

Abstract
The WD-repeat proteins Rae1 and Bub3 show extensive sequence homology, indicative of functional similarity. However, previous studies have suggested that Rae1 is involved in the mRNA export pathway and Bub3 in the mitotic checkpoint. To determine the in vivo roles of Rae1 and Bub3 in mammals, we generated knockout mice that have these genes deleted individually or in combination. Here we show that haplo-insufficiency of either Rae1 or Bub3 results in a similar phenotype involving mitotic checkpoint defects and chromosome missegregation. We also show that overexpression of Rae1 can correct for Rae1 haplo-insufficiency and, surprisingly, Bub3 haplo-insufficiency. Rae1-null and Bub3-null mice are embryonic lethal, although cells from these mice did not have a detectable defect in nuclear export of mRNA. Unlike null mice, compound haplo-insufficient Rae1/Bub3 mice are viable. However, cells from these mice exhibit much greater rates of premature sister chromatid separation and chromosome missegregation than single haplo-insufficient cells. Finally, we show that mice with mitotic checkpoint defects are more susceptible to dimethylbenzanthrene-induced tumorigenesis than wild-type mice. Thus, our data demonstrate a novel function for Rae1 and characterize Rae1 and Bub3 as related proteins with essential, overlapping, and cooperating roles in the mitotic checkpoint.
Keywords: mRNA export; Rae1/Gle2; Bub3; mitotic checkpoint; chromosomal instability
Introduction
Abnormalities of chromosome number are a hallmark of the vast majority of human cancers, but little is known about the genetic events that can initiate or promote losses or gains of whole chromosomes (Hartwell and Kastan, 1994; Lengauer et al., 1998; Cahill et al., 1999b). In a subset of human cancers with chromosomal instability, mutations have been identified in genes that encode mitotic checkpoint proteins, such as Bub1, BubR1, and Mad2 (Cahill et al., 1998; Imai et al., 1999; Gemma et al., 2001). The mitotic checkpoint is a partly resolved intricate molecular network that helps to ensure faithful segregation of duplicated chromosomes during mitosis (Burke, 2000; Shah and Cleveland, 2000; Kitagawa and Hieter, 2001; Nigg, 2001; Wassmann and Benezra, 2001). Kinetochores of chromosomes that are not yet aligned at the metaphase plate and attached to microtubules activate the mitotic checkpoint, leading to inhibition of the anaphase-promoting complex (APC)* (Morgan, 1999; Peters, 2002). When all kinetochores are attached to microtubules and the chromosomes are aligned at the metaphase plate, the checkpoint is released. The APC then enables the onset of anaphase by activation of the protease separase, which separates sister chromatids by cleaving cohesion complexes (Peters, 2002).
During prophase and prometaphase, mitotic checkpoint proteins are positioned in the mitotic cytosol and at the kinetochores of condensed chromosomes. Current models propose that checkpoint proteins associated with kinetochores act as sensors for microtubule–kinetochore attachment and kinetochore tension. In the absence of attachment or tension, they act to generate a molecular “anaphase wait” signal (Shah and Cleveland, 2000; Hoffman et al., 2001; Jallepalli and Lengauer, 2001). Although the composition of this signal is unclear, it is believed to activate mitotic checkpoint proteins in the mitotic cytosol, including BubR1, Bub3, and Mad2 (Fang et al., 1998; Gillett and Sorger, 2001; Hoyt, 2001; Sudakin et al., 2001; Fang, 2002; Yu, 2002). Activated checkpoint proteins then bind to Cdc20, thereby preventing it from activating the APC. When a chromosome has proper microtubule–kinetochore attachment and kinetochore tension, its kinetochore-associated checkpoint proteins partially detach and its emission of anaphase wait signals stops. Alignment of all chromosomes at the metaphase plate quenches all anaphase wait signals, allowing Cdc20 to bind to and activate the APC.
The mitotic checkpoint protein Bub3 shares extensive sequence homology with Rae1 (Taylor et al., 1998; Martinez-Exposito et al., 1999). Their homology is not confined to each of the four WD repeat motifs; it extends over the entire protein length and is especially high in the segment that separates WD repeats 3 and 4. Rae1 (also called Gle2 or mrnp41) is a highly conserved nuclear transport factor that is involved in the pathway for mRNA export in interphase, but whose precise role remains unclear (Brown et al., 1995; Murphy et al., 1996; Kraemer and Blobel, 1997; Taylor and McKeon, 1997; Bailer et al., 1998; Martinez-Exposito et al., 1999; Pritchard et al., 1999; Bachi et al., 2000; Yoon et al., 2000; Zenklusen et al., 2001). The nucleoporin Nup98 contains a motif named GLEBS that directs binding to Rae1 (Pritchard et al., 1999). We have recently shown that GLEBS motifs are also present in the mitotic checkpoint proteins Bub1 and BubR1, where they serve as binding sites for Bub3 (Wang et al., 2001). Bub3 exclusively binds to GLEBS sequences of mitotic checkpoint proteins. However, Rae1 binds not only to Nup98 but also to Bub1 (Wang et al., 2001). This finding led us to hypothesize that Rae1 might act as a mitotic checkpoint protein. To test this hypothesis, we have disrupted the mouse Rae1 gene by homologous recombination. We show that the loss of a single Rae1 allele causes a mitotic checkpoint defect and chromosome missegregation.
Results
Rae1 is essential for early embryogenesis
We addressed the physiological role of Rae1 through a gene-targeting approach in the mouse. We inactivated the Rae1 gene by interrupting its coding region at amino acid 50 with a lacZ-neo selection cassette (Fig. 1 A). Three independently targeted embryonic stem (ES) cell clones yielded chimeric mice that passed the disrupted Rae1 gene through the germline (Fig. 1 B). These Rae1 heterozygous mice were indistinguishable from wild-type littermates. We intercrossed heterozygous mice but found no Rae1−/− mice among 203 liveborn offspring, demonstrating that Rae1 is essential for mouse embryogenesis.
Figure 1.

Targeted disruption of the mouse Rae1 gene. (A) Indicated are part of the endogenous mouse (m)Rae1 gene (top), the targeting vector (middle), and the disrupted Rae1 allele (bottom). BamHI restriction sites and the 3′ DNA probe (solid bar) used for Southern blot identification of wild-type (WT) and knockout (KO) Rae1 alleles are shown. (B) Southern blot of genomic ES cell DNA, digested with BamH1 and hybridized to a 3′ external probe, revealing the expected 10.2-kb wild-type and 8.5-kb mutant fragments. (C) PCR genotyping of E3.5 embryos from a Rae1+/− intercross showing wild-type and knockout allele-specific amplification products of respectively 516 and 650 bp. (D) In vitro growth of representative Rae1+/+ and Rae1−/− embryos. Embryos from heterozygous crosses were recovered at E3.5 and grown on multi-well slides in DME/15% FCS. They were inspected daily and photographed at 48 (E5.5), 72 (E6.5), and 96 h (E7.5) after seeding. I, ICM; T, trophectoderm cells.
To determine when Rae1−/− embryos die, we collected embryos from heterozygous intercrosses at various stages of development and genotyped them by a PCR method (Fig. 1, A and C). Dissection of embryos at 8.5 d post-coitum (embryonic day [E]8.5) yielded 9 Rae1+/+, 22 Rae1+/−, but no Rae1−/− embryos. At 3.5 d of gestation, however, Rae1−/− embryos were present at a normal Mendelian frequency (7 +/+, 13 +/−, and 6 −/− embryos were identified among 26 blastocysts). These Rae1−/− embryos were morphologically indistinguishable from their wild-type and heterozygous counterparts.
To further investigate the developmental defects caused by Rae1 depletion, we harvested blastocysts from heterozygous intercrosses at E3.5 and then analyzed their in vitro growth properties (van Deursen et al., 1996). Regardless of the genotype, most cultured blastocysts hatched from the zona pellucida and attached to the surface of the tissue culture dish during the first 2 d of culture (Fig. 1 D). From E6.5 through E8.5, cultured Rae1+/+ and Rae1+/− blastocysts rapidly expanded their inner cell mass (ICM) on an underlying sheet of post-mitotic trophoblast cells (Fig. 1 D). We observed ICMs shortly after attachment of Rae1−/− blastocysts at E5.5, but these masses failed to expand from E6.5 through E8.5 and, instead, degenerated (34 out of 144 embryos degenerated). On the other hand, the trophoblast cells from Rae1−/− blastocysts developed normally into a flattened layer and remained viable up to and beyond E8.5 (Fig. 1 D).
Rae1 is not essential for nuclear export of mRNA
The ability to culture Rae1−/− blastocysts allowed us to further investigate the role of Rae1 in nucleocytoplasmic transport (Fig. 2 A). First, we double stained E7.5–E8.5 embryonic outgrowths with a polyclonal antibody against mouse Rae1(188–368) and monoclonal antibody mAb414, a marker of the nuclear pore complex (NPC) (Wu et al., 2001). In cells from control embryonic outgrowths, Rae1 prominently localized to the nuclear envelope (NE), although significant amounts of Rae1 were also found in the nucleus and the cytoplasm (Fig. 2 B). No Rae1 staining was detected in cells from Rae1−/− outgrowths, confirming that our gene-targeting strategy had indeed generated a null allele (Fig. 2 C). Disruption of the Rae1 homologue GLE2 in Saccharomyces cerevisiae causes severe clustering of nuclear pores (Murphy and Wente, 1996). Rae1-depleted cells showed a strong and uninterrupted rim-like labeling of the NE with mAb414, similar to that of control cells (Fig. 2 C′), indicating that knockout cells have a normal distribution of NPCs. Another feature of Gle2p-deficient yeast cells is the formation of membranous structures that seal nuclear pores (Murphy and Wente, 1996). Examination of embryonic outgrowths by transmission electron microscopy demonstrated that NPC sealing does not occur in Rae1−/− cells (unpublished data). Nup98, a nucleoporin that forms a complex with Rae1 at the NPC (Pritchard et al., 1999), exhibited a pronounced NE localization in Rae1−/− embryos (Fig. 2, D and E), demonstrating that Rae1 is not needed for binding of Nup98 to NPCs.
Figure 2.
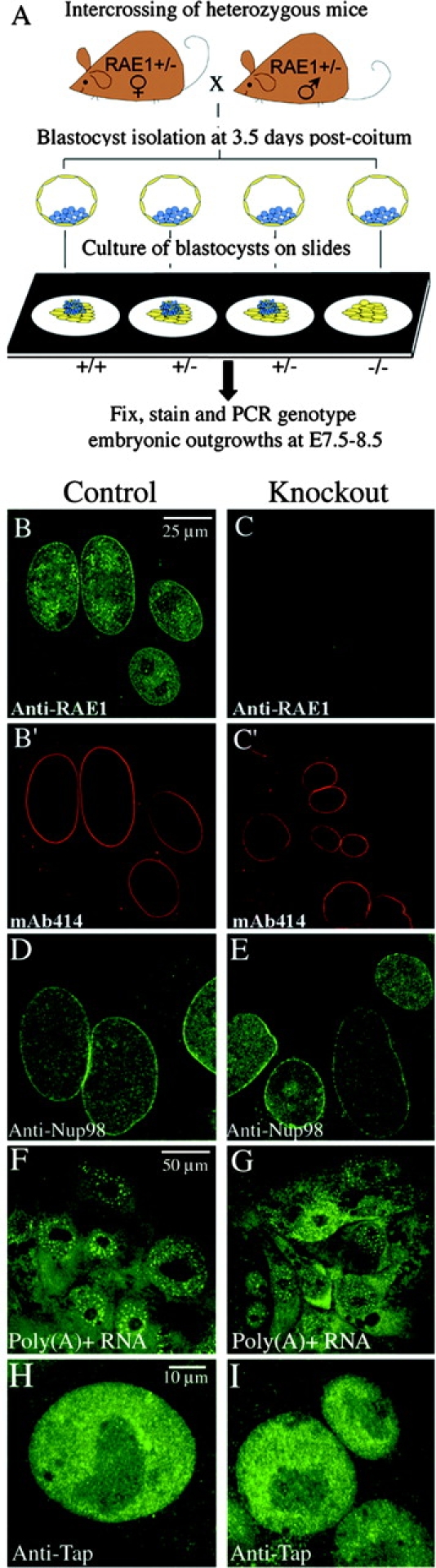
Rae1 is not essential for nuclear export of mRNA. (A) Overview of the experimental design. Blastocysts from intercrosses of Rae1+/− mice were cultured for ∼4–5 d and then analyzed by immunostaining or in situ hybridization. (B–C') Double staining of E8.5 embryonic outgrowths with a polyclonal antibody against mouse Rae1(188–368) (Pritchard et al., 1999) and monoclonal antibody mAb414, a marker of the NPC (Wu et al., 2001). Shown are representative high-resolution images of trophectoderm cells. (D and E) Immunostaining of E8.5 embryonic outgrowths with a polyclonal antibody against Nup98(151–224) (Wu et al., 2001). Shown are high-resolution images of trophectoderm cells. (F and G) Localization of poly(A)+ RNA in trophectoderm cells from E8.5 Rae1+/+ and Rae1−/− outgrowths. A FITC–oligo(dT)50 probe was used for visualization of poly(A)+ by in situ hybridization. (H and I) Trophoblast cells stained with a polyclonal antibody against human Tap (Braun et al., 1999).
Because previous studies have linked Rae1 to the pathway for nuclear export of mRNA (Brown et al., 1995; Murphy et al., 1996; Kraemer and Blobel, 1997; Pritchard et al., 1999; Bachi et al., 2000; Sabri and Visa, 2000), we asked whether cells lacking this protein accumulate mRNA in their nuclei. E8.5 embryonic outgrowths from heterozygous intercrosses were stained for poly(A)+ RNA by an in situ hybridization technique using FITC-labeled oligo(dT)50-mer probe (Pritchard et al., 1999). Surprisingly, both the level and the subcellular distribution of poly(A)+ RNA appeared normal in cells of Rae1-deficient embryonic outgrowths (Fig. 2, F and G). Similar results were obtained when earlier embryonic outgrowths (E5.5–E7.5) were stained for poly(A)+ RNA (unpublished data), revealing that ICM degeneration does not coincide with nuclear accumulation of mRNA. We also asked if the subcellular distribution of various pre-mRNA/mRNA binding proteins would be altered in the absence of Rae1. Immunolabeling experiments with antibodies against the mRNA export factors Tap (Fig. 2, H and I) and Aly, the hnRNP proteins A and C2, the SR protein SC35, and the splicing-dependent mRNA-binding protein Y14 showed normal subcellular localizations for all these proteins in Rae1−/− cells (unpublished data). Thus, it appears that the bulk of mRNAs synthesized in the nucleus can be exported to the cytoplasm when Rae1 is lacking.
Haplo-insufficiency at the Rae1 locus causes mitotic checkpoint dysfunction
Because Rae1 has a high degree of sequence similarity to Bub3 and can interact with Bub1 (Taylor et al., 1998; Martinez-Exposito et al., 1999; Wang et al., 2001), it was of interest to determine whether Rae1 would be required in mitosis. Others have recently shown that HCT116 cells with only one copy of the mitotic checkpoint gene Mad2 fail to arrest in prometaphase and exit mitosis without cytokinesis when cultured in the presence of the microtubule-depolymerizing drug nocodazole (Michel et al., 2001), a response that is typical for cells with a defective mitotic checkpoint (Wassmann and Benezra, 2001). This information prompted us to analyze the response of Rae1 haplo-insufficient cells to nocodazole. We first intercrossed heterozygous mice to derive Rae1+/+ and Rae1+/− mouse embryonic fibroblasts (MEFs) from individual 13.5-d-old fetuses. These MEFs were frozen at passages 2 and 3 and used for experimentation at passage 4 or 5. For the studies described below, at least three Rae1+/+ and three Rae1+/− clones were examined.
Western blot analysis revealed that the amount of Rae1 protein in Rae1+/− cells was consistently lower than in Rae1+/+ cell lines (Fig. 3 A); additional tests showed that Bub3 and Mad2 protein levels were similar in Rae1+/+ and Rae1+/− cells (Fig. 3 A). Growth rate measurements showed that the reduction of Rae1 protein had no significant impact on the rate of cell proliferation (Fig. 3 B). We then measured the response of the Rae1 haplo-insufficient cells to spindle damage. We treated cells with 200 ng/ml nocodazole for 0, 2, 6, 12, or 24 h, stained with phospho-histone H3 antibody to identify mitotic cells, and then measured the mitotic index (defined as the percentage of mitotic cells) at each time point using fluorescence microscopy. We found that 15% of Rae1+/+ cells were arrested by 12 h (Fig. 3 C). In contrast, only 2.5% of haplo-insufficient cells were arrested at that time point, indicating that their mitotic checkpoint was defective. Consistent with this observation, Rae1+/− cultures accumulated far fewer rounded cells in the presence of nocodazole than Rae1+/+ cultures (Fig. 3 D).
Figure 3.
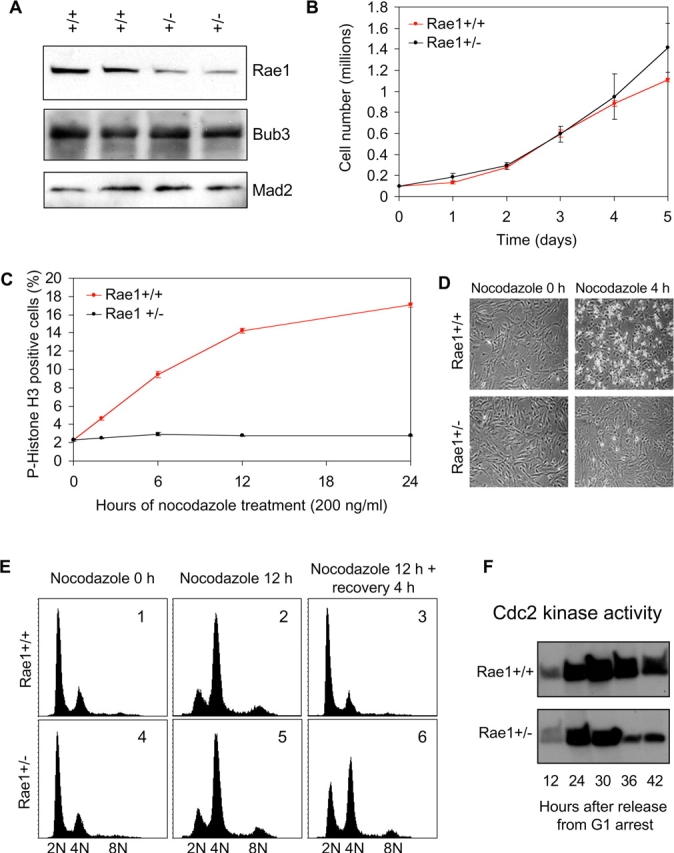
The mitotic checkpoint requires a full complement of Rae1. (A) Analysis of Rae1+/+ and Rae1+/− MEF lines for Rae1, Bub3, and Mad2 protein levels by Western blotting (4–20% polyacrylamide gel). 100 μg of total protein extract from each MEF cell line was used in the analysis. For probes, we used a rabbit antibody against mouse Rae1(188–368), a rabbit antibody against mouse Bub3(145–276), and a mouse monoclonal antibody against human Mad2 that recognizes mouse Mad2 (BD Biosciences). (B) Growth curves of primary MEFs. Data shown are means and standard deviations derived from three Rae1+/+ and three Rae1+/− MEF lines. (C) Mitotic index of nocodazole-treated Rae1+/− and Rae1+/+ MEF cell lines (n = 3 for each genotype). (D) Representative phase contrast images of Rae1+/+ and Rae1+/− MEF cultures before and after 4 h of nocodazole exposure. (E) Representative FACS® profiles of propidium-stained unsynchronized Rae1+/+ (panels 1–3) and Rae1+/− MEFs (panels 4–6). Durations of nocodazole treatment (80 ng/ml) and recovery intervals are indicated. (F) Cyclin B–associated Cdc2 kinase activity of synchronized MEF cells at indicated time points after release into nocodazole.
To confirm and extend these analyses, we examined cells by flow cytometry. Unsynchronized Rae1+/+ and Rae1+/− MEFs had similar DNA profiles (Fig. 3 E, 1 and 4). Exposure to nocodazole for 12 h resulted in accumulation of cells with 4N DNA content irrespective of the genotype (Fig. 3 E, 2 and 5). When cells were allowed to recover in medium without nocodazole for 4 h, the DNA profile of Rae1+/+ MEF cells quickly returned to normal as mitotically-arrested cells were able to complete their mitosis and enter G1 phase with 2N DNA content (Fig. 3 E, 3). In contrast, the DNA profile of Rae1+/− MEF cells showed only limited recovery (Fig. 3 E, 6), indicating that Rae1+/− cells that entered mitosis in the presence of nocodazole had exited from mitosis prematurely without dividing to produce a cell in G1 that contained 4N DNA content. When the recovery phase was extended to 12 h, essentially the same profile was obtained as with the 4-h recovery (unpublished data).
To further verify the loss of mitotic checkpoint activity, we measured the cyclin B–associated Cdc2 kinase activity of MEFs that were synchronized in G1 by low serum, released into medium with high serum and nocodazole, and collected at various time points (Wassmann and Benezra, 1998; Michel et al., 2001). At 24 h after release in nocodazole, both Rae1+/+ and Rae1+/− cultures showed a high level of Cdc2 kinase activity (indicating that the synchronized cultures had reached M phase; Fig. 3 F). Whereas Rae1+/+ cultures maintained a high level of Cdc2 kinase activity until at least 42 h after release, Rae1+/− cultures showed a marked decline in kinase activity after 30 h, confirming that Rae1 haplo-insufficient cells fail to arrest in mitosis in response to spindle damage. Taken together, the above analyses strongly suggest that the mitotic checkpoint is defective in Rae1 haplo-insufficient cells.
Rae1 haplo-insufficiency promotes chromosome missegregation
Next, we determined the effect of Rae1 haplo-insufficiency on chromosome number stability by performing chromosome counts on metaphase spreads from Rae1+/+ and Rae1+/− MEFs at passage 5. The average percentage of aneuploid metaphases was significantly higher in Rae1+/− MEFs than in Rae1+/+ MEFs (20 ± 2% vs. 9 ± 1%; Fig. 4 A; see also Fig. 8, B and D). In addition, Rae1+/− spreads showed a broader spectrum of abnormal chromosome numbers than Rae1+/+ spreads (Fig. 4 B; see also Fig. 8, B and D). Together, these findings show that loss of a single Rae1 allele leads to a significantly increased rate of chromosome missegregation.
Figure 4.

Rae1 haplo-insufficient cells exhibit increased chromosomal instability. (A) Analysis of the percentage of aneuploid cells in MEF cultures at passage 5. Values are from three independent Rae1+/+ and three independent Rae1+/− MEF cultures. 50 metaphases were counted for each MEF line. (B) Distribution of chromosome numbers of wild-type and Rae1 haplo-insufficient MEF lines. Data shown are combined values from three independent MEF lines per genotype.
Figure 8.

Rae1 synergizes with Bub3 in the development of aneuploidy. (A) Growth curves of MEFs derived from wild-type, Rae1+/−, Bub3+/−, and Rae1+/−/Bub3+/− 13.5-d-old embryos (all harvested from Rae1+/− x Bub3+/− intercrosses). Data shown are means and standard deviations derived from three independent MEF lines per genotype. (B) Chromosome analysis of MEFs (at passage 5) from 13.5-d-old embryos collected from Rae1+/− females that were fertilized by Bub3+/− males. Data shown are means and standard deviations derived from three independent MEF lines per genotype. We counted the chromosomes of 50 metaphase spreads per independent MEF line. 200 mitotic figures per MEF line were screened for premature sister chromatid separation (PMSCS). (C) Metaphase spreads showing sister chromatid cohesion in a wild-type MEF (left) and PMSCS in a double heterozygote MEF (right). (D) Comparison of chromosome number distributions between wild-type, single heterozygous, and double heterozygous MEF lines (combined values of each genotype). (E) Aneuploidy and PMSCS in primary splenocytes from 5-mo-old mice of various genotypes. Data shown are means and standard deviations derived from three mice per genotype. We counted the chromosomes of 50 metaphase spreads per mouse. 100 spreads per mouse were screened for PMSCS.
Bub3 haplo-insufficiency causes mitotic checkpoint failure and chromosome missegregation
We then asked whether a haplo-insufficiency at the Bub3 locus would also cause a mitotic phenotype. We inactivated the mouse Bub3 gene by interrupting its coding region at amino acid 191 via homologous recombination (Fig. 5, A and B). Bub3+/− mice were indistinguishable from wild-type littermates (unpublished data). Like Rae1+/− intercrosses, Bub3+/− intercrosses yielded no Bub3−/− mice at birth and at day 8.5 of embryogenesis, but produced Bub3−/− blastocysts at a Mendelian frequency (unpublished data). The growth abnormalities of these Bub3-deficient blastocysts (unpublished data) were similar to those of Rae1−/− blastocysts (Fig. 1 D) and Bub3−/− blastocysts from an earlier study (Kalitsis et al., 2000). In situ hybridization experiments on E8.5 outgrowths confirmed that disruption of Bub3 had no impact on the localization and level of poly(A)+ RNA in the cell (unpublished data).
Figure 5.
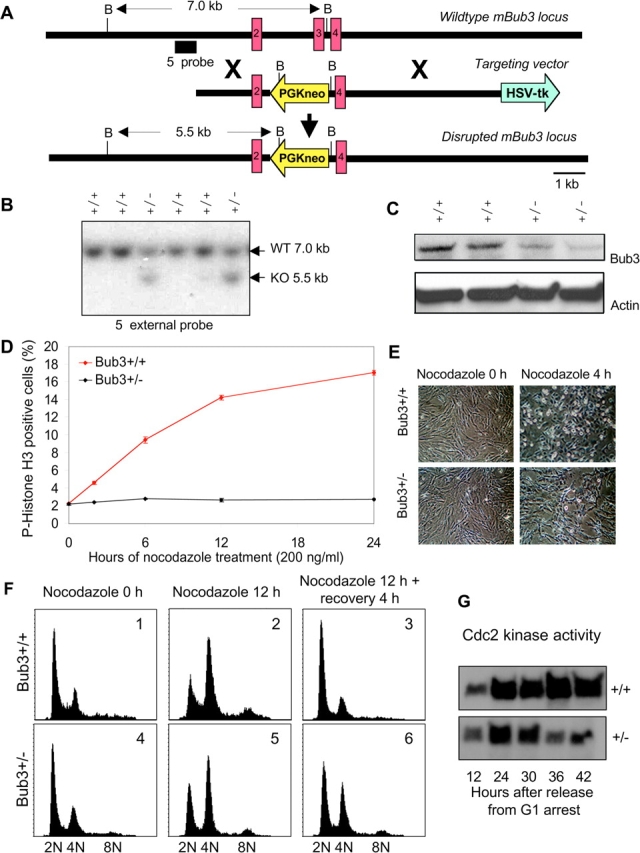
Disruption of a single Bub3 allele leads to mitotic checkpoint dysfunction. (A) Targeted inactivation of the mouse Bub3 gene. Shown is part of the endogenous mouse (m)Bub3 gene (top), the targeting vector (middle), and the disrupted mBub3 allele (bottom). BamHI restriction sites and the 5′ DNA probe (solid bar) used for Southern blot identification of wild-type (WT) and knockout (KO) Bub3 alleles are indicated. (B) Southern blot of genomic mouse tail DNA, digested with BamH1 and hybridized to a 5′ external probe, revealing the expected 7-kb wild-type and 5.5-kb mutant bands. (C) Western blot analysis of Bub3 protein levels in Bub3+/+ and Bub3+/− MEF lines using an antibody to mouse Bub3(145–276) (Wang et al., 2001). (D) Mitotic index of nocodazole-treated Bub3+/+ and Bub3+/− MEF cell lines. (E) Images of Bub3+/+ and Bub3+/− MEF cultures before and after nocodazole treatment. (F) DNA contents of asynchronous Bub3+/+ (panels 1–3) and Bub3+/− cells (panels 4–6) after the indicated treatments. (G) Cyclin B–associated Cdc2 kinase activity of synchronized MEF cultures at indicated time points after release into nocodazole.
We collected E13.5 embryos from Bub3+/− intercrosses and generated Bub3+/+ and Bub3+/− MEF lines from individual embryos. Bub3 protein levels were consistently lower in Bub3+/− MEFs than in Bub3+/+ MEFs (Fig. 5 C), but this reduction did not affect cell growth (unpublished data). Consistent with a loss of mitotic checkpoint activity, the mitotic index of Bub3+/− cultures was not much increased in the presence of nocodazole (Fig. 5, D and E). DNA profile analysis confirmed that nocodazole-exposed Bub3+/− cells leave mitosis prematurely without undergoing cytokinesis (Fig. 5 F). Furthermore, as expected, the cyclin B–associated Cdc2 kinase activity of synchronized MEFs that were released in nocodazole declined much faster in Bub3+/− cells than in Bub3+/+ cells (Fig. 5 G).
Next, we determined whether Bub3 haplo-insufficiency affects the fidelity of chromosome segregation in M phase. Chromosome counts on metaphase spreads revealed that the percentage of aneuploid cells was significantly higher in Bub3+/− cultures than in Bub3+/+ cultures (21 ± 2% vs. 9 ± 3%; Fig. 6 A; see also Fig. 8, B and D). Like Rae1+/− spreads, Bub3+/− spreads displayed a broader spectrum of nonmodal chromosome numbers than control spreads (Fig. 6 B; see also Fig. 8, B and D). Collectively, these studies reveal that the loss of a single Bub3 allele perturbs the mitotic checkpoint and establishes a higher rate of chromosome number instability.
Figure 6.

Bub3 +/− cells display increased chromosome number instability. (A) Analysis of the degree of aneuploidy in MEF cultures at passage 5. Values are from three independent Bub3+/+ and three independent Bub3+/− MEF cultures. 50 metaphase spreads were counted for each MEF culture. (B) Distribution of chromosome numbers of wild-type and Bub3 haplo-insufficient MEF lines (combined values of each genotype).
Overexpression of Rae1 rescues the checkpoint defect of Bub3 haplo-insufficient cells
Next, we cloned HA epitope–tagged mouse Rae1 cDNA into the pMSCV-Puro retroviral vector and used it to test whether ectopic expression of Rae1 would be able to correct for (a) Rae1 haplo-insufficiency (which would be expected if the mitotic phenotype is a direct consequence of the Rae1 mutation) and (b) Bub3+/− haplo-insufficiency (which would indicate that Rae1 has functional overlap with Bub3). We introduced the HA–Rae1 expression vector into two Rae1+/− and two Bub3+/− MEF lines by retroviral gene transfer (Kasper et al., 1999), selected the cultures for puromycin-resistant cells, and tested these cells for their ability to activate the mitotic checkpoint in response to nocodazole-induced spindle damage. Consistent with a reestablishment of normal mitotic checkpoint activity, the mitotic index of HA–Rae1-expressing Rae1+/− cultures showed a similar increase as Rae1+/+ cultures (Fig. 7 A). As expected, Rae1+/− cells transduced with empty pMSCV-Puro vector remained unable to sustain their mitotic checkpoint activity. Strikingly, HA–Rae1 was also capable of restoring the mitotic checkpoint defect caused by reduced Bub3 activity (Fig. 7 B). Thus, Rae1 can compensate for Bub3-mediated mitotic checkpoint activity that is affected by limited availability of Bub3 molecules.
Figure 7.
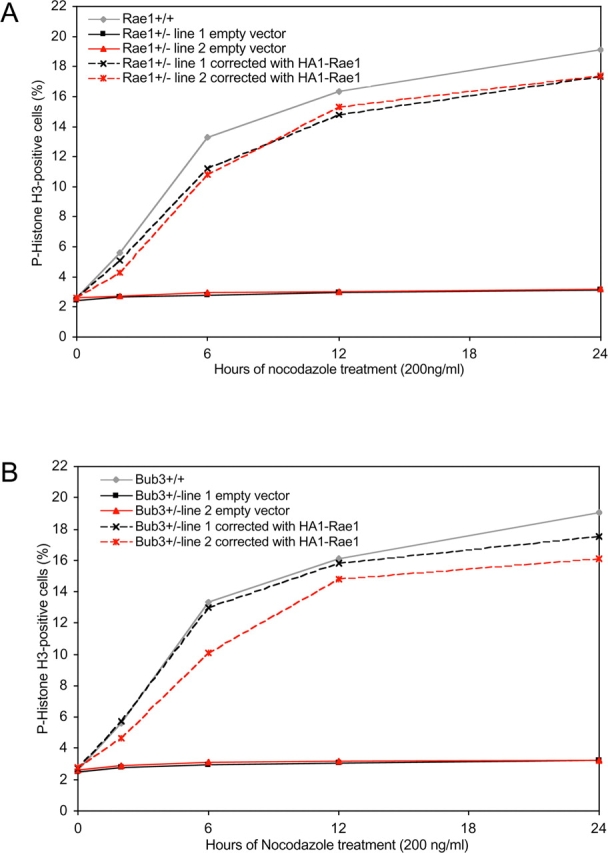
HA–Rae1 expression restores mitotic checkpoint activation in both Rae1 and Bub3 haplo-insufficient cells. (A) Mitotic index of nocodazole-treated Rae1+/−/empty vector (n = 2) and Rae1+/−/HA–Rae1 (n = 2) cell lines. (B) Mitotic index of nocodazole-treated Bub3+/−/empty vector (n = 2) and Bub3+/−/HA–Rae1 (n = 2) cell lines. Note that HA–Rae1-expressing cultures accumulate mitotic cells over time, illustrating that their mitotic checkpoint activity is restored.
Combined Rae1 and Bub3 haplo-insufficiency accelerates genetic instability
To further define the roles of Rae1 and Bub3 in mitosis, we analyzed the combined effects of Rae1 and Bub3 haplo-insufficiency. We found that crosses of Rae1+/− and Bub3+/− mice led to normal numbers of Rae1/Bub3 double heterozygous mice (53 among 214 total offspring). These compound heterozygotes had a normal appearance and displayed no reduction in body mass (unpublished data). MEFs were prepared from crosses of Rae1+/− and Bub3+/− mice. Double haplo-insufficient MEFs grew significantly slower than wild-type and single heterozygous MEFs (Fig. 8 A). Furthermore, chromosome counts at passage 5 revealed that the percentage of aneuploid cells in double haplo-insufficient cultures had increased by 32% relative to wild-type cultures (Fig. 8 B). This was significantly higher than the 10% increases measured in corresponding Rae1 and Bub3 single haplo-insufficient cultures (Fig. 8 B). Notably, the range of nonmodal chromosome numbers was much larger in double heterozygous cells than in single heterozygous cells, which in turn had a broader spectrum than wild-type cells (Fig. 8, B and D). We observed prematurely separated sister chromatids in ∼19% of mitotic figures from double haplo-insufficient cells, but essentially none in mitotic figures from single haplo-insufficient cells (Fig. 8, B and C). In addition, the percentage of anaphase figures with lagging chromosomes was much higher in Rae1+/−/Bub3+/− cells (15%) than in Rae1+/− (5%) or Bub3+/− (5%) cells, which in turn had a higher percentage than wild-type cells (2%) (n = 200 anaphases per genotype).
To investigate the degree of chromosomal instability in adult mice, we collected splenocytes from 5-mo-old wild-type, Rae1+/−, Bub3+/−, and Rae1+/−/Bub3+/− mice (n = 3 mice per genotype) and prepared metaphase spreads (Fig. 8 E). No aneuploidy was observed in wild-type splenocytes. In contrast, 9% of Rae1+/− and Bub3+/− splenocytes and 37% of Rae1+/−/Bub3+/− splenocytes showed aneuploidy. Furthermore, prematurely separated sister chromatids were present in 13% of mitotic figures from Rae1+/−/Bub3+/− mice. As expected, the other genotypes did not show this defect. Therefore, combined haplo-insufficiency of Rae1 and Bub3 accelerates chromosomal instability in cultured cells and mice.
Increased tumor formation in mice with chromosomal instability
To determine whether single and double haplo-insufficient mice have increased susceptibility to carcinogen-induced tumorigenesis, we gave pups from Rae1+/− x Bub3+/− intercrosses a single application of 50 μl of a solution of 0.5% DMBA in acetone to the dorsal surface on post-natal day 5 (Serrano et al., 1996). 5 mo after DMBA treatment, the mice were killed and screened for tumor formation. Irrespective of the mouse genotype, tumors were exclusively detectable in the lungs (Fig. 9 A). In Rae1+/−, Bub3+/−, and Rae1+/−/Bub3+/− mice, the incidence of lung tumors was increased compared with their wild-type counterparts (Fig. 9 B), as was the average number of tumors per treated animal (Fig. 9 C). As controls, we examined 10 nontreated 5-mo-old mice per genotype for spontaneous tumors. No tumors were found in the wild-type, Rae1+/−, or Bub3+/− mice, whereas 1 of the 10 Rae1+/−/Bub3+/− mice had a lung tumor. Taken together, the above data indicate that mitotic checkpoint–defective mice are predisposed to chemical-induced lung tumorigenesis. We are monitoring over 50 mice per genotype (currently ranging in age from 2 to 7 mo) for possible development of spontaneous tumors as they age.
Figure 9.
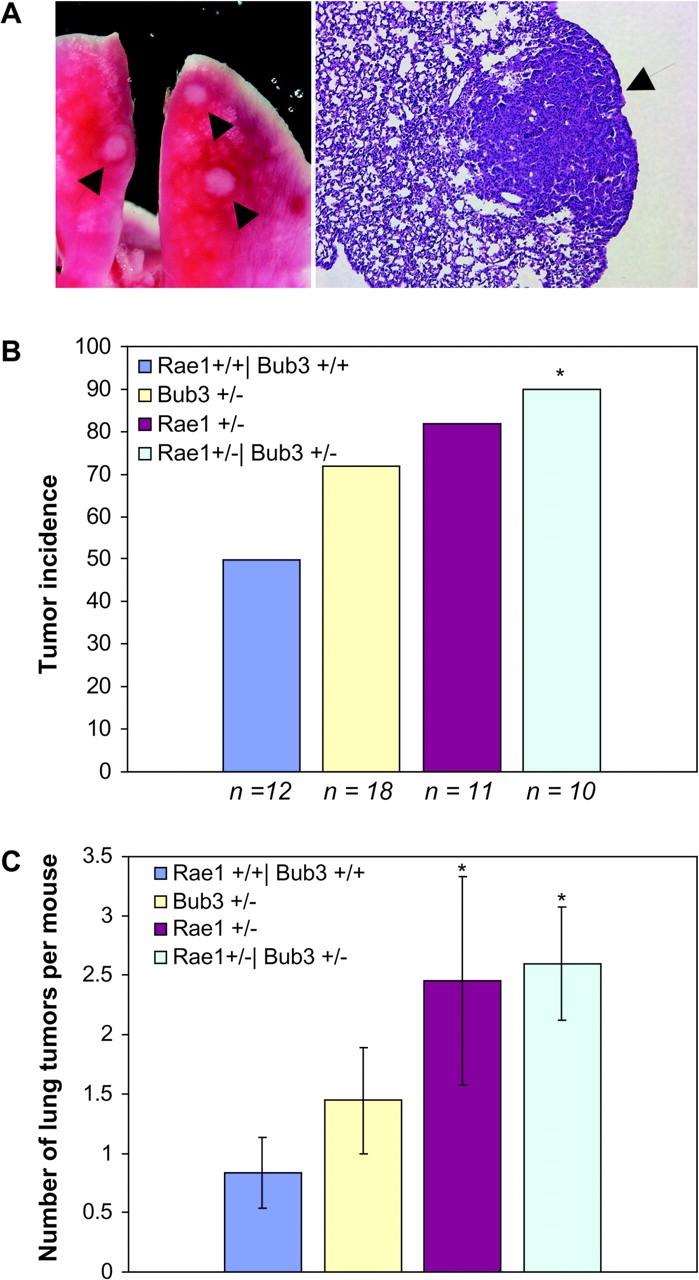
DMBA-induced tumor formation in checkpoint-defective mice. (A) Gross photograph of lung tumors of a DMBA-treated Rae1+/−/Bub3+/− mouse (left), and a section of a Rae1+/−/Bub3+/− lung tumor (right; 10×, hematoxylin + eosin). Arrows point out the tumors. (B) The occurrence of lung tumors in 5-mo-old mice plotted as percentage incidence. The asterisk indicates P < 0.05 compared with wild-type mice by Chi-squared test. We note that the tumor incidence of the checkpoint-defective group of mice as a whole is significantly increased relative to wild-type mice (P < 0.05 by Chi-squared test). (C) The average number of lung adenomas per mouse (±SEM). The asterisks indicate P < 0.05 compared with wild-type mice by Wilcoxon rank sum test.
Discussion
Rae1 and global mRNA export
Although inactivating mutations in the Schizosaccharomyces pombe and S. cerevisiae homologues of Rae1 cause nuclear accumulation of poly(A)+ RNA (Brown et al., 1995; Murphy et al., 1996), similar genetic experiments in mice do not induce such a defect, indicating that Rae1 is not essential for bulk mRNA export in mammalian cells. Consistent with this finding, no nuclear accumulation of poly(A)+ RNA occurs in human cells in which Rae1 expression is depleted by RNA interference (E. Izaurralde, personal communication). The most straightforward interpretation of the combined data would be that Rae1 is not implicated in mRNA export in higher eukaryotes. However, the previous observation that overexpression of the GLEBS motif of Nup98 (a motif that binds to and inhibits Rae1) results in nuclear accumulation of poly(A)+ RNA in HeLa cells (Pritchard et al., 1999) argues for a role of Rae1 in the mammalian mRNA export pathway. One attractive possibility is that Rae1 is an mRNA export factor that can be compensated for by another transport factor when it is absent. Perhaps such compensatory action may not be possible when Rae1 is present in the cell as a Rae1–GLEBS complex. Lastly, we would like to emphasize that our genetic study does not exclude the possibility that Rae1 is essential for export of only a small subset of mRNAs.
Mechanism of checkpoint regulation by Rae1
In this study, we show that haplo-insufficiency of Rae1 results in a mitotic checkpoint defect and chromosome missegregation. In addition, we show that Bub3 haplo-insufficient cells exhibit a strikingly similar mitotic phenotype, suggesting that Rae1 may act in an analogous way to Bub3. The finding that ectopic expression of Rae1 can correct not only for Rae1 haplo-insufficiency but also for Bub3 haplo-insufficiency strongly supports this conclusion. Although the critical functions of Rae1 and Bub3 in checkpoint activation may be redundant, the finding that compound heterozygous knockout mice are viable whereas homozygous Rae1 and Bub3 mice are embryonically lethal illustrates that both proteins have a critical, nonredundant physiological function as well.
What then could be the mechanism by which Rae1 regulates the activity of the mitotic checkpoint and how may Rae1 be able to compensate for Bub3 haplo-insufficiency? Because Rae1 has been shown to localize to unattached kinetochores at the onset of mitosis (Wang et al., 2001), it is possible that Rae1 might play a role in the production of anaphase wait signals at that position. Given that the checkpoint protein Bub1 also targets to unattached kinetochores in mitosis and can bind to Rae1 (Wang et al., 2001), it is further possible that Rae1 and Bub1 might participate in such signaling events as a protein complex. Interestingly, like Rae1, Bub3 also targets to unattached kinetochores and interacts with Bub1. We have shown that the GLEBS sequence of Bub1 serves as a shared binding motif for Rae1 and Bub3 (Wang et al., 2001). Given that both Rae1–Bub1 and Bub3–Bub1 complexes coexist in prometaphase-arrested HeLa cells (Wang et al., 2001), one possible explanation for the phenotypic similarity between Rae1 and Bub3 haplo-insufficient cells could be that Rae1–Bub1 and Bub3–Bub1 complexes fulfill redundant functions at unattached kinetochores. Correction of the mitotic checkpoint in Bub3 haplo-insufficient cells by overexpression of HA-tagged Rae1 could then simply result from the assembly of “unbound” Bub1 molecules into Rae1–Bub1 complexes that would be capable of performing the functions of the missing Bub3–Bub1 complexes (Fig. 10). Bub3 also interacts with BubR1 (Taylor et al., 1998), and Bub3–BubR1 complexes positioned in the mitotic cytosol seem to play a critical role in inhibiting the APC (Fig. 10) (Sudakin et al., 2001; Tang et al., 2001; Fang, 2002). Because Rae1 does not interact with BubR1 (Wang et al., 2001), the mechanism by which Rae1 overexpression acts to correct Bub3 haplo-insufficient cells is unlikely to involve the formation of compensatory Rae1–BubR1.
Figure 10.
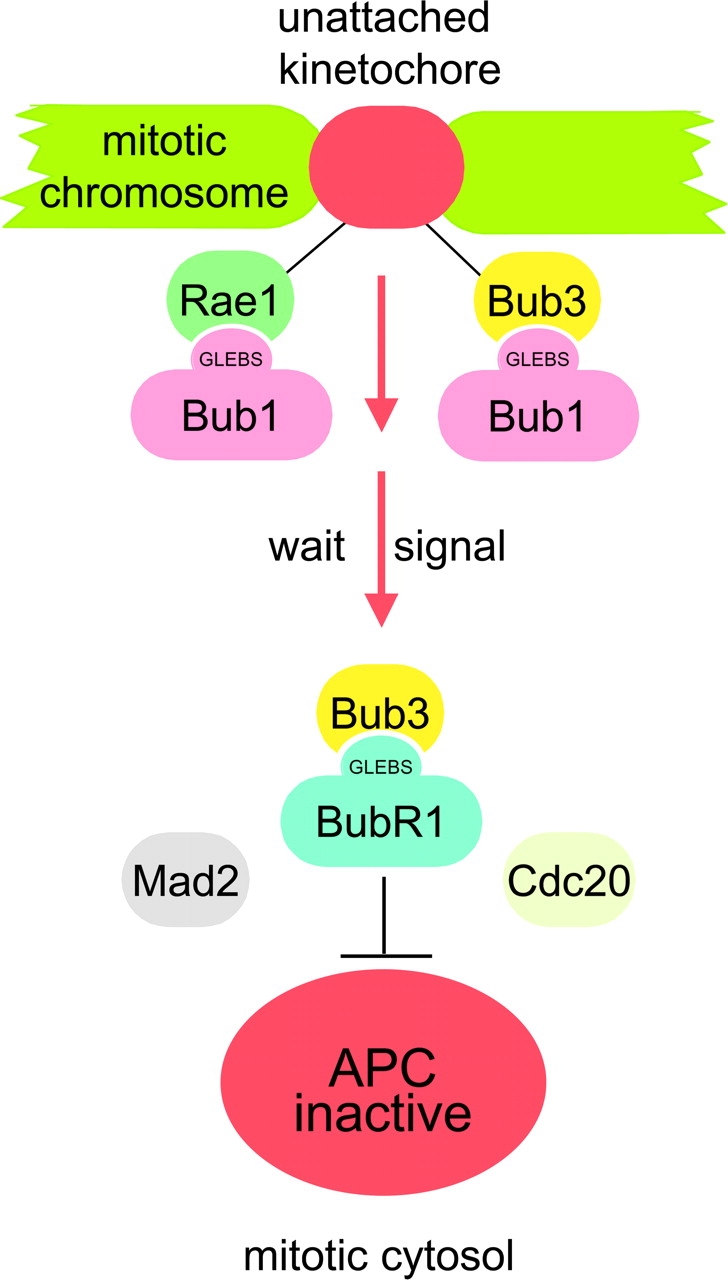
Model for Rae1 function in mitosis. We propose that Rae1 targets to unattached kinetochores at the onset of mitosis together with Bub1, just like Bub3. There, Rae1–Bub1 and Bub3–Bub1 proteins act to activate the mitotic checkpoint, potentially by producing anaphase wait signals. Unlike Rae1, Bub3 also interacts with BubR1 and localizes to the cytosolic component of the mitotic checkpoint complex that acts to inhibit the APC activator Cdc20 when anaphase wait signals are present. For simplicity, only part of the components of the checkpoint is shown in the model.
Synergy between Rae1 and Bub3
Combined haplo-insufficiency of Rae1 and Bub3 causes a much more severe chromosomal instability phenotype than single haplo-insufficiency of either gene (Fig. 8). This finding indicates that Rae1 cooperates with Bub3 and that the combination of the two mitotic checkpoint activities is critical for the prevention of chromosomal missegregation. What could be the mechanism of synergy between Rae1 and Bub3? The fact that Rae1 can compensate for Bub3 haplo-insufficiency suggests that Rae1 and Bub3 might be acting at a common step of the checkpoint pathway (Hartman et al., 2001), such as one that involves Rae1–Bub1 and Bub3–Bub1 complexes at unattached kinetochores (Fig. 10). The idea that the mechanism of synergy may involve Bub1 is supported by a previous study of Basu et al. (1999) showing that hypomorphic mutations in the Drosophila Bub1 gene cause a phenotype that has remarkable similarity to that of Rae1/Bub3 mutants. The fact that we were unable to detect protein complexes containing both Rae1 and Bub3 (Wang et al., 2001) suggests that a physical interaction between Rae1 and Bub3 may not be a requirement for their synergistic action (Herschlag and Johnson, 1993).
Mitotic checkpoint defects and tumorigenesis
Similar to Rae1 and Bub3 haplo-insufficiency, Mad2 haplo-insufficiency disturbs the mitotic checkpoint and causes chromosomal instability (Michel et al., 2001). Thus, it appears that the mammalian mitotic spindle checkpoint is a molecular backup system with exquisite sensitivity to events causing subnormal expression of its components. The present study shows that subnormal expression of Rae1 and/or Bub3 predisposes mice to carcinogen-induced lung tumors. In addition, a previous study has shown that mice with reduced expression of Mad2 are prone to develop spontaneous lung tumors after long latencies (Michel et al., 2001). Together, these data suggest that decreased mitotic checkpoint protein expression and tumor progression are causally related. Several studies have shown that inactivating point mutations in mitotic checkpoint genes are rare in human tumors with chromosomal instability (Cahill et al., 1998, 1999a; Hernando et al., 2001). However, as epigenetic events or losses of whole chromosomes are plausible mechanisms by which mitotic checkpoint genes could be down-regulated, it is conceivable that these events may actually play a more active role in cancers with chromosomal instability than point mutations. Consistent with this idea, a recent study of the mitotic checkpoint genes Bub1 and BubR1 showed that they are epigenetically down-regulated in a substantial proportion of human carcinomas (Shichiri et al., 2002). Further analysis of human tumors for decreased expression of mitotic checkpoint regulators such as Rae1 and Bub3 should provide important insights into the causal relationship between mitotic checkpoint gene activity and chromosomal instability.
Materials and methods
Generation of knockout mice and DMBA tumorigenic treatment
The Rae1 targeting vector contained a ∼9-kb Rae1 129Sv/J genomic DNA fragment in which a unique EcoRV site was used to insert a promoterless IRES-lacZ-neo selection cassette (van Deursen et al., 1996). Targeting mutagenesis of ES cells was as previously described (van Deursen et al., 1996). We identified targeted ES cell clones by Southern blot analysis using a 3′ probe on BamHI-cut genomic DNA (Fig. 1 B). The Bub3 construct contained an ∼8.5-kb Bub3 129Sv/J genomic DNA fragment in which a 1.4-kb SacI fragment was replaced by a pGKneo cassette (van Deursen et al., 1996), interrupting the Bub3 gene at amino acid 191. Targeted ES cell clones were identified by Southern blotting using a 5′ probe on BamHI-cut genomic DNA (Fig. 5, A and B). Mutant mice were derived from targeted ES cell clones through standard procedures. DMBA treatment was as previously described (Serrano et al., 1996). All major organs were screened for overt tumors using a dissection microscope. Harvested tumors were routinely processed for histopathological confirmation.
In vitro culture and PCR genotyping of blastocysts
Blastocysts were recovered from the uterine horns of plugged Rae1+/− females at 3.5 d post-coitum and cultured in DME with 15% FCS on 10-well microscope slides for 2–5 d. Processing of embryos and embryonic outgrowths for genotype analysis was performed as previously described in detail (van Deursen et al., 1996). Sequences specific for the wild-type Rae1 gene were amplified by using primer 1 (5′-GCCTTGTGCAAATGCCGCCCTTGG-3′) and primer 2 (5′-TACATCATTCGCCCATGATCCTGC-3′), those specific for the knockout Rae1 allele were amplified with primer combination 1 and 3 (5′-GGGCTGACCGCTTCCTCGTGCTTT-3′). The resulting PCR fragments measured 516 bp for the wild-type allele and 650 bp for the knockout allele.
Generation and culture of MEFs
MEFs were isolated from 13.5-d-old embryos as previously described (Kamijo et al., 1997). We counted the seeding of trypsinized embryo carcasses as passage 1 and the first replating as passage 2. Routine maintenance of MEFs occurred according to a 3T9 schedule. Growth curves were performed at passage 4. At day 0, 105 MEFs were plated per 3.5-mm-diameter dish, and duplicate cultures were counted at 24-h intervals thereafter. MEFs were synchronized as previously described (Cheng et al., 1999). In brief, confluent cultures were washed three times with PBS and then cultured in DME containing 0.1% FBS and 0.04% BSA for 18 h. Quiescent MEFs were trypsinized and reseeded in DME with 10% FBS to allow their reentry into the cell cycle.
Mitotic index and DNA profile analysis
To determine the mitotic index, MEF cells were plated in chambered microscope slides (5,000 cells per chamber of 0.4 cm2). The next day, 200 ng/ml nocodazole (Michel et al., 2001) was added to the culture medium for 0, 2, 6, 12, or 24 h. Subsequently, cells were immunostained with an antibody against phospho-histone H3 (a commonly used marker for mitotic cells) as previously described (Kasper et al., 1999). DNA was visualized by Hoechst staining. Using a fluorescence microscope, we counted both the number of Hoechst-stained nuclei and the number of anti-H3–positive cells per well to calculate the mitotic index (defined as percentage of anti–phospho-histone H3–positive cells). For DNA content analysis, adherent and nonadherent MEF cells were pooled, washed with ice-cold PBS, fixed in 95% ethanol, treated with 1 mg/ml RNase A, stained with 1 mg/ml propidium iodide, and subjected to flow cytometry as described previously (Blajeski et al., 2002). For Rae1 correction experiments, we randomly selected one wild-type, two Rae1+/−, and two Bub3+/− MEF lines, immortalized them with simian virus 40 large T antigen (this viral protein has no impact on the mitotic checkpoint activity in the MEF lines used; Fig. 7), and then transduced them with pMSCV-Puro-HA–Rae1 or pMSCV-Puro containing retroviruses, as previously described in detail (Kasper et al., 1999). Drug selection was in 1.25 μg/ml puromycin (Sigma-Aldrich) for 5 d.
Karyotype analyses
To prepare metaphase spreads from MEFs, 1–2 × 106 cells (at passage 5) were treated with 0.05 μg/ml colcemid (GIBCO BRL) for 4–5 h at 37°C. To prepare metaphase spreads from splenocytes (Rudolph et al., 1999), spleens were freshly collected and minced between two microscope slides. Released cells were suspended in 5 ml PBS, centrifuged at 1,000 rpm for 5 min, resuspended in 4 ml of RPMI containing 10% FBS, IL-2 (10 U/ml), PHA (5 μg/ml), conA (5 μg/ml), and colcemid (0.05 μg/ml), and cultured for 6–7 h at 37°C. After colcemid treatment, MEF or splenocyte cells were harvested, suspended in 5 ml 0.075 M KCl, and incubated at RT for 30 min. Cells were fixed in Carnoy's solution (75% methanol, 25% acetic acid), washed, and finally resuspended in ∼0.5 ml fixative. 25-μl aliquots were dropped onto prewetted microscope slides, stained for 10 min in 5% Giemsa solution, and analyzed on an Olympus AX70 microscope using a 100× objective.
Western blot analysis, immunoprecipitation, and in vitro kinase assay
Western blot analyses were performed as previously described (Kasper et al., 1999). Immunoprecipitation of cyclin B–associated Cdc2 and in vitro kinase assays on histone H1 substrate were as previously described (Wassmann and Benezra, 1998).
Immunofluorescence and staining for poly(A)+ RNA
Immunostainings and poly(A)+ RNA detection were as previously described (van Deursen et al., 1996). Anti-Tap antiserum (Braun et al., 1999) was used in 1:500 dilution.
Acknowledgments
We are very grateful to our colleagues Rick Bram, Paul Brindle, Tim Yen, and Fergus Couch for critically reviewing our manuscript and helpful discussions. We thank Elisa Izaurralde for providing unpublished RNAi data and anti-Tap antibody.
X. Wu, J.R. Babu, K. Jeganathan, and J. van Deursen were supported by National Institutes of Health grants RO1 CA77262-01 and RO1 CA96985-1.
Footnotes
Abbreviations used in this paper: APC, anaphase-promoting complex; E, embryonic day; ES, embryonic stem; ICM, inner cell mass; MEF, mouse embryonic fibroblast; NE, nuclear envelope; NPC, nuclear pore complex; PMSCS, premature sister chromatid separation.
References
- Bachi, A., I.C. Braun, J.P. Rodrigues, N. Pante, K. Ribbeck, C. Von Kobbe, U. Kutay, M. Wilm, D. Gorlich, M. Carmo-Fonseca, and E. Izaurralde. 2000. The C-terminal domain of TAP interacts with the nuclear pore complex and promotes export of specific CTE-bearing RNA substrates. RNA. 6:136–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailer, S.M., S. Siniossoglou, A. Podtelejnikov, A. Hellwig, M. Mann, and E. Hurt. 1998. Nup116p and nup100p are interchangeable through a conserved motif which constitutes a docking site for the mRNA transport factor gle2p. EMBO J. 17:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, J., H. Bousbaa, E. Logarinho, Z. Li, B.C. Williams, C. Lopes, C.E. Sunkel, and M.L. Goldberg. 1999. Mutations in the essential spindle checkpoint gene bub1 cause chromosome missegregation and fail to block apoptosis in Drosophila. J. Cell Biol. 146:13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blajeski, A.L., V.A. Phan, T.J. Kottke, and S.H. Kaufmann. 2002. G(1) and G(2) cell-cycle arrest following microtubule depolymerization in human breast cancer cells. J. Clin. Invest. 110:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, I.C., E. Rohrbach, C. Schmitt, and E. Izaurralde. 1999. TAP binds to the constitutive transport element (CTE) through a novel RNA-binding motif that is sufficient to promote CTE-dependent RNA export from the nucleus. EMBO J. 18:1953–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.A., A. Bharathi, A. Ghosh, W. Whalen, E. Fitzgerald, and R. Dhar. 1995. A mutation in the Schizosaccharomyces pombe rae1 gene causes defects in poly(A)+ RNA export and in the cytoskeleton. J. Biol. Chem. 270:7411–7419. [DOI] [PubMed] [Google Scholar]
- Burke, D.J. 2000. Complexity in the spindle checkpoint. Curr. Opin. Genet. Dev. 10:26–31. [DOI] [PubMed] [Google Scholar]
- Cahill, D.P., C. Lengauer, J. Yu, G.J. Riggins, J.K. Willson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1998. Mutations of mitotic checkpoint genes in human cancers. Nature. 392:300–303. [DOI] [PubMed] [Google Scholar]
- Cahill, D.P., L.T. da Costa, E.B. Carson-Walter, K.W. Kinzler, B. Vogelstein, and C. Lengauer. 1999. a. Characterization of MAD2B and other mitotic spindle checkpoint genes. Genomics. 58:181–187. [DOI] [PubMed] [Google Scholar]
- Cahill, D.P., K.W. Kinzler, B. Vogelstein, and C. Lengauer. 1999. b. Genetic instability and Darwinian selection in tumours. Trends Cell Biol. 9:M57–M60. [PubMed] [Google Scholar]
- Cheng, M., P. Olivier, J.A. Diehl, M. Fero, M.F. Roussel, J.M. Roberts, and C.J. Sherr. 1999. The p21(Cip1) and p27(Kip1) CDK “inhibitors” are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 18:1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, G. 2002. Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol. Biol. Cell. 13:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma, A., Y. Hosoya, M. Seike, K. Uematsu, F. Kurimoto, S. Hibino, A. Yoshimura, M. Shibuya, S. Kudoh, and M. Emi. 2001. Genomic structure of the human MAD2 gene and mutation analysis in human lung and breast cancers. Lung Cancer. 32:289–295. [DOI] [PubMed] [Google Scholar]
- Gillett, E.S., and P.K. Sorger. 2001. Tracing the pathway of spindle assembly checkpoint signaling. Dev. Cell. 1:162–164. [DOI] [PubMed] [Google Scholar]
- Hartman, J.L., IV, B. Garvik, and L. Hartwell. 2001. Principles for the buffering of genetic variation. Science. 291:1001–1004. [DOI] [PubMed] [Google Scholar]
- Hartwell, L.H., and M.B. Kastan. 1994. Cell cycle control and cancer. Science. 266:1821–1828. [DOI] [PubMed] [Google Scholar]
- Hernando, E., I. Orlow, V. Liberal, G. Nohales, R. Benezra, and C. Cordon-Cardo. 2001. Molecular analyses of the mitotic checkpoint components hsMAD2, hBUB1 and hBUB3 in human cancer. Int. J. Cancer. 95:223–227. [DOI] [PubMed] [Google Scholar]
- Herschlag, D., and F.B. Johnson. 1993. Synergism in transcriptional activation: a kinetic view. Genes Dev. 7:173–179. [DOI] [PubMed] [Google Scholar]
- Hoffman, D.B., C.G. Pearson, T.J. Yen, B.J. Howell, and E.D. Salmon. 2001. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at ptk1 kinetochores. Mol. Biol. Cell. 12:1995–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt, M.A. 2001. A new view of the spindle checkpoint. J. Cell Biol. 154:909–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai, Y., Y. Shiratori, N. Kato, T. Inoue, and M. Omata. 1999. Mutational inactivation of mitotic checkpoint genes, hsMAD2 and hBUB1, is rare in sporadic digestive tract cancers. Jpn. J. Cancer Res. 90:837–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli, P.V., and C. Lengauer. 2001. Chromosome segregation and cancer: cutting through the mystery. Nat. Rev. Cancer. 1:109–117. [DOI] [PubMed] [Google Scholar]
- Kalitsis, P., E. Earle, K.J. Fowler, and K.H. Choo. 2000. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14:2277–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo, T., F. Zindy, M.F. Roussel, D.E. Quelle, J.R. Downing, R.A. Ashmun, G. Grosveld, and C.J. Sherr. 1997. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 91:649–659. [DOI] [PubMed] [Google Scholar]
- Kasper, L.H., P.K. Brindle, C.A. Schnabel, C.E. Pritchard, M.L. Cleary, and J.M. van Deursen. 1999. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol. Cell. Biol. 19:764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa, K., and P. Hieter. 2001. Evolutionary conservation between budding yeast and human kinetochores. Nat. Rev. Mol. Cell Biol. 2:678–687. [DOI] [PubMed] [Google Scholar]
- Kraemer, D., and G. Blobel. 1997. mRNA binding protein mrnp 41 localizes to both nucleus and cytoplasm. Proc. Natl. Acad. Sci. USA. 94:9119–9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer, C., K.W. Kinzler, and B. Vogelstein. 1998. Genetic instabilities in human cancers. Nature. 396:643–649. [DOI] [PubMed] [Google Scholar]
- Martinez-Exposito, M.J., K.B. Kaplan, J. Copeland, and P.K. Sorger. 1999. Retention of the BUB3 checkpoint protein on lagging chromosomes. Proc. Natl. Acad. Sci. USA. 96:8493–8498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel, L.S., V. Liberal, A. Chatterjee, R. Kirchwegger, B. Pasche, W. Gerald, M. Dobles, P.K. Sorger, V.V. Murty, and R. Benezra. 2001. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 409:355–359. [DOI] [PubMed] [Google Scholar]
- Morgan, D.O. 1999. Regulation of the APC and the exit from mitosis. Nat. Cell Biol. 1:E47–E53. [DOI] [PubMed] [Google Scholar]
- Murphy, R., and S.R. Wente. 1996. An RNA-export mediator with an essential nuclear export signal. Nature. 383:357–360. [DOI] [PubMed] [Google Scholar]
- Murphy, R., J.L. Watkins, and S.R. Wente. 1996. GLE2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol. Biol. Cell. 7:1921–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg, E.A. 2001. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2:21–32. [DOI] [PubMed] [Google Scholar]
- Peters, J.M. 2002. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell. 9:931–943. [DOI] [PubMed] [Google Scholar]
- Pritchard, C.E., M. Fornerod, L.H. Kasper, and J.M. van Deursen. 1999. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J. Cell Biol. 145:237–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph, K.L., S. Chang, H.W. Lee, M. Blasco, G.J. Gottlieb, C. Greider, and R.A. DePinho. 1999. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 96:701–712. [DOI] [PubMed] [Google Scholar]
- Sabri, N., and N. Visa. 2000. The Ct-RAE1 protein interacts with Balbiani ring RNP particles at the nuclear pore. RNA. 6:1597–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano, M., H. Lee, L. Chin, C. Cordon-Cardo, D. Beach, and R.A. DePinho. 1996. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 85:27–37. [DOI] [PubMed] [Google Scholar]
- Shah, J.V., and D.W. Cleveland. 2000. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell. 103:997–1000. [DOI] [PubMed] [Google Scholar]
- Shichiri, M., K. Yoshinaga, H. Hisatomi, K. Sugihara, and Y. Hirata. 2002. Genetic and epigenetic inactivation of mitotic checkpoint genes hBUB1 and hBUBR1 and their relationship to survival. Cancer Res. 62:13–17. [PubMed] [Google Scholar]
- Sudakin, V., G.K. Chan, and T.J. Yen. 2001. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154:925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z., R. Bharadwaj, B. Li, and H. Yu. 2001. Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell. 1:227–237. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., and F. McKeon. 1997. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 89:727–735. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., E. Ha, and F. McKeon. 1998. The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J. Cell Biol. 142:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen, J., J. Boer, L. Kasper, and G. Grosveld. 1996. G2 arrest and impaired nucleocytoplasmic transport in mouse embryos lacking the proto-oncogene CAN/Nup214. EMBO J. 15:5574–5583. [PMC free article] [PubMed] [Google Scholar]
- Wang, X., J.R. Babu, J.M. Harden, S.A. Jablonski, M.H. Gazi, W.L. Lingle, P.C. de Groen, T.J. Yen, and J.M. van Deursen. 2001. The mitotic checkpoint protein hBUB3 and the mRNA export factor hRAE1 interact with GLE2p-binding sequence (GLEBS)-containing proteins. J. Biol. Chem. 276:26559–26567. [DOI] [PubMed] [Google Scholar]
- Wassmann, K., and R. Benezra. 1998. Mad2 transiently associates with an APC/p55Cdc complex during mitosis. Proc. Natl. Acad. Sci. USA. 95:11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmann, K., and R. Benezra. 2001. Mitotic checkpoints: from yeast to cancer. Curr. Opin. Genet. Dev. 11:83–90. [DOI] [PubMed] [Google Scholar]
- Wu, X., L.H. Kasper, R.T. Mantcheva, G.T. Mantchev, M.J. Springett, and J.M. van Deursen. 2001. Disruption of the FG nucleoporin NUP98 causes selective changes in nuclear pore complex stoichiometry and function. Proc. Natl. Acad. Sci. USA. 98:3191–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, J.H., D.C. Love, A. Guhathakurta, J.A. Hanover, and R. Dhar. 2000. Mex67p of Schizosaccharomyces pombe interacts with Rae1p in mediating mRNA export. Mol. Cell. Biol. 20:8767–8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. 2002. Regulation of APC-Cdc20 by the spindle checkpoint. Curr. Opin. Cell Biol. 14:706–714. [DOI] [PubMed] [Google Scholar]
- Zenklusen, D., P. Vinciguerra, Y. Strahm, and F. Stutz. 2001. The yeast hnRNP-like proteins Yra1p and Yra2p participate in mRNA export through interaction with Mex67p. Mol. Cell. Biol. 21:4219–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]