis was a striking change in cell morphology and migration “style” reminiscent of transition toward amoeboid movement. To provide maximum inhibition efficiency, five pharmacological inhibitors were used to simultaneously target different protease classes known to contribute to ECM degradation and remodeling, including MMPs, serine proteases, and cathepsins, all of which are expressed by HT-1080 and MDA-MB-231 cells. We reasoned that more specific approaches, such as genetic ablation of individual proteases, might be hampered by proteolytic redundancy and functional compensation.
The effectiveness of the protease inhibition strategy was shown by a combined biochemical and structural approach: (a) near-total inhibition of FITC release using FITC-labeled fibrillar collagen; (b) abrogation of macroscopic structural degradation of the collagen lattice by HT-1080 cells; and (c) the lack of newly generated tube-like matrix defects in the wake of the cells visualized by 4D confocal backscatter microscopy.
Inhibition of constitutive proteolysis, instead of causing the cells to become “trapped” within the fibrillar network, induced a program of morphodynamic and molecular changes that allowed the maintenance of the migratory action (Fig. 7). These changes included a reduced elongation yet increased morphodynamic flexibility, the loss of focal β1 integrin and MT1-MMP clustering at interactions with matrix fibers, and a more diffuse cortical actin distribution, including small actin-rich nucleation foci at fiber binding sites. These features are coupled to the forward flow (streaming) of cytoplasm through cell regions of narrow space confined by outside constriction and, together, result in a phenotype mimicking amoeboid moving cells (Yumura et al., 1984; Devreotes and Zigmond, 1988; Killich et al., 1993; Friedl et al., 2001). Because this induced amoeboid behavior generated significant migratory activity, yet the transmigrated collagen architecture remained intact by means of biochemical and structural analysis, our data show an alternative inhibitor-induced nonproteolytic migration type (Fig. 7).
Figure 7.

Concept on mesenchymal–amoeboid transition. A program of morphodynamic and molecular changes after abrogation of pericellular proteolysis by protease inhibitors provides a supramolecular mechanism for persistent nonproteolytic migration in 3D fibrillar collagenous tissues.
Induced protease-independent movement was observed for two different collagen-based tissue models, 3D collagen lattices in vitro and the mouse dermis in vivo. As reconstructed by intravital microscopy, the loose connective tissue of the mouse dermis exhibits strong structural similarity to 3D collagen matrices. Both models provide 3D fibrillar collagenous strands bordering matrix gaps in the range of 1–15 μm (unpublished data). Migratory alignment and shape change of tumor cells (cell diameter in the range of 15–25 μm [Friedl et al., 1997]; 10–30-fold larger in volume than amoeba or leukocytes) along preformed fiber strands may hence be sustained by constriction of the cell body and propulsion of the cytoplasm through narrow space as a nonproteolytic, physical strategy to bypass structural ECM barriers. Although the spacing of such loose connective tissue supports protease-independent cell movement, future studies will be necessary to determine under which conditions transmigration of more densely interconnected ECM, such as basement membranes, can occur by nonproteolytic means.
The finding that blocking proteases induces transition from a mesenchymal phenotype to amoeboid movement sheds light on how disseminating tumor cells may react in response to protease inhibitor–based therapy. In established tumor disease, tumor cells have already reached interstitial tissue compartments (Stetler-Stevenson et al., 1993) that are likely to support shape-driven migration independent of pericellular proteolysis. Therefore, protease-independent tumor cell dissemination may represent a candidate escape mechanism upon protease inhibitor–based treatment of progressive cancer disease (Zucker et al., 2000; Kruger et al., 2001; Coussens et al., 2002).
Molecular implications of MAT
The putative mechanistic link between loss of pericellular proteolysis and MAT can be best explained by changes in cell–matrix interaction strength. Although different experimental systems are difficult to directly relate, exaggerated mesenchymal-like phenotypes can be induced by strengthening cell–ECM contacts or reducing their turnover: by stabilizing interactions toward collagen fibers after arresting β1 integrins in a high-affinity ligand-bound state (Maaser et al., 1999) or by stabilizing stress fibers leading to spread-out, elongated morphology through activation of the small GTPase Rho (Nobes and Hall, 1999). Conversely, reduced axis length and amoeba-like morphodynamics may emerge from weakening substrate-binding strength: by reducing integrin adhesive function (Dufour et al., 1988) or by inhibiting Rho-kinase (Nobes and Hall, 1999). In addition to integrins, several of the proteases blocked in this study are known to regulate cellular morphodynamics by other mechanisms, including a mesenchymal phenotype (Lochter et al., 1997). MMP-3 overexpression in epithelial cells leads to the cleavage of E-cadherin and induces epithelial–mesenchymal transition, i.e., single cell detachment and mesenchymal invasion (Lochter et al., 1997). Proteases further control cell adhesion, cytoskeletal organization, and the release of soluble factors (Sternlicht and Werb, 2001; and references therein). These functions comprise cleavage of adhesion receptors (e.g., L-selectin); cleavage and activation of cytokines (e.g., IL-1 and TNF-α) or growth factors (e.g., EGF and bFGF); and the regulation of focal contact assembly and turnover by cleavage of talin or integrin cytoplasmic domains through, for example, calpain. The identification of pathways involved in MAT will thus require further studies on different cytoskeletal and signaling checkpoints that control cell shape and cell–matrix interactions.
MAT: comparison to other morphodynamic processes
Plasticity and adaptation in cell behavior are well established in other morphodynamic processes governed by cell–cell and cell–substrate interactions. In epithelial–mesenchymal transition, a usually nonmigrating epithelial collective abandons cell–cell cohesion after cadherin-mediated cell–cell contacts are weakened, allowing detachment of single cells and conversion toward mesenchymal morphology and migration (Birchmeier and Birchmeier, 1995). In another example, differentiating muscle precursor cells quit cell movement and fuse via N- and E-cadherin to form a multicellular contractile syncythium (Redfield et al., 1997). In conclusion, MAT may represent a novel transient morphodynamic alteration and adaptation pathway in molecular cell dynamics. Stringent, adhesive, and proteolytic cell–ECM contacts, as developed by mesenchymally moving cells, can be converted toward less well characterized, more diffuse, and less proteolytic interactions with extracellular scaffolds, reminiscent of amoeboid single cell movement. Residual amoeboid movement may hence represent a robust “salvage” pathway of cell movement in loose connective tissue after the abrogation of pericellular proteolysis.
Materials and methods
Cell migration in 3D collagen matrices: reconstruction by time-lapse video microscopy and confocal microscopy
Subconfluent HT-1080 fibrosarcoma cells stably transfected with MT1-MMP (Deryugina et al., 1997) or MDA-MB-231 breast carcinoma cells (Ishibashi et al., 1999; Bachmeier et al., 2001) were detached by EDTA (2 mM), washed, incorporated into 3-D collagen lattices (1.67 mg/ml; native dermal bovine type I collagen; Vitrogen; Cohesion Inc.), and monitored by time-lapse video microscopy (Friedl et al., 1997; Gunzer et al., 2000). The collagen was resistant to trypsin and sensitive to degradation by MT1-MMP (not depicted), confirming its native state. HT-1080/MT1 cells served as model for high constitutive collagenase expression and activity, allowing a robust visualization of surface MT1-MMP and fiber breakdown in the process of migration. In control experiments, HT1080/neo cells as well as HT-1080 wild-type cells were used for collagen degradation and migration experiments yielding similar results, including MAT (not depicted). For inhibition studies, blocking anti–β1 integrin mAb 4B4 (10 μg/ml; Coulter) or protease inhibitor cocktail (Table I) was added to the lattice before polymerization as well as to the supernatant. Protease inhibitor cocktail consisted of BB-2516 (marimastat; provided by British Biotech Inc.), leupeptin (Molecular Probes), pepstatin A, E-64, and aprotinin (all from Sigma-Aldrich). Protease inhibitor cocktail did not affect cell viability in collagen after 48 h, as assessed after cell release from the collagen lattice by collagenase digestion, propidium iodide staining, and flow cytometry, or upon liquid culture after 72 h of exposure.
For 3D time-lapse confocal microscopy (Leica-SP2 system), cells within the lattice were labeled by calcein-AM (1 μM), scanned at 2.5-min time intervals for simultaneous fluorescence and backscatter signal, and reconstructed (Friedl et al., 1997). MT1-MMP and β1 integrins were detected by rabbit AB815 (Chemicon), secondary rhodamine-conjugated goat anti–rabbit Fab' fragments (Jackson ImmunoResearch Laboratories), and a cocktail of FITC-labeled mAb 4B4 and K20 (Coulter), respectively.
Computer-assisted cell tracking and reconstruction of cell morphology
Locomotor parameters were obtained by computer-assisted cell tracking and reconstruction of the x and y coordinates of the cell paths (Maaser et al., 1999). The population speed was calculated from each 15-min tracking interval for all cells divided by the number of cells. Single cell speed represents the total length of the path divided by time. The proportion of mesenchymal to amoeboid migration was assessed 9 h (HT1080/MT1) or 18 h (MDA-MB-231) after addition of protease inhibitor cocktail by visual blinded analysis for a 60-min time interval by two independent observers. Cells of indeterminate morphology representing either transition states of partially elongated, dividing, or nonmigrating cells precluded clear categorization and were excluded from analysis (15–40% of total population). In control cells, indeterminate morphology was seen in 20% of the cells, mainly representing dividing cells and periods of retraction of otherwise elongated mobile cells (see Video 3). In the presence of inhibitor cocktail, the indeterminate fraction was increased to 40%, containing an additional subset of partially retracted cells (elongation index >2) that lacked the criteria of full amoeboid as well as mesenchymal morphodynamics (Video 3). Elongation was calculated from still frames as elongation index for each cell (axis length divided by width) and displayed as box-blot ranging from the 25th to 75th percentile including the median and whiskers from the 5th to 95th percentile. Unless indicated otherwise, statistical analysis was performed using the unpaired two-tailed Mann-Whitney U test.
RT-PCR
Primers and RT-PCR conditions were used as previously described (Hofmann et al., 1999; Konttinen et al., 1999; McCulloch et al., 2000). Additional primers were cathepsin B, 5′-GCCTGCAAGCTTCGATGCAC-3′ (direct) and 5′-CTATTGGAGACGCTGTAGGA-3′ (reverse); cathepsin D, 5′-CCAGCCCCCAATCCCAACCCCACCTCCAG-3′ (direct) and 5′-CACTGAAGCTGGGAGGCAAAGGCTACAAGC-3′ (reverse); cathepsin K, 5′-TCCATCCATAACCTTGAGGCTT-3′ (direct) and 5′-CCACAGCCATCATTCTCAGACACA-3′ (reverse); cathepsin L, 5′-CAGGCAGGTGATGAATGGCT-3′ (direct) and 5′-CAGGCCTCCATTATCCTGAA-3′ (reverse); MT1-MMP, 5′-CCCTATGCCTACATCCGTGA-3′ (direct) and 5′-TCCATCCATCACTTGGTTAT-3′ (reverse); u-PAR, 5′-GGTCACCCGCCGCTG-3′ (direct) and 5′-CCACTGCGGTACTGGACA-3′ (reverse); u-PA, 5′-AAAATGCTGTGTGCTGCTGACC-3′ (direct) and 5′-CCCTGCCCTGAAGTCGTTAGTG-3′ (reverse); and PAI-1, 5′-GAACAAGGATGAGATCAGCACC-3′ (direct) and 5′-ACTATGACAGCTGTGGATGAGG-3′ (reverse). Uniqueness of region was confirmed using the NCBI blastn program; specificity was determined from the length of each PCR product. Total RNA was isolated (QIAGEN RNA-Easy kit), transcribed to cDNA by AMV reverse transcriptase, and amplified by PCR (30 cycles). Absence of cDNA contamination was confirmed by subjecting total cell lysates to PCR in the absence of reverse transcriptase.
Collagen degradation assays
HT-1080/MT1 or MDA-MB-231 cell suspension was copolymerized with nonlabeled rat tail collagen (Becton Dickinson) containing 2% FITC-labeled collagen monomers (Molecular Probes). After migration for 40 h, solid-phase collagen containing the cells was pelleted, while FITC released into the supernatant was analyzed by spectrofluorometry. 100% values were obtained by complete collagenase digestion of cell-free collagen lattices. Background fluorescence was calculated by pelleting nondigested cell-free lattices. No autofluorescence was detected for either cells or inhibitors. FITC–collagen was sensitive to degradation by recombinant MT1-MMP, MMP-2, and trypsin. While FITC release detected both classical collagenases as well as trypsin-like activity, FITC release induced by HT-1080/MT1 cells was reduced by 80% by the MMP inhibitor BB-2516 (Fig. 2 C), indicating MMPs as primary collagenases in these cells. To exclude that FITC release merely reflected telopeptidase activity and incomplete fiber degradation, 500,000 HT1080/MT1 cells in a drop of medium were placed onto native nonlabeled 3D collagen (100 μm in depth). Fiber breakdown was detected after 40 h by Coomassie blue staining and confocal backscatter microscopy.
Intravital multiphoton microscopy
Reconstruction of HT1080/MT1 cell positioning and ECM structure were monitored by multiphoton microscopy (Maiti et al., 1997) of the mouse dermis in adult C57BL/6 mice, using a novel modification of a bone marrow intravital video microscopy model (Mazo et al., 1998). The epidermis and upper dermis, including hair follicles of the frontoparietal scalp, were removed by careful separation from the underlying connective tissue using microscissors. A fixation ring was inserted into the incision to spread the skin, and Hepes-buffered medium containing 10% fetal bovine serum was applied. Subconfluent HT-1080/MT1 cells were preincubated for 3–4 h in protease inhibitor cocktail (each 20 μM BB-2516, pepstatin A, E-64, 0.7 μM aprotinin, 2 μM leupeptin) or medium alone. Control cells and inhibitor cocktail–treated cells were labeled with calcein-AM (green) or TRITC (red), respectively, washed twice, suspended together in medium (104 cells/3 μl each), and carefully injected into the mid-dermis over the immobilized mouse scull. After injection, a temperature of 37°C was maintained by a water circulation system through the fixation ring and the use of a tempered water immersion objective. Intravital multiphoton microscopy was performed on an Olympus BX 50 WI microscope coupled to a Ti:Sapphire laser (Spectraphysics) and a Radiance 2000MP confocal multiphoton imaging system controlled by the Lasersharp software (all from Bio-Rad Laboratories). Multiphoton excitation light of 800 nm was introduced into the sample. Connective tissue structures were visualized via autofluorescence and second harmonic generation imaging at 400/40 nm. Labeled tumor cells were detected at 525/30 nm (calcein-AM) and 600/100 nm (TRITC). Image reconstruction from z-series scans up to a penetration depth of 800 μm was performed using the freeware Confocal Assistant©.
Online supplemental material
Quicktime movies of the image series from which figures were made are available at http://www.jcb.org/cgi/content/full/jcb.200209006/DC1.
Acknowledgments
We acknowledge Margit Ott for invaluable technical assistance, Jörg Geiger for expert help in fluorometric analysis, and Viven Schacht for assistance with intravital microscopy. We thank British Biotech Inc. for supplying BB-2516. Chris Overall and Eric Tam are further acknowledged for collagen cleavage analysis.
This work was supported by the Deutsche Forschungsgemeinschaft (FR 5511/2-2 and 2-3). K. Wolf was additionally supported by the Evangelisches Studienwerk, Villigst. I.B. Mazo is a recipient of the Amy Potter Fellowship and is supported by a National Institutes of Health (NIH) training grant in Transfusion Medicine from the Children's Hospital. U. von Andrian was supported by NIH grants HL56949 and HL54936 and a CHPI grant from the Dana Foundation. A.Y. Strongin was supported by NIH grants CA83017 and CA77470, California Breast Cancer Program grant 5JB0094, and Susan G. Komen Breast Cancer Foundation grant 9849.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: 2-, 3-, and 4D, two, three, and four dimensional; MAT, mesenchymal–amoeboid transition; MMP, matrix metalloproteinase; uPA, urokinase-type plasminogen activator; uPAR, receptor for uPA.
References
- Aimes, R.T., and J.P. Quigley. 1995. Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J. Biol. Chem. 270:5872–5876. [DOI] [PubMed] [Google Scholar]
- Bachmeier, B.E., A.G. Nerlich, R. Lichtinghagen, and C.P. Sommerhoff. 2001. Matrix metalloproteinases (MMPs) in breast cancer cell lines of different tumorigenicity. Anticancer Res. 21:3821–3828. [PubMed] [Google Scholar]
- Bedi, G.S., and T. Williams. 1994. Purification and characterization of a collagen-degrading protease from Porphyromonas gingivalis. J. Biol. Chem. 269:599–606. [PubMed] [Google Scholar]
- Belkin, A.M., S.S. Akimov, L.S. Zaritskaya, B.I. Ratnikov, E.I. Deryugina, and A.Y. Strongin. 2001. Matrix-dependent proteolysis of surface transglutaminase by membrane-type metalloproteinase regulates cancer cell adhesion and locomotion. J. Biol. Chem. 276:18415–18422. [DOI] [PubMed] [Google Scholar]
- Birchmeier, W., and C. Birchmeier. 1995. Epithelial-mesenchymal transitions in development and tumor progression. EXS. 74:1–15. [DOI] [PubMed] [Google Scholar]
- Birkedal-Hansen, H. 1995. Proteolytic remodeling of extracellular matrix. Curr. Opin. Cell Biol. 7:728–735. [DOI] [PubMed] [Google Scholar]
- Brakebusch, C., S. Fillatreau, A.J. Potocnik, G. Bungartz, P. Wilhelm, M. Svensson, P. Kearney, H. Korner, D. Gray, and R. Fassler. 2002. β1 integrin is not essential for hematopoiesis but is necessary for the T cell-dependent IgM antibody response. Immunity. 16:465–477. [DOI] [PubMed] [Google Scholar]
- Brooks, P.C., S. Stromblad, L.C. Sanders, T.L. von Schalscha, R.T. Aimes, W.G. Stetler-Stevenson, J.P. Quigley, and D.A. Cheresh. 1996. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell. 85:683–693. [DOI] [PubMed] [Google Scholar]
- Callas, D., P. Bacher, O. Iqbal, D. Hoppensteadt, and J. Fareed. 1994. Fibrinolytic compromise by simultaneous administration of site-directed inhibitors of thrombin. Thromb. Res. 74:193–205. [DOI] [PubMed] [Google Scholar]
- Coussens, L.M., B. Fingleton, and L.M. Matrisian. 2002. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 295:2387–2392. [DOI] [PubMed] [Google Scholar]
- Della, P.P., R. Soeltl, H.W. Krell, K. Collins, M. O'Donoghue, M. Schmitt, and A. Kruger. 1999. Combined treatment with serine protease inhibitor aprotinin and matrix metalloproteinase inhibitor Batimastat (BB-94) does not prevent invasion of human esophageal and ovarian carcinoma cells in vivo. Anticancer Res. 19:3809–3816. [PubMed] [Google Scholar]
- Deryugina, E.I., G.X. Luo, R.A. Reisfeld, M.A. Bourdon, and A. Strongin. 1997. Tumor cell invasion through matrigel is regulated by activated matrix metalloproteinase-2. Anticancer Res. 17:3201–3210. [PubMed] [Google Scholar]
- Deryugina, E.I., M.A. Bourdon, R.A. Reisfeld, and A. Strongin. 1998. Remodeling of collagen matrix by human tumor cells requires activation and cell surface association of matrix metalloproteinase-2. Cancer Res. 58:3743–3750. [PubMed] [Google Scholar]
- Devreotes, P.N., and S.H. Zigmond. 1988. Chemotaxis in eukaryotic cells: a focus on leukocytes and Dictyostelium. Annu. Rev. Cell Biol. 4:649–686. [DOI] [PubMed] [Google Scholar]
- Doane, K.J., and D.E. Birk. 1991. Fibroblasts retain their tissue phenotype when grown in three-dimensional collagen gels. Exp. Cell Res. 195:432–442. [DOI] [PubMed] [Google Scholar]
- d'Ortho, M.P., H. Stanton, M. Butler, S.J. Atkinson, G. Murphy, and R.M. Hembry. 1998. MT1-MMP on the cell surface causes focal degradation of gelatin films. FEBS Lett. 421:159–164. [DOI] [PubMed] [Google Scholar]
- Dufour, S., J.L. Duband, M.J. Humphries, M. Obara, K.M. Yamada, and J.P. Thiery. 1988. Attachment, spreading and locomotion of avian neural crest cells are mediated by multiple adhesion sites on fibronectin molecules. EMBO J. 7:2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl, P., and E.-B. Bröcker. 2000. The biology of cell locomotion within three-dimensional extracellular matrix. Cell. Mol. Life Sci. 57:41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl, P., K. Maaser, C.E. Klein, B. Niggemann, G. Krohne, and K.S. Zanker. 1997. Migration of highly aggressive MV3 melanoma cells in 3-dimensional collagen lattices results in local matrix reorganization and shedding of α2 and β1 integrins and CD44. Cancer Res. 57:2061–2070. [PubMed] [Google Scholar]
- Friedl, P., F. Entschladen, C. Conrad, B. Niggemann, and K.S. Zanker. 1998. CD4+ T lymphocytes migrating in three-dimensional collagen lattices lack focal adhesions and utilize β1 integrin-independent strategies for polarization, interaction with collagen fibers and locomotion. Eur. J. Immunol. 28:2331–2343. [DOI] [PubMed] [Google Scholar]
- Friedl, P., S. Borgmann, and E.B. Bröcker. 2001. Leukocyte crawling through extracellular matrix and the Dictyostelium paradigm of movement: lessons from a social ameba. J. Leukoc. Biol. 70:491–509. [PubMed] [Google Scholar]
- Gunzer, M., A. Schäfer, S. Borgmann, S. Grabbe, K.S. Zänker, E.-B. Bröcker, E. Kämpgen, and P. Friedl. 2000. Antigen presentation in three-dimensional extracellular matrix: interactions of T cells with dendritic cells are dynamic, short lived, and sequential. Immunity. 13:323–332. [DOI] [PubMed] [Google Scholar]
- Hiraoka, N., E. Allen, I.J. Apel, M.R. Gyetko, and S.J. Weiss. 1998. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 95:365–377. [DOI] [PubMed] [Google Scholar]
- Hofmann, U.B., J.R. Westphal, E.T. Waas, A.J. Zendman, I.M. Cornelissen, D.J. Ruiter, and G.N. van Muijen. 1999. Matrix metalloproteinases in human melanoma cell lines and xenografts: increased expression of activated matrix metalloproteinase-2 (MMP-2) correlates with melanoma progression. Br. J. Cancer. 81:774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi, O., Y. Mori, T. Kurokawa, and M. Kumegawa. 1999. Breast cancer cells express cathepsins B and L but not cathepsins K or H. Cancer Biochem. Biophys. 17:69–78. [PubMed] [Google Scholar]
- Killich, T., P.J. Plath, X. Wei, H. Bultmann, L. Rensing, and M.G. Vicker. 1993. The locomotion, shape and pseudopodial dynamics of unstimulated Dictyostelium cells are not random. J. Cell Sci. 106:1005–1013. [DOI] [PubMed] [Google Scholar]
- Konttinen, Y.T., M. Ainola, H. Valleala, J. Ma, H. Ida, J. Mandelin, R.W. Kinne, S. Santavirta, T. Sorsa, C. Lopez-Otin, and M. Takagi. 1999. Analysis of 16 different matrix metalloproteinases (MMP-1 to MMP-20) in the synovial membrane: different profiles in trauma and rheumatoid arthritis. Ann. Rheum. Dis. 58:691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger, A., R. Soeltl, I. Sopov, C. Kopitz, M. Arlt, V. Magdolen, N. Harbeck, B. Gansbacher, and M. Schmitt. 2001. Hydroxamate-type matrix metalloproteinase inhibitor batimastat promotes liver metastasis. Cancer Res. 61:1272–1275. [PubMed] [Google Scholar]
- Kurschat, P., P. Zigrino, R. Nischt, K. Breitkopf, P. Steurer, C.E. Klein, T. Krieg, and C. Mauch. 1999. Tissue inhibitor of matrix metalloproteinase-2 regulates matrix metalloproteinase-2 activation by modulation of membrane-type 1 matrix metalloproteinase activity in high and low invasive melanoma cell lines. J. Biol. Chem. 274:21056–21062. [DOI] [PubMed] [Google Scholar]
- Langholz, O., D. Rockel, C. Mauch, E. Kozlowska, I. Bank, T. Krieg, and B. Eckes. 1995. Collagen and collagenase gene expression in three-dimensional collagen lattices are differentially regulated by α1β1 and α2β1 integrins. J. Cell Biol. 131:1903–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger, D.A., and A.F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell. 84:359–369. [DOI] [PubMed] [Google Scholar]
- Laurent, V., and M. Salzet. 1995. Isolation of a renin-like enzyme from the leech Theromyzon tessulatum. Peptides. 16:1351–1358. [DOI] [PubMed] [Google Scholar]
- Lewis, W.H. 1934. On the locomotion of the polymorphonuclear neutrophiles of the rat in autoplasma cultures. Bull. Johns Hopkins Hosp. 4:273–279. [Google Scholar]
- Lochter, A., S. Galosy, J. Muschler, N. Freedman, Z. Werb, and M.J. Bissell. 1997. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 139:1861–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maaser, K., K. Wolf, C.E. Klein, B. Niggemann, K.S. Zanker, E.B. Brocker, and P. Friedl. 1999. Functional hierarchy of simultaneously expressed adhesion receptors: integrin α2β1 but not CD44 mediates MV3 melanoma cell migration and matrix reorganization within three-dimensional hyaluronan-containing collagen matrices. Mol. Biol. Cell. 10:3067–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti, S., J.B. Shear, R.M. Williams, W.R. Zipfel, and W.W. Webb. 1997. Measuring serotonin distribution in live cells with three-photon excitation. Science. 275:530–532. [DOI] [PubMed] [Google Scholar]
- Mazo, I.B., J.C. Gutierrez-Ramos, P.S. Frenette, R.O. Hynes, D.D. Wagner, and U.H. von Andrian. 1998. Hematopoietic progenitor cell rolling in bone marrow microvessels: parallel contributions by endothelial selectins and vascular cell adhesion molecule 1. J. Exp. Med. 188:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch, D.R., M. Harvey, and A.C. Herington. 2000. The expression of the ADAMs proteases in prostate cancer cell lines and their regulation by dihydrotestosterone. Mol. Cell. Endocrinol. 167:11–21. [DOI] [PubMed] [Google Scholar]
- Mignatti, P., E. Robbins, and D.B. Rifkin. 1986. Tumor invasion through the human amniotic membrane: requirement for a proteinase cascade. Cell. 47:487–498. [DOI] [PubMed] [Google Scholar]
- Montcourrier, P., P.H. Mangeat, G. Salazar, M. Morisset, A. Sahuquet, and H. Rochefort. 1990. Cathepsin D in breast cancer cells can digest extracellular matrix in large acidic vesicles. Cancer Res. 50:6045–6054. [PubMed] [Google Scholar]
- Murphy, G., and J. Gavrilovic. 1999. Proteolysis and cell migration: creating a path? Curr. Opin. Cell Biol. 11:614–621. [DOI] [PubMed] [Google Scholar]
- Nakahara, H., L. Howard, E.W. Thompson, H. Sato, M. Seiki, Y. Yeh, and W.T. Chen. 1997. Transmembrane/cytoplasmic domain-mediated membrane type 1-matrix metalloprotease docking to invadopodia is required for cell invasion. Proc. Natl. Acad. Sci. USA. 94:7959–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisbet, A.J., and P.F. Billingsley. 1999. Hydrolytic enzymes of Psoroptes cuniculi (Delafond). Insect Biochem. Mol. Biol. 29:25–32. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntayi, C., S. Lorimier, O. Berthier-Vergnes, W. Hornebeck, and P. Bernard. 2001. Cumulative influence of matrix metalloproteinase-1 and -2 in the migration of melanoma cells within three-dimensional type I collagen lattices. Exp. Cell Res. 270:110–118. [DOI] [PubMed] [Google Scholar]
- Ohuchi, E., K. Imai, Y. Fujii, H. Sato, M. Seiki, and Y. Okada. 1997. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 272:2446–2451. [DOI] [PubMed] [Google Scholar]
- Palecek, S.P., J.C. Loftus, M.H. Ginsberg, D.A. Lauffenburger, and A.F. Horwitz. 1997. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 385:537–540. [DOI] [PubMed] [Google Scholar]
- Redfield, A., M.T. Nieman, and K.A. Knudsen. 1997. Cadherins promote skeletal muscle differentiation in three-dimensional cultures. J. Cell Biol. 138:1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassi, M.L., H. Eriksen, L. Risteli, S. Niemi, J. Mansell, M. Gowen, and J. Risteli. 2000. Immunochemical characterization of assay for carboxy-terminal telopeptide of human type I collagen: loss of antigenicity by treatment with cathepsin K. Bone. 26:367–373. [DOI] [PubMed] [Google Scholar]
- Schor, S.L., T.D. Allen, and B. Winn. 1983. Lymphocyte migration into three-dimensional collagen matrices: a quantitative study. J. Cell Biol. 96:1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz, M.P., D.P. Felsenfeld, and C.G. Galbraith. 1998. Cell migration: regulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol. 8:51–54. [DOI] [PubMed] [Google Scholar]
- Sternlicht, M.D., and Z. Werb. 2001. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17:463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler-Stevenson, W.G., S. Aznavoorian, and L.A. Liotta. 1993. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 9:541–573. [DOI] [PubMed] [Google Scholar]
- Wei, Y., M. Lukashev, D.I. Simon, S.C. Bodary, S. Rosenberg, M.V. Doyle, and H.A. Chapman. 1996. Regulation of integrin function by the urokinase receptor. Science. 273:1551–1555. [DOI] [PubMed] [Google Scholar]
- Welch, M.P., G.F. Odland, and R.A. Clark. 1990. Temporal relationships of F-actin bundle formation, collagen and fibronectin matrix assembly, and fibronectin receptor expression to wound contraction. J. Cell Biol. 110:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumura, S., H. Mori, and Y. Fukui. 1984. Localization of actin and myosin for the study of ameboid movement in Dictyostelium using improved immunofluorescence. J. Cell Biol. 99:894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker, S., J. Cao, and W.T. Chen. 2000. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 19:6642–6650. [DOI] [PubMed] [Google Scholar]