

Abstract
Newly synthesized proteins that do not fold correctly in the ER are targeted for ER-associated protein degradation (ERAD) through distinct sorting mechanisms; soluble ERAD substrates require ER-Golgi transport and retrieval for degradation, whereas transmembrane ERAD substrates are retained in the ER. Retained transmembrane proteins are often sequestered into specialized ER subdomains, but the relevance of such sequestration to proteasomal degradation has not been explored. We used the yeast Saccharomyces cerevisiae and a model ERAD substrate, the cystic fibrosis transmembrane conductance regulator (CFTR), to explore whether CFTR is sequestered before degradation, to identify the molecular machinery regulating sequestration, and to analyze the relationship between sequestration and degradation. We report that CFTR is sequestered into ER subdomains containing the chaperone Kar2p, and that sequestration and CFTR degradation are disrupted in sec12 ts strain (mutant in guanine-nucleotide exchange factor for Sar1p), sec13 ts strain (mutant in the Sec13p component of COPII), and sec23 ts strain (mutant in the Sec23p component of COPII) grown at restrictive temperature. The function of the Sar1p/COPII machinery in CFTR sequestration and degradation is independent of its role in ER-Golgi traffic. We propose that Sar1p/COPII-mediated sorting of CFTR into ER subdomains is essential for its entry into the proteasomal degradation pathway. These findings reveal a new aspect of the degradative mechanism, and suggest functional crosstalk between the secretory and the degradative pathways.
Keywords: ER sorting; proteasomal degradation; CFTR; ERAD; yeast
Introduction
Quality control of newly synthesized membrane proteins occurs in the ER (Ellgaard and Helenius, 2001). Incorrectly folded proteins are targeted for ER-associated protein degradation (ERAD)* and are eliminated by the ubiquitin-proteasome pathway (Brodsky and McCracken, 1999). Because proteasomal degradation occurs in the cytoplasm, ERAD substrates must be retrotranslocated from the ER to the cytoplasm before digestion. The cellular machinery involved in the sorting and translocation of ERAD substrates is under active investigation. One of the key requirements is a fully competent early secretory pathway because mutations in proteins involved in ER-Golgi traffic cause disorganization of ER structure and inhibit ERAD (Taxis et al., 2002). The Sec61p/Sec63p translocon (Plemper et al., 1997) and auxiliary proteins Der1p (Knop et al., 1996), Der3p/Hrd1p (Bordallo and Wolf, 1999), and Hrd3 (Hampton et al., 1996) have been shown to be required for retrotranslocation of ERAD substrates. In addition, a subset of molecular chaperones such as calnexin (McCracken and Brodsky, 1996), BiP (Plemper et al., 1997; Nishikawa et al., 2001), Hsp70 (Zhang et al., 2001), and p97 (Rabinovich et al., 2002) participate in selecting ERAD substrates. Evidence indicates that two distinct mechanisms participate in directing different substrates for ERAD (Vashist et al., 2001). Soluble ERAD substrates such as carboxy peptidase Y (CPY), PrA, and KHN require functional transport between the ER and the Golgi (Caldwell et al., 2001; Vashist et al., 2001), and the degradation of these proteins is delayed when ER-Golgi traffic is inhibited in sec18–1 and sec12–4 strains. Additional components encoded by PER17 (Vashist et al., 2001), ERV14, and ERV29 (Caldwell et al., 2001) appear essential in degradation of soluble ERAD substrates, and appear to function by regulating ER-Golgi transport of specific soluble proteins. In contrast, the degradation of membrane ERAD substrates such as mutant forms of Ste6p, Yor1p, Sec61p, Vph1p, and cystic fibrosis transmembrane conductance regulator (CFTR) seem unaffected by sec18–1, sec12–4, sec21–1, per17–1, erv14Δ, or erv29Δ mutations (Loayza et al., 1998; Katzmann et al., 1999; Caldwell et al., 2001; Kiser et al., 2001; Vashist et al., 2001). These findings suggest that transmembrane ERAD substrates do not enter post-ER compartments of the secretory pathway before degradation. In agreement, soluble ERAD substrates can be detected in post-ER transport vesicles, whereas transmembrane ERAD substrates are excluded (Vashist et al., 2001).
The exact pathway for degradation of transmembrane ERAD substrates from the ER is unclear, but there is evidence to suggest that at least some retained proteins are sorted into specialized ER subdomains before degradation (Kamhi-Nesher et al., 2001; Kiser et al., 2001; Zhang et al., 2001). It is unknown what molecular mechanisms regulate sequestration of ERAD substrates, and whether such sequestration is required for ER retrotranslocation and proteasomal degradation. To explore these questions, we used the yeast Saccharomyces cerevisiae and mutant strains defective in the Sar1p/COPII sorting machinery and a known ERAD substrate, CFTR, as our model system.
Sar1p/COPII-mediated selection of ER proteins for secretory traffic is one of the best understood mechanisms for selective recruitment of proteins in the ER (Antonny and Schekman, 2001). COPII sorting is initiated by Sar1p, a small GTPase (Veldhuisen et al., 1997) that is activated by Sec12p-mediated guanine-nucleotide exchange. Sar1p in its GTP-bound state associates with the ER membrane and recruits the Sec23p/Sec24p–COPII complex and subsequently the Sec13p/Sec31p–COPII complex to the membrane (for review see Barlowe, 2000). The assembly of the COPII coat on ER budding structures is coupled to the selection of transmembrane proteins into the nascent budding structures that will eventually bud from the ER as transport vesicles. COPII function has been examined to date exclusively in the context of secretory traffic.
CFTR is a chloride channel present on the apical surface of epithelial cells lining the respiratory, intestinal, and exocrine tissues (Kirk, 2000). In mammalian cells, only ∼20% of newly synthesized wild-type CFTR folds correctly and is transported from the ER to the plasma membrane through the secretory pathway, whereas the remaining ∼80% of CFTR is degraded from the ER through the ubiquitin-proteasome pathway (Jensen et al., 1995; Ward et al., 1995; Moyer et al., 1998; Riordan, 1999). In mammalian cells, CFTR is not sequestered into ER subdomains before degradation because inhibition of proteasomal activity by chemical inhibitors or overtaxing the proteolytic capacity by overexpressing CFTR leads to retrotranslocation of CFTR from the ER and its accumulation in cytosolic aggresomes (Johnston et al., 1998). In yeast, the majority (if not all) of newly synthesized CFTR is also degraded through the ubiquitin-proteasome pathway, but in contrast to mammalian cells, CFTR in yeast is not delivered to aggresomes, but appears sequestered in ER subdomains before degradation (Kiser et al., 2001; Zhang et al., 2001).
Here, we report that the Sar1p/COPII machinery functions in sorting CFTR into ER subdomains before proteasomal degradation, and that such sorting is required for CFTR entry into the degradative pathway. This function of Sar1p/COPII does not involve ER-Golgi traffic. Our results support a model in which the Sar1p/COPII machinery participates in sorting proteins to both the anterograde secretory pathway and the degradative pathway. These findings raise the possibility that all newly synthesized proteins are subjected to a Sar1p/COPII sorting mechanism, irrespective of their ultimate secretory or degradative fate.
Results and discussion
GFP-tagged CFTR is a bona-fide ERAD substrate in yeast
To provide a substrate for our studies, we generated a construct containing EGFP fused in frame to the NH2 terminus of human CFTR in a yeast expression vector under a copper-inducible promoter. As shown in Fig. 1 A, EGFP-CFTR is detected after immunoprecipitation with anti-CFTR antibody and anti-GFP antibody after copper induction of wild-type yeast carrying the EGFP-CFTR plasmid (Fig. 1 A, lanes 1 and 4), but not from yeast carrying an empty vector (Fig. 1 A, lane 3). EGFP-CFTR is not seen in a sample precipitated with anti-F1β antibody. The mobility of EGFP-CFTR (∼175 kD) is appropriate for a chimera of EGFP (∼27 kD) and full-length human CFTR (∼145 kD).
Figure 1.
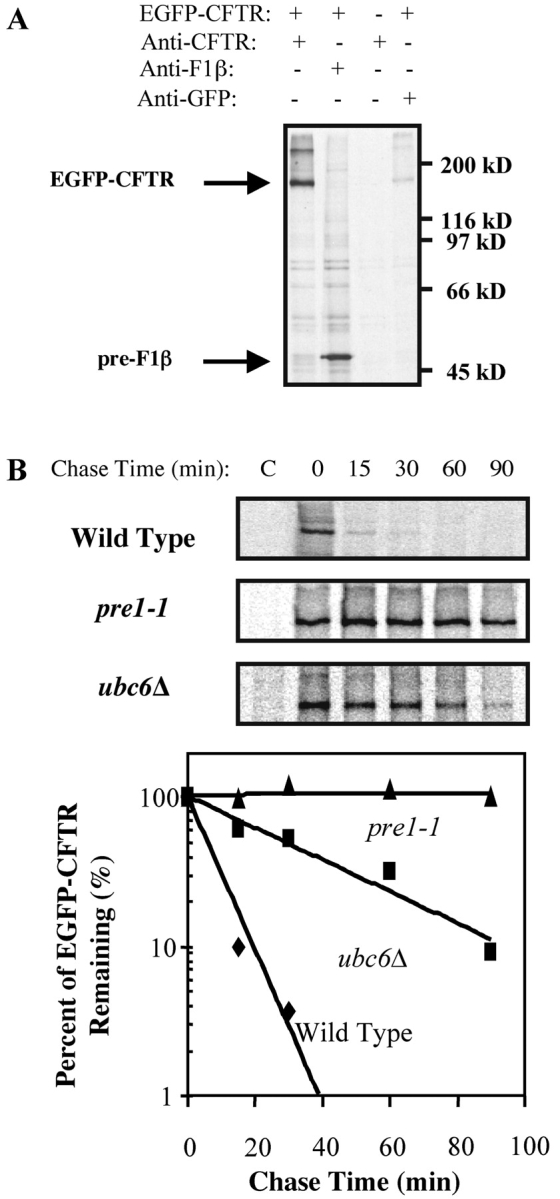
EGFP-CFTR represents yeast ERAD substrate. (A) Yeast transformed with pCU426CUP1 (EGFP-CFTR, − lane) or pCU426CUP1/EGFP-CFTR (EGFP-CFTR, + lanes) were induced, labeled with [35S]methionine for 20 min, and lysed. Lysates were immunoprecipitated with anti-CFTR, anti-F1β, or anti-GFP pAbs. A band of 175 kD, appropriate for a chimera of EGFP and CFTR, can be immunoprecipitated with anti-CFTR and anti-GFP antibodies from cells expressing EGFP-CFTR. (B) Wild-type, pre1–1, and ubc6Δ yeast were transformed with pCU426CUP1/EGFP-CFTR, induced, and pulse-labeled with [35S]methionine for 20 min. An equal amount of culture was taken at each chase time, lysed, and the lysates were immunoprecipitated with anti-CFTR antibody. Relative intensities of EGFP-CFTR bands were quantitated. EGFP-CFTR is stabilized in pre1–1 and ubc6Δ strains.
To ensure that EGFP-CFTR behaves as an ERAD substrate, we examined its degradation rate in mutant strains lacking either a functional proteasome subunit (pre1–1) or an ubiquitin-conjugation enzyme (ubc6Δ). Both proteins have been previously shown to be required for degradation of CFTR tagged at the COOH terminus with GFP (Kiser et al., 2001) or HA (Zhang et al., 2001) in yeast. As shown in Fig. 1 B, EGFP-CFTR in wild-type yeast is digested rapidly, with a half-life of ∼10–15 min (Fig. 1 B), slightly more rapidly than previously published (∼20 min, Kiser et al., 2001; ∼30 min, Zhang et al., 2001). All CFTR appears to be degraded in yeast (Kiser et al., 2001; Zhang et al., 2001), as opposed to mammalian cells where ∼20% is transported to the plasma membrane. It is possible that yeast does not have sufficient machinery to efficiently fold overexpressed CFTR, or that even correctly folded CFTR is “read” by yeast as defective. In contrast to rapid degradation in wild-type yeast, EGFP-CFTR remains stable in pre1–1 mutant yeast even after 90 min of chase, and the half-life of EGFP-CFTR in the ubc6Δ strain is extended to ∼40 min. Because our EGFP-CFTR construct is degraded through the ubiquitin-proteasome pathway in yeast, we use it as an ERAD substrate to test Sar1p/COPII involvement in proteasomal targeting.
EGFP-CFTR is sorted to subcompartments of ER before degradation
Previous studies have shown that CFTR tagged at the COOH terminus with GFP (Kiser et al., 2001) or HA (Zhang et al., 2001) is degraded as an integral membrane protein and colocalizes with the ER chaperone Kar2p in punctate ER structures before degradation. To analyze our EGFP-CFTR construct, we first tested its association with membranes. As shown in Fig. 2 A, EGFP-CFTR is detected exclusively in the membrane pellet fraction after medium speed centrifugation, indicating that it is degraded as a membrane-associated form.
Figure 2.
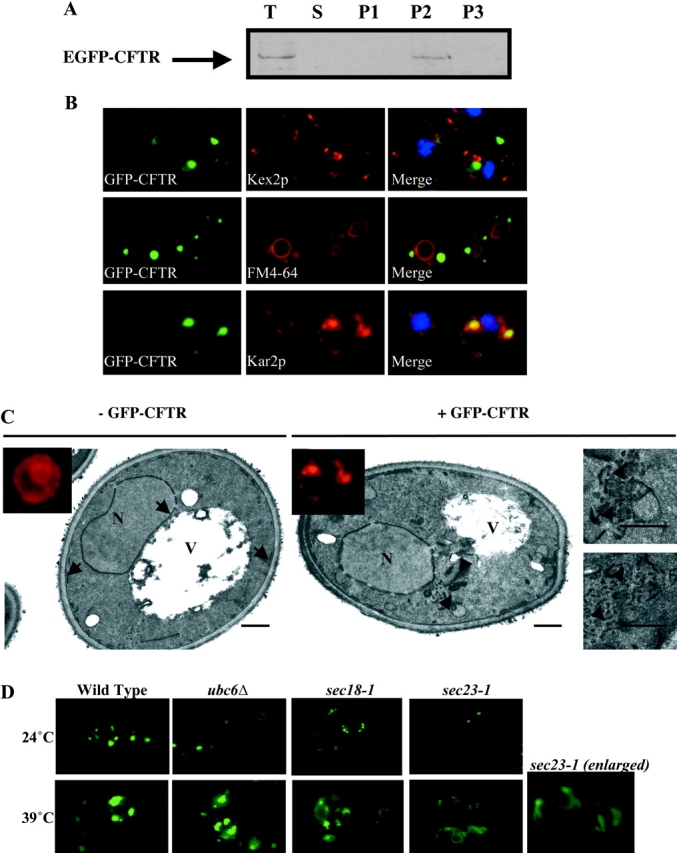
EGFP-CFTR localization depends on functional Sar1p/COPII machinery. (A) Yeast expressing EGFP-CFTR was subjected to membrane fractionation. A sample of each fraction was processed by SDS-PAGE, transferred to nitrocellulose membrane, and immunoblotted with anti-CFTR antibody. T; total lysate. S; supernatant after high speed centrifugation. P1, P2, P3; pellets after low speed, medium speed, and high speed centrifugation, respectively. EGFP-CFTR is recovered exclusively in the membrane fraction. (B) Wild-type yeast expressing EGFP-CFTR was grown to log phase and processed for immunofluorescence using anti-Kar2p or anti-Kex2p antibodies, or was incubated with the fluorescent dye FM 4–64 for 45 min at 0°C, followed by 1 h at 24°C to stain the vacuole. EGFP-CFTR colocalizes only with the ER marker Kar2p. (C) Wild-type yeast transformed with pCU426CUP1 (− EGFP-CFTR) or with pCU426CUP1/EGFP-CFTR (+ EGFP-CFTR) was grown to log phase, induced, and processed for immunofluorescence with anti-Kar2p antibodies (insets) and for electron microscopy. Kar2p shows typical ER localization in − EGFP-CFTR cells, but distributes to punctate structures in + EGFP-CFTR cells. Cells without EGFP-CFTR contain normal ER (arrows), but cells expressing EGFP-CFTR show accumulation of membranous elements (arrowheads). Bar, 0.5 μm. (D) Wild-type, ubc6Δ, sec18–1 ts, and sec23–1 ts yeast transformed with pCU426CUP1/EGFP-CFTR was grown to log phase and induced at permissive (24°C) or restrictive (39°C) temperature. Live yeast were then imaged by fluorescence microscopy. EGFP-CFTR localizes to ER subdomains in wild-type, ubc6Δ, and sec18–1 ts yeast grown at permissive or restrictive temperatures. EGFP-CFTR localizes to ER subdomains in sec23–1 ts yeast grown at permissive temperature, but is diffusely distributed throughout the ER in sec23–1 ts yeast grown under restrictive temperature.
We performed immunofluorescence with known organellar markers to localize EGFP-CFTR. As shown in Fig. 2 B, EGFP-CFTR is not detected in small punctate structures containing the late Golgi marker Kex2p (Preuss et al., 1992). This result agrees with findings that CFTR expressed in yeast undergoes ER-type N-glycosylation, but does not undergo Golgi-type oligosaccharide modifications (Kiser et al., 2001). EGFP-CFTR is not detected on the plasma membrane, and does not colocalize with the fluorescent dye FM 4–64 (Fig. 2 B) shown to be transported to endosomal (unpublished data) or vacuolar compartments of yeast as a function of incubation time and temperature (Vida and Emr, 1995; the minimal colocalization in the vacuole is due to bleed-through from the strong EGFP-CFTR signal and is not visible in cells without EGFP-CFTR). The lack of vacuolar localization is consistent with lack of degradation of CFTR via the Pep4p/Prb1p vacuolar proteases (Kiser et al., 2001; Gelman et al., 2002).
EGFP-CFTR is detected in relatively large punctate structures that contain Kar2p (Fig. 2 B). The punctate Kar2p distribution in EGFP-CFTR–expressing yeast is distinct from its normal cage-like perinuclear localization in control yeast (Paddon et al., 1996). The effects of EGFP-CFTR expression on intracellular morphology were analyzed by electron microscopy of yeast transformed with either empty or EGFP-CFTR–containing plasmids. As shown in Fig. 2 C, normal nuclear membrane and sub–plasma membrane ER structures are evident in the control cell (arrows), in agreement with published images (Zhang et al., 2001). In contrast, EGFP-CFTR cell shows clusters of membranous structures in proximity to the perinuclear ER (arrowheads). Higher magnification shows them to be accumulations of tubular and vesicular elements, morphologically analogous to ER extensions observed in yeast expressing CFTR (Zhang et al., 2001). The relative size of such clusters (average diameter ∼0.5–1 μm) corresponds to the approximate size of the fluorescent Kar2p/EGFP-CFTR puncta. Significantly, membrane amplification induced by CFTR (Zhang et al., 2001) or EGFP-CFTR (this work) is distinct from multi-layered ER “karmellae” induced by expression of HMG-CoA reductase (Koning et al., 1996). Together, the data suggest that EGFP-CFTR localizes to subdomains of the ER network that represent penultimate stations before degradation. The results also imply that sequestration into the ER domains is more efficient than subsequent removal by degradation.
Functional Sar1p/COPII machinery is required for sorting EGFP-CFTR to ER subdomains
To determine if the Sar1p/COPII machinery participates in sorting EGFP-CFTR, we examined its localization when COPII function is disrupted. First, we tested the localization of EGFP-CFTR in wild-type, ubc6Δ, and sec18–1 yeast strains when grown at 24°C (permissive temperature for strains defective in COPII components) and when grown at 39°C (restrictive temperature for strains defective in COPII components). As shown in Fig. 2 D, EGFP-CFTR in the wild-type strain remains as punctate spots at both temperatures. Similarly, the pattern of EGFP-CFTR in the ubc6Δ strain defective for proteasomal degradation is analogous to that in the wild-type strain. This strongly suggests that EGFP-CFTR is sorted into the punctate structure and maintained there before it is targeted for ubiquitination and degradation. Our data, together with the finding that CFTR remains sequestered in Kar2p-containing subdomains in yeast strain (pre1–1pre2–2) defective for proteasomal degradation (Zhang et al., 2001), suggest that the catalytic activity of ubiquitin-conjugating enzyme Ubc6p is not sufficient to remove EGFP-CFTR from ER subdomains, and that ubiquitination and proteasomal degradation are tightly coupled. The same punctate EGFP-CFTR pattern is observed in sec18–1 strain grown at either temperature, indicating that block in vesicular fusion during ER-Golgi protein transport does not significantly affect EGFP-CFTR sequestration.
Analysis of EGFP-CFTR localization in the sec23–1 ts strain (Kuehn et al., 1998) showed significant temperature-dependent changes (Fig. 2 D). At the permissive temperature, EGFP-CFTR is sequestered into ER subdomains, but at the restrictive temperature, EGFP-CFTR is diffusely distributed throughout the ER in a pattern characteristic of ER proteins (compare enlargement with Kar2p staining in inset; Fig. 2 C). Similar results were obtained when yeast strains mutant in other components of the Sar1p/COPII machinery (described below) were analyzed (unpublished data).
Functional Sar1p/COPII machinery is required for EGFP-CFTR degradation
To uncover whether the Sar1p/COPII machinery is also involved in CFTR degradation, we compared the degradation rate of EGFP-CFTR at permissive (24°C) and restrictive (39°C) temperature in yeast mutant in Sar1p/COPII components. In wild-type strain, EGFP-CFTR is degraded rapidly at both temperatures, with a half-life of 10–15 min (Fig. 3 A). The temperature-sensitive strain, sec12–4 ts, is defective in catalyzing GDP/GTP exchange on Sar1p at the restrictive temperature due to a P73L mutation (Barlowe and Schekman, 1993). EGFP-CFTR is degraded rapidly in the sec12–4 ts strain at permissive temperature; with a half-life (∼10 min) analogous to that in wild-type strain (Fig. 3 B). In contrast, the degradation rate is delayed significantly when the yeast is shifted to the restrictive temperature, with the half-life extending to 40–45 min, a threefold increase compared with the permissive temperature. The temperature-sensitive strains sec23–1 ts and sec13–1 ts (Salama et al., 1997) are defective at the restrictive temperature due to an S382L mutation (Yoshihisa et al., 1993), and a mutation that has not yet been identified, respectively. In both cases, COPII function is compromised. EGFP-CFTR is degraded rapidly in both sec23–1 ts and sec13–1 ts strains at the permissive temperature, with half-lives of <15 min (Fig. 3, C and D). In contrast, the degradation rates are significantly delayed in both strains at the restrictive temperatures, with a half-life of ∼50 min. The significant inhibition of EGFP-CFTR degradation suggests that functional COPII machinery is required for proteasomal degradation of EGFP-CFTR.
Figure 3.
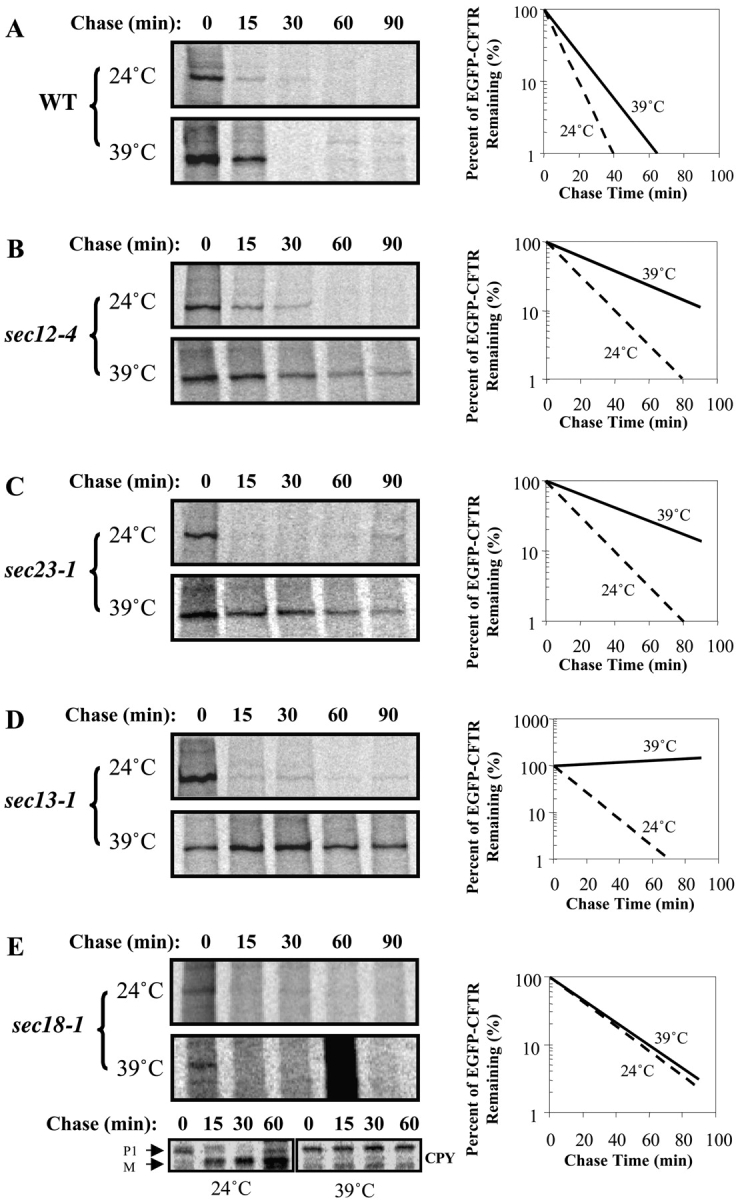
EGFP-CFTR degradation depends on functional Sar1p/COPII machinery. Wild-type (A), sec12–4 ts (B), sec23–1 ts (C), sec13–1 ts (D), and sec18–1 ts (E) yeast were transformed with pCU426CUP1/EGFP-CFTR. Yeast were grown to log phase and induced at permissive temperature. An equal amount of culture was then pulse-labeled with [35S]methionine for 20 min at either 24°C or 39°C. An equal amount of culture was taken after indicated chase times and used to prepare cell lysates. Lysates were immunoprecipitated with anti-CFTR or anti-CPY (for sec18–1 ts) antibody. Relative intensities of EGFP-CFTR bands were quantitated. EGFP-CFTR degradation is significantly inhibited by inactivation of Sec12p, Sec23p, and Sec13p.
It must be stressed that EGFP-CFTR, like other transmembrane ERAD substrates, does not enter the ER-Golgi recycling degradative pathway. Like the previously characterized CFTR (Kiser et al., 2001), EGFP-CFTR is degraded with normal kinetics at permissive and restrictive temperatures in a sec18–1 strain (Fig. 3 E). The strain is defective in Sec18p function, as shown by a block in maturation of the vacuolar CPY at the restrictive temperature (Fig. 3 E). Together, the results indicate that ER-Golgi transport and recycling are not required for EGFP-CFTR degradation. Our findings suggest a novel role for the Sar1p/COPII sorting machinery. In addition to selecting correctly folded substrates into the forward secretory pathway (Schekman and Orci, 1996; Bannykh et al., 2000) and selecting misfolded soluble ERAD substrates for the ER-Golgi recycling degradative pathway (Vashist et al., 2001), Sar1p/COPII also sorts misfolded substrates into subdomains of the ER from which they are subsequently degraded. How does the same machinery sort proteins into three distinct pathways? A common sorting mechanism for both correctly folded and misfolded membrane proteins presents a spatial problem because these classes must occupy distinct regions of the ER. It is likely that additional targeting mechanisms coordinate with Sar1p/COPII to direct the proteins to ER exit or retention. It appears that a primary distinction is made at the level of packaging into COPII vesicles; although correctly folded soluble and transmembrane proteins for secretory traffic and misfolded soluble ERAD substrates for ER-Golgi recycling degradative traffic are sorted into COPII vesicles, misfolded transmembrane ERAD substrates are excluded (Vashist et al., 2001). There is evidence that transmembrane ERAD substrates are prevented from entry into COPII vesicles and are retained in the ER through the action of luminal ER proteins. For example, mutants of the transmembrane ATPase Pma1p appear to be retained and degraded in the ER through the action of a protein disulfide isomerase–related protein, Eps1p (Wang and Chang, 1999). Similarly, the retention of a transmembrane H2a precursor in the ER is determined by a pentapeptide sequence present on the luminal side of the ER (Shenkman et al., 1997). In addition, ERAD substrates can be retained by binding to calnexin. It has been shown that calnexin binds to wild-type and folding compromised ΔF508CFTR in the ER, but the association with ΔF508CFTR is prolonged and might facilitate ER retention (Pind et al., 1994). ERAD substrates can be also retained by association with the translocon (Loayza et al., 1998; Kamhi-Nesher et al., 2001). Cytoplasmic chaperones may also participate in ER retention (Brodsky et al., 1999). The finding that strains inactivated in the cytosolic Hsp70 Ssa1p do not sort CFTR into ER subdomains supports the view that ER sequestration requires the coordinate action of Sar1p/COPII and accessory factors.
That Sar1p/COPII is likely to participate in the secretory and the degradative pathways is supported by data obtained from whole genome microarray analysis where many genes involved in secretory traffic were found to be UPR targets (Travers et al., 2000). Initially, the up-regulation was viewed as means to increase traffic in the ER-Golgi recycling degradative pathway for soluble ERAD substrates (Caldwell et al., 2001). However, our work suggests that the up-regulation is also a means to increase components required for ER sequestration of transmembrane ERAD substrates. Specifically relevant to our analysis is the up-regulation of SEC12, SEC13, and SEC24. Thus, it may be that the UPR-mediated up-regulation of these genes may not be a cellular strategy to remove protein from the ER by exporting them, but additionally might promote sorting of retained proteins toward degradation.
Materials and methods
Antibodies and plasmids
Anti-CFTR NBD1 pAb was described previously (Bebok et al., 1998) and was a gift from Dr. David Bedwell (University of Alabama at Birmingham, Birmingham, AL). Anti-Kar2p pAb was a gift from Dr. Jeffrey Brodsky (University of Pittsburgh, Pittsburgh, PA). Anti-Kex2p mAb was a gift from Dr. Vytas Bankaitis (University of North Carolina at Chapel Hill, Chapel Hill, NC). Anti–COOH-terminal CFTR mAb was purchased from USBiological. Anti-CPY antibody was purchased from Molecular Probes, Inc. Goat anti–rat and anti–mouse antibodies conjugated with fluorescein isothiocyanate or rhodamine were purchased from Jackson ImmunoResearch Laboratories.
To generate an inducible construct expressing GFP-tagged CFTR in yeast, pEGFP-CFTR plasmid (Moyer et al., 1998) was first digested with SacII, and then treated with Klenow fragment followed by digestion with NheI. The pRSETB/EGFP plasmid (provided by Dr. David Bedwell) was first digested with HindIII and then treated with Klenow fragment followed by digestion with NheI. The EGFP-containing fragment from pRSETB/EGFP and CFTR-containing fragment from pEGFP-CFTR were gel-purified, ligated, and transformed into Eschericia coli. The resulting plasmid was then digested with NheI and EcoRV, and treated with Klenow fragment. The EGFP-CFTR–containing fragment was gel-purified and cloned into the SmaI site of pCU426CUP1 (Labbe and Thiele, 1999). The final construct was sequenced to ensure the correct open reading frame.
Yeast strains
CTY182 (MATa, ura3–52, Δhis3–200, lys2–80), CTY252 (MATa, ura3–52, sec12–4 ts), CTY253 (MATa, ura3–52, sec13–1 ts), and CTY260 (MATα, ura3–52, leu2–3, 112, sec23–1 ts) were a gift from Dr. Vytas Bankaitis. YHI29/W (MATa, ura3, leu2–3, 112, his3–11, 15, cans, GAL1), YHI29/1 (MATa, pre1–1, ura3, leu2–3, 112, his3–11, 15, cans, GAL1), and YWO0346 (MATα, ura3–1, leu2–3, 112, his3–11, 15, ade2–1 ocre, trp1–1, can1–100, ubc6::LEU2) were a gift from Dr. Dieter Wolf (Universitat Stuttgart, Stuttgart, Germany). SEY6210 (MATα ura3–52 leu2–3, 112 his3-Δ200 trp1-Δ901 lys2–801 suc2-Δ9) and SEY5186 (MATα sec18–1 ura3–52 leu2–3, 112 GAL +) were a gift from Dr. David Bedwell.
Yeast media were prepared as described previously (Rose et al., 1990). Unless specified, in all experiments, cultures were grown for a minimum of 5–6 generations to an A600 of no more than 1.0.
Fractionation, immunofluorescence, and electron microscopy
Yeast expressing EGFP-CFTR was fractionated as described previously (Paddon et al., 1996). In brief, cells grown to exponential phase were converted to spheroplasts using yeast lytic enzyme (ICN Biomedicals) in the presence of 20 mM potassium phosphate buffer, pH 7.0, and containing 1.2 M sorbitol. The spheroplasts were washed and disrupted using a Dounce homogenizer in 25 mM Hepes-NaOH buffer, pH 7.4, plus protease inhibitors (Sigma-Aldrich). Cell lysates were centrifuged at 4°C at 500 g for 10 min, and the resulting supernatant was spun at 25,000 g for 15 min. The resulting supernatant was centrifuged at 150,000 g for 1 h. Pellet from each spin was washed with Hepes buffer and saved as P1, P2, and P3, respectively. Protein concentration was measured using a Detergent Compatible Protein Assay kit (Bio-Rad Laboratories).
For immunofluorescent studies, yeast were grown in synthetic complete medium lacking uracil to an OD600 of no more than 1.0. Yeast were fixed by adding formaldehyde to 3% and incubating at either 24°C or 39°C for 30 min. Cells were harvested and washed twice with 0.1 M potassium phosphate, pH 6.5, and once with potassium phosphate buffer containing 1.2 M sorbitol. The cell walls were digested by adding β-mercaptoethanol to 0.1% and zymolase 20T (U.S. Biological) to 20 μg/ml. After incubating at 30°C for 1 h, cells were washed twice with sorbitol buffer, applied to polylysine-coated coverslips, and were allowed to settle at RT for 20 min. The cells were refixed with 3% formaldehyde for 5 min followed by quenching with 50 mM of NH4Cl for 5 min. Cells were permeabilized with 0.1% Triton X-100 for 10 min and washed twice for 5 min each with blocking buffer (PBS containing 0.2% Tween 20 and 1 mg/ml BSA). Cells were incubated overnight with primary antibody diluted 1:5,000 for anti-Kar2p antibody and 1:2,000 for anti-Kex2p antibody. Coverslips were washed three times with blocking buffer. Cells were incubated with secondary antibody for 1 h and washed as above. Coverslips were mounted on slides in 9:1 glycerol/PBS with 0.1% p-phenylenediamine. Images were acquired with an inverted microscope (Axiovert 30; Carl Zeiss MicroImaging Inc.). IpLab Spectrum software (Signal Analytics Corp.) was used to control image acquisition.
Yeast containing either an empty plasmid or EGFP-CFTR–containing plasmid were grown to an A600 of ∼0.5, induced with copper for 2 h, and processed for electron microscopy as described previously (Kaiser and Schekman, 1990). Grids were examined on an electron microscope (model 100CX; JEOL USA, Inc.).
Pulse-chase labeling and immunoprecipitation
Yeast were grown overnight to log phase at 24°C in synthetic complete medium supplemented with appropriate amino acids. EGFP-CFTR expression was induced with 100 μM of CuSO4 for 2 h (Labbe and Thiele, 1999). A total of 32 OD600 U of cells was harvested (3 OD600 U for each time point) and resuspended in 4 ml synthetic complete medium plus 100 μM CuSO4. Each culture was separated into two parts and was incubated with shaking at 24°C or 39°C for 10 min. Cells were then labeled with 200 μCi of [35S]methionine (Trans35S label from ICN Biomedicals) for 20 min. The label was chased with 40 μl of chasing mixture (1 mg/ml methionine, 1 mg/ml cysteine, and 15% yeast extract). 0.4-ml samples were collected at 0, 15, 30, 60, and 90 min and added to 25 μl of 100% TCA (5% final TCA concentration). Samples were mixed, placed on ice for 20 min, and washed twice with ice-cold acetone. Cells were resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 0.5% SDS, and protease inhibitors) and broken down by vortexing with glass beads. EGFP-CFTR was immunoprecipitated from total cell lysate using a polyclonal anti-NBD1 CFTR antibody (1:250 dilutions). Immunoprecipitates were separated on 6% SDS-PAGE and analyzed by PhosphorImager (Molecular Dynamics, Inc.). EGFP-CFTR signal was quantified using ImageQuant Software (Molecular Dynamics, Inc.), and relative amounts were calculated and plotted using Excel software (Microsoft). For CPY analysis, pulse-chase was done as above, and lysates were immunoprecipitated with anti-CPY antibodies. Immunoprecipitates were resolved on 10% SDS-PAGE and analyzed as above.
SDS-PAGE and immunoblotting
Samples were resolved on 6% SDS-PAGE as described previously (Bebok et al., 1998). Some gels were electrotransferred to nitrocellulose membrane and immunoblotted with anti–COOH-terminal CFTR mAb as described previously (Bebok et al., 1998).
Acknowledgments
We thank Drs. Jeffrey Brodsky, Eric Sorscher, Dave Bedwell, and Gergely Lukacs for helpful suggestions, and Drs. Bruce Stanton, Eric Schwiebert, Dieter Wolf, and Davis Ng for essential reagents. We also thank Ratnakar Josyula and Ed Philips for help with electron microscopy.
Footnotes
Abbreviations used in this paper: CFTR, cystic fibrosis transmembrane conductance regulator; CPY, carboxy peptidase Y; ERAD, ER-associated protein degradation.
References
- Antonny, B., and R. Schekman. 2001. ER export: public transportation by the COPII coach. Curr. Opin. Cell Biol. 13:438–443. [DOI] [PubMed] [Google Scholar]
- Bannykh, S.I., G.I. Bannykh, K.N. Fish, B.D. Moyer, J.R. Riordan, and W.E. Balch. 2000. Traffic pattern of cystic fibrosis transmembrane regulator through the early exocytic pathway. Traffic. 1:852–870. [DOI] [PubMed] [Google Scholar]
- Barlowe, C. 2000. Traffic COPs of the early secretory pathway. Traffic. 1:371–377. [DOI] [PubMed] [Google Scholar]
- Barlowe, C., and R. Schekman. 1993. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 365:347–349. [DOI] [PubMed] [Google Scholar]
- Bebok, Z., C. Mazzochi, S.A. King, J.S. Hong, and E.J. Sorscher. 1998. The mechanism underlying cystic fibrosis transmembrane conductance regulator transport from the endoplasmic reticulum to the proteasome includes Sec61beta and a cytosolic, deglycosylated intermediary. J. Biol. Chem. 273:29873–29878. [DOI] [PubMed] [Google Scholar]
- Bordallo, J., and D.H. Wolf. 1999. A RING-H2 finger motif is essential for the function of Der3/Hrd1 in endoplasmic reticulum associated protein degradation in the yeast Saccharomyces cerevisiae. FEBS Lett. 448:244–248. [DOI] [PubMed] [Google Scholar]
- Brodsky, J.L., and A.A. McCracken. 1999. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 10:507–513. [DOI] [PubMed] [Google Scholar]
- Brodsky, J.L., E.D. Werner, M.E. Dubas, J.L. Goeckeler, K.B. Kruse, and A.A. McCracken. 1999. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 274:3453–3460. [DOI] [PubMed] [Google Scholar]
- Caldwell, S.R., K.J. Hill, and A.A. Cooper. 2001. Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J. Biol. Chem. 276:23296–23303. [DOI] [PubMed] [Google Scholar]
- Ellgaard, L., and A. Helenius. 2001. ER quality control: towards an understanding at the molecular level. Curr. Opin. Cell Biol. 13:431–437. [DOI] [PubMed] [Google Scholar]
- Gelman, M.S., E.S. Kannegaard, and R.R. Kopito. 2002. A principal role for the proteasome in endoplasmic reticulum-associated degradation of misfolded intracellular cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 277:11709–11714. [DOI] [PubMed] [Google Scholar]
- Hampton, R.Y., R.G. Gardner, and J. Rine. 1996. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell. 7:2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, T.J., M.A. Loo, S. Pind, D.B. Williams, A.L. Goldberg, and J.R. Riordan. 1995. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 83:129–135. [DOI] [PubMed] [Google Scholar]
- Johnston, J.A., C.L. Ward, and R.R. Kopito. 1998. Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143:1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser, C.A., and R. Schekman. 1990. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 61:723–733. [DOI] [PubMed] [Google Scholar]
- Kamhi-Nesher, S., M. Shenkman, S. Tolchinsky, S.V. Fromm, R. Ehrlich, and G.Z. Lederkremer. 2001. A novel quality control compartment derived from the endoplasmic reticulum. Mol. Biol. Cell. 12:1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann, D.J., E.A. Epping, and W.S. Moye-Rowley. 1999. Mutational disruption of plasma membrane trafficking of Saccharomyces cerevisiae Yor1p, a homologue of mammalian multidrug resistance protein. Mol. Cell. Biol. 19:2998–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk, K.L. 2000. New paradigms of CFTR chloride channel regulation. Cell. Mol. Life Sci. 57:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser, G.L., M. Gentzsch, A.K. Kloser, E. Balzi, D.H. Wolf, A. Goffeau, and J.R. Riordan. 2001. Expression and degradation of the cystic fibrosis transmembrane conductance regulator in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 390:195–205. [DOI] [PubMed] [Google Scholar]
- Knop, M., A. Finger, T. Braun, K. Hellmuth, and D.H. Wolf. 1996. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Koning, A.J., C.J. Roberts, and R.L. Wright. 1996. Different subcellular localization of Saccharomyces cerevisiae HMG-CoA reductase isozymes at elevated levels corresponds to distinct endoplasmic reticulum membrane proliferations. Mol. Biol. Cell. 7:769–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn, M.J., J.M. Herrmann, and R. Schekman. 1998. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature. 391:187–190. [DOI] [PubMed] [Google Scholar]
- Labbe, S., and D.J. Thiele. 1999. Copper ion inducible and repressible promoter systems in yeast. Methods Enzymol. 306:145–153. [DOI] [PubMed] [Google Scholar]
- Loayza, D., A. Tam, W.K. Schmidt, and S. Michaelis. 1998. Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae. Mol. Biol. Cell. 9:2767–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken, A.A., and J.L. Brodsky. 1996. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer, B.D., J. Loffing, E.M. Schwiebert, D. Loffing-Cueni, P.A. Halpin, K.H. Karlson, I.I. Ismailov, W.B. Guggino, G.M. Langford, and B.A. Stanton. 1998. Membrane trafficking of the cystic fibrosis gene product, cystic fibrosis transmembrane conductance regulator, tagged with green fluorescent protein in madin-darby canine kidney cells. J. Biol. Chem. 273:21759–21768. [DOI] [PubMed] [Google Scholar]
- Nishikawa, S.I., S.W. Fewell, Y. Kato, J.L. Brodsky, and T. Endo. 2001. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 153:1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddon, C., D. Loayza, L. Vangelista, R. Solari, and S. Michaelis. 1996. Analysis of the localization of STE6/CFTR chimeras in a Saccharomyces cerevisiae model for the cystic fibrosis defect CFTR delta F508. Mol. Microbiol. 19:1007–1017. [DOI] [PubMed] [Google Scholar]
- Pind, S., J.R. Riordan, and D.B. Williams. 1994. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 269:12784–12788. [PubMed] [Google Scholar]
- Plemper, R.K., S. Bohmler, J. Bordallo, T. Sommer, and D.H. Wolf. 1997. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 388:891–895. [DOI] [PubMed] [Google Scholar]
- Preuss, D., J. Mulholland, A. Franzusoff, N. Segev, and D. Botstein. 1992. Characterization of the Saccharomyces Golgi complex through the cell cycle by immunoelectron microscopy. Mol. Biol. Cell. 3:789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich, E., A. Kerem, K.U. Frohlich, N. Diamant, and S. Bar-Nun. 2002. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 22:626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan, J.R. 1999. Cystic fibrosis as a disease of misprocessing of the cystic fibrosis transmembrane conductance regulator glycoprotein. Am. J. Hum. Genet. 64:1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Salama, N.R., J.S. Chuang, and R.W. Schekman. 1997. Sec31 encodes an essential component of the COPII coat required for transport vesicle budding from the endoplasmic reticulum. Mol. Biol. Cell. 8:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkman, M., M. Ayalon, and G.Z. Lederkremer. 1997. Endoplasmic reticulum quality control of asialoglycoprotein receptor H2a involves a determinant for retention and not retrieval. Proc. Natl. Acad. Sci. USA. 94:11363–11368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schekman, R., and L. Orci. 1996. Coat proteins and vesicle budding. Science. 271:1526–1533. [DOI] [PubMed] [Google Scholar]
- Taxis, C., F. Vogel, and D.H. Wolf. 2002. ER-Golgi traffic is a prerequisite for efficient ER degradation. Mol. Biol. Cell. 13:1806–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers, K.J., C.K. Patil, L. Wodicka, D.J. Lockhart, J.S. Weissman, and P. Walter. 2000. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 101:249–258. [DOI] [PubMed] [Google Scholar]
- Vashist, S., W. Kim, W.J. Belden, E.D. Spear, C. Barlowe, and D.T. Ng. 2001. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol. 155:355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhuisen, G., M. Saloheimo, M.A. Fiers, P.J. Punt, R. Contreras, M. Penttila, and C.A. van den Hondel. 1997. Isolation and analysis of functional homologues of the secretion-related SAR1 gene of Saccharomyces cerevisiae from Aspergillus niger and Trichoderma reesei. Mol. Gen. Genet. 256:446–455. [DOI] [PubMed] [Google Scholar]
- Vida, T.A., and S.D. Emr. 1995. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 128:779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q., and A. Chang. 1999. Eps1, a novel PDI-related protein involved in ER quality control in yeast. EMBO J. 18:5972–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, C.L., S. Omura, and R.R. Kopito. 1995. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 83:121–127. [DOI] [PubMed] [Google Scholar]
- Yoshihisa, T., C. Barlowe, and R. Schekman. 1993. Requirement for a GTPase-activating protein in vesicle budding from the endoplasmic reticulum. Science. 259:1466–1468. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., G. Nijbroek, M.L. Sullivan, A.A. McCracken, S.C. Watkins, S. Michaelis, and J.L. Brodsky. 2001. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell. 12:1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]