

Abstract
Vacuole tethering, docking, and fusion proteins assemble into a “vertex ring” around the apposed membranes of tethered vacuoles before catalyzing fusion. Inhibitors of the fusion reaction selectively interrupt protein assembly into the vertex ring, establishing a causal assembly hierarchy: (a) The Rab GTPase Ypt7p mediates vacuole tethering and forms the initial vertex ring, independent of t-SNAREs or actin; (b) F-actin disassembly and GTP-bound Ypt7p direct the localization of other fusion factors; (c) The t-SNAREs Vam3p and Vam7p regulate each other's vertex enrichment, but do not affect Ypt7p localization. The v-SNARE Vti1p is enriched at vertices by a distinct pathway that is independent of the t-SNAREs, whereas both t-SNAREs will localize to vertices when trans-pairing of SNAREs is blocked. Thus, trans-SNARE pairing is not required for SNARE vertex enrichment; and (d) The t-SNAREs regulate the vertex enrichment of both G-actin and the Ypt7p effector complex for homotypic fusion and vacuole protein sorting (HOPS). In accord with this hierarchy concept, the HOPS complex, at the end of the vertex assembly hierarchy, is most enriched at those vertices with abundant Ypt7p, which is at the start of the hierarchy. Our findings provide a unique view of the functional relationships between GTPases, SNAREs, and actin in membrane fusion.
Keywords: membrane fusion; SNAREs; yeast vacuoles; Rab GTPase; actin
Introduction
Membrane fusion is a highly conserved step in organelle trafficking. Fusion is catalyzed by a set of proteins that are highly conserved among divergent eukaryotic organisms (Jahn and Sudhof, 1999). These include the SNARE proteins, the chaperone ATPase NSF/Sec18p and its cochaperone α-SNAP/Sec17p, the Rab/Ypt GTPases, and effector complexes. SNAREs are a superfamily of proteins that can form extended α-helical bundles, either in cis (on the same membrane) or in trans (on apposed membranes). Rab/Ypt proteins cycle between an active GTP-bound form and an inactive GDP-bound form. The GTP-bound form of these GTPases binds “effector” proteins that are needed for membrane docking and fusion. However, the hierarchy of interactions that establishes the proper spatial relationships of these proteins on membranes during docking and fusion is unknown.
Homotypic fusion of yeast vacuoles requires each of these factors and is readily studied by genetic, biochemical, and optical methods (Wickner, 2002). Genetic and biochemical studies have defined the protein factors that catalyze vacuole fusion, and optical studies have defined their spatial distributions during the reaction (Wang et al., 2002). We now combine these approaches, revealing the assembly hierarchy of the “vertex ring” domain that supports vacuole docking and fusion.
Vacuole fusion occurs in ordered steps of priming, tethering, docking, and fusion. During ATP-dependent priming, cis-complexes of SNAREs (Vti1p, Nyv1p, Ykt6p, Vam7p, and Vam3p) and Sec17p are disassembled as Sec18p displaces Sec17p from the SNAREs and from the vacuole (Mayer et al., 1996; Ungermann and Wickner, 1998; Ungermann et al., 1999). Vam7p, a homologue of the neuronal SNARE SNAP25, has a PtdIns(3)P-binding Phox homology (PX) domain, but no membrane anchor. It is transiently released from the vacuole by priming (Boeddinghaus et al., 2002). Tethering requires the GTPase Ypt7p (Mayer and Wickner, 1997). Vam7p rebinds to vacuoles through its interactions with PtdIns(3)P (Cheever et al., 2001; Boeddinghaus et al., 2002) and with Ypt7p (Ungermann et al., 2000). Two other GTPases, Rho1p and Cdc42p, are required (Eitzen et al., 2001; Muller et al., 2001) to regulate the remodeling of vacuole-bound actin (Eitzen et al., 2002). Actin remodeling consists of jasplakinolide-sensitive actin depolymerization during docking, followed by latrunculin B-sensitive repolymerization of G-actin to F-actin during the fusion stage of the reaction (Eitzen et al., 2002). After trans-SNARE pairing, these factors and others (Peters et al., 2001) catalyze lipid bilayer fusion and lumenal content mixing.
Recently, we exploited the large (>1 μm) diameter of purified vacuoles to study their membrane morphology and protein spatial dynamics during fusion (Wang et al., 2002). Vacuoles accumulate in tethered clusters due to the relative rates of docking and fusion. Clustered vacuoles have three domains: (1) “outside” membrane that is not in contact with other vacuoles; (2) “boundary” membrane that is apposed to a neighboring docked vacuole; and (3) “vertex” membrane where two boundary domains, or boundary and outside membrane domains, meet (Fig. 1 A, right). In three dimensions, the vertices circumscribe the apposed boundary membranes (Fig. 1 A, left). Fusion occurs at the vertex ring domains on docked vacuole membranes, yielding internalized membranes after the completion of fusion (Wang et al., 2002). Docking and fusion factors, such as the Ypt7p GTPase, its effector complex for homotypic fusion and vacuole protein sorting (HOPS;* Price et al., 2000; Seals et al., 2000), or class C Vps (Wurmser et al., 2000), SNARE proteins, and vacuole-bound actin (Eitzen et al., 2002), assayed by ratiometric fluorescence microscopy (Fig. 1 B), accumulate at these vertex sites before catalyzing fusion. The vertex enrichment of these proteins may follow a specific causal pathway that regulates the fusion process.
Figure 1.
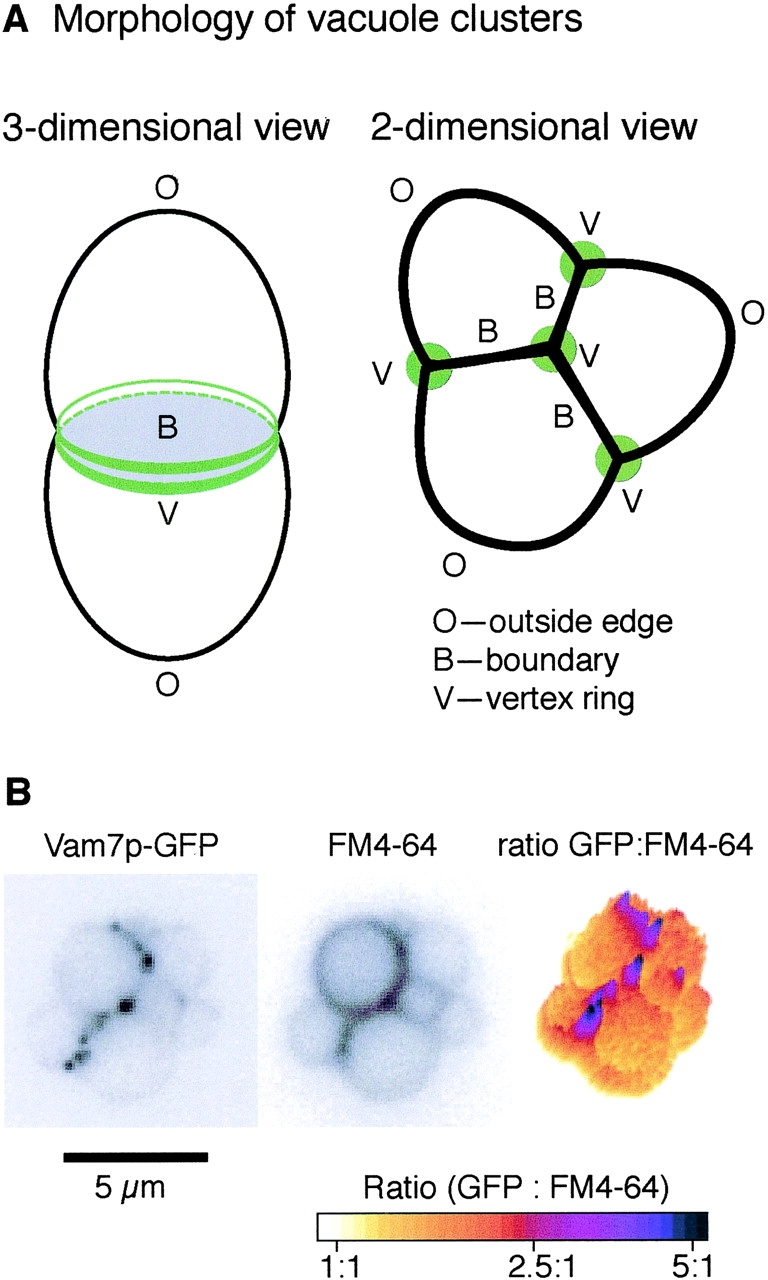
In vitro vacuole tethering. (A) A schematic of tethered vacuoles. Membrane microdomains include outside edges (O), boundary membranes (B), and vertex membranes (V). (B) Vam7p is enriched at vertices. GFP-tagged Vam7p vacuoles were tethered in vitro (see Materials and methods) and labeled with FM4–64. Images were taken in GFP (left) and rhodamine (middle) channels; ratiometric images are on the right.
To identify the assembly pathway of the vertex ring of docking and fusion proteins, we examined the effects of several well-characterized reaction inhibitors on protein localization. Their differential effects reveal a hierarchy of protein enrichment at vertices in which Ypt7p forms the initial tethering ring, Ypt7:GTP and depolymerized actin allow vertex enrichment of two t-SNAREs in an interdependent fashion, and these in turn govern the localization of HOPS and G-actin. Interfering with any step in the hierarchy of vertex enrichment prevents fusion.
Results
Vacuole fusion has been studied biochemically, using inhibitors with known targets (Wickner, 2002). For example, fusion was inhibited by antibodies against SNARE proteins (Fig. 2, lane 2; Ungermann et al., 1999) by the F-actin–stabilizing reagent jasplakinolide (Fig. 2, lane 3; Eitzen et al., 2002) and by Gyp proteins (Fig. 2, lane 4; Eitzen et al., 2000), which accelerate GTP hydrolysis by GTPases such as Ypt7p (Albert et al., 1999). Sec18p and Sec17p are cochaperones for the cis-SNARE complex and are required for priming, but the addition of excess Sec17p recaptures SNAREs into a cis-complex and thereby inhibits fusion (Fig. 2, lane 5; Wang et al., 2000). Vam7p, a homologue of neuronal SNAP-25, is a soluble SNARE protein. It is released from vacuoles during priming, but rebinds through association of its PX domain with PtdIns(3)P during docking. Exogenous recombinant PX domain inhibited fusion by competing with Vam7p for PtdIns(3)P (Fig. 2, lane 6; Boeddinghaus et al., 2002), whereas the addition of recombinant Vam7p rescued fusion from PX inhibition (Fig. 2, lane 7; unpublished data). Latrunculin B, which prevents the repolymerization of G-actin, inhibited fusion (Fig. 2, lane 8; Eitzen et al., 2002). These fusion inhibitors provide reliable tools for studying protein localization during docking.
Figure 2.
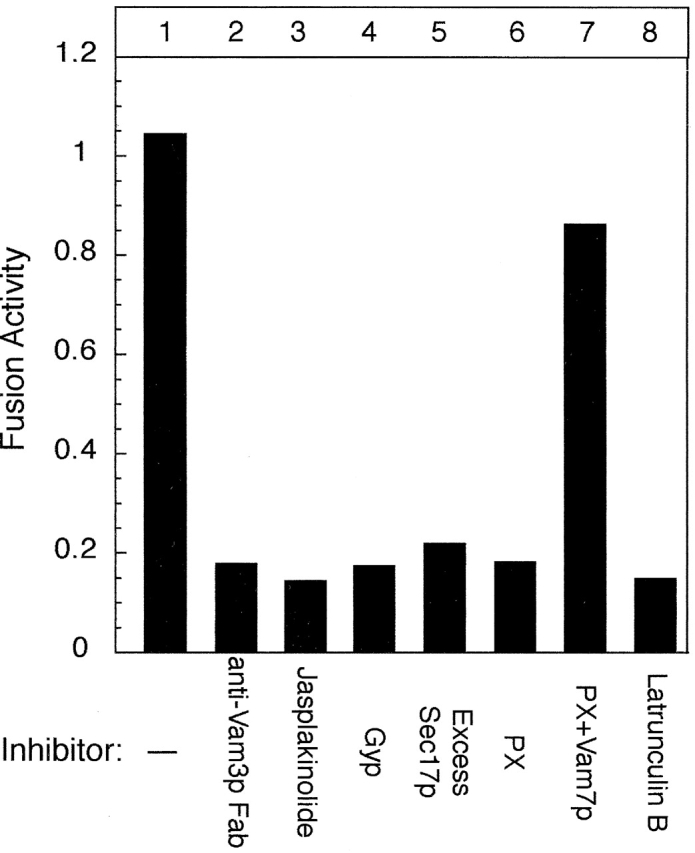
Catalytic assay of vacuole fusion. Vacuole fusion was assayed in vitro as described in Materials and methods. Fusion inhibitors, including 50 ng/μl excess Sec17p, 60 ng/μl anti-Vam3 Fab, 600 ng/μl recombinant Gyp1p, 500 μM jasplakinolide, 15 μM PX domain, 500 μM latrunculin B, and 5 μM full-length Vam7p were present where indicated.
Vacuole docking has been studied using an in vitro tethering assay (Fig. 3; Mayer and Wickner, 1997; Wang et al., 2002) in which tethered vacuoles form large clusters (Fig. 3, black bars). The tethering reaction requires the GTPase Ypt7p. Gdi1p, which extracts Ypt7p from the membrane, inhibited cluster formation (Fig. 3, open bars). Ypt7p cycles between the GTP-bound state and the GDP-bound state, though it has not been known which state catalyzes tethering. Because Gyp proteins promote GTP hydrolysis by Ypt7p, driving it to the GDP-bound state (Vollmer et al., 1999; Rak et al., 2000; Du and Novick, 2001), we analyzed the effects of recombinant Gyp1–46p (an active fragment of Gyp1p) on Ypt7p-mediated vacuole tethering. Gyp1–46p did not reduce vacuole tethering (Fig. 3, gray bars), though fusion was completely inhibited (Fig. 2, lane 4; Eitzen et al., 2000). Cluster formation was still sensitive to Gdi1p in the presence of Gyp1p (hatched bars), indicating that these vacuoles were tethered through a Ypt7p-dependent pathway. Anti-Vam3p Fab, which blocked vacuole fusion (Fig. 2, lane 2), did not affect Ypt7p dependent vacuole tethering (Fig. 3, slanted line bars), in accord with other studies showing that tethering requires Rabs but not SNAREs (Cao et al., 1998). Thus, tethering requires Ypt7p, but does not require its nucleotide exchange to Ypt7:GTP or the Vam3p t-SNARE.
Figure 3.
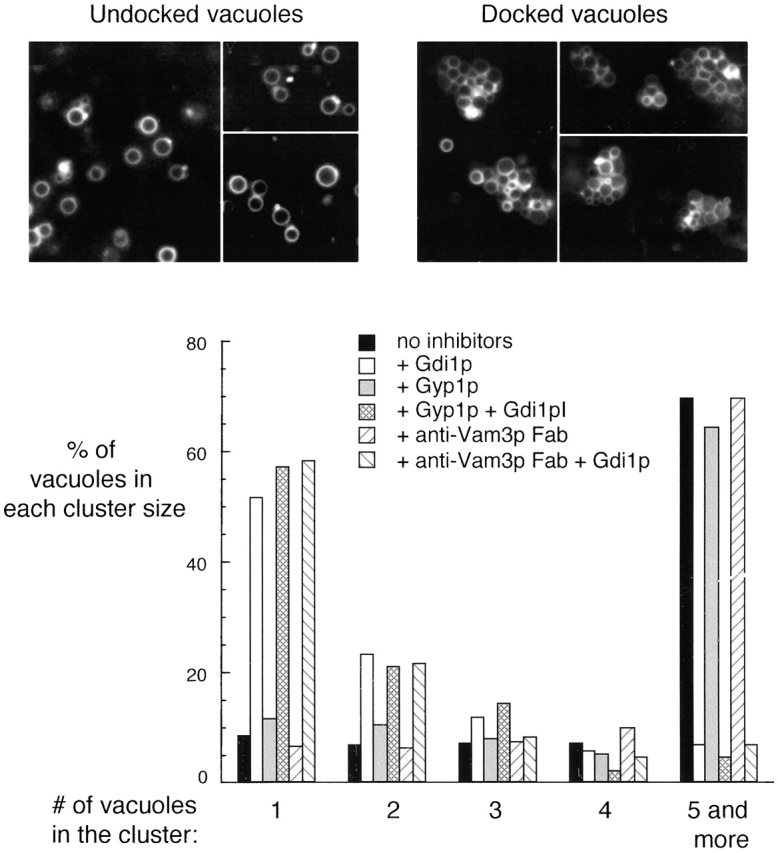
Vacuole tethering. Inhibitors, such as 300 ng/μl Gdi1p, 600 ng/μl Gyp1p, and 60 ng/μl anti-Vam3p Fab were added as indicated to an in vitro docking assay. For each condition, images were acquired from random fields and an average of 500 vacuoles were scored for cluster size.
Using ratiometric imaging, we analyzed the spatial enrichment of GFP-tagged proteins on vacuoles that were labeled with the fluorescent lipophilic dye FM4–64 (Wang et al., 2002). For example, the ratio of GFP-tagged Vam7p to FM4–64 (Fig. 1 B) showed enrichment of this protein at vertices relative to outside edges. After measuring clusters from random fields, we generated ∼200 ratio values at outside edges (black) and vertices (red), normalized them to outside edge values, and plotted them against their percentile to form a cumulative distribution plot, or CD plot (Fig. 4). Statistical analysis (Fig. 5) shows the extent and significance of spatial enrichment and its modulation by these ligands. The protein enrichments observed in this work and in our recent papers (Eitzen et al., 2002; Wang et al., 2002) are similar in magnitude to values previously obtained in other works. For example, Roberts et al. (1999) studied the localization of GFP-labeled, overexpressed Rab5, and Lang et al. (2001) examined the localization of syntaxin using GFP-fusions. In these works, the mean or median enrichments were approximately twofold. In each case, the structures studied are likely of submicron size, i.e., near or below the optical resolution limits imposed by diffraction. For this reason, the measured enrichments are expected to substantially underestimate the actual values. Like the other fusions used in this work (Wang et al., 2002), the GFP-tagged versions of Vam3p, Vti1p, Nyv1p, and Vam7p were expressed as gene replacements under their native promoters and were functional in vivo, as assayed by vacuole morphology phenotype, and in vitro, as shown by the homotypic vacuole fusion assay. Tagging the NH2 or COOH terminus of Ykt6p was not compatible with viability in our strain.
Figure 4.

Protein enrichment at vertices of tethered vacuoles. Reaction inhibitors, including 50 ng/μl excess Sec17p, 60 ng/μl anti-Vam3 Fab, 600 ng/μl recombinant Gyp1p, 500 μM jasplakinolide, 15 μM PX domain, 500 μM latrunculin B, and 5 μM full-length Vam7p were added to docking assays (see Materials and methods) with GFP-tagged proteins as indicated. After incubation, vacuoles were observed by fluorescence microscopy and images were acquired in GFP and rhodamine channels. Vacuole clusters from random fields were analyzed and ratio values of GFP:FM4–64 were generated from outside edge membranes (black) and vertex membranes (red). An average of 200 normalized ratio values for each treatment were plotted against their percentile (cumulative distribution plot).
Figure 5.

Statistical analysis of the effects of fusion inhibitors on protein enrichment at vertices. The bars show geometric means and 95% confidence intervals for the data sets that are shown as CD plots in Fig. 4. Shading indicates a treatment that causes statistically significant (P < 0.0006) partial or complete inhibition of vertex enrichment, compared with the corresponding “no inhibitor” condition (see Materials and methods).
Sequential assembly of fusion factors at vertices
Under normal reaction conditions, Ypt7p was enriched at vertices (Fig. 5 A; Wang et al., 2002) relative to its outside edge concentrations. None of the tested reaction inhibitors, which include the PX domain, excess Sec17p, jasplakinolide, anti-Vam3 Fab and Gyp1–46p, affected Ypt7p localization (Fig. 5 A). This indicates a primary role for Ypt7p in tethering, in vertex ring assembly, and (as shown below) in regulating the downstream localization of other factors to vertices.
Next, we asked how SNARE localization is regulated. The t-SNARE Vam3p was enriched at vertices on docked vacuoles (Wang et al., 2002; Fig. 5 B). The v-SNARE Vti1p and the SNAP-25 homologue Vam7p also accumulated at vertices (Fig. 5, C and D). Vertex enrichment did not occur on nonspecific vacuole clusters formed by cosedimentation (unpublished data). Excess Sec17p, which blocks fusion by reforming a cis-SNARE complex (Wang et al., 2000), inhibited vertex enrichment of each of the SNAREs (Fig. 5, line 4). Jasplakinolide, which stabilizes F-actin, also blocked the vertex localization of Vam7p, Vam3p, and Vti1p (Fig. 5, line 3), indicating that the disassembly of F-actin is required for SNARE protein localization. We have recently shown latrunculin B sensitivity of a late step in fusion, presumably actin filament assembly at vertices (Eitzen et al., 2002); however, we found that latrunculin B had no effect on the localization of Vam3 to vertices (Fig. 4 B, filled inverted triangles). Thus, actin disassembly and reassembly fulfill distinct roles in the tethering and fusion stages of the reaction, first allowing protein localization to vertices and then participating in the fusion stage of the reaction.
There are differences in the regulation of v- and t-SNARE enrichment at vertices. Added PX domain, which blocks Vam7p rebinding to vacuoles (Boeddinghaus et al., 2002), completely inhibited the localization of the t-SNARE Vam3p (Fig. 5 B). The specificity of this inhibition was shown by its reversal by recombinant Vam7p. However, PX domain had no effect on the vertex enrichment of the v-SNARE Vti1p (Fig. 5 D). Similarly, anti-Vam3p Fab fragment blocked the localization of Vam3p and had a moderate effect on Vam7p, but had no effect on the localization of the v-SNARE Vti1p (Fig. 5, B–D). Thus, Vam7p and Vam3p are interdependent in their vertex accumulation, whereas the v-SNARE follows distinct signals. Anti-Vam3 Fab had a stronger effect on Vam3p localization than on Vam7p localization, suggesting that Vam3p acts in concert with other factors to promote Vam7p localization. Further evidence for differential SNARE localization is provided by Gyp1–46, which completely inhibited the localization of the v-SNARE Vti1p, but had only intermediate effects on Vam3p and Vam7p (Fig. 5, line 2). These results show that distinct signals specify the localization of different SNAREs rather than colocalization occurring through an obligate trans-SNARE pairing.
The HOPS complex consists of six subunits (Seals et al., 2000), including the Ypt7p nucleotide exchange factor Vps39p (Wurmser et al., 2000) and Vps33p, a member of the Sec1p family. HOPS binds to both Ypt7p and SNAREs, and may serve as an effector for each. HOPS localization can be monitored by GFP-tagging either of these subunits (Wang et al., 2002); we use GFP-Vps39p in this work. HOPS is enriched at vertices on tethered vacuoles. Excess Sec17p blocked its localization (Fig. 5 E, Wang et al., 2002), indicating that SNARE complex disassembly is required for this process. HOPS localization requires Vam7p, as it was blocked by added PX domain, and this inhibition was completely reversed by recombinant Vam7p. Jasplakinolide also blocked HOPS vertex enrichment. Thus, HOPS needs F-actin disassembly and the SNARE Vam7p for its vertex localization. Anti-Vam3p Fab, which completely blocked the vertex localization of Vam3p itself, had only a modest effect on HOPS vertex localization (Fig. 5, line 5). Gyp1–46p caused a partial inhibition of HOPS vertex localization, consistent with its limited effects on the vertex enrichment of Vam3p and Vam7p. There was comparable vertex enrichment of Vps39p in the presence of both Gyp1–46p and anti-Vam3p Fab as in the presence of anti-Vam3p alone (Fig. 4 E). This suggests that SNARE associations have a primary role in maintaining HOPS vertex enrichment, though Ypt7 participates in its delivery to vertices.
Vacuole-bound actin is remodeled and participates at multiple stages of the fusion reaction (Eitzen et al., 2002). G-actin can be visualized with fluorophore-derivatized DNase I, a specific G-actin ligand (Greer and Schekman, 1982; Pollard et al., 1994). Vacuole-bound G-actin is enriched at vertices of tethered vacuoles (Fig. 5 F; Eitzen et al., 2002). This enrichment was fully blocked by either added PX domain, which prevents rebinding of the Vam7p SNARE, by excess Sec17p, which recaptures the SNAREs into a cis-complex (Wang et al., 2000), or by anti-Vam3 Fab (Fig. 5 F). Therefore, actin depends on the t-SNAREs for its vertex enrichment.
Trans-SNARE pairing is not required for SNARE protein localization
Because v-SNAREs and t-SNAREs are localized to vertices via different pathways, SNARE localization may precede the formation of the trans-SNARE complex. The vacuolar v-SNARE Nyv1p enters trans-SNARE complexes (Ungermann and Wickner, 1998) and is needed for vacuole homotypic fusion (Nichols et al., 1997). Nyv1p was also enriched at vertices on docked vacuoles (Fig. 6 A). Vacuoles lacking Nyv1p cannot fuse, and antibody to Nyv1p blocks the fusion of wild-type vacuoles (Ungermann and Wickner, 1998). Using a recently developed assay of trans-pairing of SNAREs (unpublished data), we find that the absence of Nyv1p completely blocks trans-SNARE pairing between Vti1-GFP from one partner vacuole and HA-Vam3p from the other vacuole population (Fig. 6 B). Nevertheless, the absence of Nyv1p had no effect on the vertex enrichment of the t-SNAREs Vam3p or Vam7p (Fig. 6 C). Together with the effects of fusion inhibitors on the localization of SNAREs, our results indicate that SNAREs need not pair in trans in order to be enriched at vertices. The clustered localization of SNAREs at a vertex ring might be a prerequisite for efficient trans-SNARE pairing and downstream events leading to fusion.
Figure 6.

Deletion of the v-SNARE Nyv1p has no effect on the localization of other SNAREs. (A) GFP-Nyv1p vacuoles were subject to in vitro docking assay and ratiometric fluorescence microscopy analysis, as described in Materials and methods. The GFP:FM4–64 ratio values were generated for outside edge membranes (red curves) and vertex membranes (black curves), and plotted against their percentile. (B) Trans-pairing of SNAREs. Trans-SNARE pairing was assayed directly using purified vacuoles from four strains: (1) VTI1-GFP, NYV1, pep4–3; (2) HA 3-VAM3, NYV1, pep4::HIS3; (3) VTI1-GFP, pep4::HIS3, nyv1::URA3; and (4) HA 3-VAM3, pep4::HIS3, nyv1::URA3. After a 60-min fusion reaction, membranes were dissolved in Triton X-100, and Vti1-GFP was immunoprecipitated by immobilized antibody to GFP (unpublished data). Trans-associated HA-Vam3p was detected by immunoblot and quantified by densitometry as a percentage of the total HA-Vam3p in the sample. Recombinant Gyp1–46p (0.4 μg/ml) was added where indicated (lane 3). To determine the level of background association, detergent extracts of reactions with ATP and single vacuole populations were prepared and mixed before immunoprecipitation (lane 4). (C) Vacuoles were isolated from nvy1Δ strains with GFP-tagged Vam3p or Vam7p and assayed for protein localization during docking as above (Fig. 4).
Coordinate protein enrichment at vertices
Vertex sites are heterogeneous for their enrichment of each protein (Fig. 4; Wang et al., 2002). If this represents varying degrees of activation of the assembly hierarchy we describe herein, then the HOPS complex (at the end of the hierarchy) should be most enriched at those vertices which show the greatest enrichment for Ypt7p (at the start of the hierarchy). The recent availability of a fast-maturing monomeric red fluorescent protein variant (Campbell et al., 2002) allowed us to ask whether various vertex markers colocalize. We generated a yeast strain carrying both the HOPS subunit Vps33p fused to mRFP and Ypt7p fused to GFP. The blue fluorescent lipid probe TMA-DPH was used instead of the red dye FM4–64 to monitor lipid distribution on the vacuole. In a standard docking reaction, these three fluorophores were monitored on the same vacuole clusters (Fig. 7 A). GFP-Ypt7p and Vps33-mRFP significantly colocalized (P < 0.0001) at vertex sites (Fig. 7 B). As shown above using single GFP fusions, addition of PX domain did not significantly change GFP-Ypt7p localization at vertices. However, added PX domain (Fig. 7 B) significantly decreased the amount of Vps33-mRFP at the same sites (P < 0.0001). Interestingly, Vps33p and Ypt7p intensity were similarly correlated in both the presence and absence of PX domain (r2 = 0.58 and 0.62, respectively). Although less Vps33p accumulates at vertex sites in the presence of PX domain, this result suggests that the Vps33p that does accumulate is recruited through a Ypt7p-dependent mechanism. Regression models incorporating lipid intensity effects indicate that colocalization of GFP-Ypt7p and Vps33-mRFP did not depend on TMA-DPH intensity (unpublished data). A simpler regression model that incorporates effects due to GFP-Ypt7p localization and treatment (absence or presence of PX domain) accounts for almost 70% of the variation in Vps33-mRFP localization at vertex sites (r2 = 0.68; F[3,605] = 436.0; P < 0.0001). Thus, only ∼30% of the variability in the vertex localization of Vps33p localization is explained by experimental noise and other factors. These experiments corroborate and extend our findings that HOPS localization depends on both Ypt7p function and Vam7p function, and support the interpretation that Ypt7p acts upstream of HOPS and other factors (such as Vam7p) to specify the assembly of vertex sites.
Figure 7.
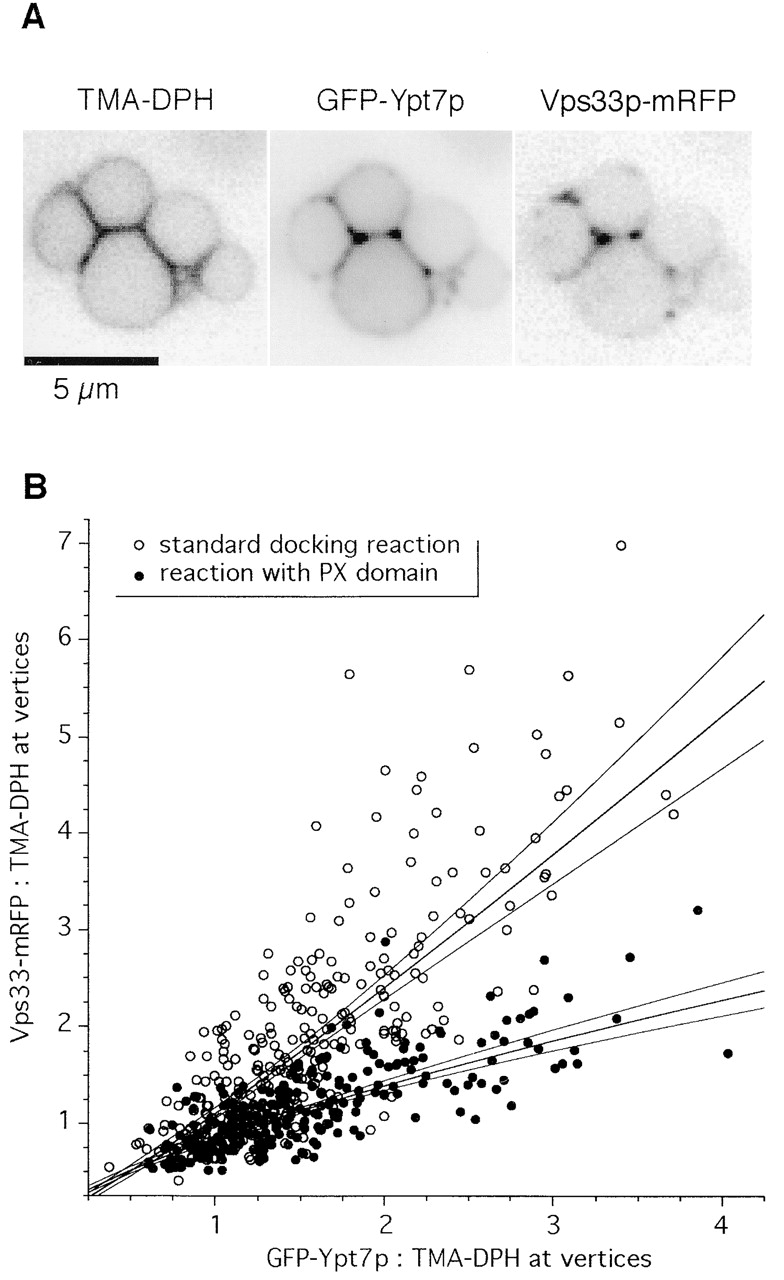
Ypt7p and Vps33p colocalization at vertex sites. GFP-Ypt7p and Vps33-mRFP were quantified at vertex sites under docking conditions in the absence (open circles) or presence (filled circles) of PX domain. Lines show linear fits to log-transformed intensity data and 99% confidence intervals for the best-fit lines. The upper fit is to the control samples (open circles) and the lower fit is to the PX domain-treated samples (filled circles). The data are pooled from three independent experiments; interexperimental variation did not significantly influence the results.
Discussion
Protein localization to membrane domains is critical for the functions of many biological systems. For large protein complexes, such as cell adhesion complexes (Martin et al., 2002; Schwartz and Ginsberg, 2002) and the immunological synapse (Bromley et al., 2001; Dustin et al., 2001), protein enrichment at specific sites on the plasma membrane relies on protein and lipid-mediated signaling networks and follows temporal and spatial hierarchies of molecular interactions. Membrane fusion is also catalyzed by a complex protein and lipid machinery that assembles at specific membrane domains. Prominent examples include Rab domains on endosomal membranes (Roberts et al., 1999; Sonnichsen et al., 2000), SNARE domains, and a RIM1a-mediated protein scaffold at neuronal synaptic junctions (Bennett et al., 1992; Lang et al., 2001; Schoch et al., 2002), and the yeast exocyst complex at the bud tip or septation ring (Finger et al., 1998; Guo et al., 2001). A vertex ring of proteins and lipids assembles on tethered vacuoles before catalyzing fusion (Wang et al., 2002). We have now identified a hierarchy in the spatial localization of these docking and fusion factors, revealing new features of the interactions between organelle-bound actin, Ypt/Rab GTPase, and SNAREs (Fig. 8). The strong correlation between enrichment of Ypt7p and HOPS at individual vertex sites, and selective loss of Vps33p enrichment by PX domain treatment provide independent support to this hierarchy. Though we have only used a select few inhibitors and GFP-tagged a subset of the proteins that catalyze vacuole fusion (Wickner, 2002), their interplay has revealed the first outlines of a vertex ring assembly hierarchy.
Figure 8.
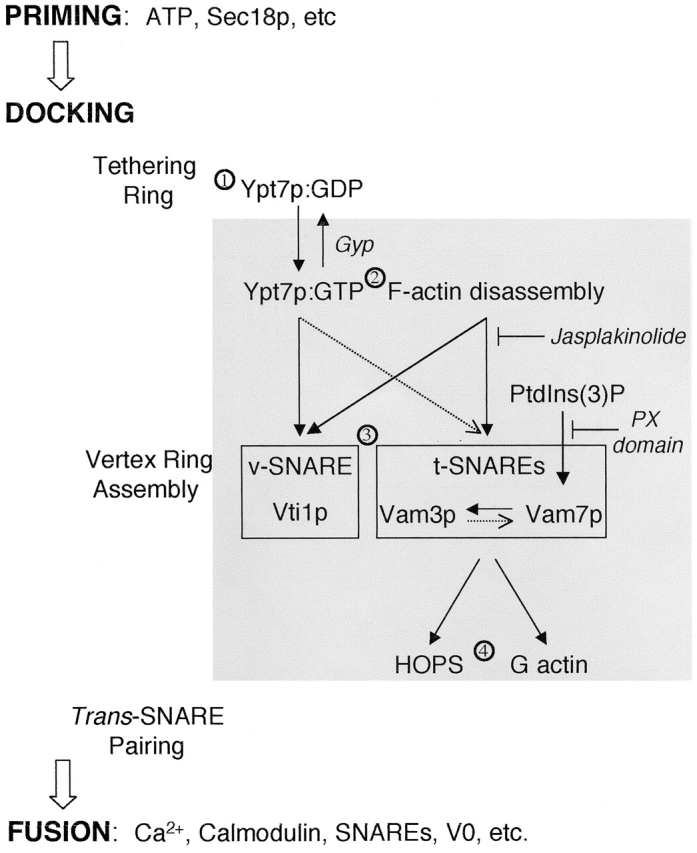
Working model of vertex ring assembly. See text for details. Solid black arrows indicate a strict hierarchical requirement for vertex enrichment. Dotted arrows indicate partial influences on vertex enrichment.
Ypt/Rab GTPases mediate tethering in each membrane fusion system (Segev, 2001). Although GTP-bound Ypt/Rab proteins bind effectors that mediate downstream functions, their GDP-bound forms may also have important functions. We now show that Ypt7p:GDP is sufficient to mediate vacuole tethering (Fig. 3), whereas Ypt7:GTP is required to localize other fusion factors, including the HOPS complex, SNAREs, and vacuole-bound actin, to the vertices of tethered vacuoles (Fig. 4). Thus, Ypt7p plays a dual role (Fig. 8). It supports tethering and forms the initial tethering ring (Fig. 8, step 1) without need for SNAREs or F-actin (Fig. 5). After exchange to its GTP form (Fig. 8, step 2), Ypt7:GTP triggers downstream assembly of SNAREs into the vertex ring (Fig. 8, step 3). Once this assembly is complete, Ypt7p is no longer needed to maintain vertex enrichment of other proteins (Wang et al., 2002) or for fusion (Eitzen et al., 2000).
Might other Ypt/Rab proteins be involved in homotypic vacuole fusion? Several lines of evidence suggest that Ypt7p is the only Ypt/Rab family member that is required for this reaction. The Ypt family GTPases of yeast have been enumerated, localized with respect to organelle, and examined for functional role in their organelle (for review see Martinez and Goud, 1998), and Ypt7p is the only member of this family to localize to the vacuole. Although antibodies to Ypt7p are potent inhibitors of vacuole fusion (Haas et al., 1995), antibodies to Ypt1p and to Ypt51p that inhibit their cognate in vitro trafficking reactions have no effect on vacuole fusion (Haas et al., 1995). This result is especially important in light of the specific localization and function of Ypt51 at the endosome (Horazdovsky et al., 1994; Singer-Kruger et al., 1994), just upstream of the vacuole, and emphasizes the specificity of the role of Ypt7p at the vacuole. Nevertheless, because one cannot simply delete large numbers of the YPT genes, it is difficult to completely rule out the involvement of other Ypt/Rab proteins.
Although Vam3p, Vam7p, and Vti1p require the disassembly of vacuole-bound F-actin for their enrichment at vertices, their movements to vertices are regulated by distinct signals (Fig. 7, step 3). Vam7p cycles on and off the membrane during the fusion reaction (Boeddinghaus et al., 2002). We find that Vam7p rebinding to the vertices is pivotal for the localization of the t-SNARE Vam3p, but not for the v-SNARE Vti1p. Similarly, anti-Vam3p Fab has no effect on Vti1p localization, but significantly reduces the enrichment of Vam7p and Vam3p. Thus, SNAREs localize to vertices by distinct pathways, presumably before trans-SNARE complex formation. Indeed, recent works (unpublished data) show that jasplakinolide, Gyp1–46p, anti-Vam3p Fab, or PX domain (Boeddinghaus et al., 2002) block trans-SNARE complex formation. The inhibitory effects of PX domain (which blocks Vam7p rebinding) on Vam3p localization and of anti-Vam3p Fab on Vam7p localization suggest an interdependent relationship between these two SNAREs. PX domain also prevents vertex enrichment of other factors, such as HOPS and G-actin, which rely on Vam7p for their localization. Thus Vam7p not only participates in trans-SNARE complex formation during docking, but is also an essential signaling molecule that regulates the localization of multiple downstream proteins.
t-SNAREs, Ypt7p:GTP, and F-actin disassembly are essential for normal HOPS enrichment at vertices (Fig. 7, step 4). HOPS can have direct physical interactions with both Ypt7p and the SNARE complex (Price et al., 2000; Seals et al., 2000; Wurmser et al., 2000). After Ypt7p establishes a vertex ring, its activation through the guanine nucleotide exchange function of the HOPS subunit Vps39 might even create an autocatalytic positive feedback loop for assembly of proteins at the vertex ring, though this concept is untested at this time. A similar feedback loop involving the nucleotide exchange factor Rabex-5 has been proposed for the recruitment of Rab5 effectors and the generation of Rab5 microdomains on endosomal membranes (Stenmark et al., 1995; Horiuchi et al., 1997; Simonsen et al., 1998; Christoforidis et al., 1999; Roberts et al., 1999; Nielsen et al., 2000; Rubino et al., 2000; Zerial and McBride, 2001).
Vacuole-bound actin participates in multiple stages of the fusion reaction (Eitzen et al., 2002). In this paper, we show that the disassembly of F-actin is required for vertex enrichment of SNAREs and the HOPS complex, but not of Ypt7p, suggesting that Ypt7p has an early role in the signaling cascade. We have previously shown that G-actin accumulates at vertices, that this enrichment relies on the function of SNAREs, and that the polymerization of G-actin at vertices at a late stage is required for fusion (Eitzen et al., 2002). Thus, actin remodeling may have two successive roles in the reaction. Actin depolymerization during tethering (Fig. 7, step 2) promotes SNARE and effector enrichment at vertices, and G-actin accumulation at vertices (Fig. 7, step 4) may allow conversion to vertex F-actin to support fusion.
Rings of membrane proteins are thought to mediate docking and fusion of apposed membranes of all sizes, from the neuronal synapse where they may create a point of membrane apposition (Weber et al., 1998) to yeast vacuoles, where the apposed boundary membrane domain is large enough to be visualized by light microscopy (Wang et al., 2002). Our current studies show that Ypt7p, actin, and SNAREs act in an ordered hierarchy to establish these vertex ring domains.
Materials and methods
Yeast strains for the in vitro vacuole fusion assay are BJ3505 and DKY6280. GFP-tagged yeast strains for the in vitro docking assay were either described in Wang et al. (2002) or generated for this work. GFP was the “Superglow” variant of Kahana and Silver (1998). For Vam7p and Vti1p, the GFP module was fused in-frame to the 3′ end of the chromosomal copy of the gene by homologous recombination (Longtine et al., 1998). For Nyv1p, the GFP module was inserted in-frame to the 5′ end of the ORF, and the fusion protein was expressed from its native promoter using an integrating plasmid PRS306. The endogenous Nyv1p ORF was deleted where indicated using homologous recombination (Longtine et al., 1998). A similar PCR-mediated approach to that described by Longtine et al. (1998) was used to generate a Vps33-mRFP fusion expressed from the native Vps33p promoter. In all cases, the parental strain is SEY6210 (MATa leu2–3 leu2–112 ura3–52 his3-D200 trp1-D901 lys2–801 suc2-D9).
Reagents were dissolved in PS buffer (20 mM Pipes-KOH, pH 6.8, and 200 mM sorbitol) unless otherwise noted. Alexa Fluor® 488-DNase I (Molecular Probes, Inc.) was dissolved at 166 μM in PBS/50% glycerol. Jasplakinolide (Molecular Probes, Inc.) was dissolved in DMSO at 10 mM. Recombinant PX domain (Boeddinghaus et al., 2002) and full-length Vam7p were purified as GST fusion proteins from Escherichia coli (unpublished data), and the GST domain was removed by thrombin cleavage. Recombinant His6-Sec17p (Haas and Wickner, 1996) and His6-Gyp1–46p were purified from E. coli using Ni-NTA chromatography (Rak et al., 2000). Anti-Vam3p IgG was precipitated from serum using 45% (NH4)2SO4 and dialyzed into 20 mM NaPi, pH 6.5 (Harlow and Lane, 1988). The dialyzed sample was loaded onto DE52 resin (Whatman) in this buffer. The flow-through was dialyzed into 10 mM Pipes/KOH, pH 6.8, and 200 mM sorbitol, and was then digested with immobilized Papain according to the manufacturer's protocol (ImmunoPure Fab Preparation Kit; Pierce Chemical Co.). The Fc fraction and undigested IgG were removed by adsorption to protein A-Sepharose. The flow-through Fab fraction was applied to a Fast Flow Q (Amersham Biosciences) column pre-equilibrated in 20 mM TrisCl, pH 8.0. The flow-through was collected and buffer was exchanged on a G25 column in 10 mM Pipes, pH 6.8, and 200 mM sorbitol. Inhibitors were used at the following final concentrations: 50 ng/μl excess Sec17p, 60 ng/μl anti-Vam3 Fab, 600 ng/μl recombinant Gyp1–46p, 500 μM jasplakinolide, 15 μM PX domain, 500 μM latrunculin B, and 5 μM full-length Vam7p.
In vitro vacuole fusion
Vacuoles were isolated from BJ3505 and DKY6281 for in vitro fusion assays (Haas, 1995). The 30-μl reaction contained 3 μg vacuole from each strain, 80 mM KCl, 0.5 mM MgCl2, 20 mM Pipes-KOH, pH 6.8, 0.6 mM ATP, 26 mM creatine phosphate, 0.3 mg/ml creatine kinase, and 10 μM coenzyme A. Reactions were incubated for 90 min at 27°C before assaying alkaline phosphatase activity.
In vitro microscopic docking assay
Docking was assayed (Wang et al., 2002) in 30-μl reactions containing 5 μg of vacuoles labeled with 3 μM of the lipophilic dye FM4–64 (Molecular Probes, Inc.). After 30 min, vacuoles were mixed with 40 μl 0.6% agarose in PS buffer. Aliquots (15 μl) were immediately mounted on slides and observed by fluorescence microscopy.
Images were acquired using a modified microscope (BX51; Olympus) equipped with a 100-W Mercury arc lamp, Plan Apochromat objective (60×, 1.4 NA) and a Sensicam QE CCD camera (Cooke). Images were acquired without pixel binning. This combination of camera and objective fulfilled the Shannon-Nyquist sampling criterion. Filters (Chroma Technology Corp.) were mounted in a motorized turret. An Endow GFP filter set was used for GFP-labeled proteins and for Alexa 488–labeled DNase I; a TRITC/Cy3 filter set was used for FM4–64 and mRFP1, and a DAPI filter set for blue lipid dye TMA-DPH. IP Lab software (Scanalytics) was used to automate microscope functions and data acquisition. Image acquisition, segmentation, and measurement were according to Wang et al. (2002). FM4–64 and TMA-DPH lipid dyes were significantly brighter than GFP and mRFP1 and were used to focus the fields. Photobleaching was performed for 30 s between each channel. The 14-bit images were then processed using Image/J v.1.21e to generate ratio images. Background was defined as the local minimum pixel value and subtracted before other analysis steps. To compare and depict relative ratios, the ratio images were normalized to the same contrast scales (fivefold range of ratio values on a logarithmic scale) and converted to 8-bit false-color format for display. Surface plots for the range-normalized images were generated using NIH Image v. 1.62.
For morphometry, 16-bit background-subtracted images were analyzed using Image/J. Ratios of protein (either GFP or mRFP1):lipid (either FM4–64 or TMA-DPH) were then calculated based on the maximum pixel values within manually defined circular regions of interest. Every vertex within a vacuole cluster was scored. For each outside edge, several locations were sampled. Vertex ratio values were then normalized by dividing by the mean ratio values of outside edges. This normalization set the mean outside-edge ratio to 1 for the different treatments; therefore, the resulting vertex ratios report enrichment relative to outside edge. Typically, for each treatment, 15–30 clusters from random fields, which contain an average of 500 vertex sites and photographed from at least two independent experiments, were analyzed. Jasplakinolide treatment caused aberrant morphology in 20–30% of vacuoles. These vacuoles had a filamentous appearance when viewed with FM 4–64 and were not scored for vertex enrichment. The normalized ratio values were then plotted as a cumulative distribution (CD) plot. For the GFP/RFP colocalization experiment (Fig. 7), the protein:lipid probe ratio data were normalized to the means previously obtained in single-probe experiments.
Image statistics were evaluated using JMP 5 (SAS Institute, Inc.). All ratio data were log-transformed before analysis; this procedure yielded near-normal distributions with comparable variances. Means and 95% confidence intervals for ratio values were computed using one-way ANOVAs for each GFP-tagged protein. Partial and complete inhibition (Fig. 5) were scored using t tests, with α initially set to 0.05 and corrected for multiple comparisons (P < 0.0006) using the Dunn-Sidak method (Sokal and Rohlf, 1994). Nonparametric tests gave indistinguishable results. For colocalization analysis (Fig. 7), multiple regression was performed. For the two-variable model (GFP ratio and PX treatment) presented in the Results, the residuals from the best-fit model were normally distributed (P > 0.8), indicating that assumptions required for the use of linear regression were not violated (Sokal and Rohlf, 1994).
Acknowledgments
We thank Dr. Charles Barlowe for fruitful discussions, Drs. Stephanie Zimsen and Deborah Chiavelli for discussions of statistical analysis, and Dr. Roger Tsien (University of California, San Diego, La Jolla, CA) for providing the clone of mRFP.
This work was supported by a grant from the National Institute of General Medical Sciences (NIGMS). A. Merz is supported by fellowship DRG1598 of the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation and K. Collins by T32GM08704 from NIGMS.
Footnotes
Abbreviations used in this paper: HOPS, homotypic fusion and vacuole protein sorting; PX, Phox homology.
References
- Albert, S., E. Will, and D. Gallwitz. 1999. Identification of the catalytic domains and their functionally critical arginine residues of two yeast GTPase-activating proteins specific for Ypt/Rab. EMBO J. 18:5216–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M.K., N. Calakos, and R.H. Scheller. 1992. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 257:255–259. [DOI] [PubMed] [Google Scholar]
- Boeddinghaus, C., A.J. Merz, R. Laage, and C. Ungermann. 2002. A cycle of Vam7p release from and PtdIns 3-P-dependent rebinding to the yeast vacuole is required for homotypic vacuole fusion. J. Cell Biol. 157:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromley, S.K., W.R. Burack, K.G. Johnson, K. Somersalo, T.N. Sims, C. Sumen, M.M. Davis, A.S. Shaw, P.M. Allen, and M.L. Dustin. 2001. The immunological synapse. Annu. Rev. Immunol. 19:375–396. [DOI] [PubMed] [Google Scholar]
- Cao, X., N. Ballew, and C. Barlowe. 1998. Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO J. 17:2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, R.E., O. Tour, A.E. Palmer, P.A. Steinbach, G.S. Baird, D.A. Zacharias, and R.Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 11:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheever, M.L., T.K. Sato, T. de Beer, T.G. Kutateladze, S.D. Emr, and M. Overduin. 2001. Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat. Cell Biol. 3:613–618. [DOI] [PubMed] [Google Scholar]
- Christoforidis, S., M. Miaczynska, K. Ashman, M. Wilm, L. Zhao, S.C. Yip, M.D. Waterfield, J.M. Backer, and M. Zerial. 1999. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1:249–252. [DOI] [PubMed] [Google Scholar]
- Du, L.L., and P. Novick. 2001. Yeast rab GTPase-activating protein Gyp1p localizes to the Golgi apparatus and is a negative regulator of Ypt1p. Mol. Biol. Cell. 12:1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin, M.L., S.K. Bromley, M.M. Davis, and C. Zhu. 2001. Identification of self through two-dimensional chemistry and synapses. Annu. Rev. Cell Dev. Biol. 17:133–157. [DOI] [PubMed] [Google Scholar]
- Eitzen, G., E. Will, D. Gallwitz, A. Haas, and W. Wickner. 2000. Sequential action of two GTPases to promote vacuole docking and fusion. EMBO J. 19:6713–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitzen, G., N. Thorngren, and W. Wickner. 2001. Rho1p and Cdc42p act after Ypt7p to regulate vacuole docking. EMBO J. 20:5650–5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitzen, G., L. Wang, N. Thorngren, and W. Wickner. 2002. Remodeling of organelle-bound actin is required for yeast vacuole fusion. J. Cell Biol. 158:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger, F.P., T.E. Hughes, and P. Novick. 1998. Sec3p is a spatial landmark for polarized secretion in budding yeast. Cell. 92:559–571. [DOI] [PubMed] [Google Scholar]
- Greer, C., and R. Schekman. 1982. Actin from Saccharomyces cerevisiae. Mol. Cell. Biol. 2:1270–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W., F. Tamanoi, and P. Novick. 2001. Spatial regulation of the exocyst complex by Rho1 GTPase. Nat. Cell Biol. 3:353–360. [DOI] [PubMed] [Google Scholar]
- Haas, A. 1995. A quantitative assay to measure homotypic vacuole fusion in vitro. Methods Cell Sci. 17:283–294. [Google Scholar]
- Haas, A., and W. Wickner. 1996. Homotypic vacuole fusion requires Sec17p (yeast a-SNAP) and Sec18p (yeast NSF). EMBO J. 15:3296–3305. [PMC free article] [PubMed] [Google Scholar]
- Haas, A., D. Scheglmann, T. Lazar, D. Gallwitz, and W. Wickner. 1995. The GTPase Ypt7p of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO J. 14:5258–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies. A Laboratory Manual. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY. 309–311.
- Horazdovsky, B.F., G.R. Busch, and S.D. Emr. 1994. VPS21 encodes a rab5-like GTP binding protein that is required for the sorting of yeast vacuolar proteins. EMBO J. 13:1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi, H., R. Lippe, H.M. McBride, M. Rubino, P. Woodman, H. Stenmark, V. Rybin, M. Wilm, K. Ashman, M. Mann, and M. Zerial. 1997. A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell. 90:1149–1159. [DOI] [PubMed] [Google Scholar]
- Jahn, R., and T.C. Sudhof. 1999. Membrane fusion and exocytosis. Annu. Rev. Biochem. 68:863–911. [DOI] [PubMed] [Google Scholar]
- Kahana, J.A., and P. Silver. 1998. The uses of green fluorescent protein in yeasts. Green Fluorescent Protein: Properties, Applications, and Protocols. M. Chalfie and S. Kain, editors. Wiley-Liss, Inc, New York. 139–151.
- Lang, T., D. Bruns, D. Wenzel, D. Riedel, P. Holroyd, C. Thiele, and R. Jahn. 2001. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 20:2202–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine, M.S., A. McKenzie, D.J. Demarini, N.G. Shah, A. Wach, A. Brachat, P. Philippsen, and J.R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 14:953–961. [DOI] [PubMed] [Google Scholar]
- Martin, K.H., J.K. Slack, S.A. Boerner, C.C. Martin, and J.T. Parsons. 2002. Integrin connections map: to infinity and beyond. Science. 296:1652–1653. [DOI] [PubMed] [Google Scholar]
- Martinez, O., and B. Goud. 1998. Rab proteins. Biochim. Biophys. Acta. 1404:101–112. [DOI] [PubMed] [Google Scholar]
- Mayer, A., and W. Wickner. 1997. Docking of yeast vacuoles is catalyzed by the Ras-like GTPase Ypt7p after symmetric priming by Sec18p (NSF). J. Cell Biol. 136:307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, A., W. Wickner, and A. Haas. 1996. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 85:83–94. [DOI] [PubMed] [Google Scholar]
- Muller, O., D.I. Johnson, and A. Mayer. 2001. Cdc42p functions at the docking stage of yeast vacuole membrane fusion. EMBO J. 20:5657–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, B.J., C. Ungerman, H.R. Pelham, W.T. Wickner, and A. Haas. 1997. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 387:199–202. [DOI] [PubMed] [Google Scholar]
- Nielsen, E., S. Christoforidis, S. Uttenweiler-Joseph, M. Miaczynska, F. Dewitte, M. Wilm, B. Hoflack, and M. Zerial. 2000. Rabenosyn-5, a novel Rab5 effector, is complexed with hVPS45 and recruited to endosomes through a FYVE finger domain. J. Cell Biol. 151:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, C., M.J. Bayer, S. Buhler, J.S. Andersen, M. Mann, and A. Mayer. 2001. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 409:581–587. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., S. Almo, S. Quirk, V. Vinson, and E.E. Lattman. 1994. Structure of actin binding proteins: insights about function at atomic resolution. Annu. Rev. Cell Biol. 10:207–249. [DOI] [PubMed] [Google Scholar]
- Price, A., D. Seals, W. Wickner, and C. Ungermann. 2000. The docking stage of yeast vacuole fusion requires the transfer of proteins from a cis-SNARE complex to a Rab/Ypt protein. J. Cell Biol. 148:1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak, A., R. Fedorov, K. Alexandrov, S. Albert, R.S. Goody, D. Gallwitz, and A.J. Scheidig. 2000. Crystal structure of the GAP domain of Gyp1p: first insights into interaction with Ypt/Rab proteins. EMBO J. 19:5105–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, R.L., M.A. Barbieri, K.M. Pryse, M. Chua, J.H. Morisaki, and P.D. Stahl. 1999. Endosome fusion in living cells overexpressing GFP-rab5. J. Cell Sci. 112:3667–3675. [DOI] [PubMed] [Google Scholar]
- Rubino, M., M. Miaczynska, R. Lippe, and M. Zerial. 2000. Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J. Biol. Chem. 275:3745–3748. [DOI] [PubMed] [Google Scholar]
- Schoch, S., P.E. Castillo, T. Jo, K. Mukherjee, M. Geppert, Y. Wang, F. Schmitz, R.C. Malenka, and T.C. Sudhof. 2002. RIM1α forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 415:321–326. [DOI] [PubMed] [Google Scholar]
- Schwartz, M.A., and M.H. Ginsberg. 2002. Networks and crosstalk: integrin signalling spreads. Nat. Cell Biol. 4:E65–E68. [DOI] [PubMed] [Google Scholar]
- Seals, D.F., G. Eitzen, N. Margolis, W.T. Wickner, and A. Price. 2000. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc. Natl. Acad. Sci. USA. 97:9402–9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev, N. 2001. Ypt and Rab GTPases: insight into functions through novel interactions. Curr. Opin. Cell Biol. 13:500–511. [DOI] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.M. Gaullier, A. Brech, J. Callaghan, B.H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Singer-Kruger, B., H. Stenmark, A. Dusterhoft, P. Philippsen, J.-S. Yoo, D. Gallwitz, and M. Zerial. 1994. Role of three rab5-like GTPases, Ypt51p, Ypt52p, and Ypt53p, in the endocytic and vacuole sorting pathways of yeast. J. Cell Biol. 125:283–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal, R.R., and F.J. Rohlf. 1994. Biometry: The Principles and Practice of Statistics in Biological Research. 3rd ed. W.H. Freeman, New York. 859 pp.
- Sonnichsen, B., S. De Renzis, E. Nielsen, J. Rietdorf, and M. Zerial. 2000. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark, H., G. Vitale, O. Ullrich, and M. Zerial. 1995. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell. 83:423–432. [DOI] [PubMed] [Google Scholar]
- Ungermann, C., and W. Wickner. 1998. Vam7p, a vacuolar SNAP-25 homolog, is required for SNARE complex integrity and vacuole docking and fusion. EMBO J. 17:3269–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann, C., G.F. von Mollard, O.N. Jensen, N. Margolis, T.H. Stevens, and W. Wickner. 1999. Three v-SNAREs and two t-SNAREs, present in a pentameric cis-SNARE complex on isolated vacuoles, are essential for homotypic fusion. J. Cell Biol. 145:1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann, C., A. Price, and W. Wickner. 2000. A new role for a SNARE protein as a regulator of the Ypt7/Rab-dependent stage of docking. Proc. Natl. Acad. Sci. USA. 97:8889–8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer, P., E. Will, D. Scheglmann, M. Strom, and D. Gallwitz. 1999. Primary structure and biochemical characterization of yeast GTPase- activating proteins with substrate preference for the transport GTPase Ypt7p. Eur. J. Biochem. 260:284–290. [DOI] [PubMed] [Google Scholar]
- Wang, L., C. Ungermann, and W. Wickner. 2000. The docking of primed vacuoles can be reversibly arrested by excess Sec17p (alpha-SNAP). J. Biol. Chem. 275:22862–22867. [DOI] [PubMed] [Google Scholar]
- Wang, L., E.S. Seeley, W. Wickner, and A.J. Merz. 2002. Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell. 108:357–369. [DOI] [PubMed] [Google Scholar]
- Weber, T., B.V. Zemelman, J.A. McNew, B. Westermann, M. Gmachl, F. Parlati, T.H. Sollner, and J.E. Rothman. 1998. SNAREpins: Minimal machinery for membrane fusion. Cell. 92:759–772. [DOI] [PubMed] [Google Scholar]
- Wickner, W. 2002. Yeast vacuoles and membrane fusion pathways. EMBO J. 21:1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser, A.E., T.K. Sato, and S.D. Emr. 2000. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J. Cell Biol. 151:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial, M., and H. McBride. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2:107–117. [DOI] [PubMed] [Google Scholar]