

Abstract
Epithelial tight junctions regulate paracellular permeability, restrict apical/basolateral intramembrane diffusion of lipids, and have been proposed to participate in the control of epithelial cell proliferation and differentiation. Previously, we have identified ZO-1–associated nucleic acid binding proteins (ZONAB), a Y-box transcription factor whose nuclear localization and transcriptional activity is regulated by the tight junction–associated candidate tumor suppressor ZO-1. Now, we found that reduction of ZONAB expression using an antisense approach or by RNA interference strongly reduced proliferation of MDCK cells. Transfection of wild-type or ZONAB-binding fragments of ZO-1 reduced proliferation as well as nuclear ZONAB pools, indicating that promotion of proliferation by ZONAB requires its nuclear accumulation. Overexpression of ZONAB resulted in increased cell density in mature monolayers, and depletion of ZONAB or overexpression of ZO-1 reduced cell density. ZONAB was found to associate with cell division kinase (CDK) 4, and reduction of nuclear ZONAB levels resulted in reduced nuclear CDK4. Thus, our data indicate that tight junctions can regulate epithelial cell proliferation and cell density via a ZONAB/ZO-1–based pathway. Although this regulatory process may also involve regulation of transcription by ZONAB, our data suggest that one mechanism by which ZONAB and ZO-1 influence proliferation is by regulating the nuclear accumulation of CDK4.
Keywords: polarized epithelia; tight junctions; cell cycle; MAGUK; CDK4
Introduction
Epithelial cells interact with each other via specialized intercellular junctions that are critical for the development and function of epithelial tissues. Tight junctions are the most apical intercellular junction, and form a morphological border between the apical and basolateral cell surface domains (Farquhar and Palade, 1963). They regulate paracellular permeability and prevent intermixing of apical and basolateral membrane components (Cereijido et al., 1998). Tight junctions have also been proposed to function in the regulation of epithelial cell proliferation and differentiation; however, the involved components and affected signaling pathways are not well understood (Mitic and Anderson, 1998; Stevenson and Keon, 1998; Tsukita et al., 1999; Benais-Pont et al., 2001).
Tight junctions are composed of different types of membrane proteins that mediate cell–cell adhesion and form the diffusion barriers. The membrane proteins interact with a complex of cytoplasmic plaque that contains different types of proteins and is linked to the actin cytoskeleton (Cereijido et al., 2000; Gonzalez-Mariscal et al., 2000; Zahraoui et al., 2000; Fanning, 2001; Tsukita et al., 2001; D'Atri and Citi, 2002). ZO-1, one of these tight junction-associated cytosolic proteins, is a member of the membrane-associated guanylate kinases (MAGUK) family of proteins, which possess several PDZ domains, one SH3 domain, and a domain homologous to yeast guanylate kinase (Stevenson et al., 1986; Itoh et al., 1993; Willott et al., 1993; Woods and Bryant, 1993). Although ZO-1 specifically associates with tight junctions in epithelial cells (Stevenson et al., 1986), the protein has been reported to appear in the nucleus during proliferation (Gottardi et al., 1996). ZO-1 is down-regulated in breast cancer tissues and during corneal wound repair, suggesting that expression levels of ZO-1 are related to the proliferation state of epithelial cells (Hoover et al., 1998; Cao et al., 2002). This is also supported by the observations that ZO-1 becomes stabilized in confluent MDCK cells and that its expression levels increase with cell density (Gumbiner et al., 1991; Balda and Matter, 2000).
We recently identified ZO-1–associated nucleic acid binding protein (ZONAB),* a Y-box transcription factor that specifically binds to the SH3 domain of ZO-1 and regulates the expression of the proto-oncogene erbB-2 in a cell density–dependent manner (Balda and Matter, 2000). ZO-1 functions as an inhibitor of ZONAB and controls accumulation of ZONAB in the nucleus by cytoplasmic sequestration. MDCK epithelial cells express two isoforms of ZONAB (ZONAB-A and -B) that differ in a 68-amino acid insertion. Both isoforms bind to ZO-1 and associate with intercellular junctions (Balda and Matter, 2000).
Y-box transcription factors are multifunctional regulators of gene expression and have been proposed to play a general role in promoting proliferation (Bargou et al., 1997; Matsumoto and Wolffe, 1998). Interestingly, low density epithelial cells grow as islands of cells that are forming intercellular junctions while they are still proliferating. Cell cycle arrest is consequently not triggered by simple cell–cell contact, but when monolayers reach a critical cell density. Nuclear ZONAB levels are high in proliferating cells in peripheral regions of growing islands that have still a low cell density; but in central regions of islands where the cell density is high, nuclear ZONAB levels are low (Balda and Matter, 2000). Here, we present data indicating that this differential distribution of ZONAB reflects a role in the regulation of epithelial proliferation and cell density. Our results show that inhibition of ZONAB function by partial depletion of total cellular pools or by increased expression of ZO-1 inhibits Go/G1 to S-phase transition and suggest that reduced proliferation is at least in part due to an inhibition of the CDK4/retinoblastoma protein pathway.
Results
Regulation of epithelial proliferation by ZONAB
ZONAB expression is high in proliferating but low in growth-arrested MDCK cells (Fig. 1 A), suggesting that high expression levels might be required for cell proliferation (Balda and Matter, 2000). To test this, we used an antisense RNA approach to reduce ZONAB expression. Stable expression of ZONAB antisense RNA resulted in a reduction of cellular ZONAB levels by about >50% (Fig. 1 B), whereas expression of a 50% homologous Y-box factor, YB-1, was not affected (unpublished data).
Figure 1.
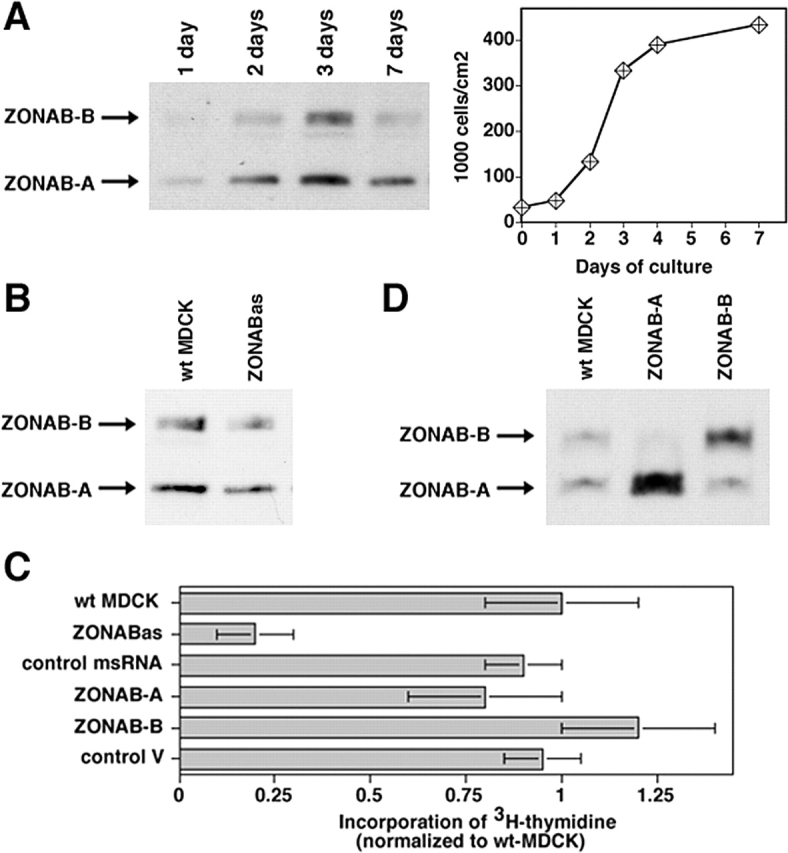
Regulation of proliferation by ZONAB. (A) MDCK cells were grown for the indicated amounts of days. Cultures were harvested and equal amounts of protein were loaded on SDS-PAGE gels for analysis of ZONAB expression by immunoblotting. The bands representing the ZONAB-A and -B isoforms are indicated. Parallel cultures were trypsinized, and the cells were counted to determine the cell density. (B) Reduction of ZONAB expression in low density cells by ZONAB antisense RNA. Proliferating wild-type MDCK cells (wt MDCK) and cells expressing ZONAB antisense RNA (ZONABas) were harvested, and equal amounts of protein were loaded on SDS-PAGE gels for analysis of ZONAB expression by immunoblotting. Based on densitometric scanning of the immunoblots, expression of ZONAB was reduced by >50% in cells transfected with the antisense construct. (C) Proliferation of low density MDCK cells expressing ZONAB antisense RNA (ZONABas), a control missense RNA (msRNA), overexpressing either one of the ZONAB isoforms, or transfected with a control cDNA were analyzed by measuring incorporation of [3H]thymidine. Data were normalized to wild-type cells (shown are means ± 1 SD of at least three independent clones per construct that were analyzed in three independent experiments with quadruplicate cultures). Note that incorporation of [3H]thymidine by ZONABas cells was reduced by >70% (t test; P < 0.01). (D) Overexpression of ZONAB isoforms in low density MDCK cells. Proliferating wild-type MDCK cells (wt MDCK) and cells overexpressing ZONAB-A or ZONAB-B were harvested, and equal amounts of protein were loaded on SDS-PAGE gels for analysis of ZONAB expression by immunoblotting.
Next, we assessed proliferation by measuring [3H]thymidine incorporation in low density cultures. Fig. 1 C shows that expression of ZONAB antisense RNA resulted in a fourfold reduction of [3H]thymidine incorporation, indicating that high levels of ZONAB are indeed required for efficient proliferation. Further increases in ZONAB expression levels by overexpression of either ZONAB-A or ZONAB-B did not result in increased incorporation of [3H]thymidine, suggesting that the endogenous expression levels are sufficient for efficient proliferation (Fig. 1, C and D). These data indicate that ZONAB participates in the regulation of epithelial proliferation.
ZONAB localizes to the nucleus as well as tight junctions in proliferating cells, but is not detectable in the nucleus of nonproliferating high density cells (Balda and Matter, 2000), suggesting that accumulation of ZONAB in the nucleus may be required for efficient proliferation. ZO-1 levels are low in proliferating cells and increase with cell density, and overexpression of ZO-1 inhibits the nuclear accumulation of ZONAB (Balda and Matter, 2000); hence, ZO-1 may regulate proliferation by preventing ZONAB from accumulating in the nucleus. To test whether high levels of ZO-1 expression reduce proliferation, we used cell lines in which ZO-1 was three- to fivefold overexpressed in low confluent cells; an increase that is comparable to the up-regulation of the protein in wild-type cells once they reach high cell densities (Fig. 2, A and B). Next, we assessed proliferation by measuring [3H]thymidine incorporation in low density cultures. Fig. 2 C shows that exogenous expression of ZO-1 resulted in a fourfold reduction of [3H]thymidine incorporation, indicating that high levels of ZO-1 expression indeed reduced proliferation.
Figure 2.

Regulation of proliferation by ZO-1. (A and B) Expression of ZO-1 in wild-type and transfected MDCK cells. Wild-type MDCK cells (A) or wild-type (wt MDCK) and ZO-1–overexpressing (ZO-1 1/21 and ZO-1 2/3) cells (B) were grown for the indicated number of days as described in Fig. 1 A. Cells were then harvested, and equal amounts of protein were loaded on SDS-PAGE gels for analysis of ZO-1 expression by immunoblotting. Note that ZO-1 was overexpressed in transfected proliferating cells to a similar extent as it was up-regulated in mature monolayers. (C) Incorporation of [3H]thymidine by low density MDCK cells stably transfected with ZO-1 or HA-tagged ZO-1 with (HA-ZO-1) or without (HA-ZO-1ΔSH3) the SH3 domain, or constructs containing specified domains. ZO-1/ZONAB indicates data obtained from double transfected cells overexpressing both proteins. Data were normalized to wild-type cells (shown are means ± 1 SD of at least three independent clones per construct that were analyzed in three independent experiments with quadruplicate cultures). Note that all cell lines expressing constructs containing the SH3 domain of ZO-1 exhibited significantly reduced [3H]thymidine incorporation (t test; P < 0.05). (D) Domain structure of ZO-1. PDZ, PSD95-DlgA-ZO-1 homology domain; SH3, src homology domain 3; GUK, guanylate kinase homology domain.
ZO-1 interacts with ZONAB via its SH3 domain (Balda and Matter, 2000); hence, if ZO-1 inhibits proliferation via ZONAB, the antiproliferative effect of ZO-1 should depend on its SH3 domain. To determine whether the ZONAB binding site of ZO-1 is needed for the regulation of proliferation, we generated MDCK cells expressing HA-tagged full-length or truncated ZO-1 and measured [3H]thymidine incorporation in low density cultures. Fig. 2 C shows that HA-ZO-1 inhibited [3H]thymidine incorporation, indicating that the epitope did not interfere with ZO-1's role in proliferation control. Overexpression of ZO-1 lacking the SH3 domain, however, did not affect [3H]thymidine incorporation even though these cell lines expressed similar amounts of transfected protein (unpublished data). The SH3 domain is thus critical for ZO-1 to control proliferation. None of the other domains of ZO-1 was found to be required for inhibiting [3H]thymidine incorporation (Fig. 2, C and D).
If the overexpression of ZO-1 indeed reduces proliferation due to sequestration of ZONAB, one would expect that coexpression of ZONAB neutralizes the effect of ZO-1. Fig. 2 C shows that the inhibitory effect of ZO-1 was indeed neutralized by coexpression of ZONAB-A, supporting the conclusion that ZO-1 regulates proliferation by interacting with ZONAB.
Y-box transcription factors have been proposed to regulate cell cycle entry (Bargou et al., 1997; Matsumoto and Wolffe, 1998), and ZONAB cannot be detected in the nucleus in nonproliferating high density cells. To test whether ZONAB regulates cell cycle entry, we synchronized low density cultures of MDCK cells in Go/G1 phase by serum starvation. Based on FACS analysis, around 75% of the cells were arrested in Go/G1 after the 48-h incubation in 0.1% serum (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200210020/DC1). Entry into S-phase on addition of serum was then quantified by determining the incorporation of [3H]thymidine. Fig. 3 A shows that ZO-1 overexpression indeed reduced DNA synthesis, suggesting that entry into S-phase was affected. The ZO-1 effect depended on the presence of the SH3 domain, and overexpression of ZONAB again reversed the levels of [3H]thymidine incorporation to control levels. This suggests that ZO-1 indeed regulates cell cycle entry.
Figure 3.

Regulation of G1/S-phase transition by ZO-1 and ZONAB. Wild-type (wt MDCK) and transfected MDCK cells expressing the indicated cDNAs were plated at low confluence and then synchronized in low serum. Cell cycle entry was then triggered by serum addition, and progression to S-phase was monitored by measuring incorporation of [3H]thymidine (A) or BrdU (B). Note, whereas in A total DNA synthesis was measured, B shows the fraction of cells in S-phase. Expression of full-length ZO-1 significantly inhibited entry into S-phase (t test; A, P < 0.05; B, P < 0.01). Shown are means ± 1 SD of three independent clones per construct that were analyzed in at least three different experiments performed in quadruplicate.
To confirm the [3H]thymidine data and to determine the number of cells that actually entered S-phase, we counted the cells incorporating BrdU. Fig. 3 B shows that exogenous expression of ZO-1 resulted in reduced numbers of cells entering S-phase, and that coexpression of ZONAB again counteracted the inhibition by ZO-1. Reduced proliferation was not found to be paralleled by increased rates of apoptosis or senescence (Fig. S1). These data suggest that overexpression of ZO-1 regulates entry into S-phase.
Next, we wanted to test whether reduction of total cellular ZONAB levels also inhibits cell cycle entry. Because expression of long antisense RNAs as the one used in the experiment described in Fig. 1 can cause nonspecific cellular responses due to activation of antiviral defense mechanisms (Williams, 1999), we used an alternative approach to reduce ZONAB expression. This second approach was based on the expression of small ZONAB-directed RNA duplexes to induce RNA interference using a recently described expression vector containing an RNA polymerase III promoter (Yu et al., 2002). Fig. 4 A shows that stable transfection of two different vectors targeting different regions of the ZONAB sequence resulted in efficient reduction of ZONAB expression, but a vector expressing a control RNA duplex with a random sequence did not affect ZONAB expression.
Figure 4.

Depletion of ZONAB by RNA interference inhibits G1/S-phase transition. (A) Wild-type MDCK cells or cells stably expressing RNA duplexes corresponding to region I (ZONAB-RD-I c1 and c2) or II (ZONAB-RD-II c8 and c9) of ZONAB, or control RNA duplexes (control-RD c1 and c3) were grown for 2 d and then lysed and processed for electrophoresis. The samples were immunoblotted with anti-ZONAB and anti–α-tubulin antibodies. (B) Wild-type and transfected MDCK cells expressing the indicated RNA duplexes were plated at low confluence and synchronized in low serum. Cell cycle entry was triggered by serum addition, and progression to S-phase was monitored by determining the fraction of cell incorporation of BrdU. Shown are means ± 1 SD of at least two independent clones per type of RNA duplex that were analyzed in two independent experiments. Both ZONAB-directed RNA duplexes significantly inhibited entry into S-phase (t test; P < 0.02).
Next, we used these cell lines to determine whether reduced ZONAB expression affects cell cycle entry. Cells were arrested in Go/G1 and cell cycle entry was stimulated by the addition of serum. Fig. 4 B shows that reduction of ZONAB expression by RNA interference reduced the number of cells that had entered S-phase after serum stimulation by ∼50%. Entry into S-phase of clones expressing control RNAs was not affected. These data support the conclusion that high levels of ZONAB expression are required for efficient proliferation, and suggest that ZONAB participates in the regulation of Go/G1–S-phase transition.
Regulation of cell density by ZONAB
Because reduced nuclear expression of ZONAB in low density cells reduces proliferation and ZONAB becomes down-regulated with increasing cell density (Fig. 1 A; Balda and Matter, 2000), we used cell lines overexpressing either one of the ZONAB isoforms to test whether overexpression of ZONAB affects either cell cycle arrest in high density cells or the final cell density (Fig. 5 A). First, we labeled high density cells with BrdU to determine whether overexpression of ZONAB affects cell cycle arrest in mature monolayers.
Figure 5.
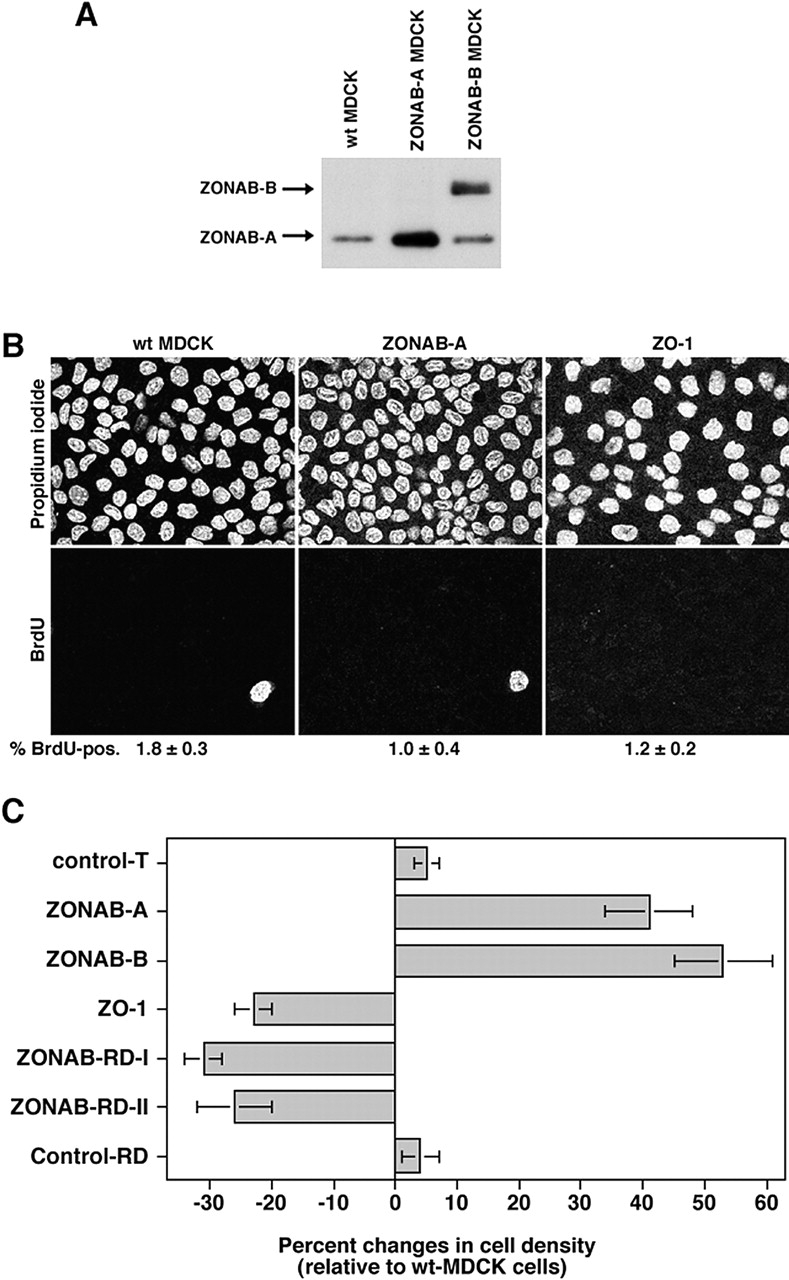
Regulation of final cell density by ZONAB. (A) Expression of ZONAB in high density wild-type and transfected MDCK cells. Wild-type (wt MDCK) and ZONAB-A or -B overexpressing cells were grown for 7 d. Cells cultured for 7 d had reached maximal cell density. Cultures were harvested, and equal amounts of protein were loaded on SDS-PAGE gels for analysis of ZONAB expression by immunoblotting. (B) Cell cycle arrest of high density MDCK cells. Wild-type or transfected MDCK cells overexpressing ZO-1 or ZONAB were grown to full density, and cell cycle arrest was determined by BrdU incorporation. Shown are representative images of propidium iodide and BrdU labelings, and the indicated numbers are the percentage of BrdU-positive cells (averages of two independent experiments are shown). Note the different cell densities in the propidium iodide staining. (C) Cell density of mature monolayers. Cells grown as those in B were harvested and counted. The cell number per cm2 of culture was calculated and expressed as percent changes relative to wild-type MDCK cells. The value shown for control transfections (control T) represents cell densities obtained from cell lines expressing three different control cDNAs generated with the same expression vector. Shown are averages ± 1 SD of independent clones that were analyzed twice independently using triplicate cultures. Analyzed were three different clones expressing ZONAB-A, three ZONAB-RD-I clones, and two independent clones each for ZONAB-B, ZO-1, ZONAB-RD-II, and Control-RD. All cell lines with the exception of the two types of control transfection were significantly different from wild-type cells (t test; P < 0.05).
Quantification of BrdU-positive cells revealed that only a small percentage of ZONAB-overexpressing cells still synthesized DNA comparable to nontransfected cells or cell lines overexpressing ZO-1 (Fig. 5 B). Thus, ZONAB overexpression did not cause continued proliferation of high density MDCK cells. However, visualization of nuclei with propidium iodide suggested that cells reached different final cell densities.
Next, we quantified the cell density of mature monolayers that were grown as those in Fig. 5 B. These experiments revealed that cells expressing exogenous ZONAB grew to cell densities that were ∼40% higher than the density reached by wild-type cells. Overexpression of ZO-1 consequently produced monolayers with a decreased final cell density similar to the reduction of ZONAB expression levels by RNA interference. These results suggest that ZONAB and ZO-1 regulate the cell density at which cell cycle arrest occurs during monolayer formation.
Interaction of ZONAB and CDK4
The human Y-box factor DbpA has been shown to interact in a two-hybrid screen as well as in vitro with cell division kinases (CDKs), suggesting that Y-box factors may regulate the activity or subcellular localization of CDKs (Moorthamer et al., 1999). This interaction involves a domain that is highly conserved in all Y-box factors and that is also thought to participate in DNA binding and regulation of transcription. We hypothesized that ZONAB also interacts with a CDK and, hence, may affect proliferation by influencing either the activity or subcellular localization of a CDK involved in the regulation of entry into S-phase.
First, we immunoprecipitated extracts generated from wild-type MDCK cells with a stringent buffer (i.e., containing deoxylcholate, SDS, Empigen BB, and Triton X-100), and monitored the presence of CDKs by immunoblotting using a γ-chain–specific secondary antibody. Under stringent extraction conditions, CDK4 was present in immunoprecipitates generated with anti-ZONAB antibodies, but absent from control precipitates (Fig. 6 A). Other tested CDKs (e.g., CDK6) were not detected in ZONAB precipitates (unpublished data).
Figure 6.

Interaction of ZONAB with CDK4. MDCK cells were extracted under stringent conditions (A) or in PBS-Triton X-100 (B), and cell extracts were subjected to immunoprecipitation with the indicated antibodies. CDK4 and cyclin D1 were then detected by immunoblotting using a γ-chain–specific secondary antibody. (C) In vitro interaction between recombinant His-tagged ZONAB and GST-tagged CDK4. Glutathione beads loaded with either GST or GST-CDK4 were incubated with His-tagged ZONAB fusion protein. The beads were then washed, and pull-down was monitored by immunoblotting. A His-tagged control fusion protein was not detected in the precipitates (not depicted). (D) Kinase activity of recombinant CDK4/cyclin D1 was assayed in the presence of recombinant GST-ZONAB or GST using GST-retinoblastoma (GST-Rb) as a substrate. Note that CDK4 activity was not affected by GST-ZONAB.
To determine whether ZONAB-associated CDK4 is complexed to cyclin D1, we used more gentle conditions to extract the cells (i.e., 1% Triton X-100 in PBS) that do not break the interaction between the cyclin and the CDK. Both CDK4 and cyclin D1 could be detected in ZONAB immunoprecipitates generated from such extracts, suggesting that at least part of the ZONAB-bound CDK4 is complexed to cyclin D1 (Fig. 6 B). The interaction between ZONAB and CDK4 could be reconstituted with recombinant proteins, indicating that this is indeed a direct interaction (Fig. 6 C).
CDK4 is a key regulator of G1/S transition (Sherr, 2000; Malumbres and Barbacid, 2001). Hence, ZONAB could regulate proliferation by controlling the activity or the localization of CDK4. To test whether ZONAB influences the kinase activity of CDK4, we measured the activity of recombinant CDK4/cyclin D1 using GST-retinoblastoma as a substrate in the absence or presence of purified GST-ZONAB (Fry et al., 2001). Fig. 6 D shows that in vitro CDK4 activity was not significantly affected by ZONAB, suggesting that ZONAB does not affect the activity of CDK4. This is also supported by the finding that ZONAB immunoprecipitates were found to contain an active kinase activity that phosphorylates recombinant retinoblastoma protein (unpublished data).
Because CDK4 was found to coimmunoprecipitate with ZONAB, one would expect that the two proteins have a similar subcellular distribution. To test this, proliferating MDCK cells, in which ZONAB localizes to the nucleus as well as tight junctins, were fixed and processed for immunofluorescence using an antibody against CDK4. Fig. 7 A shows that endogenous CDK4 was indeed detected in the nucleus as well as at cell–cell junctions, which were visualized with anti-ZO-1 antibodies (Fig. 7 B). Similar stainings were obtained with two different CDK4 antibodies. To test CDK4 association with cell–cell junctions in an independent manner, we transiently expressed HA-tagged CDK4 in MDCK cells. Fig. 7 C shows that HA-CDK4 also localizes to intercellular junctions that were labeled with anti-ZO-1 antibody (Fig. 7 D).
Figure 7.

Association of CDK4 with intercellular junctions. (A and B) MDCK cells were plated on coverslips and grown for 3 d. The cells were then fixed with methanol and processed for double immunofluorescence using rabbit anti-CDK4 and rat anti–ZO-1 antibodies. Shown are confocal XY-sections (A, CDK4; B, ZO-1 staining). (C and D) Confocal XY-sections of MDCK cells transiently transfected with a cDNA coding for HA-tagged CDK4. The cells were stained with rabbit anti-HA and rat anti–ZO-1 antibodies (C, HA-CDK4; D, ZO-1 staining). Bars, 10 μm.
Overexpression of ZO-1 in low density cells causes a redistribution of ZONAB from the nucleus to the cytoplasm (Balda and Matter, 2000) and, as shown in Fig. 2, reduced proliferation. Because ZONAB binds CDK4, the nuclear pools of the two proteins may decrease in parallel. To test this, we isolated nuclear fractions from proliferating wild-type and ZO-1–transfected cells and measured the amount of nuclear ZONAB and CDK4. Fig. 8 A shows that the nuclear pools of CDK4 were strongly reduced by overexpression of ZO-1, as were ZONAB levels. Quantification of the immunoblots revealed that the nuclear pools of both proteins were reduced by >60% compared with wild-type cells. This effect was specific for CDK4 because the nuclear accumulation of CDK6, a kinase that does not coprecipitate with ZONAB, was not affected. This indicates that ZO-1 expression levels regulate the subcellular distribution of CDK4. This is also supported by the observation that in high density MDCK cells, which express high levels of ZO-1, CDK4 is primarily cytoplasmic (unpublished data).
Figure 8.
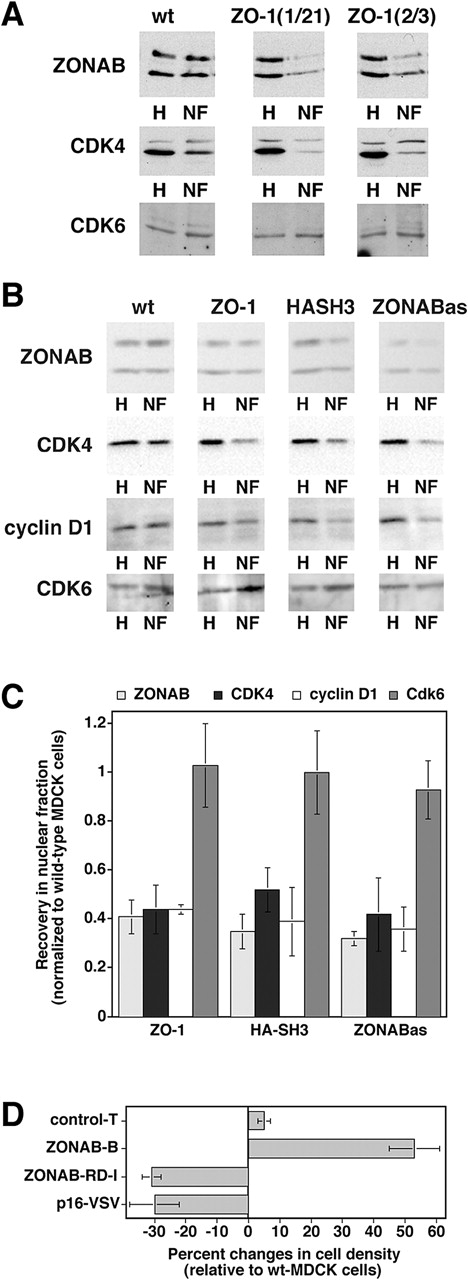
Regulation of CDK4 accumulation in the nucleus. (A) Nuclear fractions isolated from low density wild-type and ZO-1–overexpressing MDCK cells were immunoblotted for the indicated proteins. Equal amounts of protein were loaded for all homogenates and for all nuclear fractions. The weak upper band in the CDK4 blot is a background band that was only detected by early batches of the commercial anti-CDK4 antibody, but not by later ones (see panel B for comparison). (B) Wild-type cells, clones transfected with cDNAs for either ZO-1 or the HA-tagged SH3 (HASH3), or cells expressing antisense ZONAB RNA (ZONABas) were grown, franctionated, and analyzed as in the experiment shown in A. To increase the sensitivity and accuracy of quantification, higher amounts of nuclear protein were loaded in B as compared with A to obtain bands with a similar intensity. (C) Quantification of nuclear accumulation. Immunoblots such as those shown in B were quantified by densitometric scanning, and the ratio of nuclear versus total signal was calculated. For each experiment, the ratio obtained from wild-type cells was set to 1 and the other cell lines were normalized to this value. Shown are averages ± 1 SD from at least three independent experiments. The values obtained for ZONAB, CDK4, and cyclin D1 in ZO-1, HA-SH3, and ZONABas cells were significantly different from those of wild-type cells with a confidence level of P < 0.02. (D) Reduced cell density in cells expressing a CDK4 inhibitor. Wild-type and transfected MDCK cells were grown as in Fig. 5 C, and were then harvested and counted. The cell number per cm2 of culture was calculated and expressed as percent changes relative to wild-type MDCK cells. Two independent clones expressing VSV-tagged p16-INK4a were analyzed in two independent experiments performed in triplicate. For comparison, the values of ZONAB-overexpressing or depleted clones from Fig. 5 are also shown. Note that expression of p16-INK4a significantly reduced the final cell density (t test; P < 0.05).
If the redistribution of CDK4 in ZO-1 overexpressing cells is ZONAB-dependent and causes the reduction of proliferation, one would expect that expression of the SH3 domain of ZO-1 is sufficient to induce the redistribution of ZONAB and CDK4 observed in ZO-1 overexpressing cells. Fig. 8 B shows that the nuclear levels of ZONAB and CDK4 in MDCK cells expressing HA-SH3 were reduced to a similar extent as in ZO-1–overexpressing cells (see Fig. 8 C for quantification). Moreover, reduction of the cellular ZONAB level by expression of ZONAB antisense RNA resulted in strongly reduced nuclear ZONAB levels. A smaller proportion of the remaining ZONAB was nuclear than in wild-type cells, suggesting that the expression level of ZONAB determines its relative distribution. Consequently, this was paralleled by a reduction in the nuclear pool of CDK4, indicating that high levels of ZONAB are required for efficient accumulation of CDK4 in the nucleus. In all cases, the reduction of CDK4 was paralleled by a reduction of nuclear cyclin D1, the main cyclin binding CDK4. These observations suggest that inhibition of the nuclear accumulation of ZONAB results in less nuclear CDK4 activity. This is supported by the observation that such cell lines exhibited reduced hyperphosphorylation of retinoblastoma protein, a nuclear CDK substrate (unpublished data).
Reduced expression of ZONAB and overexpression of ZO-1 were shown to decrease the final cell density of MDCK monolayers (Fig. 5). If this effect is at least in part due to reduced CDK4 function, one would expect that inhibition of CDK4 by other means would also affect the final cell density; hence, we generated MDCK cells overexpressing the CDK4 inhibitor p16INK4a tagged with a COOH-terminal VSV epitope (p16-VSV). Fig. 8 D shows that monolayers expressing p16-VSV also stopped growing at lower cell densities similar to ZO-1–overexpressing cells, suggesting that regulation of CDK4 is sufficient to modulate the cell density of mature epithelial monolayers.
Discussion
Our results indicate that ZONAB functions in the regulation of epithelial proliferation and cell density. Reduction of ZONAB expression levels were found to reduce proliferation rates and final cell densities, whereas overexpression resulted in monolayers with increased cell densities. Premature increases in expression of ZO-1 in low density cells, which results in decreased nuclear ZONAB levels, also resulted in reduced proliferation and, in mature monolayers, decreased cell density. These observations suggest that ZONAB and ZO-1 are part of a pathway by which tight junctions can regulate epithelial cell proliferation and density.
ZONAB was found to interact with CDK4, and manipulations that resulted in lowered nuclear pools of ZONAB also decreased nuclear CDK4 levels and reduced hyperphosphorylation of retinoblastoma protein. These data suggest that regulation of the nuclear accumulation of CDK4 represents one way by which ZONAB and ZO-1 can influence epithelial proliferation. Increased ZO-1 and decreased ZONAB expression not only resulted in reduced proliferation, but also in lowered cell densities in mature monolayers, and overexpression of ZONAB had the opposite effect on cell density. Interestingly, experiments in mice linked CDK4 to the regulation of cell number in certain tissues (Malumbres and Barbacid, 2001), suggesting that regulation of CDK4 function by ZONAB and ZO-1 may be important for the regulation of cell density. This possibility is further supported by the observation that inhibition of CDK4 by expression p16INK4a was sufficient to reduce cell density in mature MDCK monolayers.
CDK4 is a central regulator of cellular proliferation, and its activity is tightly controlled by several interacting proteins (Serrano, 1997; Ekholm and Reed, 2000; Sherr, 2000; Malumbres and Barbacid, 2001). Generally, these proteins either activate or inhibit the kinase. Inhibitory proteins can be transcription factors that are up-regulated during terminal differentiation (Zhang et al., 1999; Wang et al., 2001). The here-described mechanism of regulating CDK4 is different in two aspects. First, ZONAB does not directly affect the enzymatic activity of CDK4, but links it to a mechanism that controls the nuclear accumulation of the kinase. Second, ZONAB is up-regulated during proliferation and down-regulated in differentiated cells, resulting in nuclear accumulation at times of high nuclear CDK4 activity.
ZONAB is a transcription factor and may therefore not only control proliferation by regulating CDK4 localization, but also by modulating the expression of genes coding for proteins involved in proliferation. Y-box transcription factors bind to inverted CCAAT boxes, which occur frequently in promoters of cell cycle regulators (Ladomery and Sommerville, 1995; Bargou et al., 1997; Chen, 1997), and ZONAB can bind to some of them in gel shift assays (Balda and Matter, 2000). Direct evidence for ZONAB functioning in the suppression or activation of promoters of cell cycle regulators has not yet been obtained, however, and will require a comprehensive search for ZONAB target genes. The only currently known gene that is regulated by ZONAB is erbB-2 (Balda and Matter, 2000). However, neither overexpression nor down-regulation of ErbB-2 with a KDEL-tagged single chain antibody affected the proliferation rates of MDCK cells (unpublished data), indicating that regulation of ErbB-2 expression is not part of the mechanism by which ZONAB regulates proliferation of these cells. Nevertheless, it is attractive to speculate that ZONAB forms a complex with CDK4, and that nuclear accumulation of the two proteins is co-regulated because they both have nuclear functions that are critical for proliferation.
ZONAB function can be regulated by at least two different mechanisms; the overall expression level and the subcellular distribution. ZONAB is expressed at high levels in proliferating cells and at low levels in confluent cells (Balda and Matter, 2000); the mechanisms that control up-regulation and down-regulation are not known yet, but may involve potential PEST sequences residing in the COOH-terminal half of the protein.
The subcellular distribution of ZONAB is regulated by the expression level of ZO-1 and the cell density (Balda and Matter, 2000). If ZO-1 is overexpressed in low confluent cells or monolayers reach full cell density, the nuclear ZONAB pools and proliferation are reduced. ZO-1 levels increase with cell density and become down-regulated when proliferation is required (e.g., during wound repair), suggesting that up-regulation of ZO-1 contributes to cell cycle arrest in mature monolayers. It will be important to determine how expression of ZO-1 is regulated. Because the half-life of ZO-1 has been demonstrated to increase with cell density, stabilization of the protein at forming intercellular junctions appears to be important for the up-regulation (Gumbiner et al., 1991). Nevertheless, it is likely that the contribution of transcriptional mechanisms is also important for the regulation of ZO-1 expression. This is suggested by the observation that ZO-1 mRNA is down-regulated during corneal wound repair (Cao et al., 2002).
The interaction of ZO-1 with tight junctions may only be important for the stabilization of ZO-1, but not to anchor ZO-1 to the plasma membrane to inhibit nuclear accumulation of associated proteins. This is suggested by the observation that a truncated protein consisting only of the HA-tagged SH3 domain accumulated in the cytosol (unpublished data), but was sufficient to reduce proliferation and nuclear accumulation of ZONAB. Hence, binding of the SH3 domain to ZONAB may compete with another interaction required for nuclear transport by, for example, restricting access to a nuclear localization signal. Because reduction of cellular ZONAB levels resulted in reduced nuclear accumulation of CDK4, it is possible that ZONAB plays a role in nuclear transport of CDK4. Alternatively, the reduced but preferentially cytoplasmic pool of ZONAB may have been sufficient to retain a large proportion of CDK4 in the cytoplasm.
Our data indicate that ZONAB and ZO-1 regulate proliferation and the final cell density of MDCK cells. The observations that ZO-1 accumulates with increasing cell density, and increased expression of ZO-1 in transfected cells reduces the final cell density suggest a model in which ZO-1 functions as a gauge for cell density that, on reaching a threshold level, signals growth arrest by cytoplasmic sequestration of ZONAB and the associated cell cycle kinase CDK4. Consequently, reduction of ZONAB expression also reduced cell density, whereas overexpression resulted in monolayers with more cells.
Multiple signaling pathways regulate epithelial proliferation and can become activated if epithelial sheets are disturbed and intercellular junctions are disrupted (Behrens, 1999; Li and Mrsny, 2000; Reichert et al., 2000; Ryeom et al., 2000; Gottardi and Gumbiner, 2001; Savagner, 2001; Vietor et al., 2001). Overexpression of ZONAB resulted in increased cell density, but did not cause overgrowth of MDCK cells, suggesting that ZONAB becomes either inactivated or overridden by other signaling pathways. Such mechanisms may be cell type-dependent as ZONAB overexpression accelerates proliferation of a mammary epithelial cell line (unpublished data). It will be crucial to determine how the ZO-1/ZONAB pathway cross talks with other signaling systems that influence proliferation. For example, β-catenin as well as ECM signaling regulate the expression of cyclin D1, which activates CDK4 (Zhao et al., 1998; Shtutman et al., 1999; Tetsu and McCormick, 1999; D'Amico et al., 2000; Assoian and Schwartz, 2001). Thus, it is striking that multiple pathways deriving from structures mediating cell adhesion can regulate cell cycle progression via regulation of CDK4, and it will be important to determine how these different pathways interact with each other during epithelial differentiation and development.
Materials and methods
Cell lines and cDNAs
MDCK cells overexpressing ZO-1 and ZONAB isoforms were generated with the CMV-based expression vector pCB6 and were described previously (Balda and Matter, 2000). Cells expressing epitope-tagged ZO-1 constructs and ZONAB antisense RNA were generated using the bicistronic CMV-based pCIN4 expression vector (Rees et al., 1996). p16-INK4a was tagged with a COOH-terminal VSV epitope and cloned into pCB6 for stable expression.
The mU6pro vector was used for the expression of RNA duplexes (Yu et al., 2002). Two regions of ZONAB were targeted that are part of the sequence coding for different protein domains (region I, 5′-AAGTTCTCGCCACCAAAGTCC-3′; region II, 5′ AACTTACCGCCCAAGGTACCG-3′). Derivatives of the mU6pro vector were constructed as described previously (Yu et al., 2002), and were cotransfected with a plasmid carrying a puromycin resistance gene for the selection of stable cell lines. All functional assays with transfected cells were performed with 2–5 independent clones for each construct.
A cDNA coding for CDK4 was generated by RT-PCR from total RNA isolated from Caco-2 cells and cloned into the pCB6 expression vector containing an HA-tag (Balda et al., 1996). The resulting plasmid pCB6-HA-CDK4 was transfected into MDCK cells by calcium phosphate and expression was analyzed after 48 h.
Antibodies
Rabbit anti-CDK4, goat anti-CDK6, and anti-retinoblastoma antibodies were obtained from Santa Cruz Biotechnology, Inc., and the rabbit anti-cyclin D1 antibody was purchased from MBL International Corporation. An additional anti-CDK4 antibody was generated against a peptide with the sequence NH2–A F R A L Q H S Y L H K D E–COOH and was affinity-purified using the same peptide coupled to sepharose beads. The peptide NH2–S S E A E T Q Q P P A A P P–COOH was used to generate a rabbit anti-YB1 antibody. All other antibodies were as described previously (Balda and Matter, 2000). For α-tubulin, mAb 1A2 was used (Kreis, 1987).
Biochemical assays and immunofluorescence
Cells were solubilized in either 10 mM Hepes, pH 7.4, 150 mM NaCl, 1% Empigen BB, 1% Triton X-100, 0.5% sodium deoxylcholate, and 0.2% sodium dodecylsulfate for stringent immunoprecipitates, or in PBS containing 1% Triton X-100 for less stringent conditions. Both extraction buffers contained protease and phosphatase inhibitor cocktails. ZO-1 and ZONAB were then immunoprecipitated as described previously (Balda and Matter, 2000). Recombinant His-tagged ZONAB, GST-tagged CDK4, and GST were produced in Escherichia coli, and interaction between the proteins was tested in PBS containing 0.5% Triton X-100 (Balda and Matter, 2000). GST and GST-CDK4 were pulled down using glutathione-agarose, and His-ZONAB was visualized by immunoblotting with an antibody specific for the COOH terminus of ZONAB. Enzymatic assays for CDK4 were performed as described previously (Fry et al., 2001). Nuclear fractions were isolated essentially as described previously (Balda and Matter, 2000), except that the centrifugation step was performed with an S80AT3 rotor in an ultracentrifuge (40,000 rpm for 50 min; model RC M150, Sorvall). Equal amounts of proteins of all homogenates and equal amounts of proteins of all nuclear fractions were loaded on SDS-PAGE gels, and the presence of particular proteins was tested by immunoblotting. For immunofluorescence, cells were plated on glass coverslips and cultured for at least 3 d. Processing for immunofluorescence and confocal microscopy was performed as described previously (Balda and Matter, 2000).
Analysis of proliferation and cell density
Confluent cells were trypsinized and plated (5,600 cells/cm2) in 10% FCS for 2 d. At such an early stage of proliferation, all cultures were still at a low density and formed only small islands of cells that, based on BrdU labelings, did not yet have a significant proportion of arrested cells in their center. 24-well plates were used for all experiments, and all determinations were done in quadruplicate. For synchronized cultures, the cells were washed with PBS and transferred to 0.1% of serum for 2 d. For [3H]thymidine incorporation, the cells were then incubated for 24 h with 0.1 or 10% serum-containing medium including 1 μCi/ml [3H]thymidine and 0.24 μg/ml thymidine. The incorporation was stopped with two washes with cold PBS, precipitation with 5% TCA, and two washes with cold ethanol. The samples were solubilized with SDS in sodium carbonate and counted. For BrdU incorporation, the cells were incubated with 100 μM BrdU for the last 2 h of the incubation with 10% serum. The cells were washed with PBS, fixed with methanol for 10 min at 4°C, washed with PBS, and incubated with 1.5 M HCl for 30 min at RT. After four washes with threefold PBS (5–10 min each) and blocking with 1% BSA, the cells were incubated with mouse anti-BrdU with nuclease (Amersham Biosciences). After 2 h, the samples were washed and were stained with a fluorescently labeled goat anti–mouse antibody and Hoechst 33258 or propidium iodide. For quantification, pictures of 4–6 different fields were taken and counted (400–500 cells per sample).
For the flow cytometric analysis, MDCK cells were washed with PBS/EDTA, trypsinized, washed with cold PBS, and counted. 2 × 106 cells resuspended in 300 μl PBS were then slowly added to 700 μl of −20°C ethanol (on a vortex at low speed) and were subsequently left at 4°C for 2 h (Urbani et al., 1995). The mix was vortexed four times during this incubation. After washing twice with cold PBS, the cells were incubated with 400 μl of 1% Triton X-100 in PBS containing 0.2 mg/ml RNase A (DNase free) and 20 ug/ml propidium iodide for 2 h at 37°C. During this time, the cells were resuspend every 20–30 min by vortexing. Flow cytometric analysis was performed on a Becton Dickinson flow cytometer (University Medical Center, University of Geneva, Geneva, Switzerland).
Apoptosis was determined by fluorescent detection of active caspase-3 using the CaspACE™ FITC-VAD-FMK in situ marker (Promega). In brief, cells were incubated for 20 min with the marker, washed twice with PBS, fixed, mounted, and observed with a fluorescence microscope. Senescence-associated β-galactosidase staining was performed at pH 6.0 for senescence-associated activity, at pH 4.0 for lysosomal (positive control), and at pH 7.5 for bacterial β-galactosidase (negative control) essentially as described previously (Dimri et al., 1995). Cells were washed in PBS, fixed in 3% PFA (3–5 min at RT), washed, and incubated at 37°C with fresh substrate solutions (1 mg/ml of 5-bromo-4-chloro-3-indolyl-b-d-galactoside in corresponding buffer). Staining was evident within 2–4 h and maximal after 12–16 h. Before observation, the cells were rinsed twice with PBS and washed with methanol.
For cell density experiments, the cells were grown to high cell densities (controlled by visual inspection) in 12-well plates and were then cultured for seven more days. After this time, the analyzed MDCK cell lines were arrested and did not exhibit any differences in BrdU incorporation. The cells were then trypsinized and subsequently resuspended, and counted with a hematocytometer.
Online supplemental material
Results illustrating synchronization and viability of wild-type and transfected MDCK cells can be found at http://www.jcb.org/cgi/content/full/jcb.200210020/DC1.
Supplemental Material
Acknowledgments
We thank Drs. J.M. Anderson and A.S. Fanning (University of North Carolina, Chapel Hill, NC) for the ZO-1 cDNA, Dr. N. Hynes (Friederich Miescher Institute, Basel, Switzerland) for ErbB-2 reagents, and Dr. C.J. Marshall (Institute for Cancer Research, London, UK) for the p16INK4a cDNA.
This research was supported by The Wellcome Trust (063661 and 066100 to M.S. Balda and K. Matter), Cancer Research UK (CRC), Fight for Sight, and the Swiss National Science Foundation.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: CDK, cell division kinase; ZONAB, ZO-1–associated nucleic acid binding protein.
References
- Assoian, R.K., and M.A. Schwartz. 2001. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr. Opin. Genet. Dev. 11:48–53. [DOI] [PubMed] [Google Scholar]
- Balda, M.S., and K. Matter. 2000. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. 19:2024–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balda, M.S., J.A. Whitney, C. Flores, S. González, M. Cereijido, and K. Matter. 1996. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 134:1031–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargou, R.C., K. Jurchott, C. Wagener, S. Bergmann, S. Metzner, K. Bommert, M.Y. Mapara, K.J. Winzer, M. Dietel, B. Dorken, and H.D. Royer. 1997. Nuclear localization and increased levels of transcription factor YB-1 in primary human breast cancers are associated with intrinsic MDR1 gene expression. Nat. Med. 3:447–450. [DOI] [PubMed] [Google Scholar]
- Behrens, J. 1999. Cadherins and catenins: role in signal transduction and tumor progression. Cancer Metastasis Rev. 18:15–30. [DOI] [PubMed] [Google Scholar]
- Benais-Pont, G., K. Matter, and M.S. Balda. 2001. Intracellular signaling in classical and new tight junction functions. Tight Junctions. J.M. Anderson and M. Cereijido, editors. CRC Press, Boca Raton, FL. 367–394.
- Cao, Z., H.K. Wu, A. Bruce, K. Wollenberg, and N. Panjwant. 2002. Detection of differentially expressed genes in healing mouse corneas using cDNA microarrays. Invest. Ophthalmol. Vis. Sci. 43:2897–2904. [PubMed] [Google Scholar]
- Cereijido, M., J. Valdes, L. Shoshani, and R.G. Contreras. 1998. Role of tight junctions in establishing and maintaining cell polarity. Annu. Rev. Physiol. 60:161–177. [DOI] [PubMed] [Google Scholar]
- Cereijido, M., L. Shoshani, and R.G. Contreras. 2000. Molecular physiology and pathophysiology of tight junctions. I. Biogenesis of tight junctions and epithelial polarity. Am. J. Physiol. Gastrointest. Liver. Physiol. 279:G477–G482. [DOI] [PubMed] [Google Scholar]
- Chen, K.Y. 1997. Transcription factors and the down-regulation of G1/S boundary genes in human diploid fibroblasts during senescence. Front. Biosci. 2:d417–d426. [DOI] [PubMed] [Google Scholar]
- D'Amico, M., J. Hulit, D.F. Amanatullah, B.T. Zafonte, C. Albanese, B. Bouzahzah, M. Fu, L.H. Augenlicht, L.A. Donehower, K.I. Takemaru, et al. 2000. The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3beta and cAMP-responsive element-binding protein-dependent pathways. J. Biol. Chem. 275:32649–32657. [DOI] [PubMed] [Google Scholar]
- D'Atri, F., and S. Citi. 2002. Molecular complexity of vertebrate tight junctions. Mol. Membr. Biol. 19:103–112. [DOI] [PubMed] [Google Scholar]
- Dimri, G.P., X. Lee, G. Basile, M. Acosta, G. Scott, C. Roskelley, E.E. Medrano, M. Linskens, I. Rubelj, O. Pereira-Smith, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 92:9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekholm, S.V., and S.I. Reed. 2000. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 12:676–684. [DOI] [PubMed] [Google Scholar]
- Fanning, A.S. 2001. Organization and the regulation of the tight junction by the actin-myosin cytoskeleton. Tight Junctions. J.M. Anderson and M. Cereijido, editors. CRC Press, Boca Raton, FL. 265–284.
- Farquhar, M.G., and G.E. Palade. 1963. Junctional complexes in various epithelia. J. Cell Biol. 17:375–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry, D.W., D.C. Bedford, P.H. Harvey, A. Fritsch, P.R. Keller, Z. Wu, E. Dobrusin, W.R. Leopold, A. Fattaeyi, and M.D. Garrett. 2001. Cell cycle and biochemical effects of PD 0183812. J. Biol. Chem. 276:16617–16623. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal, L., A. Betanzos, and A. Avila-Flores. 2000. MAGUK proteins: structure and role in the tight junction. Semin. Cell Dev. Biol. 11:315–324. [DOI] [PubMed] [Google Scholar]
- Gottardi, C.J., and B.M. Gumbiner. 2001. Adhesion signaling: how beta-catenin interacts with its partners. Curr. Biol. 11:R792–R794. [DOI] [PubMed] [Google Scholar]
- Gottardi, C.J., M. Arpin, A.S. Fanning, and D. Louvard. 1996. The junction-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc. Natl. Acad. Sci. USA. 93:10779–10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner, B., T. Lowenkopf, and D. Apatira. 1991. Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc. Natl. Acad. Sci. USA. 88:3460–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover, K.B., S.Y. Liao, and P.J. Bryant. 1998. Loss of the tight junction MAGUK ZO-1 in breast cancer: relationship to glandular differentiation and loss of heterozygosity. Am. J. Pathol. 153:1767–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, M., A. Nagafuchi, S. Yonemura, T. Kitani-Yasuda, and S. Tsukita. 1993. The 220-kD protein colocalizing with cadherins in non-epithelial cells is identical to ZO-1, a tight junction-associated protein in epithelial cells: cDNA cloning and immunoelectron microscopy. J. Cell Biol. 121:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis, T.E. 1987. Microtubules containing detyrosinated tubulin are less dynamic. EMBO J. 6:2597–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladomery, M., and J. Sommerville. 1995. A role for Y-box proteins in cell proliferation. Bioessays. 17:9–11. [DOI] [PubMed] [Google Scholar]
- Li, D., and R.J. Mrsny. 2000. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J. Cell Biol. 148:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres, M., and M. Barbacid. 2001. To cycle or not to cycle: a critical decision in cancer. Nat. Rev. Cancer. 1:222–231. [DOI] [PubMed] [Google Scholar]
- Matsumoto, K., and A.P. Wolffe. 1998. Gene regulation by Y-box proteins: coupling control of transcription and translation. Trends Cell Biol. 8:318–323. [DOI] [PubMed] [Google Scholar]
- Mitic, L.L., and J.M. Anderson. 1998. Molecular architecture of tight junctions. Annu. Rev. Physiol. 60:121–142. [DOI] [PubMed] [Google Scholar]
- Moorthamer, M., S. Zumstein-Mecker, and B. Chaudhuri. 1999. DNA binding protein dbpA binds Cdk5 and inhibits its activity. FEBS Lett. 446:343–350. [DOI] [PubMed] [Google Scholar]
- Rees, S., J. Coote, J. Stables, S. Goodson, S. Harris, and M.G. Lee. 1996. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. Biotechniques. 20:102–104. [DOI] [PubMed] [Google Scholar]
- Reichert, M., T. Muller, and W. Hunziker. 2000. The PDZ domains of zonula occludens-1 induce an epithelial to mesenchymal transition of Madin-Darby canine kidney I cells. Evidence for a role of beta-catenin/Tcf/Lef signaling. J. Biol. Chem. 275:9492–9500. [DOI] [PubMed] [Google Scholar]
- Ryeom, S.W., D. Paul, and D.A. Goodenough. 2000. Truncation mutants of the tight junction protein ZO-1 disrupt corneal epithelial cell morphology. Mol. Biol. Cell. 11:1687–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner, P. 2001. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 10:912–923. [DOI] [PubMed] [Google Scholar]
- Serrano, M. 1997. The tumor suppressor protein p16INK4a. Exp. Cell Res. 237:7–13. [DOI] [PubMed] [Google Scholar]
- Sherr, C.J. 2000. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 60:3689–3695. [PubMed] [Google Scholar]
- Shtutman, M., J. Zhurinsky, I. Simcha, C. Albanese, M. D'Amico, R. Pestell, and A. Ben-Ze'ev. 1999. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA. 96:5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson, B.R., and B.H. Keon. 1998. The tight junction: morphology to molecules. Annu. Rev. Cell Dev. Biol. 14:89–109. [DOI] [PubMed] [Google Scholar]
- Stevenson, B.R., J.D. Siliciano, M.S. Mooseker, and D.A. Goodenough. 1986. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol. 103:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu, O., and F. McCormick. 1999. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 398:422–426. [DOI] [PubMed] [Google Scholar]
- Tsukita, S., M. Furuse, and M. Itoh. 1999. Structural and signalling molecules come together at tight junctions. Curr. Opin. Cell Biol. 11:628–633. [DOI] [PubMed] [Google Scholar]
- Tsukita, S., M. Furuse, and M. Itoh. 2001. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2:285–293. [DOI] [PubMed] [Google Scholar]
- Urbani, L., S.W. Sherwood, and R.T. Schimke. 1995. Dissociation of nuclear and cytoplasmic cell cycle progression by drugs employed in cell synchronization. Exp. Cell Res. 219:159–168. [DOI] [PubMed] [Google Scholar]
- Vietor, I., T. Bader, K. Paiha, and L.A. Huber. 2001. Perturbation of the tight junction permeability barrier by occludin loop peptides activates β-catenin/TCF/LEF-mediated transcription. EMBO Rep. 2:306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., P. Iakova, M. Wilde, A. Welm, T. Goode, W.J. Roesler, and N.A. Timchenko. 2001. C/EBPα arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol. Cell. 8:817–828. [DOI] [PubMed] [Google Scholar]
- Williams, B.R. 1999. PKR; a sentinel kinase for cellular stress. Oncogene. 18:6112–6120. [DOI] [PubMed] [Google Scholar]
- Willott, E., M.S. Balda, A.S. Fanning, B. Jameson, C. van Itallie, and J.M. Anderson. 1993. The tight junction protein ZO-1 is homologous to the Drosophila discs-large tumor suppressor protein of septate junctions. Proc. Natl. Acad. Sci. USA. 90:7834–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods, D.F., and P.J. Bryant. 1993. ZO-1, DlgA and PSD-95/SAP90: homologous proteins in tight, septate and synaptic junctions. Mech. Dev. 44:85–89. [DOI] [PubMed] [Google Scholar]
- Yu, J.Y., S.L. DeRuiter, and D.L. Turner. 2002. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl. Acad. Sci. USA. 99:6047–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahraoui, A., D. Louvard, and T. Galli. 2000. Tight junction, a platform for trafficking and signaling protein complexes. J. Cell Biol. 151:F31–F36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J.M., X. Zhao, Q. Wei, and B.M. Paterson. 1999. Direct inhibition of G(1) cdk kinase activity by MyoD promotes myoblast cell cycle withdrawal and terminal differentiation. EMBO J. 18:6983–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J.H., H. Reiske, and J.L. Guan. 1998. Regulation of the cell cycle by focal adhesion kinase. J. Cell Biol. 143:1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}