

Abstract
The protective antigen (PA) of the anthrax toxin binds to a cell surface receptor and thereby allows lethal factor (LF) to be taken up and exert its toxic effect in the cytoplasm. Here, we report that clustering of the anthrax toxin receptor (ATR) with heptameric PA or with an antibody sandwich causes its association to specialized cholesterol and glycosphingolipid-rich microdomains of the plasma membrane (lipid rafts). We find that although endocytosis of ATR is slow, clustering it into rafts either via PA heptamerization or using an antibody sandwich is necessary and sufficient to trigger efficient internalization and allow delivery of LF to the cytoplasm. Importantly, altering raft integrity using drugs prevented LF delivery and cleavage of cytosolic MAPK kinases, suggesting that lipid rafts could be therapeutic targets for drugs against anthrax. Moreover, we show that internalization of PA is dynamin and Eps15 dependent, indicating that the clathrin-dependent pathway is the major route of anthrax toxin entry into the cell. The present work illustrates that although the physiological role of the ATR is unknown, its trafficking properties, i.e., slow endocytosis as a monomer and rapid clathrin-mediated uptake on clustering, make it an ideal anthrax toxin receptor.
Keywords: anthrax toxin; rafts; microdomains; clustering; protective antigen
Introduction
Anthrax toxin is one of the two dominant virulence factors produced by Bacillus anthracis (Leppla, 1991). The toxin is composed of three subunits; edema factor (EF),* lethal factor (LF), and protective antigen (PA). EF is a calmodulin-dependent adenylate cyclase that elevates intracellular levels of cAMP (Leppla, 1982). LF is a metalloprotease that targets all MAPK kinases (Duesbery et al., 1998; Vitale et al., 1998) with the exception of MEK5 (Vitale et al., 2000), and is responsible for macrophage cell death (Chaudry et al., 2002; Mourez et al., 2002). Although LF and EF are ultimately responsible for the toxicity of the anthrax toxin, these two subunits cannot exert their effects in the absence of PA because they are unable to reach their cytoplasmic targets. Their recognition of the target cell and transport from the extracellular space to the cytoplasm absolutely requires PA.
PA is an 83-kD protein (PA83) that binds to a widely expressed, 368 amino acid, type 1 membrane protein termed anthrax toxin receptor (ATR; Bradley et al., 2001). Receptor-bound PA is then cleaved by members of the furin family of proteases, causing release of an NH2-terminal 20-kD fragment and leaving the COOH-terminal 63-kD moiety (PA63) bound to ATR. It is important to stress that furin cleavage occurs at the cell surface, even though this enzyme is more abundant intracellularly and in particular, in the TGN (Chaudry et al., 2002; Mourez et al., 2002). Unlike PA83, PA63 can oligomerize to form ring-shaped heptamers (Petosa et al., 1997). Interaction of LF and EF with PA63 occurs at the cell surface after heptamerization has occurred (Singh et al., 1994; Mogridge et al., 2001; Cunningham et al., 2002). The complex of PA63 with LF and/or EF is then internalized and transported to endosomes where the low pH triggers membrane insertion of the PA63 heptamer and channel formation (Milne et al., 1994; Mourez et al., 2002). Delivery of EF and LF to the cytosol is concomitant with PA63 channel formation and may involve passage of these proteins through the channel. Once in the cytoplasm, LF and EF modify their respective targets.
A crucial step in the mode of action of anthrax toxin that has received surprisingly little attention is the initial entry. Interestingly, PA63 is endocytosed, whereas the precursor PA83 remains at the cell surface (Beauregard et al., 2000). Here, we have analyzed the mechanism that triggers the specific cellular uptake of PA63 and thereby of the enzymatic units, LF and EF.
Results and discussion
We investigated whether the selective uptake of PA63, and not of PA83, was due to a change in surface distribution on conversion of PA83 to PA63. The similarity between the structure and the mode of action of PA and that of certain bacterial pore-forming toxins such as aerolysin (Abrami et al., 2000) prompted us to determine whether PA63 was associated with raftlike lipid microdomains of the plasma membrane. These domains are thought to form through lateral movement and assembly of cholesterol and glycosphingolipids. A specific subclass of rafts form flasklike invaginations at the plasma membrane and are then called caveolae (Simons and Ikonen, 1997; Brown and London, 1998). Rafts act as surface platforms in signal transduction, cholesterol homeostasis, and endocytosis (Brown and London, 1998; Simons and Toomre, 2000). Lipid rafts have also been implicated in various infectious processes (Fivaz et al., 1999), and in particular, were shown to favor heptamerization of the pore-forming toxin aerolysin (Abrami and van der Goot, 1999) via mechanisms that could well apply to PA. One biochemical characteristic of rafts is their resistance to nonionic detergents at 4°C, which allows their purification on density gradients. Native, full-size PA83 was associated with detergent-soluble domains of the plasma membrane (Fig. 1 A) in agreement with previous observations (Beauregard et al., 1999), as was a variant of PA mutated in the consensus furin cleavage site (PASNKE; Gordon et al., 1995). In contrast, PA63 (which can be obtained either in vitro by trypsin cleavage or in vivo by cell surface furin processing) was almost exclusively associated with detergent-resistant membranes (DRMs; Fig. 1 A). To confirm that the presence of PA63 in more buoyant, upper fractions of the gradient, is indeed due to its association with lipid rafts, we investigated whether the distribution of PA63 was altered by removal of plasma membrane cholesterol using β-methyl cyclodextrin (β-MCD) or sequestration of cholesterol using filipin (Simons and Toomre, 2000). As shown in Fig. 1 B, PA63 was partially (β-MCD) or completely (filipin) redistributed to the bottom of the gradient, in a manner similar to that of the normally raft-associated protein flotillin-1 (Bickel et al., 1997), but in contrast to the caveolar protein caveolin-1 (Kurzchalia and Parton, 1999). Because rafts are also rich in sphingomyelin, we analyzed whether reduction of sphingomyelin levels would affect the surface distribution of PA63. In sphingomyelin-deficient mutant CHO cells (Hanada et al., 1998), PA63 was no longer associated with DRMs (Fig. 1 B). Flotillin-1 was also partially redistributed to detergent-soluble fractions, in contrast to caveolin-1, which remained at the top of the gradient. Thus, PA63 is associated with noncaveolar cholesterol and sphingolipid-rich domains of the plasma membrane.
Figure 1.

Proteolytic processing of PA triggers partitioning of the anthrax toxin into lipid rafts. (A) Wild-type CHO cells were incubated for 1 h at 4°C with 500 ng/ml of either a mixture of native and trypsin-nicked PA83 (PA63), or PASNKE. DRMs were prepared and analyzed by Western blotting against PA and caveolin-1. The load (L) corresponds to 1/10 of the total material on the gradient. (B) Wild-type, sphingomyelin-deficient (CHO SPB), and recomplemented (CHO SPB(lcb1)) CHO cells were treated or not with β-MCD or filipin, then incubated with 500 ng/ml PA83 for 1 h at 4°C followed by 30 min at 37°C. DRMs were prepared and fractions were probed for the presence of PA, caveolin-1, and flotillin-1 by Western blotting. (C) ATR-deficient CHO cells were stably transfected with human ATR having an HA-tag at the COOH terminus. DRMs were prepared and fractions were probed using a biotinylated anti-HA antibody. The two upper bands are endogenous biotinylated CHO cell proteins recognized by the streptavidin-HRP, even in cells not expressing ATR-HA. (D) CHO cells were incubated with 500 ng/ml of either trypsin-nicked PA83 or of PASNKE for 1 h at 4°C followed by 30 min at 37°C, washed at 4°C, and further incubated with 1 μg/ml LF for 1 h at 4°C before preparation of DRMs. Fractions were analyzed by Western blot with anti-LF pAb.
The fact that PA83, which is the form of the toxin that initially encounters the cell, was not raft-associated suggested that the PA receptor (ATR) is not itself a raft resident protein. Because antibodies against ATR are not available, we stably expressed a COOH-terminal HA epitope–tagged version of ATR (ATR-HA) in CHO PR230, a spontaneous ATR-deficient mutant derived from CHO-K1 cells (Liu and Leppla, 2003). As predicted, the receptor was solubilized by the detergent (Fig. 1 C), indicating that ATR has little, if any, affinity for rafts. Therefore, although aerolysin and other toxins owe their rafts association to the fact that their receptors reside in these microdomains (Abrami and van der Goot, 1999), PA appears to force the raft association of its receptor that is normally in the glycerophospholipidic, nonraft region of the membrane.
The fact that only the mature PA63 form can promote raft association suggests that it might be heptamerization of PA63 that leads to clustering of ATR, which in turn would increase raft partitioning of the receptor. If so, PA63 heptamers should be associated with cell surface rafts.
To probe for cell surface heptamers, we measured binding of LF to cells treated with PA63 at 4°C because LF interacts with heptameric, but not with monomeric PA63 (Mogridge et al., 2001; cell surface PA63 heptamers cannot be revealed by SDS-PAGE because they dissociate in SDS). As shown in Fig. 1 D, surface-bound LF was indeed associated with DRMs of PA63-treated cells (but was not detectable on cells incubated with PASNKE), indicating that heptameric PA63 and its ligand LF are predominantly present in lipid rafts.
Our hypothesis also predicts that clustering ATR by means other than PA heptamerization, for example by cross-linking ATR using a sandwich of primary and secondary antibodies (Harder et al., 1998), should lead to raft association. Not having anti-ATR antibodies, ATR was labeled with PA83, which cannot heptamerize unless processed, and thus clustering at 4°C was induced with antibodies against PA. Whether a monoclonal or a polyclonal antibody was used, the clustered PA83 was predominantly present in DRMs, in contrast to the nonclustered PA83 (Fig. 2 A). That antibody clustering modified the surface distribution of ATR was confirmed by fluorescence microscopy. As shown in Fig. 2 B, a very punctate and intense signal was observed on cross-linking, whereas on cells that had been fixed before the addition of antibodies, the staining was weaker and more diffuse (note that some punctate structures can be observed that could well be due to cross-linking after PFA fixation as previously observed for GPI-anchored proteins; Maxfield and Mayor, 1997). The antibody-induced DRM association was cholesterol-dependent, as shown by the sensitivity to filipin and β-MCD (Fig. 2 C), strengthening the notion that lipid rafts control this process. Importantly, clustering of PA83 also led to the redistribution of a population of ATR-HA to DRMs (Fig. 2 D ; the majority of ATR-HA remains in detergent soluble fractions because of the vast excess of receptor with respect to PA, and because intracellular receptors are unavailable for PA63 binding). We further confirmed the raft association of clustered PA83 by cellular localization studies. As a typical raft marker, we used antibody-clustered GPI-anchored proteins (Harder et al., 1998) that were labeled using an inactive mutant of aerolysin, a specific probe for this class of proteins (Fivaz et al., 2002). As shown in Fig. 2 E (arrows), we found strong colocalization with GPI-anchored proteins not only for PA63, as expected, but also for clustered wild-type PA83 and PASNKE, consistent with our biochemical analysis. However, we could not see any significant colocalization at the cell surface of clustered PA83 with caveolin-1 (Fig. 2 F), strengthening the notion that PA associates with noncaveolar rafts (Fig. 1). The above experiments show that clustering of ATR, either by PA63 heptamerization or by antibody cross-linking, promotes its partitioning into rafts (Fig. 2 G).
Figure 2.
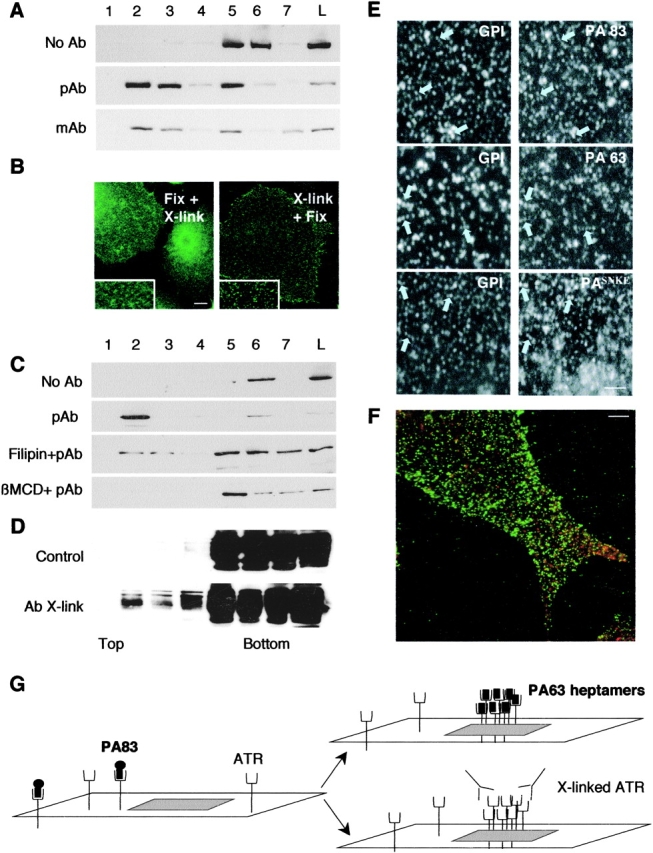
Antibody cross-linking promotes raft association of PA83 in a cholesterol-dependent manner. (A) CHO cells were incubated at 4°C with 500 ng/ml PA83 for 20 min. Surface PA was then clustered by an antibody sandwich at 4°C. DRMs were prepared and fractions were probed for PA by Western blotting. (B) CHO cells were treated for 1 h at 4°C with 500 ng/ml PA83, and then were either fixed with PFA followed by incubation with primary and secondary antibodies to detect PA (Fix+Xlink), or incubated for 30 min at 4°C with rabbit polyclonal anti-PA, or for 30 min at 4°C with the secondary antibodies and then fixed (Xlink+Fix). Bar, 10 μm. (C) CHO cells were treated or not with β-MCD or filipin, then treated as in A. DRMs were prepared and fractions were probed for the presence of PA as in A. (D) ATR-HA–expressing CHO cells were incubated or not with 500 ng/ml PA83 for 20 min and submitted to antibody (polyclonal) clustering as in A. DRMs were prepared and fractions were probed for the presence of ATR-HA by HA antibodies. (E) CHO cells were treated for 1 h at 4°C with 500 ng/ml PA (either native PA83, trypsin nicked PA83, or PASNKE) and 500 ng/ml of an inactive aerolysin mutant (ASSP), a monovalent probe for GPI-anchored proteins. Cells were then successively incubated for 30 min at 4°C with rabbit polyclonal anti-PA and chicken polyclonal anti-aerolysin antibodies for 30 min at 4°C with corresponding fluorescent secondary antibodies. Cells were fixed and visualized. For ease of analysis, only a small region of the plasma membrane is shown for each condition. Bar, 2.5 μm. (F) CHO cells were treated as in B (Xlink+Fix), and were then permeabilized with saponin and double labeled for the presence of caveolin-1. Bar, 5 μm. (G) Schematic behavior of ATR at the cell surface. ATR and the ATR–PA83 complex are present in the glycerophospholipid area of the plasma membrane. On clustering of ATR either via heptamerization of PA63 or by anti-body cross-linking, the transmembrane protein is stabilized within lipid rafts.
Next, we investigated whether ATR clustering is also a trigger for internalization. Again, we made use of the PA variant that cannot be processed by furin because such mutants remain at the cell surface (Beauregard et al., 2000), and investigated whether antibody clustering would promote internalization. Cells were incubated with PASNKE at 4°C, and a sandwich of primary and secondary antibodies was then added (Fig. 3 A, middle and bottom rows) or not (Fig. 3 A, top row) at 4°C. Cells were then shifted to 37°C to allow internalization (Fig. 3 A, top and middle rows) or kept on ice (Fig. 3 A, bottom row). Control cells (PASNKE with no antibodies; Fig. 3 A, top row) were shifted back to 4°C and were then treated with primary and secondary antibodies. Finally, cells were submitted or not to an acid wash in order to remove surface-bound antibodies and to reveal only the intracellular PASNKE. Intracellular PASNKE could only be detected when antibody cross-linking was performed before the internalization step (Fig. 3 A, middle row), demonstrating that clustering of PA83 is necessary and sufficient to promote its cellular uptake. When cells were treated with β-MCD before PA and antibody addition, intracellular accumulation was significantly reduced (Fig. 3 B).
Figure 3.

Raft association of PA triggers cellular uptake. (A) Antibody cross-linking leads to the appearance of intracellular PA83. As schematized in the right panel, CHO cells were incubated for 20 min at 4°C with 500 ng/ml PASNKE. Cells were then either (1) submitted to antibody cross-linking at 4°C and then further incubated at 4°C (condition: X-link+4°C) or 37°C (condition: X-link+37°C) for 30 min or (2) incubated at 37°C for 30 min and then submitted to the antibody sandwich at 4°C (condition: 37°C+X-link). For all conditions, cells were submitted or not to an acid wash before fixation. Bar, 10 μm. (B) Cholesterol depletion inhibits intracellular accumulation of PA63. CHO cells were treated or not with β-MCD, then incubated for 20 min at 4°C with 500 ng/ml PASNKE followed by an antibody sandwich (X-link+37°C). Internalization was allowed to proceed for 30 min at 37°C and cells were then submitted to a cold acid wash, fixed, and visualized using a fluorescent microscope.
To evaluate the physiological relevance of the role of rafts in the delivery of PA to endosomes, we measured the effect of cholesterol depletion on the appearance of the SDS-resistant PA63 heptamer because this form only appears in an acidic environment. As shown in Fig. 4 A, formation of the membrane-inserted SDS-resistant heptamer was strongly delayed on β-MCD treatment.
Figure 4.

Disruption of lipid rafts blocks formation of the PA channel and intracellular delivery of LF. (A) CHO cells were treated or not with β-MCD, incubated for 1 h at 4°C with 500 ng/ml trypsin-nicked PA83, and were transferred to 37°C for different periods of time. Aliquots of 80 μg total cell extract proteins were loaded on a 7.5% SDS-gel and probed for PA by Western blotting. (B) CHO cells were treated or not with β-MCD, incubated for 1 h at 4°C with a mixture of 1 μg/ml trypsin-nicked PA83 and 1 μg/ml LF, and transferred to 37°C for different periods of time. 40 μg of total cell extracts were analyzed by Western blotting (12.5% SDS-gel) for the presence of LF, total MEK1 (anti-COOH–terminal antibody), LF-processed MEK1 (anti-NH2–terminal antibody), and PA (using 7.5% SDS-gels for the latter).
Finally, to emphasize the physiological relevance of the association of the PA63–LF complex with lipid rafts, we analyzed the effects of β-MCD on the kinetics of cleavage of MEK1, one of the intracellular MAPK kinases targeted by LF. Processing of MEK1 by LF can be easily followed by Western blotting because it is accompanied by the loss of NH2-terminal epitopes due to the removal of seven amino acids form the NH2 terminus (Duesbery et al., 1998). In control cells, NH2-terminal cleavage of MEK1 was observed with time, whereas total MEK1, detected with an anti-COOH-terminal antibody, remained constant (Fig. 4 B). In marked contrast, MEK1 remained intact within the time frame studied in β-MCD–treated cells (Fig. 4 B). We confirmed, as a control, that similar amounts of LF were associated with control and β-MCD–treated cells (Fig. 4 B). Thus, LF cannot reach its intracellular target if lipid rafts are disrupted with the drug.
The fact that internalization of the ATR was both ligand-triggered and raft-dependent raised the possibility that caveolin-dependent endocytosis might be involved (Pelkmans and Helenius, 2002). However, the observation that PA63 and caveolin-1 showed different behaviors on cells at 4°C (Fig. 1 B and Fig. 2 F) and the fact that caveosomes are neutral and do not appear to communicate with any acidic organelle (Pelkmans and Helenius, 2002; knowing that the PA63 heptamer requires an acidic pH for membrane insertion) argue against a caveolar mediated uptake. To clarify this issue, we investigated whether expression of a dominant-negative mutant of caveolin-1, namely caveolin-1 with the GFP fused to its NH2 terminus (GFP-Cav1; Pelkmans et al., 2001), would affect anthrax toxin entry. The kinetics of appearance of the SDS-resistant PA63 heptamer (Fig. 5 A) as well as the kinetics of MEK1 cleavage (unpublished data) were very similar in control and GFP-Cav1–expressing cells. Also, we found little colocalization between 30-min internalized antibody-clustered PA83 and caveolin-1-GFP, used here as a marker of caveolae and caveosomes (large green structures seen in Fig. 5 B, which were never positive for PA; note that fusion of GFP to the COOH terminus of caveolin-1 does not lead to a dominant-negative phenotype; Pelkmans et al., 2001). Finally, to further rule out a caveolin-mediated entry pathway and transport to caveosomes, we found that intracellular SDS-resistant PA63 heptamers also form in macrophages RAW264.7 and FRT cells; two cell types that do not express caveolin-1 (Zurzolo et al., 1994; Kiss et al., 2000; unpublished data), in agreement with the fact that macrophages are sensitive to LF (Tang and Leppla, 1999).
Figure 5.
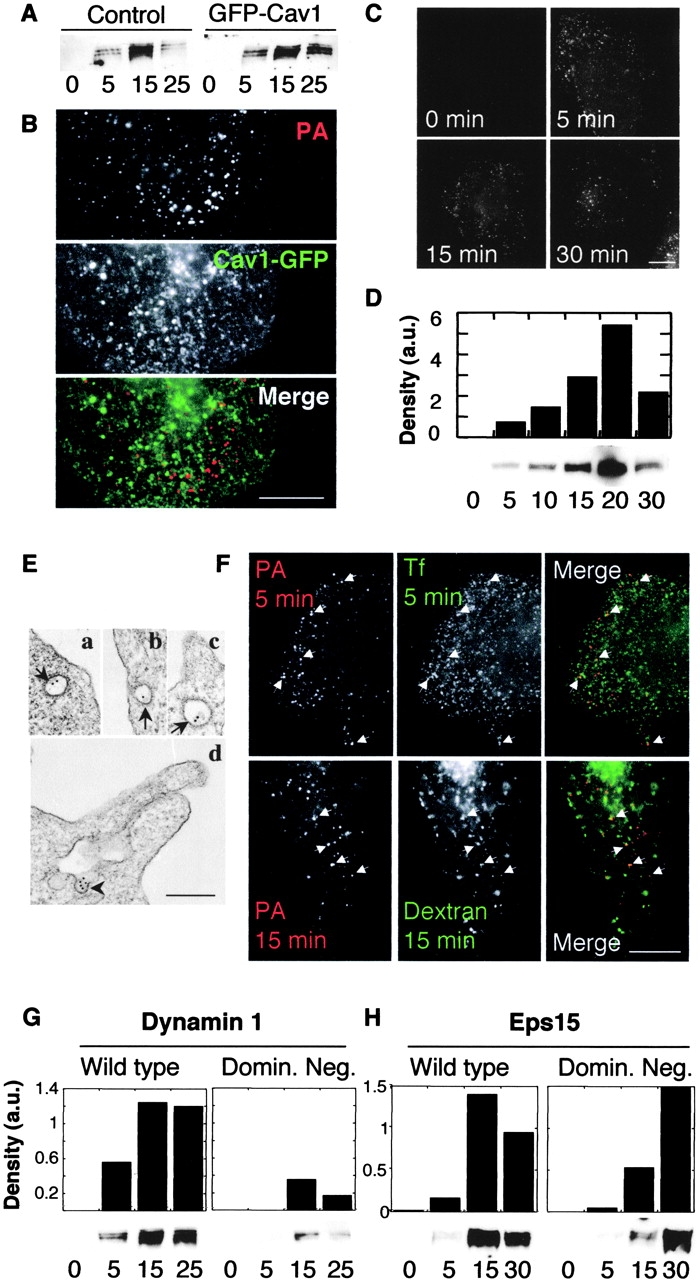
The anthrax toxin enters preferentially via a clathrin-mediated pathway. (A) HeLa cells were transiently transfected with the dominant-negative caveolin-1 mutant (GFP-Cav1), incubated for 1 h at 4°C with1 μg/ml trypsin-nicked PA83, and transferred to 37°C for different periods of time. 40 μg of total cell extracts were analyzed by Western blotting for the presence of heptameric PA63. (B) CHO cells were transiently transfected with caveolin-1-GFP (which has a wild-type phenotype), then incubated for 20 min at 4°C with 500 ng/ml PASNKE followed by an antibody sandwich as in Fig. 3 (X-link+37°C). Internalization was allowed to proceed for 30 min at 37°C, and cells were then fixed and visualized using a fluorescent microscope. A blow-up of a region of the plasma membrane is shown. Bar, 10 μm. (C) CHO cells were incubated for 20 min at 4°C with 500 ng/ml PASNKE followed by an antibody sandwich as in Fig. 3 (X-link+37°C). Internalization was allowed to proceed for 0, 5, 15, or 30 min at 37°C, cells were then submitted to a cold acid wash, fixed, and visualized. Bar, 10 μm. (D) CHO cells were incubated for 1 h at 4°C with1 μg/ml trypsin-nicked PA83, transferred to 37°C for different periods of time. 40 μg of total cell extracts were analyzed by SDS-PAGE and Western blotting for the presence of SDS-resistant heptameric PA63. The band intensity was quantified by densitometry (expressed in arbitrary units, a.u.), and is shown as a histogram to clearly illustrate the appearance and the degradation of the heptamer. (E) Cells were treated as in C, then incubated with protein A coupled to 10 nm gold, fixed, and processed for embedding in Epon and sectioning. Three examples are shown on which the clathrin coat is clearly visible (a–c, arrows); d is an example showing PASNKE in an invagination with no apparent coat (arrowhead). Bar, 200 nm. (F) CHO cells were incubated for 20 min at 4°C with 500 ng/ml PASNKE followed by an antibody sandwich (X-link+37°C). Internalization was allowed to proceed either for 5 min at 37°C in the presence of FITC-transferrin (Tf) or for 15 min in the presence of FITC-dextran. Cells were then submitted to a cold acid wash, fixed, and visualized. Examples of colocalization are indicated by arrowheads. Bar, 10 μm. (G) HeLa cells induced to express either wild-type or K44A dominant-negative dynamin-1 were treated and analyzed as in A. Numbers below lanes represent the incubation times at 37°C in min. The band intensities were quantified and plotted as in C. (H) HeLa cells transiently transfected with wild-type or dominant-negative EΔ95/295 Eps15 mutant were treated and analyzed as in G.
As shown in Fig. 5 C, significant internalization of antibody-clustered PASNKE occurred within 5 min, and a similarly short time was required to observe the SDS-resistant PA63 heptamer (Fig. 5 D). Subsequent degradation of PA63 heptamers was apparent within 30 min (Fig. 5 D). These rapid kinetics suggest that initial entry of PA might be clathrin-mediated. First, we investigated whether we could detect clustered PASNKE in clathrin-coated structures by electron microscopy, after warming the cells for 5 min at 37°C. As shown in Fig. 5 E, gold particles could be detected both in clathrin-coated pits and vesicles (Fig. 5 E, a–c) as well as sometimes in nonclathrin-coated invaginations of the plasma membrane (Fig. 5 E, d), but never in grape-like caveosomes (not depicted). Next, we investigated whether PASNKE was transported to endosomes. As shown in Fig. 5 F, colocalization was found between intracellular PASNKE and internalized transferrin (5 min), as well as internalized dextran (15 min), indicating early endosomal localization. Next, we investigated whether the GTPase dynamin was involved in anthrax toxin uptake. Dynamin has been implicated in both internalization of caveolae and in clathrin-mediated endocytosis (for review see McNiven et al., 2000). We made use of a well-described HeLa cell line that can be induced to express either wild-type dynamin-1 or a mutant defective in GTP binding and hydrolysis (DynK44A; Damke et al., 1994). As shown in Fig. 5 G, appearance of the SDS-resistant PA63 heptamer was severely delayed in DynK44A-expressing cells when compared with DynWT, as was the cleavage of MEK1 (unpublished data), showing that PA is preferentially internalized via a dynamin-dependent pathway. Because we have excluded the caveolar uptake pathway, these results suggest that clathrin is involved. To inhibit the clathrin-dependent pathway more specifically, we overexpressed a dominant-negative mutant of Eps15 (EΔ95/295), a protein required for clathrin-mediated endocytosis that binds directly to the plasma membrane adaptor AP2 (Benmerah et al., 1999). Transient transfection of HeLa cells was performed using either wild-type or dominant-negative Eps15 fused to GFP. Transfection rates of ∼75% were reached, and it was apparent when measuring uptake of rhodamine transferrin that very high expression levels of dominant-negative Eps15 (followed by GFP fluorescence) were required to inhibit clathrin-mediated uptake (unpublished data); levels that were reached in only few cells. Despite this, we have analyzed biochemically the kinetics of appearance of the SDS-resistant PA63 heptamer. As shown in Fig. 5 H, appearance of the SDS-resistant heptamer was significantly delayed, as was the subsequent degradation of the complex, even within this population of HeLa cells with mixed levels of inhibition of the clathrin pathway. Together, the inhibitory effects of dominant-negative mutants of dynamin and Eps15 show that the anthrax toxin preferentially enters the cell via a clathrin-dependent pathway.
The behavior of ATR is reminiscent of that of the B cell receptor, which undergoes ligand-dependent clustering and raft association (Cheng et al., 1999), and is subsequently internalized via a clathrin-dependent mechanism (Stoddart et al., 2002). Further studies will be required to determine whether ATR moves laterally out of rafts before endocytosis or, more likely, whether clathrin directly associates with ATR-containing rafts as for the B cell receptor.
The physiological role of ATR, as well as that of its splicing variant tumor endothelial marker 8 (St. Croix et al., 2000), is unknown. However, the present work highlights why anthrax toxin has highjacked this surface protein in particular. It indeed appears that independently of its function, ATR has a number of properties that make it the ideal anthrax toxin receptor. For a successful intoxication, PA requires a receptor that keeps it at the cell surface as long as it is monomeric, but drags it into the cell as soon as heptamerization, with concomitant binding of LF and/or EF, has occurred. Indeed, if on binding, PA83 were rapidly internalized by the cell, processing by furin could still efficiently occur in endosomes (Chapman and Munro, 1994), but binding of extracellular LF and/or EF could never take place, and the toxin would remain inactive. The initial surface localization of ATR and its low endocytosis rate ensure that cleavage by furin (which is also in the glycerophospholipidic, nonraft region of the membrane; Abrami and van der Goot, 1999) and heptamerization occur before uptake. The ability of ATR to redistribute to lipid rafts and to then be rapidly internalized via a clathrin-dependent mechanism on clustering guarantees the intracellular delivery of the PA heptamer–LF complex. This study clearly shows that subtle changes in the surface distribution of ATR are crucial for its function as an efficient anthrax toxin receptor, and might also be required for its physiological function. Lipid rafts appear to play an important role in regulating the assembly and the intracellular delivery of the anthrax toxin, and therefore constitute a potential therapeutic target.
Materials and methods
Reagents
Proteins produced as described previously (Leppla, 1988) include wild-type PA, PASNKE (a non-furin cleavage variant of PA in which the furin cleavage site was replaced by SNKE; Gordon et al., 1995), and LF. The aerolysin mutant (ASSP) was obtained as described previously (Fivaz et al., 2002). pAbs against PA (Liu et al., 2001) and LF were generated in the Leppla lab (Liu and Leppla, 2003). Anti-COOH-terminal MEK1 antibodies were obtained from Santa Cruz Biotechnology, Inc., anti-NH2-terminal MEK1 antibodies obtained from Upstate Biotechnology, anti-caveolin-1 and anti-flotillin-1 mAbs were obtained from Transduction laboratories, and FITC-conjugated secondary antibodies obtained from Molecular Probes, Inc. β-MCD and filipin were obtained from Sigma-Aldrich.
Sphingomyelin-deficient (CHO SPB) and recomplemented (CHO SPB(lcb1)) CHO cells were provided by K. Hanada (National Institute of Infectious Diseases, Tokyo, Japan) and maintained as described previously (Hanada et al., 1997). CHO PR230 is a previously described spontaneous PA receptor–deficient mutant (Liu and Leppla, 2003). Transfection of CHO PR230 with the ATR cDNA restored PA binding. Stable transfectants of CHO PR230 were used expressing human ATR having an HA-tag at the COOH terminus (Liu and Leppla, 2003). HeLa cell lines expressing wild-type and K44A dynamin mutant were maintained as described previously (Damke et al., 1994).
Biochemical methods
DRMs were prepared from CHO cells using OptiPrep gradients as described previously (Lafont et al., 2002). 7–8 fractions were collected from the top, the total protein content of each fraction was precipitated with 6% TCA in the presence of 375 μg sodium deoxylcholate as a carrier, and were analyzed by SDS-PAGE. For Western blot analysis, gels were transferred onto a nitrocellulose membrane either using a semi-dry system or, in order to detect the PA heptamer, by wet transfer for 5 h at 86 V.
Drug treatments
CHO cells were treated with 10 mM β-MCD or 1 μg/ml filipin for 1 h at 37°C as described previously (Abrami and van der Goot, 1999). The β-MCD treatment led to a 59.3 ± 3.8% decrease in total cholesterol as measured by one-dimensional TLC (Abrami and van der Goot, 1999).
Immunofluorescence
Coverslip-grown CHO cells were incubated with 500 ng/ml PA. Cell surface–bound PA was submitted to clustering by an antibody sandwich. In brief, cells were put on ice and incubated for 20 min with polyclonal anti-PA antibodies followed by 20 min with secondary antibody FITC-anti–rabbit. To remove surface-bound antibodies, live cells were submitted to an acid wash, i.e., incubated for 5 min at 23°C with 140 mM NaCl, 100 mM citrate, pH 2.0. For all experiments, cells were fixed with 3% PFA and then mounted on microscopy slides.
Electron microscopy
Cells were incubated with 1 μg/ml PASNKE for 1 h at 4°C followed by a sandwich of polyclonal anti-PA, mouse anti–rabbit IgG and 10-nm gold–labeled protein A. Cells were then washed, shifted 5 min at 37°C, and fixed with 2% glutaraldehyde in culture medium buffered with Hepes for 10 min at 37°C and with 2% glutaraldehyde in phosphate buffer for 30 min at RT. After washes, the cells were pelleted in agar and incubated with 2% osmium for 1 h at 4°C and overnight with 0.25% uranylacetate before degradation and Epon embedding.
Acknowledgments
We would like to thank Kentaro Hanada for providing us with the sphingomyelin-deficient cells, S.L. Schmidt (Scripps Research Institute, La Jolla, CA) for the dynamin mutant cells, A. Dautry-Varsat (Pasteur Institute, Paris, France) for the Eps15 constructs, A. Helenius (ETH Zurich, Zurich, Switzerland) for the Cav1-GFP and GFP-Cav1 constructs, Dana Hsu (National Institutes of Health) for making toxins, and Dan Kolakovsky and Jean Gruenberg for critical reading of the manuscript.
This work was supported by grant 31-61 507.00 of the Swiss National Science Foundation (G. van der Goot).
Footnotes
Abbreviations used in this paper: ATR, anthrax toxin receptor; ATR-HA, COOH-terminal HA epitope-tagged version of ATR; β-MCD, β-methyl cyclodextrin; DRM, detergent-resistant membrane; EF, edema factor; LF, lethal factor; PA, protective antigen; PA63, COOH-terminal 63-kD moiety; PA83, protective antigen of 83-kD; PASNKE, PA mutated in the consensus furin cleavage site.
References
- Abrami, L., and F.G. van der Goot. 1999. Plasma membrane microdomains act as concentration platforms to facilitate intoxication by aerolysin. J. Cell Biol. 147:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami, L., M. Fivaz, and F.G. van Der Goot. 2000. Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol. 8:168–172. [DOI] [PubMed] [Google Scholar]
- Beauregard, K.E., S. Wimer-Mackin, R.J. Collier, and W.I. Lencer. 1999. Anthrax toxin entry into polarized epithelial cells. Infect. Immun. 67:3026–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauregard, K.E., R.J. Collier, and J.A. Swanson. 2000. Proteolytic activation of receptor-bound anthrax protective antigen on macrophages promotes its internalization. Cell Microbiol. 2:251–258. [DOI] [PubMed] [Google Scholar]
- Benmerah, A., M. Bayrou, N. Cerf-Bensussan, and A. Dautry-Varsat. 1999. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J. Cell Sci. 112:1303–1311. [DOI] [PubMed] [Google Scholar]
- Bickel, P.E., P.E. Scherer, J.E. Schnitzer, P. Oh, M.P. Lisanti, and H.F. Lodish. 1997. Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J. Biol. Chem. 272:13793–13802. [DOI] [PubMed] [Google Scholar]
- Bradley, K.A., J. Mogridge, M. Mourez, R.J. Collier, and J.A. Young. 2001. Identification of the cellular receptor for anthrax toxin. Nature. 414:225–229. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- Chapman, R.E., and S. Munro. 1994. Retrieval of TGN proteins from the cell surface requires endosomal acidification. EMBO J. 13:2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudry, G.J., M. Moayeri, S. Liu, and S.H. Leppla. 2002. Quickening the pace of anthrax research: three advances point towards possible therapies. Trends Microbiol. 10:58–62. [DOI] [PubMed] [Google Scholar]
- Cheng, P.C., M.L. Dykstra, R.N. Mitchell, and S.K. Pierce. 1999. A role for lipid rafts in B cell antigen receptor signaling and antigen targeting. J. Exp. Med. 190:1549–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, K., D.B. Lacy, J. Mogridge, and R.J. Collier. 2002. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc. Natl. Acad. Sci. USA. 99:7049–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damke, H., T. Baba, D.E. Warnock, and S.L. Schmid. 1994. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127:915–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duesbery, N.S., C.P. Webb, S.H. Leppla, V.M. Gordon, K.R. Klimpel, T.D. Copeland, N.G. Ahn, M.K. Oskarsson, K. Fukasawa, K.D. Paull, and G.F. Vande Woude. 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 280:734–737. [DOI] [PubMed] [Google Scholar]
- Fivaz, M., L. Abrami, and F.G. van der Goot. 1999. Landing on lipid rafts. Trends Cell Biol. 9:212–213. [DOI] [PubMed] [Google Scholar]
- Fivaz, M., F. Vilbois, S. Thurnheer, C. Pasquali, L. Abrami, P.E. Bickel, R.G. Parton, and and F.G. van der Goot. 2002. Different sorting and fate of endocytosed GPI-anchored proteins. EMBO J. 21:3989–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, V.M., K.R. Klimpel, N. Arora, M.A. Henderson, and S.H. Leppla. 1995. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 63:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada, K., T. Hara, M. Nishijima, O. Kuge, R.C. Dickson, and M.M. Nagiec. 1997. A mammalian homolog of the yeast LCB1 encodes a component of serine palmitoyltransferase, the enzyme catalyzing the first step in sphingolipid synthesis. J. Biol. Chem. 272:32108–32114. [DOI] [PubMed] [Google Scholar]
- Hanada, K., T. Hara, M. Fukasawa, A. Yamaji, M. Umeda, and M. Nishijima. 1998. Mammalian cell mutants resistant to a sphingomyelin-directed cytolysin. Genetic and biochemical evidence for complex formation of the LCB1 protein with the LCB2 protein for serine palmitoyltransferase. J. Biol. Chem. 273:33787–33794. [DOI] [PubMed] [Google Scholar]
- Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss, A.L., A. Turi, N. Mullner, and J. Timar. 2000. Caveolin isoforms in resident and elicited rat peritoneal macrophages. Eur. J. Cell Biol. 79:343–349. [DOI] [PubMed] [Google Scholar]
- Kurzchalia, T.V., and R.G. Parton. 1999. Membrane microdomains and caveolae. Curr. Opin. Cell Biol. 11:424–431. [DOI] [PubMed] [Google Scholar]
- Lafont, F., G. Tran Van Nhieu, K. Hanada, P.J. Sansonetti, and F.G. van der Goot. 2002. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44-IpaB interaction. EMBO J. 21:4449–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppla, S.H. 1982. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA. 79:3162–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppla, S.H. 1988. Production and purification of anthrax toxin. Methods Enzymol. 165:103–116. [DOI] [PubMed] [Google Scholar]
- Leppla, S.H. 1991. The anthrax toxin complex. Sourcebook of Bacterial Protein Toxins. J.E. Alouf and J.H. Freer, editors. Academic Press, New York. 277–302.
- Liu, S., and S.H. Leppla. 2003. Cell surface tumor endothelium marker 8 cytoplasmic tail-independent anthrax toxin binding, proteolytic processing, oligomer formation and internalization. J. Biol. Chem. In press. [DOI] [PubMed] [Google Scholar]
- Liu, S., T.H. Bugge, and S.H. Leppla. 2001. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J. Biol. Chem. 276:17976–17984. [DOI] [PubMed] [Google Scholar]
- Maxfield, F.R., and S. Mayor. 1997. Cell surface dynamics of GPI-anchored proteins. Adv. Exp. Med. Biol. 419:355–364. [DOI] [PubMed] [Google Scholar]
- McNiven, M.A., L. Kim, E.W. Krueger, J.D. Orth, H. Cao, and T.W. Wong. 2000. Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J. Cell Biol. 151:187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne, J.C., D. Furlong, P.C. Hanna, J.S. Wall, and R.J. Collier. 1994. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 269:20607–20612. [PubMed] [Google Scholar]
- Mogridge, J., M. Mourez, and R.J. Collier. 2001. Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J. Bacteriol. 183:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourez, M., D.B. Lacy, K. Cunningham, R. Legmann, B.R. Sellman, J. Mogridge, and R.J. Collier. 2002. 2001: a year of major advances in anthrax toxin research. Trends Microbiol. 10:287–293. [DOI] [PubMed] [Google Scholar]
- Pelkmans, L., and A. Helenius. 2002. Endocytosis via caveolae. Traffic. 3:311–320. [DOI] [PubMed] [Google Scholar]
- Pelkmans, L., J. Kartenbeck, and A. Helenius. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3:473–483. [DOI] [PubMed] [Google Scholar]
- Petosa, C., R.J. Collier, K.R. Klimpel, S.H. Leppla, and R.C. Liddington. 1997. Crystal structure of the anthrax toxin protective antigen. Nature. 385:833–838. [DOI] [PubMed] [Google Scholar]
- Simons, K., and E. Ikonen. 1997. Functiomal rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- Singh, Y., K.R. Klimpel, N. Arora, M. Sharma, and S.H. Leppla. 1994. The chymotrypsin-sensitive site, FFD315, in anthrax toxin protective antigen is required for translocation of lethal factor. J. Biol. Chem. 269:29039–29046. [PubMed] [Google Scholar]
- St. Croix, B., C. Rago, V. Velculescu, G. Traverso, K.E. Romans, E. Montgomery, A. Lal, G.J. Riggins, C. Lengauer, B. Vogelstein, and K.W. Kinzler. 2000. Genes expressed in human tumor endothelium. Science. 289:1197–1202. [DOI] [PubMed] [Google Scholar]
- Stoddart, A., M.L. Dykstra, B.K. Brown, W. Song, S.K. Pierce, and F.M. Brodsky. 2002. Lipid rafts unite signaling cascades with clathrin to regulate BCR internalization. Immunity. 17:451–462. [DOI] [PubMed] [Google Scholar]
- Tang, G., and S.H. Leppla. 1999. Proteasome activity is required for anthrax lethal toxin to kill macrophages. Infect. Immun. 67:3055–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale, G., R. Pellizzari, C. Recchi, G. Napolitani, M. Mock, and C. Montecucco. 1998. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 248:706–711. [DOI] [PubMed] [Google Scholar]
- Vitale, G., L. Bernardi, G. Napolitani, M. Mock, and C. Montecucco. 2000. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 352:739–745. [PMC free article] [PubMed] [Google Scholar]
- Zurzolo, C., W. van't Hof, G. van Meer, and E. Rodriguez-Boulan. 1994. VIP21/caveolin, glycosphingolipid clusters and the sorting of glycosylphosphatidylinositol-anchored proteins in epithelial cells. EMBO J. 13:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]