

Abstract
Afilopodium protrudes by elongation of bundled actin filaments in its core. However, the mechanism of filopodia initiation remains unknown. Using live-cell imaging with GFP-tagged proteins and correlative electron microscopy, we performed a kinetic-structural analysis of filopodial initiation in B16F1 melanoma cells. Filopodial bundles arose not by a specific nucleation event, but by reorganization of the lamellipodial dendritic network analogous to fusion of established filopodia but occurring at the level of individual filaments. Subsets of independently nucleated lamellipodial filaments elongated and gradually associated with each other at their barbed ends, leading to formation of cone-shaped structures that we term Λ-precursors. An early marker of initiation was the gradual coalescence of GFP-vasodilator-stimulated phosphoprotein (GFP-VASP) fluorescence at the leading edge into discrete foci. The GFP-VASP foci were associated with Λ-precursors, whereas Arp2/3 was not. Subsequent recruitment of fascin to the clustered barbed ends of Λ-precursors initiated filament bundling and completed formation of the nascent filopodium. We propose a convergent elongation model of filopodia initiation, stipulating that filaments within the lamellipodial dendritic network acquire privileged status by binding a set of molecules (including VASP) to their barbed ends, which protect them from capping and mediate association of barbed ends with each other.
Keywords: actin; Arp2/3; VASP; fascin; lamellipodia
Introduction
The crawling movement of a cell involves protrusion of its leading edge coordinated with translocation of its cell body. Protrusion is driven by polymerization of actin within two organelles, lamellipodia and filopodia, which have strikingly different designs of the actin polymerization machinery and are regulated by different signaling pathways (Hall, 1998; Svitkina and Borisy, 1999b).
In lamellipodia, which are broad, flat protrusions, actin filaments form a branched network (Svitkina et al., 1997; Svitkina and Borisy, 1999a). The current model for lamellipodial dynamics (Borisy and Svitkina, 2000; Pollard et al., 2000) suggests that treadmilling of the branched actin filament array consists of repeated cycles of dendritic nucleation, elongation, capping, and depolymerization of filaments. Dendritic nucleation is mediated by the Arp2/3 complex, which is activated by members of WASP family (Higgs and Pollard, 2001). During a period of elongation after nucleation, the filament pushes the membrane. When a filament elongates beyond the efficient length for pushing, its growth is thought to be terminated by capping protein (Cooper and Schafer, 2000). Depolymerization is assisted by proteins of the ADF/cofilin family (Bamburg, 1999). Other proteins play supporting roles in this process. Profilin targets filament elongation to barbed ends (Carlier and Pantaloni, 1997), enabled/vasodilator-stimulated phosphoprotein (Ena/VASP)*family proteins protect elongating barbed ends from capping (Bear et al., 2002), cortactin stabilizes branches (Weaver et al., 2001), and filamin A (Flanagan et al., 2001) and α-actinin stabilize and consolidate the whole network.
In filopodia, which are thin cellular processes, actin filaments are long, parallel, and organized into tight bundles (Small, 1988; Lewis and Bridgman, 1992; Small et al., 2002). Other cellular structures, such as microspikes and retraction fibers, bear similarities to filopodia and may be related to them. Microspikes are parallel actin bundles within the lamellipodium. Retraction fibers are long, thin cellular processes that remain attached to the substratum after cell withdrawal. They also contain a parallel bundle of actin filaments (Small, 1988; Lewis and Bridgman, 1992; Svitkina et al., 1997). Filopodial protrusion is thought to occur by a filament treadmilling mechanism, which was originally proposed for both filopodia and lamellipodia (Small, 1994). According to this model, all actin filaments within a bundle elongate at their barbed ends and release subunits from their pointed ends. Existing experimental data support this model of filopodial elongation. Structurally, actin filaments in filopodia are long and unbranched (Svitkina and Borisy, 1999a), suggesting that assembly occurs by elongation, not by branched nucleation. Dynamic observations (Mallavarapu and Mitchison, 1999) revealed that labeled actin incorporated at the filopodial tips, moved backward, and dissipated at the rear (as predicted by a treadmilling mechanism), and that actin turnover in filopodia was slow; consistent with the idea of long filaments adding or losing subunits only at their ends. A frequent event in filopodial behavior is their fusion, which frequently occurs as elongating oblique bundles collide and subsequently grow as a single unit (Katoh et al., 1999b; Small et al., 2002).
A set of molecules essential for filopodial protrusion has not been explicitly determined. However, some proteins are enriched in filopodia, suggesting that they play important roles. One of them is a cross-linking protein (fascin) that mediates filament bundling (Bartles, 2000; Kureishy et al., 2002). Many different proteins are enriched at filopodial tips (Small et al., 2002), including Ena/VASP proteins (Lanier et al., 1999; Rottner et al., 1999), N-WASP and CR16 (Ho et al., 2001), myosin X (Berg and Cheney, 2002), talin (DePasquale and Izzard, 1991), syndapin I (Qualmann and Kelly, 2000), Abl interactor proteins (Stradal et al., 2001), and Vav (Kranewitter et al., 2001). Roles for these proteins remain largely unknown with the exception of Ena/VASP proteins. In lamellipodia, GFP-VASP forms a thin line along the extreme leading edge (Rottner et al., 1999), and in filopodia it appears as a bright dot at filopodial tips (Lanier et al., 1999; Rottner et al., 1999). Ena/VASP proteins bind barbed ends of actin filaments and protect them from capping at the leading edge of lamellipodia, which results in formation of longer filaments within the lamellipodial dendritic network (Bear et al., 2002). These data suggest that Ena/VASP proteins that are enriched at filopodial tips may mediate continuous elongation of filopodial actin filaments.
The major gap in our understanding of filopodia behavior is the mechanism of their initiation. The filament treadmilling model refers to the steady state of an established organelle, but does not explain how it arose in the first place. A small GTPase, Cdc42, is a well-known signaling molecule inducing filopodia in cells (Kozma et al., 1995; Nobes and Hall, 1995). One of its downstream effectors, N-WASP, is an ubiquitous activator of the Arp2/3 complex (Rohatgi et al., 1999, 2000), which significantly facilitates Cdc42-induced filopodial formation (Miki et al., 1998), suggesting that the Arp2/3 complex may be involved in filopodial protrusion. This suggestion has been supported experimentally by perturbing Arp2/3 function in permeabilized platelets with an inhibitory antibody (Li et al., 2002), and in HeLa cells by expressing VCA domain of N-WASP (Qualmann and Kelly, 2000). Because the Arp2/3 complex is absent from established filopodia (Svitkina and Borisy, 1999a), we hypothesized that it plays a role during filopodia initiation. One possibility for how the Arp2/3 complex induces filopodial bundles is that it forms a “nucleation center” which starts a bundle and subsequently dissociates. Another possibility is that the normal dendritic array produced by Arp2/3-mediated nucleation is rearranged into parallel bundles. In this work, we investigated the mechanism of filopodia initiation in B16F1 mouse melanoma cells and found that filopodial bundles were initiated by reorganization of the dendritic network in a process that involved elongation and convergence of subsets of privileged barbed ends.
Results
Filopodia, microspikes, and retraction fibers
Crawling cells elaborate filopodia, microspikes, and retraction fibers in the course of cycles of protrusion and withdrawal. These have been considered as distinct entities, but because of their structural similarities, we investigated whether they were truly distinct or interconvertible. Determining whether they were functionally related was important to define the scope of our study.
We followed the kinetics of peripheral actin bundles by phase contrast or fluorescence microscopy in untransfected or GFP-actin–expressing cells, respectively. We observed many examples of transition between filopodia, microspikes, and retraction fibers (Fig. 1). The predominant order of transitions was from microspike to filopodium to retraction fiber. Transitions in the opposite direction were also observed. For each type of structure, the filament bundle was able to protrude, suggesting that the actin polymerizing machinery was functional in each morphological state. The protrusive activity of the surrounding lamellipodium seemed to be an important factor determining the transitions between these organelles. Depending on whether the lamellipodium advanced as fast as or slower than an actin bundle elongated, the bundle appeared as a microspike or a filopodium. If the lamellipodium withdrew while the actin bundle remained stable or elongated, the bundle appeared as a retraction fiber. Increased net protrusion of an actin bundle also contributed to the transition from microspikes to filopodia, especially after fusion of two microspikes. Thus, filopodia, microspikes, and retraction fibers are interconvertible organelles, which may transform one into another because of a disparity in the protrusion velocity of the bundles themselves and of the surrounding lamellipodium. Therefore, in this paper, we will consider these types of peripheral actin bundles together and will refer to them collectively as “filopodia,” because this is the most commonly used term.
Figure 1.

Interconversion between microspikes, filopodia, and retraction fibers. Time-lapse sequences of untransfected (A, phase contrast) or GFP-actin expressing (B and C, fluorescence) cells. Time in seconds. (A) Lamellipodium containing several microspikes (triple arrow and arrowhead in first frame) retracts leaving microspikes in the form of retraction fibers (240 s), some of which continue to protrude (240–400 s, arrow). At ∼400 s, lamellipodium resumes protrusion and absorbs retraction fibers, some of which disappear, one becoming a filopodium (560 s, arrow), and another becoming a microspike (560 s, arrowhead). (B) Transition of a microspike (0 and 20 s) to filopodium (20–60 s) to retraction fiber (60–140 s). Actin bundle displayed continuous elongation, whereas surrounding lamellipodium initially kept up with the bundle (0–20 s), paused (40 s), and then withdrew (60–140 s). (C) Transition of microspike (0–30 s) to filopodium (45–75 s) and back to microspike (90–105 s) as a result of uncoordinated protrusive behavior of the bundle and the lamellipodium. Bars, 2 μm.
Kinetics of filopodia initiation
To approach the central question of the mechanism of filopodia initiation, we first investigated the kinetics of spontaneous filopodia initiation using GFP-tagged structural proteins. If filopodia were initiated by an Arp2/3-containing nucleation center, one would expect a nascent filopodium to arise from a distinct fluorescent dot of actin or Arp2/3 complex, whereas the rearrangement model predicts a gradual condensation of actin fluorescence into a filopodial bundle.
Filopodia in GFP-actin–expressing cells displayed a broad range of lengths and fluorescence intensities. Histories of large filopodia revealed that they were formed by fusion of smaller filopodia, which in turn were the result of fusion at an even finer scale (Fig. 2 A). Events of true filopodia initiation were recognized as the appearance of thin, faint nascent filopodia contained within the lamellipodial network. In most cases (81%, n = 124), they arose from fishtail-shaped actin densities within the lamellipodium (Fig. 2, B and C). These densities, which we will call Λ-precursors because of their shape, were just slightly brighter than the surrounding lamellipodium at their vertices, but gradually diffused into a lamellipodial network at their bases (Fig. 2 D). Although hardly distinguishable from the rest of the lamellipodial network, Λ-precursors could be consistently recognized, after contrast enhancement, by tracing back in time the sequences of newly formed filopodia. In the remaining cases of filopodial initiation (19%), Λ-precursors were not visible, possibly because of insufficient temporal resolution or contrast. Nascent filopodia subsequently fused with each other (Fig. 2, B and C) or with other filopodia, and thus increased in size. Fusion produced Λ-configurations of filopodial bundles reminiscent of the shape of Λ-precursors, but with more distinct individual branches. Over time, these Λ-shaped bundles treadmilled backward at the root of the fused filopodium (Fig. 2 C) until they disappeared in the course of depolymerization. The observed actin kinetics appears more consistent with the idea of network reorganization as a mechanism of filopodia initiation.
Figure 2.

Actin kinetics during filopodia initiation. (A–C) Time-lapse sequences of GFP-actin–expressing B16F1 cells. Time in seconds; individual features marked by arrows and arrowheads. Nascent filopodia are marked starting from the frame preceding the appearance of the recognizable precursor. (A) Three established filopodia (0 s, arrowheads) fuse with each other (0–60 s). Two Λ-precursors (arrows) appear (20 s), and fuse with each other (40 s), forming a nascent filopodium that subsequently (60 s) joins the fusing older filopodia. (B) Several nascent filopodia form from Λ-precursors that appear within lamellipodium. (C) Two Λ-precursors existing at 0 s (arrows) fuse with each other (12 s), producing a nascent filopodium with a Λ-shaped root. The fusion point treadmills backward while the filopodium protrudes forward. (D) Gallery of Λ-precursors. First four examples represent enlarged and enhanced Λ-precursors from A (20 s), B (8 s), B (24 s), and C (0 s), respectively. Remaining examples are from other sequences. Bars, 2 μm.
The Arp2/3 complex is predicted to be enriched in the hypothetical filopodial nucleation center. Therefore, we performed kinetic analysis of GFP-Arp3–expressing cells (Fig. 3 A). Because filopodial bundles were invisible in GFP-Arp3 images, we acquired phase contrast images at the beginning and at the end of the sequence to detect nascent filopodia initiated during the sequence. Filopodia were observed to appear by phase microscopy and GFP-Arp3 was present throughout the lamellipodium, but no increase in GFP-Arp3 intensity was detected to spatially and temporally correlate with filopodial initiation. The essentially uniform distribution of the GFP-Arp3 signal does not support the hypothesis of an Arp2/3-based nucleation center for filopodial initiation.
Figure 3.

Kinetics of marker proteins during filopodia initiation. Time-lapse sequences of GFP-Arp3 (A), GFP-fascin (B and C), or GFP-VASP (D–F). (A) Two flanking phase contrast frames (0 and 164 s) demonstrate formation of a new filopodium during the sequence. Positions of preexisting (arrow) and nascent (arrowhead) filopodia are indicated. No focal enrichment of Arp3 was seen during formation of the nascent filopodium. (B and C) Most nascent filopodia arise from bright GFP-fascin dots without obvious Λ-precursors. In some cases, a bright dot localizes to the tip of a faint Λ-shaped density, as in the inset in 16 s frame in C, which shows the region indicated by arrowhead in this frame after enlargement and adjustment of contrast to reveal weak fluorescence. Filopodia fusion occurs in C (arrows). (D–F) Bright GFP-VASP dots corresponding to nascent filopodia arise by gradual coalescence of the initially even line of leading-edge fluorescence. Brackets indicate regions of brighter GFP-VASP fluorescence that shrink into dots over time. In F, two smaller dots (24 s) are formed during coalescence of the shrinking region, which subsequently fuse with each other (32 s) and with the adjacent preexisting dot (40 s). Bars, 2 μm.
To obtain insight into the actual mechanism of filopodia initiation, we next analyzed the kinetics of proteins enriched in filopodia, i.e., fascin and VASP. In GFP-fascin–expressing cells, a majority of nascent filopodia (66%, n = 207) first appeared as a bright dot or short rod on a dark background (Fig. 3, B and C). In other cases (34%), a bright dot of GFP-fascin rather suddenly appeared at the tip of a very faint Λ-shaped density in lamellipodia (Fig. 3 C, inset in 16 s frame). Both kinds of nascent fascin dots subsequently elongated to form a filopodium. Fusion of mature fascin-containing filopodia was also frequently seen (Fig. 3 C). Because fascin is present in lamellipodia, albeit at much lower concentration than in filopodia, the faint Λ-shaped fascin densities might correspond to Λ-precursors, suggesting that in the course of filopodia initiation, fascin initially appears at the tips of the Λ-precursors (see next section). In GFP-VASP sequences, we followed the formation of brighter dots corresponding to filopodial tips among the weaker fluorescence of the lamellipodial edge. The major pathway for filopodia initiation (Fig. 3, D–F) was a gradual coalescence of VASP to ultimately produce a bright dot. The first sign detected was a slight elevation of GFP-VASP fluorescence intensity within a small domain (2–4 μm) along the leading edge. Then, this region gradually shrank into a dot concurrently increasing its intensity. In some cases, the condensing region became discontinuous during shrinkage (Fig. 3 F), suggesting intermediate formation of smaller filopodia. Because Ena/VASP proteins bind barbed ends and protect them from capping (Bear et al., 2002), these data suggest that elongating barbed ends from the lamellipodial network gradually segregate into a small region, which becomes a filopodial tip.
Protein composition of Λ-precursors
Our kinetic analysis identified Λ-precursors as intermediates in filopodial initiation. Next, we examined whether molecular markers that discriminate between lamellipodia and filopodia are present in Λ-precursors (Fig. 4). Expression of GFP-actin or staining with labeled phalloidin was used to visualize actin. Putative Λ-precursors were identified in the actin channel based on their characteristic shape, and slightly increased actin density. Immunostaining or expression of GFP-tagged proteins was used to localize the second protein.
Figure 4.

Localization of filopodial and lamellipodial markers in Λ-precursors. Left column; actin revealed by Texas Red-phalloidin (A, B, and F) or by GFP-actin expression (C–E). Central column; actin-binding proteins (as indicated) revealed by expression of GFP-fusion proteins (A, B, and F) or by immunostaining (C–E). Right column; merged images. Λ-precursors are indicated by arrowheads. (A) Fascin is strongly enriched in established filopodia and localizes to the distal section of some Λ-precursors (wide arrowheads), but not others (narrow arrowheads). (B) VASP forms bright dots at the tips of filopodia and Λ-precursors. Additional dots can be seen along the leading edge (arrows), which apparently do not correspond to Λ-precursors. (C–E) Lamellipodial markers, Arp2/3 complex (C), cortactin (D), and capping protein (E), are excluded from filopodia and are partially depleted from Λ-precursors. (F) α-Actinin localizes to proximal parts of lamellipodia and filopodia. Bars, 2 μm.
Fascin and VASP were used as filopodial markers. In GFP-fascin–expressing cells, putative Λ-precursors either did not contain significant amount of fascin (Fig. 4 A, arrows; also see Fig. 6), or they had fascin enriched only at the tip (Fig. 4 A, arrowheads), suggesting that fascin was recruited to the tips of the preformed Λ-precursors. In GFP-VASP–expressing cells (Fig. 4 B), VASP fluorescence at the leading edge became less uniform after extraction, perhaps revealing more strongly associated protein. The brightest VASP dots corresponded to established filopodia. The majority of Λ-precursors contained a distinct VASP dot at their vertex (Fig. 4 B, arrowheads). Weak VASP dots were also evident without recognizable Λ-precursors (Fig. 4 B, arrows). These may represent fluctuations in density of actin filament barbed ends within the lamellipodium or Λ-precursors not detected in the actin channel.
Figure 6.
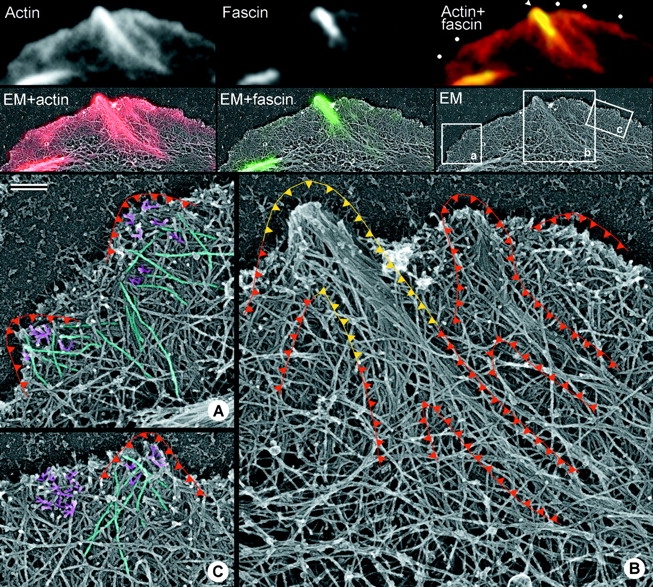
Actin filament organization in Λ-precursors. Correlative fluorescence and EM of the same cell. First row; fluorescence microscopy of the leading edge showing Texas Red-phalloidin labeled actin (left), GFP-fascin (middle), and merged image (right). White dots in the merged image mark putative Λ-precursors, and the arrowhead indicates a filopodium, which have been analyzed by EM. Second row; EM of the same region (right) overlaid with fluorescence images of actin in red (left) and fascin in green (middle). Boxed regions (a–c) are enlarged in the bottom panels, labeled respectively. Red outlines in enlarged panels denote areas of increased actin density in the fluorescence image that represent putative Λ-precursors or filopodia. Λ-precursors contain relatively long filaments, some of which are highlighted in cyan in A and C, along with short branching filaments highlighted in magenta; adjacent lamellipodium (C) contains mostly short branching filaments highlighted in magenta. Yellow outline denotes region enriched in fascin in the established filopodium. This region contains tightly bundled filaments. Bar, 0.2 μm.
As lamellipodial markers, we used Arp2/3 complex, cortactin, and capping protein. Previously, we have shown that Arp2/3 complex is excluded from filopodial bundles (Svitkina and Borisy, 1999a). Here, we report that cortactin and capping protein are also excluded from filopodia (Fig. 4, D and E). In Λ-precursors, these proteins were partially depleted, especially close to the vertex, but not completely absent (Fig. 4, C–E), suggesting that gradual depletion of lamellipodial proteins occurs during formation of Λ-precursors and filopodia initiation. No enrichment of Arp2/3 complex was detected at filopodial roots.
We also investigated the ability of α-actinin to discriminate between filopodia and lamellipodia. Previously, α-actinin has been shown to localize to lamellipodia (Langanger et al., 1984), but its localization in filopodia is unclear. GFP-tagged α-actinin associated with both lamellipodia and filopodia (Fig. 4 F), but appeared in these organelles with delay and, consequently, localized to the base of the fast-protruding lamellipodia and filopodia. Thus, α-actinin is a late marker for actin bundling in filopodia and is apparently not involved in filopodia initiation.
Together, the molecular marker analysis suggests that Λ-precursors represent a transitional structure displaying enrichment of filopodial markers and partial depletion of lamellipodial proteins. Dual-channel correlation of actin distribution with that of filopodial markers suggested that VASP accumulation occurred early in the process of formation of Λ-precursors, and that fascin appeared at the tips of established Λ-precursors.
Structure of Λ-precursors
Light microscopic analysis suggested a gradual reorganization of the lamellipodial network into bundles through intermediate formation of Λ-precursors. This hypothesis was analyzed using a higher resolution technique; platinum replica electron microscopy (EM). Treadmilling behavior of filopodia has a remarkable consequence in that the history of the actin array is imprinted in its structure (Katoh et al., 1999a), so that moving from the leading edge in a proximal direction in space is analogous to traveling back in time. To understand the mechanism of filopodia initiation, we first focused on the analysis of filopodial roots. In this work, we were most interested in analyzing young filopodia, which are usually thin and short according to our kinetic study.
The majority of apparently young filopodial bundles were splayed apart at their roots into smaller bundles or individual filaments (Fig. 5), suggesting that the bundles were formed by convergence of the composing elements. Filopodial roots consisting of two or more smaller bundles are consistent with an event of filopodial fusion in the recent history of that filopodium (Fig. 5 B). More importantly, we observed many examples of filopodial bundles whose roots suggested the convergence of individual filaments originating from distant places in the surrounding lamellipodial network and entering the bundle at different levels. In some cases, it was possible to track filaments back from the bundle toward their origin as a branch on another filament in the surrounding network (Fig. 5 C). These findings suggest that filaments comprising filopodial bundles were asynchronously recruited from the dendritic network. Remarkably, filaments entering filopodial bundles were long compared with the branched network near the leading edge (Fig. 5, inset in A). Older filopodia, which could be recognized by their length and thickness, either had their actin bundles rooted deeply in the cytoplasm, which impeded visualization, or had tapered (not splayed) roots. This is consistent with depolymerization from the pointed ends of the composing filaments causing progressive elimination of the original splayed roots.
Figure 5.
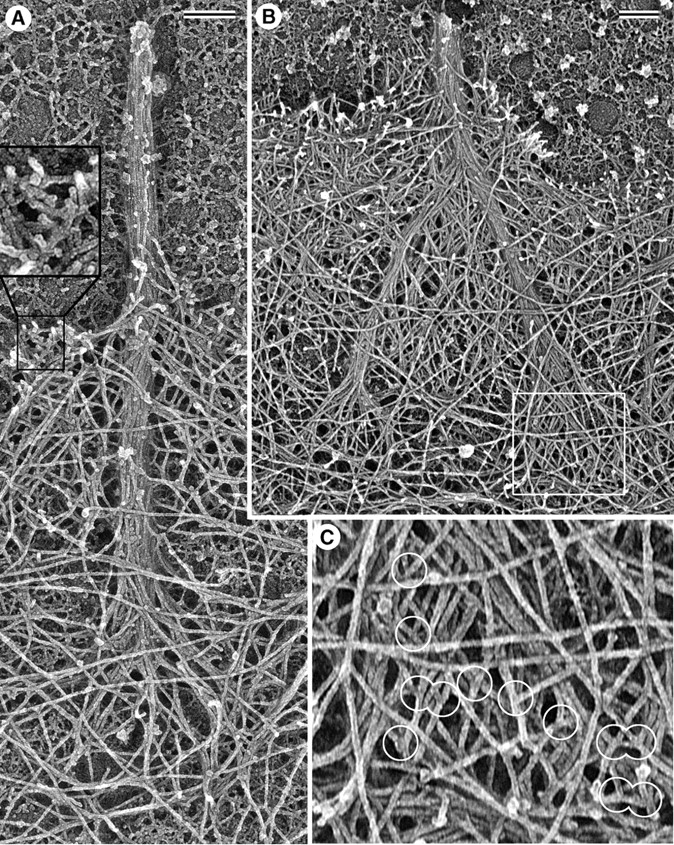
Filopodial filaments originate from the surrounding dendritic network. Platinum replica EM. (A) Filopodium contains a tight bundle of actin filaments that splays apart at its root and becomes an integral part of the surrounding network. Filaments in the roots are long compared with the branching network of the adjacent lamellipodium (inset). (B) Recently fused filopodium consists of two sub-bundles, each of which has a splayed root; the boxed region at the root of the right sub-bundle is enlarged in C and shows many branches (encircled) at which filopodial filaments originate. Rough background outside the cell edge is due to laminin coating of the glass coverslip. Bars, 0.2 μm.
Splayed filopodial roots apparently corresponded to aged Λ-precursors that treadmilled backward during filopodium growth. To identify Λ-precursors at a stage when they had not yet produced a filopodium, we performed correlative light microscopy and EM (Fig. 6). Putative Λ-precursors were identified in cells by fluorescence microscopy and relocalized after EM processing of the same cells. For these experiments, we used cells expressing GFP-fascin, which allowed us to compare parts of the Λ-precursor containing and not containing fascin.
Λ-precursors lacking fascin clearly displayed features of dendritic organization, such as short filaments, branching filaments, and numerous free filament ends (Fig. 6). Also, consistent with the idea of the transitional character of Λ-precursors, we found many rather long filaments within Λ-precursors, whereas long filaments were not frequent in the dendritic network outside Λ-precursors (Fig. 6, A and C). These long filaments apparently became enriched during transition of Λ-precursors into splayed filopodial roots, perhaps because of faster depolymerization of short filaments. The actin array in fascin-positive parts (Fig. 6 B) had a clearly bundled organization with densely packed filaments. The more proximal parts of actin bundles were not significantly enriched in fascin and displayed long, loosely aligned filaments (Fig. 6 B), suggesting that fascin-mediated bundling was delayed compared with accumulation of long filaments in the forming bundle. Thus, structural analysis of Λ-precursors and filopodial roots demonstrated enrichment of long filaments in these structures that apparently occurred before fascin-mediated bundling.
Structural organization of filopodia with known history
Because not every Λ-precursor produced a filopodium in kinetic studies, we performed correlative EM for cells with known history. For this purpose, we acquired time-lapse sequences of GFP-actin–expressing cells. After extraction and fixation, we prepared those cells for EM and analyzed filopodia formed in the course of the sequence (Fig. 7). Fig. 7 A illustrates the correlation between the last live image of one such cell, the image of the lysed cell, and the EM image of the same cell taken at low magnification comparable with that of light microscopy. During the 19-s interval between the last live image and the image of the lysed cell, the lamellipodium protruded ∼0.9 μm, which is evident in the superimposed image. The subsequent processing for EM did not introduce significant distortions into the structure of the lamellipodium because extracted light and low power EM images could be almost perfectly overlapped. Coincidence of light and EM features could also be seen at higher magnification, where brighter areas in fluorescence corresponded to denser actin arrays in EM (Fig. 7 B).
Figure 7.

Structural organization of filopodia with known history. Correlative live imaging and EM. (A) Overview of the cell lamellipodium at different stages of sample processing. GFP-actin fluorescence images taken just before (live, 16 s) and immediately after (lysed, 35 s) cell lysis were merged (live + lysed) in green and red channels, respectively; cell advance during the 19 s between images appears as red strip at the leading edge. Low magnification EM image of the same region (EM) is overlaid with actin fluorescence image of the lysed cell (EM + actin). (B) Boxed region in A is enlarged in B to show correlation between light and EM in more detail. Brighter areas in fluorescence image correspond to denser actin arrays in EM. (C) Detailed history of the boxed region in A. Time in seconds. The 35 s frame is taken from the lysed cell. Arrows of different color indicate position of individual nascent filopodia. (D) Enlarged EM of the boxed region in B showing the structure of nascent filopodia, whose history is presented in C; individual nascent filopodia are outlined in colors corresponding to colors of arrows in C. Some filaments converging into the bundle of the “blue” filopodium are highlighted in shades of blue. Boxes 1 and 2 are further enlarged in corresponding insets and show organization of the filopodial tip (1) and of the root of “green” filopodium (2); branching filaments are highlighted in green in the inset (2). See detail in text. Bars, 2 μm (A) and 0.2 μm (D).
Several filopodia were spontaneously formed during the total period of 35 s of this sequence. Fig. 7 C illustrates the history of three nascent filopodia of different age, which are color coded in yellow, blue, and green for convenience of description. The filopodium marked with a yellow arrow was the oldest one. This filopodium was formed in the course of fusion of two faint, converging linear densities existing at the beginning of the sequence. A bright spot of actin fluorescence, which appeared in the filopodium shaft in the second frame, allows one to recognize treadmilling and retrograde flow in this filopodium. The filopodium marked with the blue arrow was not visible in the first frame; at 4 s, it appeared as a Λ-precursor, which at 8 s could be seen near the edge, and which produced a faint filopodium by 12 s. The filopodium marked with the green arrow is the youngest one. It was formed from a Λ-precursor first visible at 12 s. Tips of all three filopodia converged by the end of the sequence, suggesting that they began fusing at the moment of extraction.
The detailed structural organization of the region containing all three filopodia is shown in Fig. 7 D. The root of the “yellow” filopodium consisted of two thin, fusing bundles corresponding to two converging lines in the first frame of the sequence. Each of these sub-bundles, when followed backward, splayed into individual filaments originating from the surrounding dendritic network (not depicted). The Λ-precursor of the “blue” filopodium that treadmilled backward during the sequence was identified with the splayed root of the blue filopodium in the EM image, indicating that splayed roots of filopodia indeed represent former Λ-precursors. Like in other EM images, filaments were collected into the bundle of the blue filopodium from the wide surrounding area (Fig. 7 D). In fluorescence images (Fig. 7 C), the splayed root became invisible already in 12 s frame, probably because of low fluorescence intensity. The root of the youngest “green” filopodium displayed many features of the dendritic organization of Λ-precursors. It had relatively high network density, many short filaments, frequent branching, and numerous free barbed ends (Fig. 7, inset 2 in D). Some filaments originating as a branch on another filament could be seen to enter the bundle of the green filopodium. These data support the idea that Λ-precursors initially represent a part of the dendritic network, but lose short filaments with age. An interesting feature of the green filopodium was that it was barely recognizable in the EM image because its filaments, although long, were not well-aligned, suggesting that filament cross-linking is not an early event during filopodial formation, in agreement with delayed recruitment of fascin to Λ-precursors.
In contrast to delayed bundling, the filament barbed ends at filopodial tips were in register, suggesting they were interacting with each other, even though they supposedly encountered each other just a few seconds before extraction. A substantial amount of granular material was associated with the tip of the fused filopodium (Fig. 7, inset 1 in D). The presence of tip-associated material may play an important role in filopodial formation, and we therefore investigated it in more detail.
Filopodial tip complex
In EM images, many filopodial tips were associated with a distinct structure, which had a rough granular surface and variable shape and size (Fig. 8 A). To test whether this tip complex was involved in physical association of filopodial barbed ends with each other, we incubated lysed cells overnight in phalloidin-containing buffer. Phalloidin prevented depolymerization of actin filaments during incubation, whereas dissociation of other proteins was allowed. We monitored dissociation of fascin or VASP using cells expressing GFP-fusion proteins. Although lysis removed soluble and weakly bound proteins, lysed cells initially retained most of filopodia-associated fascin and VASP. However, fascin was completely gone after incubation, whereas VASP remained (Fig. 8 B). EM of incubated cells revealed that filopodial bundles became loose, consistent with the loss of fascin, but the tip complexes were mostly preserved, consistent with retention of VASP, and filament barbed ends remained associated with each other and with the filopodial tip complex (Fig. 8 C). These results suggest that the tip complex physically links barbed ends in filopodia independently of fascin. Occasionally, the tip complex detached partially or completely during incubation. In such cases, released filopodial filaments completely splayed apart.
Figure 8.
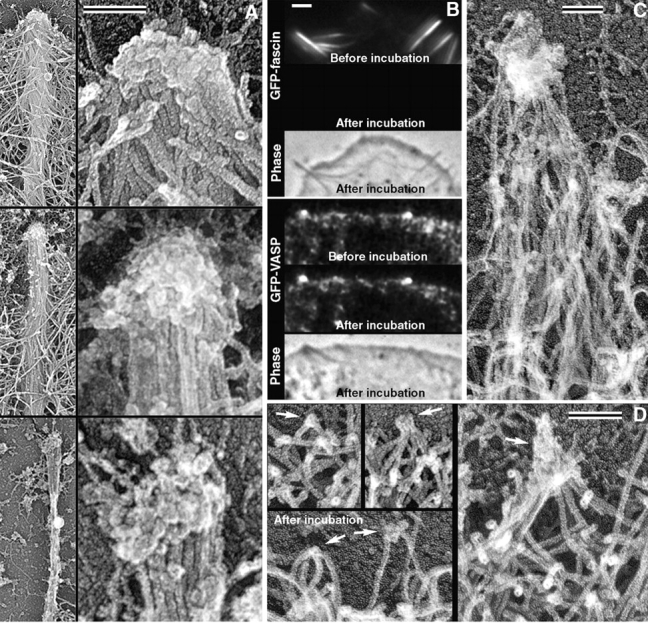
Filopodial tip complex. (A) Structure of tip complex in three filopodia seen by replica EM. Lower magnification, left; higher magnification, right. (B) Incubation of lysed cells in buffer causes dissociation of fascin (top) but not VASP (bottom) from filopodia. Phase contrast and GFP fluorescence images of cells transiently expressing indicated fusion proteins were taken immediately after cell lysis (“before incubation” phase images are not depicted) and after overnight incubation in phalloidin-containing buffer (“after incubation”). Acquisition and processing of fluorescence images was identical for each pair. (C) Filopodial bundle and tip complex after overnight incubation of the lysed cell in phalloidin-containing buffer. Replica EM. Filopodial tip complex keeps barbed ends of filopodial filaments together, whereas filaments within the bundle become loose. (D) Junctions between barbed ends of lamellipodial filaments (arrows) immediately after lysis or after incubation in buffer like in B or C (“after incubation”). Bars: 0.1 μm (A, C, and D), and 2 μm (B).
The next question we addressed was the origin of the tip complex. Because filopodia formation seemed to occur by gradual convergence of filaments from the dendritic network, we wondered whether smaller tip complexes existed in lamellipodia. Careful examination of the leading edge of lamellipodia indeed revealed junctions between barbed ends of two or more lamellipodial filaments (Fig. 8 D). Occasionally, additional material could be seen at the junction points, which may correspond to the tip complex of established filopodia. Association between filament-barbed ends was retained even after overnight incubation in the phalloidin-containing buffer (Fig. 8 D).
Discussion
Our kinetic and structural investigation of filopodial initiation in B16F1 melanoma cells demonstrated that filopodial bundles were formed by gradual reorganization of the lamellipodial dendritic network in a process that involved elongation of a subset of lamellipodial filaments, self-segregation of these filaments into filopodial precursors, and initiation of bundling at the tips of the precursors (Fig. 9). We propose that the mechanism of filopodia initiation is analogous to filopodial fusion, but that it begins at the level of individual filaments and gradually propagates to the fusion of thick bundles. We now discuss this hypothesis in detail.
Figure 9.

Convergent elongation model for filopodia initiation. (1) Lamellipodial network is formed by Arp2/3-mediated dendritic nucleation. Elongation of some barbed ends in the network is terminated by capping protein, but other barbed ends acquire a privileged status by binding a complex of molecules (tip complex) that allows them to elongate continuously. Ena/VASP proteins are likely members of the tip complex mediating protection from capping. (2) Privileged barbed ends drift laterally during elongation and collide with each other. Tip complex mediates clustering of privileged barbed ends upon collision. (3) Converged filaments with linked barbed ends continue to elongate together. Other laterally drifting barbed ends encounter and join the initial cluster of tip complexes. Multiple collisions of barbed ends during elongation lead to gradual clustering of their barbed ends, multimerization of associated tip complexes, and convergence of filaments. (4) The multimeric filopodial tip complex initiates filament cross-linking by recruiting and/or activating fascin, which allows the bundling process to keep up with the elongation and guarantee efficient pushing. (5) In the nascent filopodium, the filopodial tip complex retains its functions of promoting coordinated filament elongation and bundling, as well as fusion with other filopodia.
Dendritic network as a source of filopodial filaments
Arp2/3 complex has been suspected to play a role in filopodia initiation, but paradoxically, is not present in filopodial bundles. Our data do not support the idea of filopodial bundles arising from a nucleation center, but indicate that Arp2/3 complex produces a normal dendritic array which then becomes rearranged into parallel bundles through intermediate formation of Λ-precursors. Considering the ability to fuse displayed by filopodial bundles of all sizes, one can deduce that fusion may also occur below the resolution limit of the light microscope. Fusion of submicroscopic bundles or individual filaments may represent a mechanism of formation of Λ-precursors and explain their Λ-shape. The structure of Λ-precursors and filopodial roots was consistent with this idea, and showed that filaments composing bundles were collected from the surrounding area in an asynchronous manner, suggesting that Arp2/3-mediated actin filament nucleation provides a source of filaments to be rearranged into a bundle by gradual fusion.
Formation of long filaments as a prerequisite of filopodia initiation
The major difference between actin filament arrays in filopodia and lamellipodia is that filopodial filaments elongate continuously, reaching lengths of many micrometers, whereas lamellipodial filaments become capped after a brief period of elongation, and consequently, are relatively short (∼100 nm). Extensive elongation of filopodial filaments would seem to require a way to antagonize or protect against capping activity in the cytoplasm. A mechanism for protection is likely to involve proteins of the Ena/VASP family because these proteins are enriched at filopodial tips (Lanier et al., 1999; Rottner et al., 1999), they antagonize the terminating activity of capping protein in vitro, and their depletion from or targeting to the membrane leads to shorter or longer filaments, respectively (Bear et al., 2002). Our data support an idea that distributed filaments with prebound Ena/VASP proteins are gradually brought together to form a filopodium. The kinetics of GFP-VASP demonstrated a correlation between coalescence of VASP and filopodia initiation. At present, it is not clear whether VASP remains associated with the same barbed end for a long time or frequently switches its protégé. The persistence of VASP at the leading edge and filopodial tips after overnight incubation in buffer is evidence that at least some actin filaments can remain tightly associated with VASP. However, in a living cell, the association of VASP with barbed ends may be subject to regulation that affects the degree of protection it confers against capping.
On the assumption that GFP-VASP reports on the behavior of the population of privileged barbed ends, the gradual coalescence of VASP to the tips of Λ-precursors reflects the gradual segregation of longer filaments into Λ-precursors. Consistent with this coalescence process, actin arrays within Λ-precursors were enriched in long filaments and depleted in the lamellipodial markers, Arp2/3 complex, cortactin, and capping protein. Long filaments became more clearly visible when Λ-precursors treadmilled away from the leading edge and became filopodial roots. Long filaments in lamellipodia have been also observed by other EM techniques (Small, 1988; Lewis and Bridgman, 1992; Resch et al., 2002), although no distinction has been made between Λ-precursors and other parts of lamellipodia.
Other studies have indicated that Ena/VASP family members contribute to the formation of filopodia, but the mechanism of their involvement has not been elucidated. In Dictyostelium, the VASP family homologue (DdVASP) is essential for filopodia formation (Han et al., 2002). Induction of filopodia by Irs53 involved recruitment of a member of the Ena/VASP family, Mena (Krugmann et al., 2001). Our data suggest that the mechanism of action of Ena/VASP proteins is through the formation of privileged filaments that can elongate persistently.
Clustering of barbed ends as a mechanism of segregation of filopodial filaments
The persistent elongation of filaments by itself would not result in their local accumulation unless they were able to associate with each other. Consistent with this idea, we found structural interaction between filament barbed ends that was mediated by the filopodial tip complex in a fascin-independent manner. This result suggests that bundling and barbed-end interaction are mediated by different molecules. In addition, we detected formation of junctions between filament barbed ends at the extreme leading edge. Similar to filopodial tips, these junctions were stable even after overnight incubation in buffer and were frequently associated with additional material. The filopodial tip complexes described here may be similar to the material associated with distal filament ends in filopodia and lamellipodia of fibroblasts (Small et al., 1982) and neuronal growth cones (Lewis and Bridgman, 1992). Interaction between barbed ends appears to be responsible for the Λ-configuration of actin arrays at different scales from junctions between individual filaments to Λ-precursors to fusing bundles.
Privileged barbed ends seem to combine the ability for continuous elongation with multimerization potential. The cross-linking molecules mediating junction formation between barbed ends remain unclear, but they are likely to be components of the filopodial tip complex. The molecular composition of the filopodial tip complex remains to be established. However, proteins previously found to localize specifically to filopodial tips (see Introduction), including Ena/VASP proteins, are predicted to be members of this complex. One possibility is that Ena/VASP proteins, which mediate protection of barbed ends from capping, may also work as barbed end “glue” because of their ability to oligomerize (Bachmann et al., 1999). In support of this idea, a domain mediating oligomerization of Mena has been shown to be required for full function of Mena in cell motility (Loureiro et al., 2002). Another possibility is that additional (yet unidentified) molecules within the filopodial tip complex mediate interaction between barbed ends. These possibilities are not mutually exclusive, and the hypothetical barbed end linking molecules may act indirectly through Ena/VASP proteins, which would have the benefit of rendering the anti-capping and clustering capabilities to the same subset of filaments.
The combination of continuous elongation and self-association properties of privileged barbed ends allows one to explain how the privileged filaments in the dendritic network become gradually self-segregated during filopodia initiation. The lamellipodial filaments, on average, have a diagonal orientation of about ±35 degrees with respect to the leading edge (Maly and Borisy, 2001). During elongation, the barbed ends of diagonally oriented filaments drift laterally along the edge, which increases chances of their collision. Such lateral drift of lamellipodial filaments was proposed to mediate formation of filopodia due to activity of bundling proteins (Small et al., 1982, 1998). We propose that the cross-linking molecules at the barbed ends of colliding privileged filaments cause them to associate with each other and travel together. Multiple collisions lead to clustering of barbed ends of filaments and multimerization of individual barbed-end–associated molecular complexes, producing a filopodial tip complex.
Filament bundling as a mechanism of stabilization
Individual long actin filaments are not efficient at pushing. Because of their low persistence length, they bend rather than push. Filament cross-linking along the length is thought to be a solution for this problem (Mogilner and Oster, 1996). The existing evidence suggests that fascin is the major actin cross-linking protein in filopodia (Bartles, 2000; Kureishy et al., 2002) that is required for filopodia maintenance. In support of this idea, we found that fascin enrichment in actin arrays correlated with tight bundling of actin filaments, and fascin dissociation from filopodial bundles resulted in filament unbundling. To allow for efficient pushing, cross-linking of growing filaments is predicted to occur soon in the course of actin polymerization so that the effective length of the filament after the last cross-link remains short. Indeed, fascin is enriched in the distal section of filopodia (Cohan et al., 2001, and this paper) suggesting that its association with the growing actin bundles occurs in parallel with actin assembly. In contrast, association of α-actinin with fast-protruding filopodia and lamellipodia is detected toward their rear, indicating that the role of α-actinin is not to provide the pushing efficiency to growing filaments.
Fascin recruitment during filopodia initiation was not the earliest event. Instead, GFP-fascin rather abruptly appeared at the vertex of a preformed Λ-precursor. We suggest that recruitment and/or activation of fascin to tips of Λ-precursors cross-links the long filaments accumulated there, thus completing the initiation of filopodial bundle formation.
Convergent elongation mechanism of filopodium initiation
Based on our findings, we propose a convergent elongation model for filopodial initiation (Fig. 9), which stipulates that filopodia are formed by reorganization of the dendritic network formed in an Arp2/3-dependent manner. The key assumption of this model is that some filaments within the lamellipodial dendritic network acquire privileged status by binding a set of molecules to their barbed ends, which protect them from capping, and mediate association of barbed ends with each other on collision. Ena/VASP proteins are likely candidates for the role of protection from capping. The glue molecule remains to be established. Multiple collisions of privileged filaments during elongation lead to gradual clustering of their barbed ends and multimerization of associated barbed-end complexes. A set of privileged filaments originating from distant sites of the dendritic network and converging to the same spot forms a Λ-precursor, and aggregated barbed-end complexes form the tip complex of the future filopodium. The filopodial tip complex initiates filament cross-linking by recruiting and/or activating fascin, which allows the bundling process to keep up with elongation and guarantee efficient pushing. Initiated filopodia elongate and attain steady-state by the filament treadmilling mechanism. The filopodial tip complex remains associated with the growing tip, allowing for continuous elongation of filopodial filaments and mediating filopodia fusion on collision.
Initiation of filopodial bundles within the lamellipodial network necessarily leads to their birth in the form of microspikes. They die either during ruffling or in the form of retraction fibers, when a cell decides to move in a different direction. In between, the microspikes may at some point elongate faster than lamellipodial advance to form a conventional filopodium. Additional evidence for the relatedness of these structures comes from the fact that Cdc42 induces cell retraction and formation of retraction fibers along with formation of genuine filopodia (Kozma et al., 1995). Consequently, the three morphological types of peripheral actin bundle, microspikes, filopodia, and retraction fibers, may be considered as transient states of the same core structure differing primarily in their relationship to the membrane and in the state of their cycle. Also, they may differ in protein composition and dynamics. Filopodia and retraction fibers are predicted to contain actin–membrane linkers, like ERM proteins, which are unnecessary for microspikes. The remote position of barbed ends in filopodia and retraction fibers may require a myosin motor, for example, myosin X (Berg and Cheney, 2002), to deliver building components to the tip.
The pathway of filopodia initiation established in this paper has a remarkable similarity to the mechanism of formation of filopodial-like bundles in vitro (unpublished data). In that work, we found that beads coated with Arp2/3-activating proteins induced formation of radial actin bundles when capping activity in cytoplasmic extracts was decreased. In vitro bundles displayed many filopodial characteristics; they had uniform polarity, grew at the barbed end, were enriched in fascin, and lacked Arp2/3 complex, capping protein, and α-actinin. As in the present paper, individual filaments in bundles in vitro originated from the dendritic network near the bead, and a decreased rate of capping in the extracts allowed them to elongate and be bundled by fascin.
A tightly packed parallel actin bundle, which is a hallmark of filopodia, can also be found in other organelles across tissues and organisms (Bartles, 2000; DeRosier and Tilney, 2000). Our findings that filopodia in B16F1 cells during normal locomotion were formed by reorganization of the dendritic network raises the possibility of a similar pathway for initiation of other parallel bundles, but does not exclude other mechanisms of bundle formation in other cells types or under other circumstances. For example, proteins of the formin family have recently been shown to nucleate actin filaments in vitro (Pruyne et al., 2002; Sagot et al., 2002b) and induce actin cables in yeast in vivo in the absence of the Arp2/3 complex (Evangelista et al., 2002; Sagot et al., 2002a). Thus, it remains an open question whether Arp2/3- or formin-dependent mechanisms operate in other cases, and whether they are exclusive or can cooperate.
In conclusion, we investigated the pathway of filopodia initiation in B16F1 cells and formulated the convergent elongation model for filopodia formation. Although many assumptions of this model remain to be tested, it provides a conceptual framework for further studies aimed at explicitly identifying participating molecules and their precise roles.
Materials and methods
Cells and reagents
Mouse melanoma B16F1 cell line stably expressing EGFP-β-actin, as well as untransfected B16F1 cells, were provided by Dr. C. Ballestrem (Weizmann Institute of Science, Rehovot, Israel) and were cultured as described previously (Ballestrem et al., 1998). For experiments, cells were plated onto coverslips coated with 10–25 μg/ml laminin (Invitrogen) and blocked with 0.1 μg/ml heat-inactivated BSA. For live imaging, cells were transferred from DME into L-15 medium at least 2 h before observation.
EGFP-VASP–expressing construct was obtained from Drs. J. Bear and F. Gertler (Massachusetts Institute of Technology, Cambridge, MA). EGFP-fascin (Adams and Schwartz, 2000) was provided by Dr. J. Adams (Cleveland Clinic Foundation, Cleveland, OH). EGFP-α-actinin (Edlund et al., 2001) was obtained from Dr. C. Otey (University of North Carolina, Chapel Hill, NC). For transient protein expression, cells were transfected with FuGENE™ 6 (Roche) according to the manufacturer's recommendation.
The following primary antibodies were used for immunostaining: affinity-purified rabbit antibody to ARPC5 subunit of Arp2/3 (unpublished data), capping protein antibody (Schafer et al., 1994; provided by Dr. D.A. Schafer, University of Virginia, Charlottesville, VA), and mouse monoclonal cortactin antibody, clone 4F11 (Upstate Biotechnology). Secondary TRITC-conjugated antibodies were purchased from Sigma-Aldrich.
Microscopy
Light microscopy was performed using an inverted microscope (Eclipse or Diaphot 300; Nikon) equipped with a Plan 100×, 1.3 NA objective and a back-illuminated cooled CCD camera (model CH250; Roper Scientific) or a slow-scan cooled CCD camera (model CH350; Photometrics), respectively, driven by MetaMorph® imaging software (Universal Imaging Corp.). FITC filter set was used for GFP-fusion protein observations, and Cy3 and Texas Red filter sets were used for rhodamine and Texas Red imaging, respectively. For live imaging, cells were kept on the microscope stage at 36–37°C during observation.
Immunostaining was performed after cell extraction for 3–10 min with 1% Triton X-100 in PEM buffer (100 mM Pipes, pH 6.9, 1 mM MgCl2, and 1 mM EGTA), containing 4% polyethylene glycol, mol wt 40,000 (SERVA), and 2 μM phalloidin (Sigma-Aldrich), followed by fixation with 0.2% glutaraldehyde and quenching with NaBH4. For phalloidin staining, 0.033 μM Texas Red phalloidin (Molecular Probes, Inc.) was added to the extraction solution instead of unlabeled phalloidin. GFP-fascin–expressing cells were stained with Texas Red phalloidin after fixation of unextracted cells, followed by permeabilization with 1% Triton X-100 in PBS.
For determination of protein dissociation from the cytoskeletons, GFP-fascin or GFP-VASP cells were grown on locator coverslips, extracted as for immunostaining, and images of cells expressing fusion proteins were acquired within 20 min after extraction. Then cells were washed twice with PEM containing 2 μM phalloidin and left in this buffer overnight at RT. After incubation, another set of images of the same cells was acquired. Platinum replica EM and correlative light EM were performed as described previously (Svitkina and Borisy, 1998).
Acknowledgments
We thank Drs. C. Ballestrem, J. Bear, F. Gertler, J. Adams, C. Otey, and D.A. Schafer for gifts of reagents and Dr. M. Mejillano, Dr. A. Biyasheva, and I. Maly for critical reading of the manuscript.
Supported by NIH Grants GM 62431 and IU 54 GM 63126 to G.G. Borisy.
Footnotes
Abbreviations used in this paper: EM, electron microscopy; Ena/VASP, enabled/vasodilator-stimulated phosphoprotein.
References
- Adams, J.C., and M.A. Schwartz. 2000. Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J. Cell Biol. 150:807–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann, C., L. Fischer, U. Walter, and M. Reinhard. 1999. The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding, and actin bundle formation. J. Biol. Chem. 274:23549–23557. [DOI] [PubMed] [Google Scholar]
- Ballestrem, C., B. Wehrle-Haller, and B.A. Imhof. 1998. Actin dynamics in living mammalian cells. J. Cell Sci. 111:1649–1658. [DOI] [PubMed] [Google Scholar]
- Bamburg, J.R. 1999. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 15:185–230. [DOI] [PubMed] [Google Scholar]
- Bartles, J.R. 2000. Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 12:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear, J.E., T.M. Svitkina, M. Krause, D.A. Schafer, J.J. Loureiro, G.A. Strasser, I.V. Maly, O.Y. Chaga, J.A. Cooper, G.G. Borisy, and F.B. Gertler. 2002. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 109:509–521. [DOI] [PubMed] [Google Scholar]
- Berg, J.S., and R.E. Cheney. 2002. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat. Cell Biol. 4:246–250. [DOI] [PubMed] [Google Scholar]
- Borisy, G.G., and T.M. Svitkina. 2000. Actin machinery: pushing the envelope. Curr. Opin. Cell Biol. 12:104–112. [DOI] [PubMed] [Google Scholar]
- Carlier, M.F., and D. Pantaloni. 1997. Control of actin dynamics in cell motility. J. Mol. Biol. 269:459–467. [DOI] [PubMed] [Google Scholar]
- Cohan, C.S., E.A. Welnhofer, L. Zhao, F. Matsumura, and S. Yamashiro. 2001. Role of the actin bundling protein fascin in growth cone morphogenesis: localization in filopodia and lamellipodia. Cell Motil. Cytoskeleton. 48:109–120. [DOI] [PubMed] [Google Scholar]
- Cooper, J.A., and D.A. Schafer. 2000. Control of actin assembly and disassembly at filament ends. Curr. Opin. Cell Biol. 12:97–103. [DOI] [PubMed] [Google Scholar]
- DePasquale, J.A., and C.S. Izzard. 1991. Accumulation of talin in nodes at the edge of the lamellipodium and separate incorporation into adhesion plaques at focal contacts in fibroblasts. J. Cell Biol. 113:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosier, D.J., and L.G. Tilney. 2000. F-actin bundles are derivatives of microvilli: What does this tell us about how bundles might form? J. Cell Biol. 148:1–6. [PMC free article] [PubMed] [Google Scholar]
- Edlund, M., M.A. Lotano, and C.A. Otey. 2001. Dynamics of alpha-actinin in focal adhesions and stress fibers visualized with alpha-actinin-green fluorescent protein. Cell Motil. Cytoskeleton. 48:190–200. [DOI] [PubMed] [Google Scholar]
- Evangelista, M., D. Pruyne, D.C. Amberg, C. Boone, and A. Bretscher. 2002. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat. Cell Biol. 4:32–41. [DOI] [PubMed] [Google Scholar]
- Flanagan, L.A., J. Chou, H. Falet, R. Neujahr, J.H. Hartwig, and T.P. Stossel. 2001. Filamin A, the Arp2/3 complex, and the morphology and function of cortical actin filaments in human melanoma cells. J. Cell Biol. 155:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. [DOI] [PubMed] [Google Scholar]
- Han, Y.H., C.Y. Chung, D. Wessels, S. Stephens, M.A. Titus, D.R. Soll, and R.A. Firtel. 2002. Requirement of a vasodilator-stimulated phosphoprotein family member for cell adhesion, the formation of filopodia, and chemotaxis in Dictyostelium. J. Biol. Chem. 277:49877–49887. [DOI] [PubMed] [Google Scholar]
- Higgs, H.N., and T.D. Pollard. 2001. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu. Rev. Biochem. 70:649–676. [DOI] [PubMed] [Google Scholar]
- Ho, H.Y., R. Rohatgi, L. Ma, and M.W. Kirschner. 2001. CR16 forms a complex with N-WASP in brain and is a novel member of a conserved proline-rich actin-binding protein family. Proc. Natl. Acad. Sci. USA. 98:11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K., K. Hammar, P.J. Smith, and R. Oldenbourg. 1999. a. Arrangement of radial actin bundles in the growth cone of Aplysia bag cell neurons shows the immediate past history of filopodial behavior. Proc. Natl. Acad. Sci. USA. 96:7928–7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K., K. Hammar, P.J. Smith, and R. Oldenbourg. 1999. b. Birefringence imaging directly reveals architectural dynamics of filamentous actin in living growth cones. Mol. Biol. Cell. 10:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma, R., S. Ahmed, A. Best, and L. Lim. 1995. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 15:1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranewitter, W.J., C. Danninger, and M. Gimona. 2001. GEF at work: Vav in protruding filopodia. Cell Motil. Cytoskeleton. 49:154–160. [DOI] [PubMed] [Google Scholar]
- Krugmann, S., I. Jordens, K. Gevaert, M. Driessens, J. Vandekerckhove, and A. Hall. 2001. Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr. Biol. 11:1645–1655. [DOI] [PubMed] [Google Scholar]
- Kureishy, N., V. Sapountzi, S. Prag, N. Anilkumar, and J.C. Adams. 2002. Fascins, and their roles in cell structure and function. Bioessays. 24:350–361. [DOI] [PubMed] [Google Scholar]
- Langanger, G., J. de Mey, M. Moeremans, G. Daneels, M. de Brabander, and J.V. Small. 1984. Ultrastructural localization of alpha-actinin and filamin in cultured cells with the immunogold staining (IGS) method. J. Cell Biol. 99:1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier, L.M., M.A. Gates, W. Witke, A.S. Menzies, A.M. Wehman, J.D. Macklis, D. Kwiatkowski, P. Soriano, and F.B. Gertler. 1999. Mena is required for neurulation and commissure formation. Neuron. 22:313–325. [DOI] [PubMed] [Google Scholar]
- Lewis, A.K., and P.C. Bridgman. 1992. Nerve growth cone lamellipodia contain two populations of actin filaments that differ in organization and polarity. J. Cell Biol. 119:1219–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., E.S. Kim, and E.L. Bearer. 2002. Arp2/3 complex is required for actin polymerization during platelet shape change. Blood. 99:4466–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro, J.J., D.A. Rubinson, J.E. Bear, G.A. Baltus, A.V. Kwiatkowski, and F.B. Gertler. 2002. Critical roles of phosphorylation and actin binding motifs, but not the central proline-rich region, for Ena/vasodilator-stimulated phosphoprotein (VASP) function during cell migration. Mol. Biol. Cell. 13:2533–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallavarapu, A., and T. Mitchison. 1999. Regulated actin cytoskeleton assembly at filopodium tips controls their extension and retraction. J. Cell Biol. 146:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maly, I.V., and G.G. Borisy. 2001. Self-organization of a propulsive actin network as an evolutionary process. Proc. Natl. Acad. Sci. USA. 98:11324–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki, H., T. Sasaki, Y. Takai, and T. Takenawa. 1998. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 391:93–96. [DOI] [PubMed] [Google Scholar]
- Mogilner, A., and G. Oster. 1996. Cell motility driven by actin polymerization. Biophys. J. 71:3030–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 81:53–62. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., L. Blanchoin, and R.D. Mullins. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576. [DOI] [PubMed] [Google Scholar]
- Pruyne, D., M. Evangelista, C. Yang, E. Bi, S. Zigmond, A. Bretscher, and C. Boone. 2002. Role of formins in actin assembly: nucleation and barbed-end association. Science. 297:612–615. [DOI] [PubMed] [Google Scholar]
- Qualmann, B., and R.B. Kelly. 2000. Syndapin isoforms participate in receptor-mediated endocytosis and actin organization. J. Cell Biol. 148:1047–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resch, G.P., K.N. Goldie, A. Krebs, A. Hoenger, and J.V. Small. 2002. Visualisation of the actin cytoskeleton by cryo-electron microscopy. J. Cell Sci. 115:1877–1882. [DOI] [PubMed] [Google Scholar]
- Rohatgi, R., L. Ma, H. Miki, M. Lopez, T. Kirchhausen, T. Takenawa, and M.W. Kirschner. 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 97:221–231. [DOI] [PubMed] [Google Scholar]
- Rohatgi, R., H.Y. Ho, and M.W. Kirschner. 2000. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4, 5-bisphosphate. J. Cell Biol. 150:1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner, K., B. Behrendt, J.V. Small, and J. Wehland. 1999. VASP dynamics during lamellipodia protrusion. Nat. Cell Biol. 1:321–322. [DOI] [PubMed] [Google Scholar]
- Sagot, I., S.K. Klee, and D. Pellman. 2002. a. Yeast formins regulate cell polarity by controlling the assembly of actin cables. Nat. Cell Biol. 4:42–50. [DOI] [PubMed] [Google Scholar]
- Sagot, I., A.A. Rodal, J. Moseley, B.L. Goode, and D. Pellman. 2002. b. An actin nucleation mechanism mediated by Bni1 and profilin. Nat. Cell Biol. 4:626–631. [DOI] [PubMed] [Google Scholar]
- Schafer, D.A., Y.O. Korshunova, T.A. Schroer, and J.A. Cooper. 1994. Differential localization and sequence analysis of capping protein beta-subunit isoforms of vertebrates. J. Cell Biol. 127:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, J.V. 1994. Lamellipodia architecture: actin filament turnover and the lateral flow of actin filaments during motility. Semin. Cell Biol. 5:157–163. [DOI] [PubMed] [Google Scholar]
- Small, J.V. 1988. The actin cytoskeleton. Electron Microsc. Rev. 1:155–174. [DOI] [PubMed] [Google Scholar]
- Small, J.V., G. Rinnerthaler, and H. Hinssen. 1982. Organization of actin meshworks in cultured cells: the leading edge. Cold Spring Harb. Symp. Quant. Biol. 46:599–611. [DOI] [PubMed] [Google Scholar]
- Small, J.V., K. Rottner, I. Kaverina, and K.I. Anderson. 1998. Assembling an actin cytoskeleton for cell attachment and movement. Biochim. Biophys. Acta. 1404:271–281. [DOI] [PubMed] [Google Scholar]
- Small, J.V., T. Stradal, E. Vignal, and K. Rottner. 2002. The lamellipodium: where motility begins. Trends Cell Biol. 12:112–120. [DOI] [PubMed] [Google Scholar]
- Stradal, T., K.D. Courtney, K. Rottner, P. Hahne, J.V. Small, and A.M. Pendergast. 2001. The Abl interactor proteins localize to sites of actin polymerization at the tips of lamellipodia and filopodia. Curr. Biol. 11:891–895. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1998. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol. 298:570–592. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. a. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. b. Progress in protrusion: the tell-tale scar. Trends Biochem. Sci. 24:432–436. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., A.B. Verkhovsky, K.M. McQuade, and G.G. Borisy. 1997. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J. Cell Biol. 139:397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, A.M., A.V. Karginov, A.W. Kinley, S.A. Weed, Y. Li, J.T. Parsons, and J.A. Cooper. 2001. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr. Biol. 11:370–374. [DOI] [PubMed] [Google Scholar]