

Abstract
Mitochondrial morphology is determined by a dynamic equilibrium between organelle fusion and fission, but the significance of these processes in vertebrates is unknown. The mitofusins, Mfn1 and Mfn2, have been shown to affect mitochondrial morphology when overexpressed. We find that mice deficient in either Mfn1 or Mfn2 die in midgestation. However, whereas Mfn2 mutant embryos have a specific and severe disruption of the placental trophoblast giant cell layer, Mfn1-deficient giant cells are normal. Embryonic fibroblasts lacking Mfn1 or Mfn2 display distinct types of fragmented mitochondria, a phenotype we determine to be due to a severe reduction in mitochondrial fusion. Moreover, we find that Mfn1 and Mfn2 form homotypic and heterotypic complexes and show, by rescue of mutant cells, that the homotypic complexes are functional for fusion. We conclude that Mfn1 and Mfn2 have both redundant and distinct functions and act in three separate molecular complexes to promote mitochondrial fusion. Strikingly, a subset of mitochondria in mutant cells lose membrane potential. Therefore, mitochondrial fusion is essential for embryonic development, and by enabling cooperation between mitochondria, has protective effects on the mitochondrial population.
Keywords: membrane fusion; mitochondria; GTPase; mice; knockout
Introduction
Mitochondria are remarkably dynamic organelles. Time-lapse microscopy of living cells reveals that mitochondria undergo constant migration and morphological changes (Bereiter-Hahn and Voth, 1994; Nunnari et al., 1997; Rizzuto et al., 1998). Even in cells with a seemingly “stable” network of mitochondrial tubules, there are frequent and continual cycles of mitochondrial fusion and fission, opposing processes that exist in equilibrium and serve to maintain the overall architecture of these organelles (Bereiter-Hahn and Voth, 1994; Nunnari et al., 1997).
In both yeast and flies, mitochondrial fusion is controlled by the nuclearly encoded mitochondrial transmembrane GTPase, fuzzy onions (Fzo).* In Drosophila, Fzo is specifically and transiently expressed in spermatids. Disruption of Fzo prevents developmentally regulated mitochondrial fusion in postmeiotic spermatids and results in male sterility (Hales and Fuller, 1997). In budding yeast, deletion of FZO1 disrupts the highly branched, tubular mitochondrial network typical of normal cells and results in numerous small spherical mitochondria. fzo1Δ yeast form “petite” colonies that lack mitochondrial DNA (mtDNA) (Hermann et al., 1998; Rapaport et al., 1998). Furthermore, there is a disruption of mating-induced mitochondrial fusion (Hermann et al., 1998). Humans contain two Fzo homologues, termed mitofusin (Mfn)1 and Mfn2, that can alter mitochondrial morphology when overexpressed in cell lines (Santel and Fuller, 2001; Rojo et al., 2002). Both mitofusins are broadly expressed (Rojo et al., 2002), and therefore their functional redundancy is unclear.
Despite our increasing knowledge about the importance of mitochondrial fusion in lower eukaryotes, there is disagreement about its physiological role in vertebrates. Doubts have been raised about the frequency and importance of mitochondrial fusion in cultured mammalian cells (Enriquez et al., 2000). However, several observations suggest that this basic cellular process indeed plays a significant role in vertebrate cells. First, ultrastructural studies of mitochondria show that dramatic transitions occur during the development of certain tissues. For example, the mitochondria of rat cardiac muscle and diaphragm skeletal muscle appear as isolated ellipses or tubules in embryonic stages but then reorganize into reticular networks in the adult (Bakeeva et al., 1978, 1981, 1983). It is likely that these progressive morphological changes occur through mitochondrial fusion. Second, time-lapse fluorescence microscopy of cultured HeLa cells shows that the mitochondria are organized into extensive tubular networks that undergo frequent fusion and fission (Rizzuto et al., 1998). Third, experimentally induced cell hybrids demonstrate rapid mtDNA mixing (Hayashi et al., 1994) and complementation of mtDNA gene products (Nakada et al., 2001b; Ono et al., 2001). Finally, mitochondrial dynamics have been implicated in the regulation of apoptosis. Induction of cell death is sometimes associated with fragmentation of the mitochondrial network. This fragmentation requires dynamin-related protein (Drp)1, which is involved in mitochondrial fission (Frank et al., 2001).
To assess the physiological role of mitochondrial fusion in vertebrates, we have generated knockout mice for Mfn1 and Mfn2. Our analysis reveals that Mfn1 and Mfn2 are each essential for embryonic development and mitochondrial fusion. These mitofusins can exist as both homotypic and heterotypic oligomers and therefore can cooperate as well as act individually to promote mitochondrial fusion. In addition, our results suggest that mitochondrial fusion functions to allow cooperation between mitochondria, thereby protecting mitochondria from respiratory dysfunction.
Results
Identification of murine Fzo homologues
We identified two murine homologues of fzo from a mouse cDNA library. In accordance with the nomenclature for the human mitofusins (Santel and Fuller, 2001), we designate these murine homologues as Mfn1 and Mfn2. Linkage analysis placed Mfn1 at the proximal end of mouse chromosome 3 (∼12–13 cM) in a region syntenic to human 3q25-26. Mfn2 was localized to the distal end of mouse chromosome 4 (∼70–80 cM) in a region syntenic to human 1p36.
As with the fzo genes from Drosophila melanogaster (Santel and Fuller, 2001; Hwa et al., 2002), Homo sapiens (Santel and Fuller, 2001), and Saccharomyces cerevisiae (Hermann et al., 1998; Rapaport et al., 1998), each murine Mfn gene encodes a predicted transmembrane GTPase. The transmembrane segment is flanked by two regions containing hydrophobic heptad repeats, hallmarks of coiled-coil regions (Lupas, 1996). Mfn1 and Mfn2 are 81% similar to each other and are both 52% similar to Drosophila Fzo.
Generation of knockout mice deficient in Mfn1 and Mfn2
We constructed gene replacement vectors for Mfn1 and Mfn2 using the neomycin resistance gene for positive selection and the diphtheria toxin subunit A gene for negative selection. In both cases, a stop codon was engineered at the very beginning of the GTPase domain near the NH2 terminus (Fig. 1, A and E). In addition, the resulting genomic loci each contain a replacement of the G1 and G2 motifs of the GTPase domain with the neomycin expression cassette. These universal GTPase motifs are crucial for binding of the α and β phosphates of GTP and for Mg+2 coordination (Bourne et al., 1991; Sprang, 1997). Genetic analyses in Drosophila and Saccharomyces cerevisiae (Hales and Fuller, 1997; Hermann et al., 1998), as well as our own studies (see Fig. 7 C and Fig. 8 C), demonstrate that an intact GTPase domain is essential for Fzo function. Therefore, the disrupted Mfn1 and Mfn2 alleles described here should be null alleles. Both Southern blot and PCR analysis confirmed germline transmission of the targeted alleles (Fig. 1, B, C, F, and G). Importantly, Western blot analysis using affinity-purified antisera raised against Mfn1 or Mfn2 confirmed loss of the targeted protein in homozygous mutant lysates (Fig. 1, D and H).
Figure 1.

Construction and verification of knockout mice. (A) Genomic targeting of Mfn1. The top bar indicates the wild-type Mfn1 genomic locus with exons aligned above. The dark gray segment contains coding sequences for the G1 and G2 motifs of the GTPase domain. A double crossover with the targeting construct (middle bar) results in a targeted allele (bottom bar) containing a premature stop codon (asterisk) in exon 3 and a substitution of the G1 and G2 encoding genomic sequence with a neomycin- resistance gene (light gray segment labeled Neo; flanking loxP sites indicated by triangles). PGK-DTA, diphtheria toxin subunit A driven by the PGK promoter; Xb, XbaI. (E) Genomic targeting of Mfn2. Drawn as in A. RI, EcoRI. (B and F) Southern blot analyses of targeted embryonic stem clones and offspring. Genomic DNAs were digested with XbaI (B) for Mfn1 and EcoRI (F) for Mfn2 and analyzed with the probes indicated in A and E. The wild-type and knockout bands are indicated as are genotypes. (C and G) PCR genotyping. Three primers (labeled 1, 2, and 3) were used simultaneously to amplify distinct fragments from the wild-type and mutant loci. The DNA samples are identical to those in B and F, respectively. (D and H) Western analyses of wild-type and mutant lysates. Postnuclear embryonic lysates were analyzed with affinity-purified antibodies directed against Mfn1 (D) and Mfn2 (H). β-Actin was used as a loading control.
Figure 7.
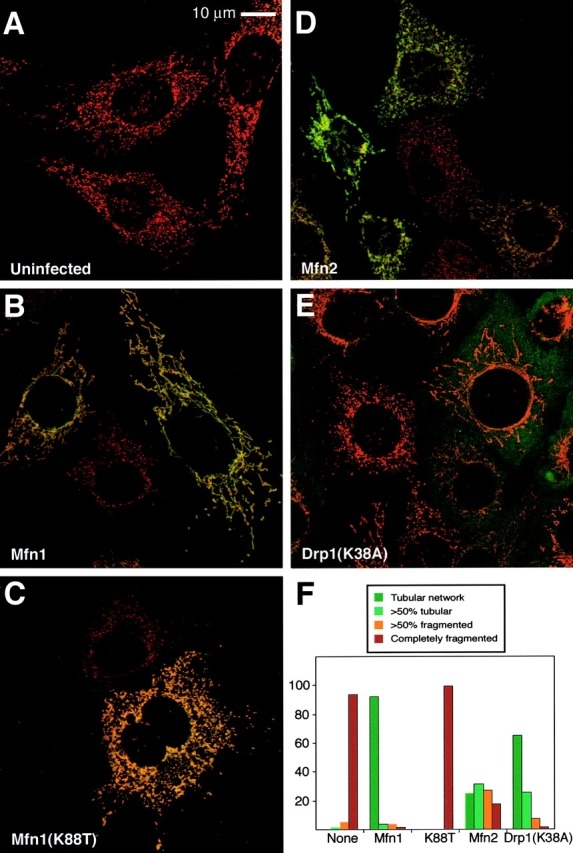
Rescue of Mfn1-deficient cells Mfn1 mutant cells. Mfn1 mutant cells (A) were infected with a retrovirus expressing Myc epitope-tagged versions of Mfn1 (B), Mfn1(K88T) (C), Mfn2 (D), or dominant- negative Drp1(K38A) (E). In the merged images, mitochondrial morphology is revealed by MitoTracker Red staining, and infected cells are identified by immunofluorescence with an anti-Myc antibody (green). In E, the signals are largely nonoverlapping because most of the Drp1 resides in a cytosolic pool. The results are summarized in F, which depicts the percentage of infected cells belonging to each of four morphological classifications. 600 cells were scored for each infection.
Figure 8.
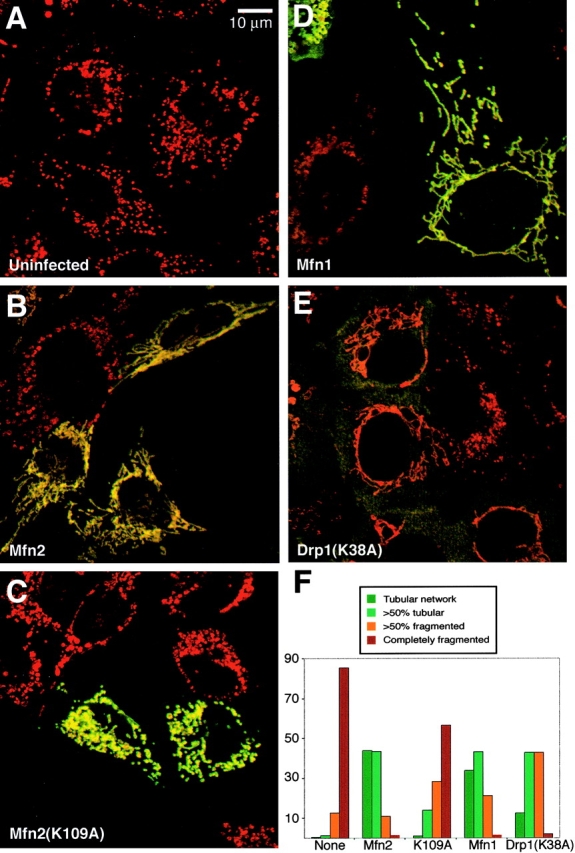
Rescue of Mfn2-deficient cells Mfn2 mutant cells. Mfnz mutant cells (A) were infected with a retrovirus expressing Myc epitope-tagged versions of Mfn2 (B), Mfn2(K109A) (C), Mfn1 (D), or dominant-negative Drp1(K38A) (E). The cells were stained as in Fig. 7. The results are summarized in F. 200 cells were scored for each infection.
Embryonic lethality in homozygous mutant mice
Both the Mfn1 and Mfn2 knockouts demonstrate full viability and fertility in heterozygous animals but result in embryonic lethality of homozygous mutants. For the Mfn1 knockout line, normal frequencies of live mutant embryos were obtained up to embryonic day (e)10.5 (noon of the day a copulatory plug was detected is designated e0.5). However, by e11.5, 20% of the mutant embryos were resorbed, indicating inviability. At e12.5, most identifiable mutant embryos were resorbed (86%), and additional resorptions were so advanced that they could not be genotyped. Although resorptions were not evident until e11.5, by e8.5 all mutant embryos were significantly smaller and showed pronounced developmental delay. In addition, mutant embryos were often deformed.
In the Mfn2 knockout line, normal numbers of live homozygous mutant embryos were recovered up to e9.5. However, starting at e10.5, 29% of homozygous mutant embryos were in the process of resorption. By e11.5, 87% of homozygous mutant embryos were resorbed. The live homozygous mutant embryos at e9.5 and e10.5 were slightly smaller than their littermates but were otherwise well developed and showed no obvious malformations.
Preliminary studies indicate that double homozygous mutant embryos die earlier than either single mutant and show greater developmental delay (unpublished results). This observation suggests that Mfn1 and Mfn2 are both required, and may have a cooperative relationship, in a particular developmental event early in embryogenesis.
Placental defects in Mfn2 mutant mice
We examined placental development in detail because placental insufficiency is one of the most common causes of midgestation lethality (Copp, 1995). Hematoxylin-eosin–stained histological sections of placentae from wild-type and heterozygous embryos showed the typical trilaminar structure composed of a proximal labyrinthine layer, a middle spongiotrophoblast layer, and a distal, circumferential giant cell layer (Fig. 2 E). Trophoblast giant cells are polyploid cells (derived from endoreplication, a process where DNA replication proceeds repeatedly without associated cytokinesis) that lie at the critical interface between fetal and maternal tissues and play important roles in hormone production, recruitment of blood vessels, and invasion of the conceptus into the uterine lining (Cross, 2000). Deficiencies in these cells lead to midgestation lethality (Kraut et al., 1998; Riley et al., 1998; Hesse et al., 2000; Scott et al., 2000). Strikingly, placentae from Mfn2 mutant embryos reproducibly show an impaired giant cell layer with two defects. The giant cells are deficient in quantity, and the few that are observed contain smaller nuclei, implying a reduction in the number of endoreplication cycles (Fig. 2 F). These observations were confirmed with DAPI staining, which highlights the giant cells because of their high DNA content (Fig. 2, A–D). No defects in placental development were detected in Mfn1 mutant embryos (unpublished data) despite expression of Mfn1 in the placenta (Fig. 1 D).
Figure 2.
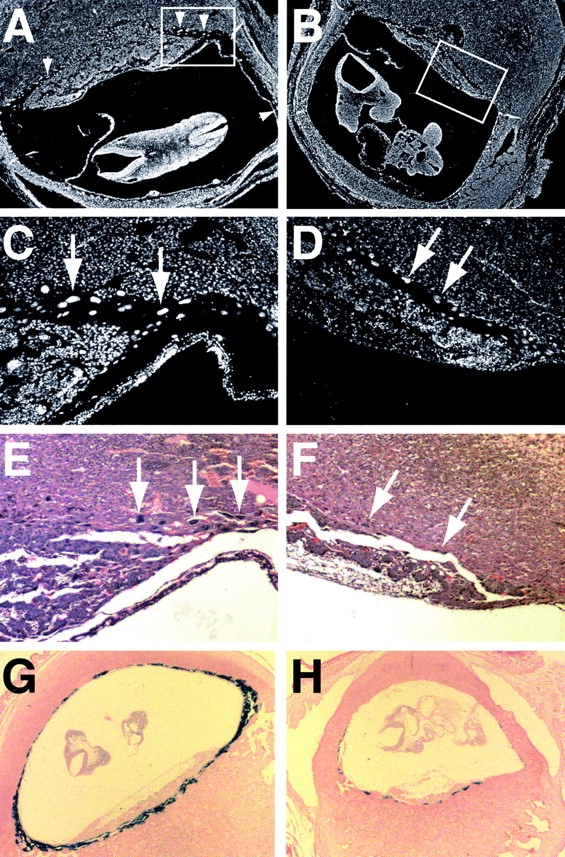
Defective giant cell layer of mutant placentae. (A–D) DAPI-stained sections of placentae from e10.5 wild-type (A and C) and mutant (B and D) littermate embryos. The boxed areas of A and B are enlarged in C and D. Arrows and arrowheads indicate trophoblast giant cells. Note that the giant cells in D are sparser and have smaller nuclei. (E and F) Hematoxylin-eosin–stained sections from the placentae above. (G and H) PL-I (giant cell marker) RNA in situ analysis of placentae from e9.5 wild-type (G) and mutant (H) littermates.
Using RNA in situ hybridization, we examined the expression of molecular markers specific for each of the three placental layers. With the giant cell marker PL-I (Faria et al., 1991), wild-type placentae showed an intense full ring of giant cell staining with multiple layers of giant cells packed in the region distal to the spongiotrophoblast layer (Fig. 2 G). In contrast, Mfn2 mutant placentae showed an incomplete ring of weakly staining giant cells (Fig. 2 H). Moreover, the giant cell layer was generally only one cell layer thick. Similar results were seen in placentae from e8.5 through e10.5. These observations were confirmed with RNA in situ hybridizations (unpublished data) using the additional giant cell markers, proliferin (Lee et al., 1988) and Hand1 (Riley et al., 1998). Interestingly, our in situ hybridizations also revealed somewhat weaker staining with the ectoplacental cone and spongiotrophoblast marker 4311 (Lescisin et al., 1988), suggesting an additional but more subtle defect in this placental layer (unpublished data). No differences in staining between wild-type and Mfn2 mutant sections were observed using TEF5 (unpublished data), a marker for the labyrinthine layer (Jacquemin et al., 1998). Together, these findings suggest that the Mfn2 mutant embryos are dying in midgestation secondary to an inadequate placenta.
Both Mfn1- and Mfn2-deficient cells have aberrant mitochondrial morphology
To examine mitochondrial morphology in Mfn-deficient cells, we derived mouse embryonic fibroblasts (MEFs) from e10.5 embryos. The MEF cultures were infected with a retrovirus expressing mitochondrially targeted enhanced yellow fluorescent protein (EYFP). Wild-type MEFs showed a range of mitochondrial morphologies. The major morphological class, which encompassed >90% of wild-type cells, was characterized by a network of extended wavy tubules distributed in a roughly radial manner throughout the cytoplasm. Such cells had no or only a few spherical mitochondria. The length of these mitochondrial tubules ranged from several microns to >10 μm (Fig. 3, A and B, arrow). A much smaller morphological class, which encompassed only ∼6% of wild-type cells, had mitochondria that were mostly spherical and were termed as “fragmented.”
Figure 3.
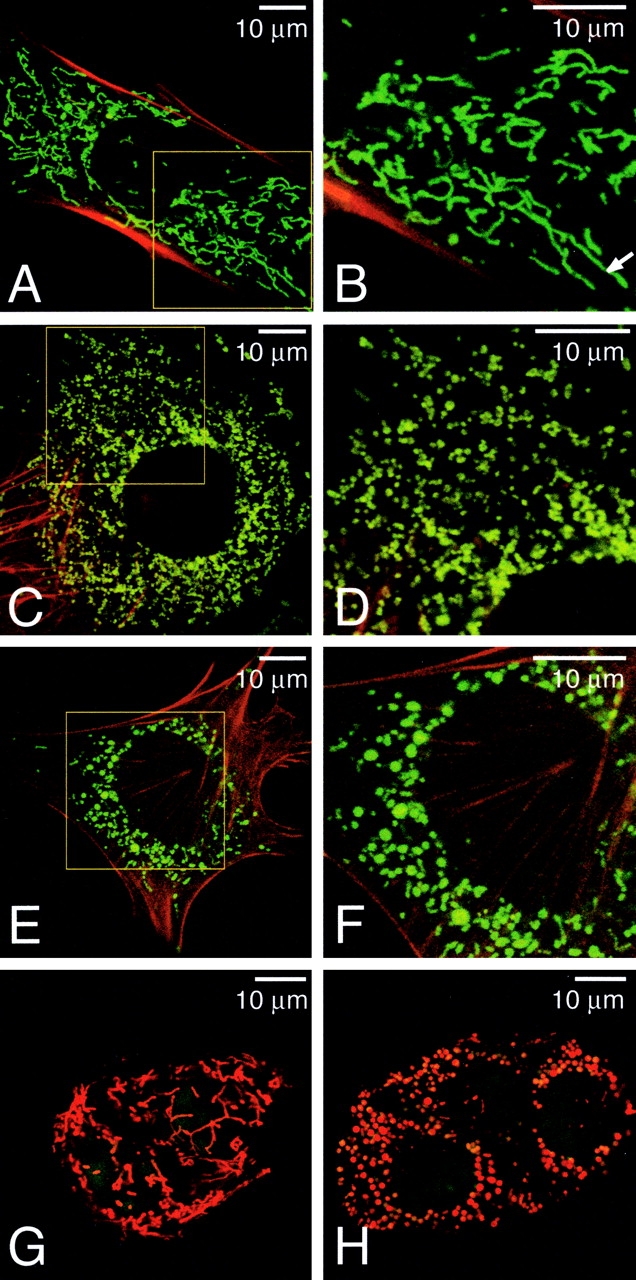
Morphological defects in mitochondria of mutant cells. (A–F) Mitochondrial morphology in wild-type (A and B), Mfn1 mutant (C and D), and Mfn2 mutant (E and F) MEF cells. MEFs expressing mitochondrial EYFP (green) were counterstained with rhodamine-phalloidin (red). (B, D, and F) Higher magnification images of the boxed areas in A, C, and E, respectively. Arrow indicates a tubule >10 μm in length. (G and H) Mitochondrial morphology in live wild-type (G) and mutant (H) TS cells. The mitochondria were stained with MitoTracker Red, and the nuclei were stained with Syto16 (green). Several cells are tightly clustered.
In contrast, Mfn1 mutant MEFs had dramatically fragmented mitochondria (Fig. 3, C and D). Greater than 95% of these cells contained only severely fragmented mitochondria, whereas only 1–2% of cells contained any significant tubules. Similarly, in 85% of Mfn2 mutant fibroblasts the mitochondria appeared mostly as spheres or ovals (Fig. 3, E and F). Only a small minority of mutant cells (4.5%) contained significant tubules. Neither Mfn1 nor Mfn2 mutant cells ever displayed the networks of long extended tubules that characterize the largest morphological class in wild-type cells. Thin section EM revealed that in spite of their aberrant dimensions the spherical mitochondria in these mutant cells contained both cristae and the typical double membrane (unpublished data). These results are consistent with the hypothesis that Mfn1 and Mfn2 play major roles in mitochondrial fusion.
To determine whether mitochondrial defects underlie the placental insufficiency of Mfn2 mutant embryos, we derived trophoblast stem (TS) cell lines from wild-type and mutant blastocysts. With MitoTracker Red staining, the cells within wild-type TS colonies displayed a network of long mitochondrial tubules (Fig. 3 G). In contrast, Mfn2 mutant TS cells showed only spherical mitochondria (Fig. 3 H).
Although loss of either Mfn1 or Mfn2 results in fragmentation of mitochondrial tubules, the two mutations lead to characteristic mitochondrial morphologies that are readily distinguishable. Loss of Mfn1 leads to a greater degree of fragmentation, resulting in either very short mitochondrial tubules or very small spheres that are uniform in size, with diameters no larger than that of a normal tubule. In contrast, many Mfn2 mutant cells exhibit mitochondrial spheres of widely varying sizes. Interestingly, the diameters of some of the spherical mitochondria in both Mfn2 mutant MEF and TS cells are several times larger than the diameters of mitochondrial tubules in wild-type cells. Because a simple defect in mitochondrial fusion would be expected to result in shorter tubules and small spheres with the diameter of normal tubules, these observations suggest that in addition to controlling mitochondrial fusion, Mfn2 is involved in maintaining tubular shape. Together, these results suggest that even though both Mfn1 and Mfn2 are expressed in fibroblasts, each protein is essential for mitochondrial fusion and may have some distinct functions.
Altered mitochondrial dynamics in Mfn-deficient cells
Because mitochondria are such dynamic organelles, we used time-lapse confocal microscopy to determine whether mutant MEFs display aberrations in mitochondrial dynamics. In wild-type cells, the mitochondria are highly motile, and at least one apparent fusion or fission event was observed for most mitochondria during 20-min recordings (Fig. 4 A; video 1, available at http://www.jcb.org/cgi/content/full/jcb.200211046/DC1). However, in both Mfn1 and Mfn2 mutant cells the ovoid or spherical mitochondria undergo fusion events much less frequently (videos 2 and 3, available at http://www.jcb.org/cgi/content/full/jcb.200211046/DC1), although a few such events can be found (Fig. 4 C).
Figure 4.

Dynamics of mitochondria in wild-type and mutant cells. Still frames from time-lapse confocal microscopy. (A) In a wild-type cell, two pairs of mitochondria can be seen moving toward each other. These pairs contact end-to-end and fuse immediately. Note that mitochondria move along their long axes. (B) In a Mfn1 mutant cell, the mitochondria move in an undirected manner. (C) In a Mfn2 mutant cell, two ovoid mitochondria contact each other but do not fuse until much later. Note also the lack of directed movement in most mitochondria. (D) One spherical Mfn2-deficient mitochondrion protrudes a tubular extension that separates and then migrates away along its long axis. Images were processed in Adobe Photoshop® with the emboss filter, and selected mitochondria were manually highlighted in blue. See also videos 1–3 available at http://www.jcb.org/cgi/content/full/jcb.200211046/DC1.
In addition to reduced fusion, our time-lapse videos revealed striking defects in the mobility of mitochondria in mutant cells. Most mitochondria in wild-type cells were tubular, were directed radially, and moved back and forth along their long axis on radial tracks (Fig. 4 A). This movement along defined tracks is consistent with reports that mitochondria can be anchored along microtubule or actin filaments, depending on the cell type (Nangaku et al., 1994; Morris and Hollenbeck, 1995; Tanaka et al., 1998b). The mitochondria of Mfn-deficient cells display a dramatic alteration in mobility; the spherical or ovoid mitochondria in mutant cells show random “Brownian-like” movements (Fig. 4, B and C). This lack of directed movement is not due to disorganization of the cytoskeleton because staining with an antitubulin mAb or rhodamine-phalloidin revealed no defects (unpublished data; Fig. 3). Strikingly, in Mfn2 mutant cells with both spherical and short tubular mitochondria, the tubular mitochondria can still move longitudinally along radial tracks, whereas the spherical mitochondria do not. However, the two morphological classes are interchangeable because tubules can project out of spheres and subsequently move away (Fig. 4 D), or conversely tubules can merge with spheres and lose directed movement. These observations suggest that there is no intrinsic defect in mobilizing Mfn2-deficient mitochondria. Instead, the hampered mobility of spherical mitochondria is secondary to their altered morphology resulting from reduced fusion.
Decreased mitochondrial fusion rates in cells lacking Mfn1 or Mfn2
To definitively show that lack of fusion is the basis for the fragmented mitochondria in mutant cells, we measured mitochondrial fusion activity using a polyethylene glycol (PEG) cell fusion assay. A cell line expressing mitochondrially targeted dsRed was cocultured with a cell line expressing mitochondrially targeted GFP, and PEG was transiently applied to fuse the cells. Cycloheximide was included to prevent synthesis of new fluorescent molecules in the fused cells. When wild-type cells were examined 7 h after PEG treatment, all of the fused cells (n = 200) contained extensively fused mitochondria (Fig. 5 A) as demonstrated by colocalization of red and green fluorescent signals. In contrast, when Mfn1 mutant cells were examined 7 h after PEG fusion 57% (n = 364) of the fused cells contained predominantly unfused mitochondria (Fig. 5, B and C) even when red and green mitochondria were dispersed throughout the fused cell. 35% of cells showed extensive mitochondrial fusion, and 8% showed partial fusion. Similarly, 69% (n = 202) of fused Mfn2 mutant cells showed predominantly unfused mitochondria after 7 h (Fig. 5, E and F). 1% showed extensive fusion, and 30% showed partial fusion. Mfn1 and Mfn2 mutant cells with unfused mitochondria were observed even 24 h after PEG treatment (unpublished data). Thus, mutant cells have severely reduced levels of mitochondrial fusion. Interestingly, in 10% of fused Mfn1 mutant cells, the mitochondria did not readily spread throughout the cytoplasm as shown by discrete sectors of red and green fluorescence (Fig. 5 D). Only 1% of fused Mfn2 mutant cells exhibited this sectoring effect. Therefore, it seems that the mobility of Mfn1-deficient mitochondria is impaired to a greater extent than that of Mfn2-deficient mitochondria, perhaps due to their more severely fragmented morphology.
Figure 5.
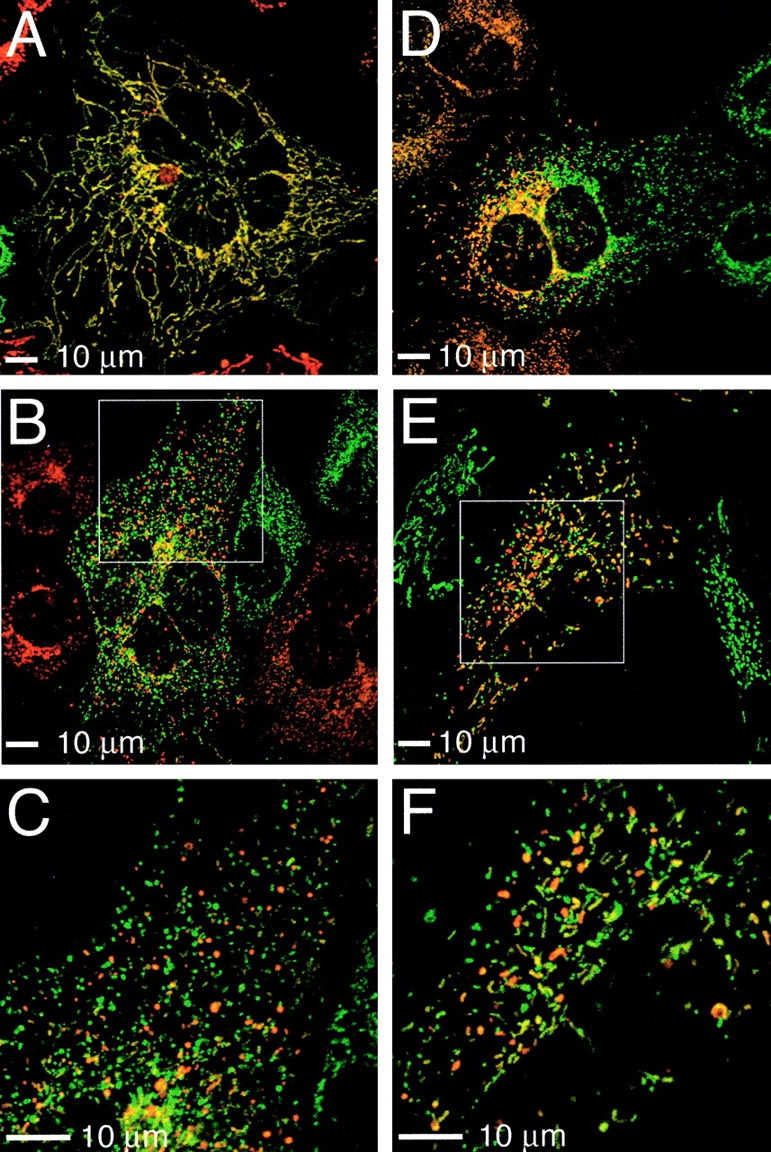
Mitochondrial fusion assay. PEG fusion of cells containing mitochondrially targeted dsRed and GFP. (A) Wild-type cell showing extensive mitochondrial fusion. (B and E) Mfn1 (B) and Mfn2 (E) mutant cells displaying predominantly unfused mitochondria. (C and F) Magnified views of boxed portions in B and E, respectively. (D) Sectoring effect in Mfn1 mutant cell.
Stochastic defects in mitochondrial membrane potential
We tested whether Mfn1 and Mfn2 mutant cells lose mtDNA because there is complete loss of mtDNA in fzo1Δ yeast (Hermann et al., 1998; Rapaport et al., 1998). Southern blot and PCR analysis showed that both mutant cell lines contain normal levels of mtDNA (Fig. 6 A; unpublished data). In addition, the mitochondria in mutant cells expressed COX1, a mitochondrial protein encoded by mtDNA (Fig. 6, B and C). Therefore, unlike fzo1Δ yeast, Mfn-deficient cells clearly retain mtDNA, and this feature allows a critical assessment of the relationship between respiration and mitochondrial fusion. Like wild-type cells, both Mfn1 and Mfn2 mutant cultures showed high rates of endogenous and coupled respiration (unpublished data), indicating no gross defects in respiration and further confirming that mtDNA products are intact and functional.
Figure 6.
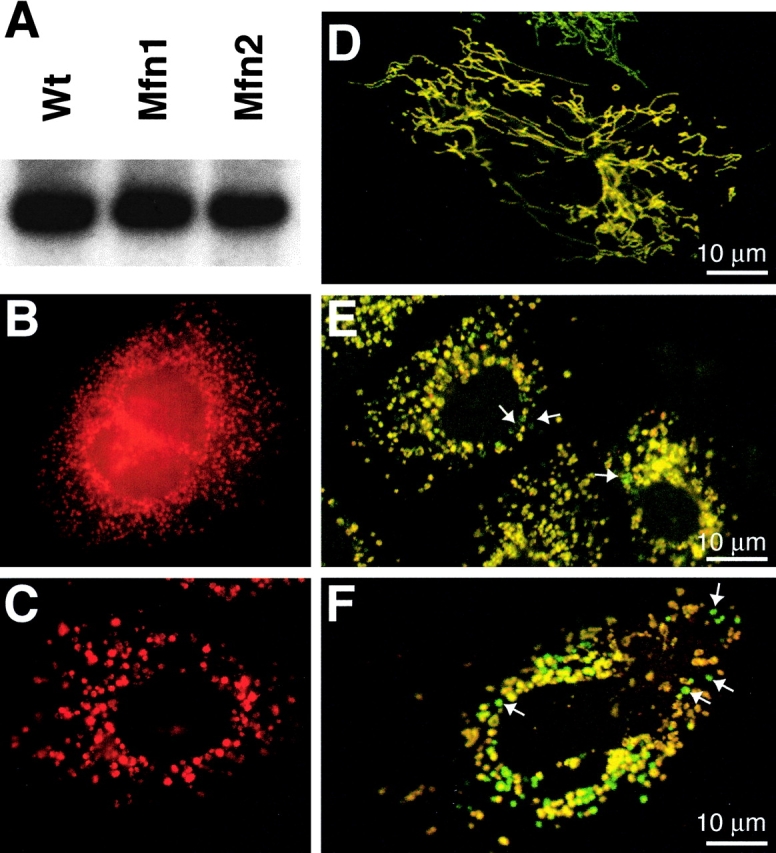
Stochastic loss of membrane potential in mitochondria of mutant cells. (A) mtDNA is detected by Southern blot analysis using a COX1 probe. (B and C) COXI expression in Mfn1 (B) and Mfn2 (C) mutant cells. (D–F) Staining of mitochondria using dyes sensitive to membrane potential. Wild-type (D), Mfn1 mutant (E), and Mfn2 mutant (F) cells expressing mitochondrially targeted EYFP (green) were stained with the dye MitoTracker Red, whose sequestration into mitochondria is sensitive to membrane potential. In these merged images, note that in the mutant cells (E and F) a subset of mitochondria (arrows) stain poorly with MitoTracker Red and thus appear green.
Although the mutant cells are capable of respiration, we reasoned that functional defects may exist at the level of individual mitochondria. To test this hypothesis, we used MitoTracker Red, a lipophilic cationic dye which is sensitive to the mitochondrial membrane potential, to stain MEFs expressing EYFP targeted to the mitochondrial matrix. In wild-type cells, EYFP-marked mitochondria were typically uniformly stained with MitoTracker Red, resulting in colocalization of the two fluorophores (Fig. 6 D). In 7% of wild-type cells, there existed isolated mitochondria that were labeled with EYFP but failed to sequester MitoTracker Red dye. Such mitochondria were invariably small and spherical as opposed to the tubular mitochondria that predominate in these cells. In Mfn1 mutant cultures, almost every cell contained a subset of mitochondria that showed poor staining with MitoTracker Red (Fig. 6 E). Likewise, in Mfn2 mutant cultures over one third of the cells showed some mitochondria that were marked by EYFP but not by MitoTracker Red (Fig. 6 F). It is likely that the compromised mitochondria detected by this assay initially contained membrane potential because the matrix-targeted EYFP requires an intact membrane potential for import. Because of the high stability of EYFP, mitochondria that subsequently lose or reduce their membrane potential would still contain EYFP fluorescence. These results indicate that while bulk cultures display respiratory activity, the function of individual mitochondria is compromised in the absence of either Mfn1 or Mfn2.
Rescue of mitochondrial tubules by modulation of fusion or fission
To unequivocally demonstrate that the mitochondrial morphological defects observed in mutant cells are due to loss of Mfn, we tested whether these defects could be rescued by restoration of Mfn function and whether such rescue depended on an intact GTPase domain. In uninfected Mfn1 mutant cultures or mutant cultures infected with an empty virus, >95% of the cells showed highly fragmented mitochondria that lacked interconnections (Fig. 7, A and F). However, when Mfn1 mutant cultures were infected with a retrovirus expressing Mfn1-Myc >90% of the infected cells showed extensive mitochondrial networks consisting of long tubules (Fig. 7, B and F). Similarly, >80% of Mfn2 mutant cells infected with a Mfn2-expressing retrovirus showed predominantly tubular mitochondria (Fig. 8, B and F). In contrast, constructs carrying mutations in the GTPase G1 motif (Mfn1[K88T] and Mfn2[K109A]) had greatly reduced ability to restore tubular structure (Fig. 7, C and F, and Fig. 8, C and F). These results show that both Mfn1 and Mfn2 promote mitochondrial fusion in a GTPase-dependent manner.
Although both Mfn1 and Mfn2 are clearly required for normal mitochondrial tubules in MEFs, we were able to rescue the morphological defects by overexpression of a single mitofusin. Overexpression of Mfn1 in Mfn2-deficient cells was sufficient for rescue of mitochondrial morphology (Fig. 8, D and F). In addition, overexpression of Mfn2 in Mfn1-deficient cells was sufficient to restore mitochondrial tubules (Fig. 7, D and F). These results show that although both Mfn1 and Mfn2 are required in vivo for normal mitochondrial morphology in MEFs, when overexpressed, each protein is capable of acting alone to promote mitochondrial fusion. Interestingly, these experiments also reveal differential activities of the two homologues. With Mfn2-deficient cells, both Mfn1-Myc and Mfn2-Myc were equally effective in restoring extensive mitochondrial networks, resulting in rescue in 75–80% of expressing cells (Fig. 8 F). In contrast, with Mfn1-deficient cells, 91% of Mfn1-Myc–infected cells showed entirely tubular networks, but only 25% of Mfn2-Myc–infected cells showed such a phenotype (Fig. 7 F).
Because the morphological defects in mutant cells were clearly reversible, we tested whether inhibition of mitochondrial fission could also restore tubular structure. Drp1 plays a key role in mediating mitochondrial fission. To inhibit fission, we constructed an allele of mouse Drp1 with the same mutation (K38A) as in a dominant-negative allele of human Drp1 (Smirnova et al., 2001). Infection of either Mfn1 or Mfn2 mutant cells with a retrovirus expressing Drp1(K38A) resulted in a striking restoration of mitochondrial tubules in 90% (Fig. 7, E and F) and 50% (Fig. 8, E and F) of the cells, respectively. Although the mitochondrial networks in these infected cells were tubular, they were imperfect compared with the networks obtained by rescue with Mfn1 or Mfn2. First, the thickness of the tubules promoted by Drp1(K38A) were less uniform, often showing regions of thinning attached to thicker knobs. Second, the mitochondrial networks were generally less dispersed, with an increase in density near the cell nucleus that often gave the appearance of circumferential rings. Nevertheless, these results indicate that a reduction in fusion caused by loss of Mfn1 or Mfn2 can be compensated, at least partially, by a reduction in fission.
Mfn1 and Mfn2 form homotypic and heterotypic oligomers
Because both Mfn1 and Mfn2 are essential for normal mitochondrial morphology, it is possible that they act in concert to promote mitochondrial fusion. To explore this idea, we tested whether these two proteins form a complex when expressed in wild-type MEF cells (Fig. 9 A). Our results revealed three types of intermolecular interactions. First, both Mfn1 and Mfn2 can form homotypic complexes. Mfn1-HA is coimmunoprecipitated with Mfn1-Myc; analogously, Mfn2-HA is coimmunoprecipitated with Mfn2-Myc. Second, Mfn1 and Mfn2 form heterotypic complexes. Mfn2-HA is coimmunoprecipitated with Mfn1-Myc; conversely, Mfn1-HA is coimmunoprecipitated with Mfn2-Myc. All of these interactions are specific because no HA-tagged protein is found in the anti-Myc immunoprecipitate when the Myc-tagged protein is omitted (unpublished data) or when a control Drp1-Myc is used.
Figure 9.

Immunoprecipitation of Mfn complexes. (A) Wild-type cells were infected with retroviruses expressing Myc- or HA-tagged Mfn1 (labeled 1), Mfn2 (labeled 2), or Drp1 (labeled D) as indicated on top. Anti-Myc immunoprecipitates (top) and total cell lysates (bottom) were analyzed by Western blotting against the HA epitope. The total cell lysate samples contain one sixth cell equivalents compared with the immunoprecipitates. (B) Anti-Myc immunoprecipitates (top) and total cell lysates (bottom) from Mfn1 or Mfn2 mutant cells (indicated on top) were used in an analysis similar to A.
It is a formal possibility that the homotypic interactions detected might not be strictly homotypic due to the expression of endogenous Mfn1 and Mfn2 in the parental cells. To relieve these concerns, we also performed the immunoprecipitation assay in mutant MEFs (Fig. 9 B). We find that Mfn1-Mfn1 homotypic complexes are formed in Mfn2-deficient cells, thereby showing that this interaction is strictly homotypic. Likewise, we also detect Mfn2-Mfn2 homotypic complexes in Mfn1-deficient cells.
Discussion
An essential role for mitochondrial fusion in mouse development
Although mitochondrial dynamics involving fusion and fission has been previously demonstrated in vertebrates, the physiological importance of these processes has remained unclear. Our analysis of Mfn1 and Mfn2 mutant mice demonstrates an essential role for mitochondrial fusion in vertebrate development. Both lines of mice die in midgestation. For Mfn2-deficient mice, we observe a dramatic disruption in placental development, most obviously in the paucity of trophoblast giant cells. Trophoblast cell lines cultured from Mfn2 mutant blastocysts show fragmentation of mitochondrial tubules, consistent with a defect in mitochondrial fusion. These results show that despite its broad expression pattern (unpublished data) Mfn2 is only required for selective developmental transitions.
Trophoblast giant cells are polyploid cells that arise from endoreplication, a process often associated with highly metabolically active cells (Edgar and Orr-Weaver, 2001). Because of their high metabolic rate, we speculate that, in vivo, trophoblast giant cells may be particularly vulnerable to perturbations in mitochondrial dynamics and presumably function. In fact, preeclampsia, the leading cause of fetal and maternal morbidity in the United States, is marked by shallow trophoblast invasion, often resulting in fetal growth retardation. Genetic and cellular evidence suggests that the underlying cause may be a mitochondrial defect (Widschwendter et al., 1998; Talosi et al., 2000).
We anticipate that there are other developmental processes in which the precise regulation of mitochondrial dynamics is essential. Thus, we have constructed conditional alleles of Mfn1 and Mfn2 that should facilitate the analysis of mitochondrial dynamics in adult tissues under both physiological and experimental conditions.
A model for the role of mitochondrial fusion in protecting mitochondrial function
It has been paradoxical why eukaryotes have invested in fusion and fission pathways in cells that have “stable” mitochondrial networks. Given that mitochondrial tubules can be maintained by reducing fusion and fission simultaneously (Bleazard et al., 1999; Sesaki and Jensen, 1999; Fekkes et al., 2000; Mozdy et al., 2000; Tieu and Nunnari, 2000), what is the advantage of maintaining mitochondria in a highly dynamic state?
Our analysis of Mfn-deficient cells suggests an answer to this question. Although bulk cultures of Mfn-deficient fibroblasts show normal levels of endogenous and coupled respiration by oxygen electrode measurements, when individual mitochondria are examined we find that many cells contain a percentage of nonfunctional mitochondria as evidenced by loss of membrane potential. In other cell culture systems, the use of dyes sensitive to mitochondrial membrane potential has revealed occasional and transient losses of membrane potential within small regions of a single mitochondrial tubule (Loew, 1999). We suggest that the dynamic nature of mitochondria protects these organelles by ensuring that regional losses of membrane potential, caused perhaps by local depletion of metabolic substrates or mtDNA, are always transient (Fig. 10 A). Mitochondrial fusion enables intermitochondrial cooperation by allowing exchange of both membrane and matrix contents and therefore may help to restore local depletions and maintain mitochondrial function (Nakada et al., 2001a). Although there is no gross loss of mtDNA in mutant cells, we currently do not know if the individual, defective mitochondria have lost mtDNA.
Figure 10.

Models. (A) The protective role of mitochondrial fusion. At a low rate, individual mitochondria stochastically lose function. In wild-type cells, a defective mitochondrion (shaded) undergoes fusion with functional mitochondria and regains activity. In Mfn- deficient cells, such rescue occurs at a much reduced rate. (B) Three modes of mitofusin action. Mitofusins form homotypic and heterotypic complexes that lead to three activities (I, II, III) involving fusion. See Discussion for details. Mfn1 mutant cells contain only activity III; Mfn2 mutant cells contain only activity I. Since disruption of either Mfn1 or Mfn2 fragments mitochondria and results in distinct phenotypes, MEFs appear to use all three activities (indicated by asterisks). In contrast, trophoblast giant cells predominantly use activity III because they are affected in Mfn2 mutants and not Mfn1 mutants.
Multiple molecular modes of mitofusin action
Given their broad and overlapping expression, it has been unclear why there are two separate mammalian mitofusins. Our results show that the two mitofusins form three distinct molecular complexes that are capable of promoting mitochondrial fusion–Mfn1 homotypic oligomers, Mfn2 homotypic oligomers, and Mfn1-Mfn2 heterotypic oligomers. We propose that the relative importance of each of these modes of mitofusin action can change depending on the cell type (Fig. 10 B). In mouse fibroblasts, all three oligomeric complexes are likely to play important roles because disruption of either Mfn1 or Mfn2 leads to severe mitochondrial fragmentation. Nevertheless, overexpression of Mfn1 homotypic oligomers or Mfn2 homotypic oligomers is sufficient to restore mitochondrial tubules, clearly demonstrating that the homotypic complexes are functional for fusion. In trophoblast giant cells, it appears that Mfn2 homotypic oligomers are most important. As a result, this cell type is affected in Mfn2-deficient embryos but not Mfn1-deficient embryos.
It remains to be determined whether these three complexes function in the same or distinct types of mitochondrial fusion. It is also interesting to note that loss of Mfn2 leads to the formation of both large and small mitochondrial spheres. This phenotype is easily distinguished from loss of Mfn1. The larger mitochondrial fragments may be due to a higher residual fusion activity in Mfn2 mutant cells than in Mfn1 mutant cells. The loss of tubular shape may simply reflect the loss of cytoskeletal interactions as noted in Fig. 4. Alternatively, it may be that Mfn2, and by extension Mfn2 homotypic complexes, play a more direct role in maintaining mitochondrial tubular shape. Thus, although each of the Mfn complexes is involved in fusion, it is possible that they have distinct functions in addition to fusion.
In conclusion, in cells with continual cycles of fusion and fission, the mitochondrial population is essentially functionally homogeneous. However, at any given time point individual mitochondria are functionally distinct entities (Collins et al., 2002). Therefore, without mitochondrial fusion, the stochastic differences between distinct mitochondria can accumulate to affect the well-being of the cell. The mammalian mitofusins, Mfn1 and Mfn2, function in three distinct molecular complexes to promote mitochondrial fusion and thus protect mitochondrial function.
Materials and methods
Cloning of Mfn cDNA and genomic constructs
Homology searches using the Drosophila Fzo sequence (Hales and Fuller, 1997) identified a murine EST (IMAGE Consortium Clone ID 733269) that encodes a highly homologous polypeptide. This cDNA was used to screen a mouse kidney cDNA library, resulting in isolation of cDNAs for both Mfn1 and Mfn2. These sequences are available from GenBank/EMBL/DDBJ under accession nos. AY174062 and AY123975.
Genomic clones of Mfn1 and Mfn2 were retrieved from a lambda 129/SvJ mouse genomic library (Stratagene). Genomic fragments were subcloned into the targeting vector pPGKneobpAlox2PGKDTA (a gift from F. Gertler and L. Jackson-Grusby, Massachusetts Institute of Technology, Cambridge, MA) as the right arms. To insert the left arms, genomic fragments were amplified by PCR, which also introduced a stop codon just before the conserved GKS sequence in each GTPase domain. All constructs were verified by DNA sequence analysis. The sequences of all oligonucleotides used in this study are available from the authors.
To identify the chromosomal locations of Mfn1 and Mfn2, the Jackson Laboratory (C57BL/6JEi × SPRET/Ei) F1 × SPRET/Ei backcross panel was used. Linkage analysis was performed by the Jackson Laboratory Mapping Resource.
Construction of knockout mice
Each gene replacement vector was linearized with SacII and electroporated into low passage 129/SvEv embryonic stem cells using established procedures (Chester et al., 1998). Neomycin-resistant colonies were screened by Southern blot analysis. Two independently isolated clones for each targeted allele were injected into C57BL/6 blastocysts to generate chimeric mice. Excision of the neomycin cassette had no effect on phenotypes.
Confirmation of targeting event
For Southern blot analysis of targeted alleles, genomic DNAs were digested with XbaI (Mfn1 mutant line) or EcoRI (Mfn2 mutant line) and hybridized with a flanking genomic probe. PCR was used for routine genotyping of offspring. A forward genomic primer, a reverse genomic primer, and a reverse neomycin primer were used to detect both the targeted and wild-type alleles in a single reaction.
For Western blot analysis, chicken antisera was generated against Mfn1 (residues 348–579) or Mfn2 (residues 369–598) fused to an NH2-terminal histidine tag. IgY purified from chicken eggs was affinity purified on a column coupled to a maltose-binding protein fusion protein containing either Mfn1 residues 348–579 or Mfn2 residues 369–598.
In situ hybridization
Individual implantation sites were fixed overnight at 4°C in 4% paraformaldehyde, dehydrated through an ethanol series, treated with xylenes, and embedded into paraffin blocks. For staining, 10-μm sections were cut in a transverse plane with respect to the placenta. Slides were processed for hematoxylin-eosin staining or in situ hybridization as described previously (Vortkamp et al., 1996). All riboprobe templates were derived from RT-PCR using e11.5 placental RNA as template. No specific staining was detected using sense probes. For genotyping, embryonic tissue was scraped off unstained slides. DNA was recovered and genotyped by PCR.
Retroviral and plasmid constructs
To construct Mfn1 or Mfn2 with Myc epitope tags, a BamHI site was introduced immediately before the stop codon. The cDNA was subcloned into the vector pcDNA3.1(−)/Myc-His A (Invitrogen). Myc epitope tag cassettes derived from pMMHb-3×Myc were then inserted into the BamHI site to form pMfn-Myc. Mfn1(K88T) and Mfn2(K109A) were constructed by site-directed mutagenesis (Kunkel et al., 1991). To make the Mfn-HA constructs, Mfn1 and Mfn2 cDNAs were subcloned into a pcDNA3.1(−) vector containing a COOH-terminal 3×HA tag. Murine Drp1 was amplified from mouse placental RNA, and Drp1(K38A) was constructed by site-directed mutagenesis. The Drp1(K38A) insert was subcloned into a modified pcDNA3.1(−)/Myc-HisA vector containing seven Myc epitope tags at the COOH terminus.
To generate retroviral expression constructs for each of the above, the epitope-tagged cDNAs were recloned into the retroviral vector pCLβW (a gift from C. Lois, California Institute of Technology). The retroviral expression vectors were cotransfected with the ecotropic retroviral packaging vector pCLEco (a gift from C. Lois, California Institute of Technology) into 293T cells. Retroviral stocks were harvested 48 h after transfection and used to infect MEF cultures. Fragments encoding mitochondrially targeted GFP and dsRed (from plasmids pHS1 and pHS51; gifts from H. Sesaki and R. Jensen, Johns Hopkins University, Baltimore, MD) were subcloned into pCLβW to generate retroviral expression vectors.
MEF and TS cell lines
MEFs were derived from e10.5 embryos. Embryos were mechanically dispersed by repeated passage through a P1000 pipette tip and plated with MEF media (DME, 10% FCS, 1× nonessential amino acids, 1 mM l-glutamine, penicillin/streptomycin [Life Technologies/GIBCO BRL]).
For visualization of mitochondria, the MEFs were either stained with 150 nM MitoTracker Red CMXRos (Molecular Probes) or infected with a retrovirus expressing EYFP fused to the presequence from subunit VIII of human cytochrome c oxidase, which directs EYFP to the mitochondrial matrix (a gift from R. Lansford, California Institute of Technology, Pasadena, CA) (Okada et al., 1999). To facilitate immortalization, the MEFs were later infected with a retrovirus expressing SV40 large T antigen (a gift from L. Jackson-Grusby, Massachusetts Institute of Technology) (Jat et al., 1986). Neither retroviral infection nor immortalization affected mitochondrial morphology. To label actin filaments, cells were fixed in 4% PFA and stained with 2.5 U/ml rhodamine-phalloidin (Molecular Probes). The stained cells were postfixed in 4% PFA.
For time-lapse confocal microscopy, cells were plated at low density onto chambered glass coverslips. Cells with culture medium were overlaid with light mineral oil and imaged in a 37°C chamber. EYFP-optimized filters and dichroics (q497lp, HQ500lp; Chroma) were used on a Zeiss 410 laser scanning confocal microscope (Carl Zeiss MicroImaging, Inc.)
TS cells from e3.5 blastocysts were derived using established protocols (Tanaka et al., 1998a). Live cells were stained with MitoTracker Red (150 nM) and Syto16 (100 nM; Molecular Probes).
PEG fusion
40,000 cells expressing mitochondrially targeted GFP were cultured overnight on 25-mm coverslips with 40,000 cells expressing mitochondrially targeted dsRed. The next morning, cells were fused for 60 s with 50% PEG 1500 (Roche). The cells were washed and grown for 7 h or 24 h in medium containing 30 μg/ml cycloheximide before fixation.
Immunofluorescence
Cells grown on polylysine-coated coverslips were fixed with prewarmed (37°C) 3.7% formaldehyde and permeabilized in PBS/0.1% Triton X-100. In some experiments, cells were incubated with 150 nM MitoTracker Red for 30 min before fixation and then permeabilized with acetone at −20°C. Cells were blocked with 5% bovine calf serum and incubated with primary antibody. For Myc epitope–tagged proteins, the mouse monoclonal antibody 9E10 was used. For COX1, mouse monoclonal 1D6-E1-A8 (Molecular Probes) was used. For detection, Cy3- or Alexa Fluor 488–conjugated secondary antibodies (Jackson ImmunoResearch Laboratories and Molecular Probes) were used. Cells were imaged with a Plan NeoFluar 63× objective on a Zeiss 410 laser scanning confocal microscope (Carl Zeiss MicroImaging, Inc.).
Analysis of mtDNA
Southern analysis of mtDNA was performed by linearizing with XhoI and probing with a radio-labeled PCR fragment containing the COX1 gene.
Immunoprecipitation
Cell lines were infected with various combinations of retroviruses expressing Myc- and HA-tagged Mfn1, Mfn2, and Drp1(K38A). Monolayers were resuspended in lysis buffer (150 mM NaCl, 50 mM Tris, pH 8.0, 1% Triton X-100, and a protease inhibitor cocktail) and immunoprecipitated with 9E10 antibody coupled to protein A–Sepharose beads. After washing, samples were immunoblotted with HA.11 (Covance).
Online supplemental material
Confocal time-lapse videos of EYFP-labeled mitochondria in wild-type (video 1), Mfn1 mutant (video 2), and Mfn2 mutant (video 3) MEFs are available at http://www.jcb.org/cgi/content/full/jcb.200211046/DC1. Videos 1–3 show mitochondrial dynamics in wild-type and mutant MEFs. Videos 1 and 3 are 20-min recordings; video 2 is a 10-min recording. The microscopic field is 96 μm × 96 μm in video 1, 68 μm × 68 μm in video 2, and 75 μm × 75 μm in video 3.
Acknowledgments
We thank Dr. Philip Leder for his support in the early stages of this work. We are grateful to M. Michelman for embryonic stem cell culture assistance and A. Harrington for blastocyst injections. We thank Drs. M. Rojo and A. Lombes for stimulating discussions about the PEG fusion assay.
H. Chen is supported by an Alcott Postdoctoral fellowship. S.A. Detmer and E.E. Griffin are supported by a National Institutes of Health training grant NIHGM07616 and E.E. Griffin is funded by a Ferguson fellowship. A.J. Ewald is a participant in the Initiative in Computational Molecular Biology funded by the Burroughs Wellcome Fund Interfaces program. D.C. Chan is a Bren scholar, Rita Allen scholar, Beckman Young investigator, and recipient of a Burroughs Wellcome Fund Career Development award in Biomedical Sciences. This research was supported by the National Institutes of Health (grant 1 RO1 GM62967-01).
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: Drp, dynamin-related protein; e, embryonic day; EYFP, enhanced YFP; Fzo, fuzzy onions; MEF, mouse embryonic fibroblast; Mfn, mitofusin; mtDNA, mitochondrial DNA; PEG, polyethylene glycol; TS, trophoblast stem.
References
- Bakeeva, L.E., S. Chentsov Yu, and V.P. Skulachev. 1978. Mitochondrial framework (reticulum mitochondriale) in rat diaphragm muscle. Biochim. Biophys. Acta. 501:349–369. [DOI] [PubMed] [Google Scholar]
- Bakeeva, L.E., Y.S. Chentsov, and V.P. Skulachev. 1981. Ontogenesis of mitochondrial reticulum in rat diaphragm muscle. Eur. J. Cell Biol. 25:175–181. [PubMed] [Google Scholar]
- Bakeeva, L.E., S. Chentsov Yu, and V.P. Skulachev. 1983. Intermitochondrial contacts in myocardiocytes. J. Mol. Cell. Cardiol. 15:413–420. [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn, J., and M. Voth. 1994. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc. Res. Tech. 27:198–219. [DOI] [PubMed] [Google Scholar]
- Bleazard, W., J.M. McCaffery, E.J. King, S. Bale, A. Mozdy, Q. Tieu, J. Nunnari, and J.M. Shaw. 1999. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne, H.R., D.A. Sanders, and F. McCormick. 1991. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 349:117–127. [DOI] [PubMed] [Google Scholar]
- Chester, N., F. Kuo, C. Kozak, C.D. O'Hara, and P. Leder. 1998. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom's syndrome gene. Genes Dev. 12:3382–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, T.J., M.J. Berridge, P. Lipp, and M.D. Bootman. 2002. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21:1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp, A.J. 1995. Death before birth: clues from gene knockouts and mutations. Trends Genet. 11:87–93. [DOI] [PubMed] [Google Scholar]
- Cross, J.C. 2000. Genetic insights into trophoblast differentiation and placental morphogenesis. Semin. Cell Dev. Biol. 11:105–113. [DOI] [PubMed] [Google Scholar]
- Edgar, B.A., and T.L. Orr-Weaver. 2001. Endoreplication cell cycles: more for less. Cell. 105:297–306. [DOI] [PubMed] [Google Scholar]
- Enriquez, J.A., J. Cabezas-Herrera, M.P. Bayona-Bafaluy, and G. Attardi. 2000. Very rare complementation between mitochondria carrying different mitochondrial DNA mutations points to intrinsic genetic autonomy of the organelles in cultured human cell. J. Biol. Chem. 275:11207–11215. [DOI] [PubMed] [Google Scholar]
- Faria, T.N., L. Ogren, F. Talamantes, D.I. Linzer, and M.J. Soares. 1991. Localization of placental lactogen-I in trophoblast giant cells of the mouse placenta. Biol. Reprod. 44:327–331. [DOI] [PubMed] [Google Scholar]
- Fekkes, P., K.A. Shepard, and M.P. Yaffe. 2000. Gag3p, an outer membrane protein required for fission of mitochondrial tubules. J. Cell Biol. 151:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, S., B. Gaume, E.S. Bergmann-Leitner, W.W. Leitner, E.G. Robert, F. Catez, C.L. Smith, and R.J. Youle. 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell. 1:515–525. [DOI] [PubMed] [Google Scholar]
- Hales, K.G., and M.T. Fuller. 1997. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 90:121–129. [DOI] [PubMed] [Google Scholar]
- Hayashi, J., M. Takemitsu, Y. Goto, and I. Nonaka. 1994. Human mitochondria and mitochondrial genome function as a single dynamic cellular unit. J. Cell Biol. 125:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann, G.J., J.W. Thatcher, J.P. Mills, K.G. Hales, M.T. Fuller, J. Nunnari, and J.M. Shaw. 1998. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 143:359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse, M., T. Franz, Y. Tamai, M.M. Taketo, and T.M. Magin. 2000. Targeted deletion of keratins 18 and 19 leads to trophoblast fragility and early embryonic lethality. EMBO J. 19:5060–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa, J.J., M.A. Hiller, M.T. Fuller, and A. Santel. 2002. Differential expression of the Drosophila mitofusin genes fuzzy onions (fzo) and dmfn. Mech. Dev. 116:213–216. [DOI] [PubMed] [Google Scholar]
- Jacquemin, P., V. Sapin, E. Alsat, D. Evain-Brion, P. Dolle, and I. Davidson. 1998. Differential expression of the TEF family of transcription factors in the murine placenta and during differentiation of primary human trophoblasts in vitro. Dev. Dyn. 212:423–436. [DOI] [PubMed] [Google Scholar]
- Jat, P.S., C.L. Cepko, R.C. Mulligan, and P.A. Sharp. 1986. Recombinant retroviruses encoding simian virus 40 large T antigen and polyomavirus large and middle T antigens. Mol. Cell. Biol. 6:1204–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraut, N., L. Snider, C.M. Chen, S.J. Tapscott, and M. Groudine. 1998. Requirement of the mouse I-mfa gene for placental development and skeletal patterning. EMBO J. 17:6276–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel, T.A., K. Bebenek, and J. McClary. 1991. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 204:125–139. [DOI] [PubMed] [Google Scholar]
- Lee, S.J., F. Talamantes, E. Wilder, D.I. Linzer, and D. Nathans. 1988. Trophoblastic giant cells of the mouse placenta as the site of proliferin synthesis. Endocrinology. 122:1761–1768. [DOI] [PubMed] [Google Scholar]
- Lescisin, K.R., S. Varmuza, and J. Rossant. 1988. Isolation and characterization of a novel trophoblast-specific cDNA in the mouse. Genes Dev. 2:1639–1646. [DOI] [PubMed] [Google Scholar]
- Loew, L.M. 1999. Potentiometric membrane dyes and imaging membrane potential in single cells. Fluorescent and Luminescent Probes for Biological Activity. W.T. Mason, editor. Academic Press, London. 210–221.
- Lupas, A. 1996. Coiled coils: new structures and new functions. Trends Biochem. Sci. 21:375–382. [PubMed] [Google Scholar]
- Morris, R.L., and P.J. Hollenbeck. 1995. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 131:1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy, A.D., J.M. McCaffery, and J.M. Shaw. 2000. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 151:367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada, K., K. Inoue, and J. Hayashi. 2001. a. Interaction theory of mammalian mitochondria. Biochem. Biophys. Res. Commun. 288:743–746. [DOI] [PubMed] [Google Scholar]
- Nakada, K., K. Inoue, T. Ono, K. Isobe, A. Ogura, Y.I. Goto, I. Nonaka, and J.I. Hayashi. 2001. b. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 7:934–940. [DOI] [PubMed] [Google Scholar]
- Nangaku, M., R. Sato-Yoshitake, Y. Okada, Y. Noda, R. Takemura, H. Yamazaki, and N. Hirokawa. 1994. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 79:1209–1220. [DOI] [PubMed] [Google Scholar]
- Nunnari, J., W.F. Marshall, A. Straight, A. Murray, J.W. Sedat, and P. Walter. 1997. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell. 8:1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, A., R. Lansford, J.M. Weimann, S.E. Fraser, and S.K. McConnell. 1999. Imaging cells in the developing nervous system with retrovirus expressing modified green fluorescent protein. Exp. Neurol. 156:394–406. [DOI] [PubMed] [Google Scholar]
- Ono, T., K. Isobe, K. Nakada, and J.I. Hayashi. 2001. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 28:272–275. [DOI] [PubMed] [Google Scholar]
- Rapaport, D., M. Brunner, W. Neupert, and B. Westermann. 1998. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 273:20150–20155. [DOI] [PubMed] [Google Scholar]
- Riley, P., L. Anson-Cartwright, and J.C. Cross. 1998. The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat. Genet. 18:271–275. [DOI] [PubMed] [Google Scholar]
- Rizzuto, R., P. Pinton, W. Carrington, F.S. Fay, K.E. Fogarty, L.M. Lifshitz, R.A. Tuft, and T. Pozzan. 1998. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 280:1763–1766. [DOI] [PubMed] [Google Scholar]
- Rojo, M., F. Legros, D. Chateau, and A. Lombes. 2002. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 115:1663–1674. [DOI] [PubMed] [Google Scholar]
- Santel, A., and M.T. Fuller. 2001. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114:867–874. [DOI] [PubMed] [Google Scholar]
- Scott, I.C., L. Anson-Cartwright, P. Riley, D. Reda, and J.C. Cross. 2000. The HAND1 basic helix-loop-helix transcription factor regulates trophoblast differentiation via multiple mechanisms. Mol. Cell. Biol. 20:530–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki, H., and R.E. Jensen. 1999. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J. Cell Biol. 147:699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova, E., L. Griparic, D.L. Shurland, and A.M. van Der Bliek. 2001. Dynamin-related protein drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 12:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprang, S.R. 1997. G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66:639–678. [DOI] [PubMed] [Google Scholar]
- Talosi, G., E. Endreffy, S. Turi, and I. Nemeth. 2000. Molecular and genetic aspects of preeclampsia: state of the art. Mol. Genet. Metab. 71:565–572. [DOI] [PubMed] [Google Scholar]
- Tanaka, S., T. Kunath, A.K. Hadjantonakis, A. Nagy, and J. Rossant. 1998. a. Promotion of trophoblast stem cell proliferation by FGF4. Science. 282:2072–2075. [DOI] [PubMed] [Google Scholar]
- Tanaka, Y., Y. Kanai, Y. Okada, S. Nonaka, S. Takeda, A. Harada, and N. Hirokawa. 1998. b. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 93:1147–1158. [DOI] [PubMed] [Google Scholar]
- Tieu, Q., and J. Nunnari. 2000. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J. Cell Biol. 151:353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortkamp, A., K. Lee, B. Lanske, G.V. Segre, H.M. Kronenberg, and C.J. Tabin. 1996. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 273:613–622. [DOI] [PubMed] [Google Scholar]
- Widschwendter, M., H. Schrocksnadel, and M.G. Mortl. 1998. Pre-eclampsia: a disorder of placental mitochondria? Mol. Med. Today. 4:286–291. [DOI] [PubMed] [Google Scholar]