

Abstract
Glycosylphosphatidylinositol (GPI)-anchored proteins exit the ER in distinct vesicles from other secretory proteins, and this sorting event requires the Rab GTPase Ypt1p, tethering factors Uso1p, and the conserved oligomeric Golgi complex. Here we show that proper sorting depended on the vSNAREs, Bos1p, Bet1p, and Sec22p. However, the t-SNARE Sed5p was not required for protein sorting upon ER exit. Moreover, the sorting defect observed in vitro with bos1–1 extracts was also observed in vivo and was visualized by EM. Finally, transport and maturation of the GPI-anchored protein Gas1p was specifically affected in a bos1–1 mutant at semirestrictive temperature. Therefore, we propose that v-SNAREs are part of the cargo protein sorting machinery upon exit from the ER and that a correct sorting process is necessary for proper maturation of GPI-anchored proteins.
Keywords: membrane trafficking; SNARE; protein sorting; GPI; ER to Golgi
Introduction
In eukaryotic cells, transport along the secretory and endocytic pathways is mediated by vesicular carriers, which bud from one donor compartment, are targeted to, and fuse with the next compartment in the pathway (Schekman and Orci, 1996). In yeast, GPI-anchored proteins are transported from the ER to the Golgi apparatus in distinct vesicles from those containing non-GPI–anchored proteins (Muniz et al., 2001). This finding demonstrated that cargo protein sorting into different vesicles occurs upon exit from the ER, a function that was previously only attributed to the Golgi compartment for the exocytic route. Genetic evidence is consistent with two pathways for ER to Golgi transport because mutants that are defective in the first step of ceramide synthesis or in the α-subunit of coatomer show a specific defect in ER to Golgi transport of GPI-anchored proteins (Sütterlin et al., 1997).
The mechanism involved in protein sorting upon ER exit is poorly understood. Recently, we showed that the Rab GTPase Ypt1p and the tethering factors Uso1, Sec34p, and Sec35p (two members of the conserved oligomeric Golgi [COG] complex) but not Bet3p (a member of the TRAPP complex) are necessary for sorting of GPI-anchored proteins upon ER exit (Morsomme and Riezman, 2002). Rab GTPases have also been proposed to facilitate cargo packaging on other donor compartments. The Rab9 GTPase seems to facilitate mannose-6-phosphate receptor recruitment for endosome to Golgi traffic (Carroll et al., 2001), and Rab5 seems to be required for ligand sequestration into clathrin-coated pits (McLauchlan et al., 1998). More recently, direct interactions between Rab GTPases and cargo were found, indicating that a cargo protein can bind to a specific Rab protein, which controls its trafficking (Seachrist et al., 2002; van IJzendoorn et al., 2002).
In addition to its function in sorting, the Rab-GTPase Ypt1p, in combination with the tethering factors Uso1p and Sec34/35p, is also involved in tethering of ER-derived vesicles to the Golgi membranes (Cao et al., 1998; VanRheenen et al., 1998, 1999), suggesting that Rab-mediated cargo sorting on donor membranes may be a general mechanism to couple protein sorting and packaging into vesicles to targeting, docking, and fusion. Consistent with this hypothesis, Rab GTPases were shown to recruit tethering factors and interact with SNARE molecules in order to ensure specificity of vesicle fusion with the acceptor membrane. For example, Rab5 was shown to regulate the recruitment of the tethering factor EEA1 into a complex with syntaxin13 (Simonsen et al., 1998; Christoforidis et al., 1999; McBride et al., 1999), and the mammalian homologue of Uso1p, p115, was shown to be recruited at the ER exit sites via Rab1 and to form a cis complex with v-SNAREs (Allan et al., 2000). These findings suggest that v-SNAREs can also participate in the same packaging and/or sorting function as Rab proteins.
Therefore, we decided to test the function of v-SNAREs in the sorting of GPI-anchored proteins using an in vitro assay that reconstitutes the ER-derived vesicle budding into distinct classes of vesicles upon exit from the ER. We found that sorting of GPI-anchored proteins from other secretory proteins was defective when we used membrane extracts from the v-SNARE mutants, bos1–1, bet1–1, or sec22–3, with the bos1–1 mutant membranes showing the strongest defect. In contrast, the t-SNARE Sed5p was not required for protein sorting upon ER exit. Importantly, the packaging of cargo proteins into distinct vesicles budding from the ER and the Bos1p requirement for this process could be visualized by EM. Moreover, transport and maturation of the GPI-anchored protein Gas1p was specifically affected in vivo in the bos1–1 mutant. Therefore, we propose that v-SNAREs are essential for protein sorting upon exit from the ER and that a correct sorting process is necessary for proper maturation of GPI-anchored proteins.
Results
The v-SNAREs Sec22p, Bet1p, and Bos1p are required for protein sorting upon ER exit
To analyze cargo protein sorting upon exit from the ER, we used an in vitro assay that reconstitutes a single round of budding from the ER by incubation of permeabilized spheroplasts with exogenous cytosol and energy (Kuehn et al., 1996; Muniz et al., 2000). After the incubation, vesicles were purified by flotation into a Nycodenz® gradient. To allow vesicle immunoisolation, we used a strain expressing an HA-tagged general amino acid permease (Gap1HA). With wild-type or sec18 extracts, we found 64% of Gap1pHA in the pellet after vesicle immunoisolation and only a small amount of the GPI-anchored protein, Gas1p, was coprecipitated (Fig. 1 A; Muniz et al., 2001). The immunoisolation required addition of HA antibody. This result shows that Gas1p is not incorporated into Gap1pHA-containing vesicles.
Figure 1.
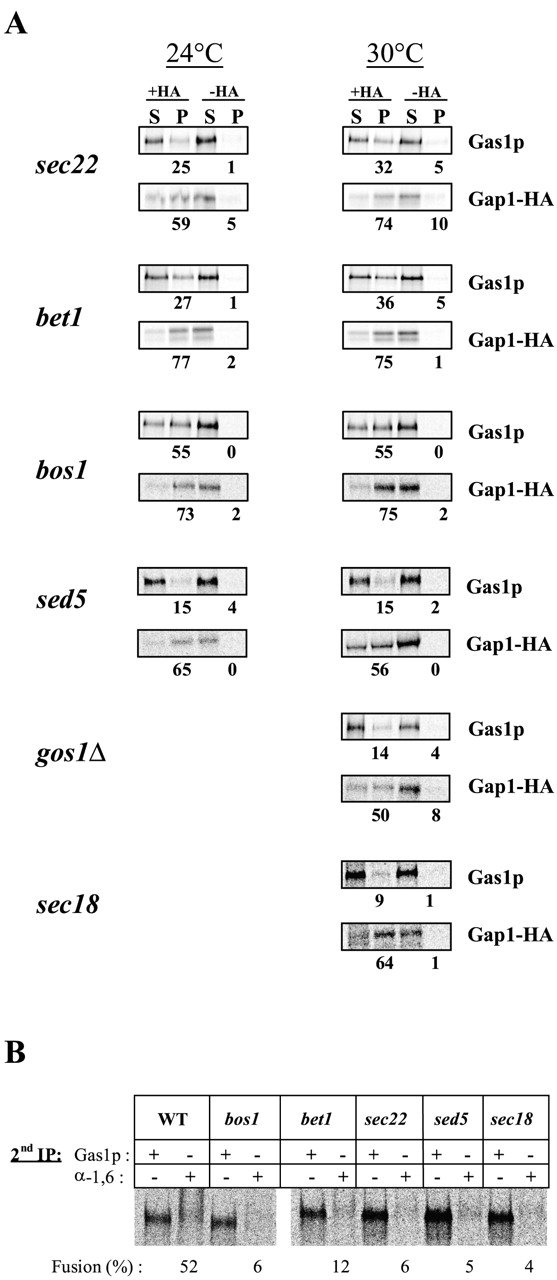
ER v-SNAREs are necessary for protein sorting upon ER exit. (A) Vesicles were generated from sec18–20 membranes and cytosol or from bos1–1, bet1–1, sec22–3, sed5–1, or gos1Δ membranes and wild-type cytosol. The vesicles were immunoisolated with or without monoclonal anti-HA antibody. The supernatants (S) and pellets (P) were processed for immunoprecipitation, and samples were analyzed by SDS-PAGE and quantified using a phosphorimager. The total recovery, S plus P, was set to 100%. Numbers represent the percentage of recovery in the pellet. This experiment is representative of at least two independent experiments. (B) ER budding and fusion with the Golgi compartment from wild-type, bos1–1, bet1–1, sec22–3, sed5–1, and sec18–20 membranes. Purified vesicles were processed for two consecutive immunoprecipitations. Samples were immunoprecipitated first with antibodies against Gas1p, then reprecipitated with antibodies against Gas1p or α-1,6 mannose, analyzed by SDS-PAGE, and quantified using a phosphor- imager. The percentage of fusion is the percentage of Gas1p after two immunoprecipitations that was recovered after precipitation with antibodies against α-1,6 mannose.
To test the role of SNAREs in cargo protein sorting, we performed in vitro budding and sorting experiments with permeabilized spheroplasts and cytosol prepared from temperature-sensitive sec22–3, bet1–1, and bos1–1 mutants. We found that at 24°C sorting of cargo proteins upon ER exit was partially defective using sec22–3 and bet1–1 mutant extracts and almost completely defective in the bos1–1 mutant (Fig. 1 A). A slightly stronger defect in sorting was observed using sec22–3 and bet1–1 extracts when the experiments were performed at 30°C. The bos1–1 defect was not further increased at 30°C. Functional v-SNAREs are thus necessary for correct sorting of GPI-anchored proteins upon ER exit. In contrast, the t-SNARE, Sed5p, is not required for this process since sorting was not affected using sed5–1 mutant extracts (Fig. 1 A). We also performed the sorting experiment using the Golgi SNARE deletion mutant gos1Δ and found that, as expected, sorting was normal under these conditions. Shorter incubation times for the in vitro budding reaction do not modify the sorting defect. We repeated the budding and sorting assay with only a 15-min incubation of bos1 membranes. 67% of the Gap1p containing ER-derived vesicles were immunoisolated in this experiment, and 47% of the Gas1p signal was coprecipitated.
ER-derived vesicle fusion with the cis-Golgi compartment can be measured by following the α-1,6 mannosylation of Gas1p. This fusion step is Sec18p dependent (Muniz et al., 2001). Here, we tested whether the vesicles produced from SNARE mutants were also deficient for fusion with the Golgi compartment. After a 1-h incubation of SNARE mutant extracts at 30°C, only a very low amount of α-1,6 mannose modification was detected on Gas1p, where >50% of it was modified with wild-type membranes and cytosol (Fig. 1 B). This result suggests that the vesicles we analyzed in our assay with the SNARE mutants are primary ER-derived vesicles because they were virtually incapable of fusion with the Golgi compartment.
We considered the possibility that sec22–3, bet1–1, or bos1–1 membranes are fragile and that the apparent lack of protein sorting was due to the isolation of ER fragments rather than bona fide vesicles that have budded from the ER. To test this, we determined the specificity and nucleotide dependence of vesicle formation using mutant membranes. The sorting defect observed with the sec22–3, bet1–1, or bos1–1 mutants was not due to a budding defect since the budding efficiency of Gap1p and Gas1p were similar to wild-type or sec18 membranes (Fig. 2 A). Packaging of Gas1p and Gap1p into ER-derived vesicles was dependent on cytosol (unpublished data) and on the presence of nucleotides in the assay (Fig. 2 A). Furthermore, resident ER proteins, like Sec61p, were not packaged into vesicles (Fig. 2 B). Finally, we treated the floated fraction of vesicles generated from bos1–1 membranes with proteinase K and observed that Gas1p was protected from protease digestion in absence of detergent but was digested when detergent was present (Fig. 2 C). This confirms that Gas1p exits the ER in vesicles in our in vitro assay even when using SNARE mutant extracts.
Figure 2.
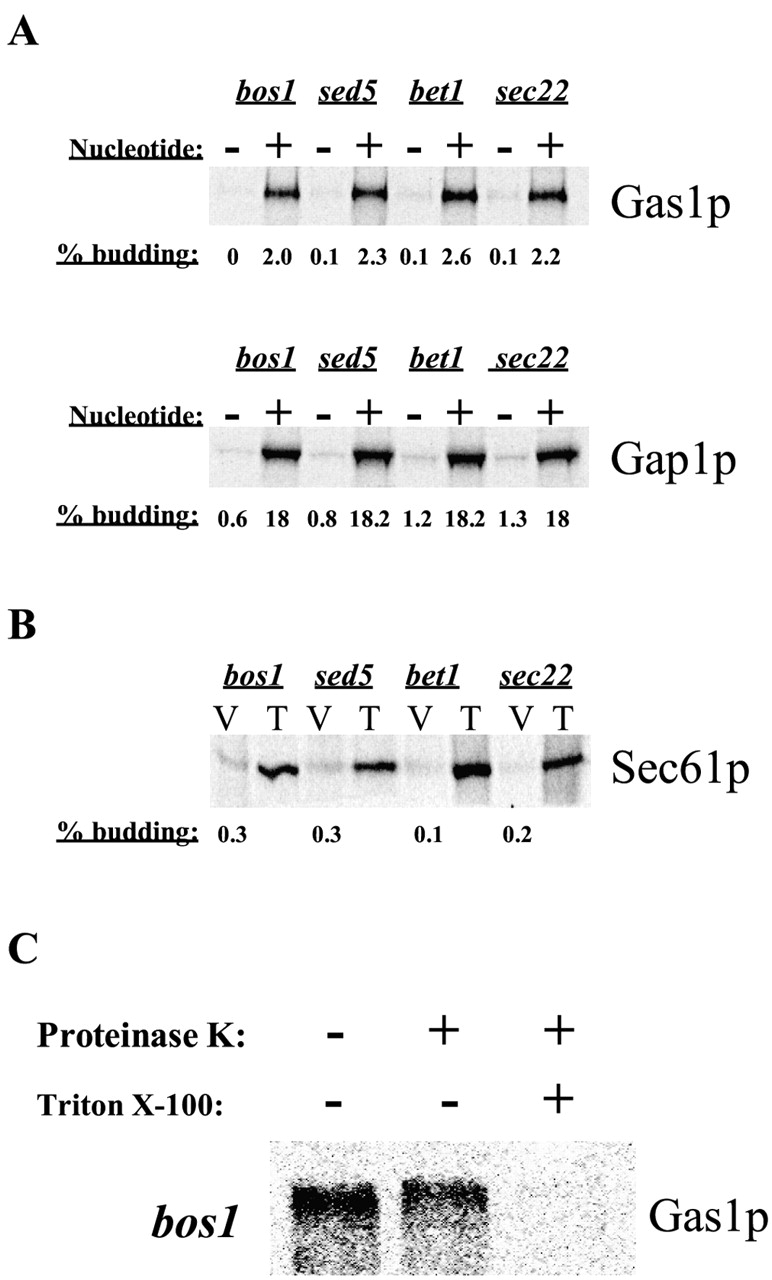
In vitro budding of ER-derived vesicles containing secretory proteins from SNARE mutants. (A) In vitro budding reactions were performed using sec18–20 membranes and cytosol or from bos1–1, bet1–1, sec22–3, sed5–1, or gos1Δ membranes and wild-type cytosol. The nucleotide requirement was tested in the absence of exogenous ATP and GTP and in presence of apyrase (1.5 U/reaction). Budding efficiencies were calculated as the percentage of the total input that was recovered in the purified vesicles for each individual protein. (B) Budding efficiencies determined for Sec61p packaging into ER-derived vesicles generated from bos1–1, bet1–1, sec22–3, and sed5–1 extracts. Numbers represent the percentage of the total input (T) that was recovered in the purified vesicles (V). (C) Integrity of vesicles generated from bos1–1 membranes. Purified vesicles produced from bos1–1 membranes were incubated on ice for 30 min with or without 0.5 mg/ml proteinase K in the presence or absence of 1% Triton X-100. Samples were processed for Gas1p immunoprecipitation, analyzed by SDS-PAGE, and visualized using a phosphorimager.
Anti–v-SNAREs antibodies can inhibit cargo sorting upon ER exit
Another way to test the function of the SNAREs in sorting of GPI-anchored proteins from other secretory proteins is to block their function by adding antibodies specific for the different SNARE proteins during the in vitro budding assay. We thus performed the budding assay using wild-type membranes and cytosol in the presence of antibodies against Sed5p, Bos1p, or Sec22p (Fig. 3). The budding was still specific because it was cytosol and nucleotide dependent and was not the result of ER fragmentation (unpublished data). Budding was somewhat more efficient in the presence of antiserum, but the efficiency was improved even with control antibodies or antibodies against plasma membrane proteins (unpublished data). The presence of anti-Sed5p antibodies during budding did not perturb sorting because only 17% of Gas1p was coimmunoisolated with 72% of Gap1HA. In contrast, the presence of anti-Bos1p or anti-Sec22p antibodies during budding caused a significant sorting defect because 30 and 37% of Gas1p, respectively, was coimmunoisolated with Gap1HA. The presence of unrelated antibodies directed against the plasma membrane protein Ste2p did not affect sorting (Fig. 3 A). These data suggest a direct role of the v-SNAREs in sorting GPI-anchored proteins into distinct vesicles.
Figure 3.

Anti–v-SNARE antibodies affect sorting. (A) Vesicles were generated from wild-type membranes and cytosol in the absence or presence of 5 μl of anti-Bos1p, anti-Sed5p, anti-Sec22p, or anti-Ste2p antibodies. The vesicles were immunoisolated with or without monoclonal anti-HA antibody. The supernatants (S) and pellets (P) were processed for immunoprecipitation, and samples were analyzed as described above. The total recovery, S plus P, was set to 100%. Numbers represent the percentage of recovery in the pellet from which the background was subtracted. The data are the mean ± SD of three independent experiments for anti-Bos1p, anti-Sed5p, and anti-Sec22p antibodies and two independent experiments for anti-Ste2p antibodies. (B) Immunoblot of floated vesicles generated from wild-type membranes and cytosol in absence or in presence of 5 μl of anti-Bos1p, anti-Sed5p, anti-Sec22p, or anti-Ste2p antibodies.
To further analyze the mechanism of action of the anti-SNARE antibodies during the in vitro budding reaction, we checked whether their presence could inhibit the packaging of the corresponding SNARE protein into the ER-derived vesicles. Addition of anti-Bos1p antibodies inhibited Bos1p packaging into ER-derived vesicles but did not inhibit the packaging of Sec22p nor Sed5p. Similarly, anti-Sec22 antibodies inhibited the budding of Sec22p but not of Bos1p or Sed5p, and anti-Sed5 antibodies inhibited the budding of Sed5p but not of Bos1p nor Sec22p. Presence of unrelated anti-Ste2p antibodies did not affect the budding of any of the SNARE proteins (Fig. 3 B). Therefore, the presence of anti-SNARE antibodies in the reaction inhibits the packaging of the corresponding SNARE protein but not of the others.
The presence of anti-SNARE antibodies also reduced the fusion competence of ER-derived vesicles. When the budding reaction was performed with wild-type membranes and cytosol, 62% of Gas1p received α-1,6 mannose modification. In contrast, in presence of anti-Bos1p, anti-Sec22p, or anti-Sed5p antibodies only 6 to 15% of Gas1p received α-1,6 mannose modification (unpublished data). This indicates that the anti-SNARE antibodies also blocked fusion of Gas1p-containing vesicles with the Golgi, even in the case of Sed5p where there was no effect on protein sorting.
Bos1p requirement for Gas1p sorting upon ER exit can be visualized by EM
Thus far, there is strong genetic and biochemical evidence that GPI-anchored proteins exit the ER in distinct vesicles from other secretory proteins (Sütterlin et al., 1997; Muniz et al., 2001; Morsomme and Riezman, 2002). Although this evidence is solid, we sought to strengthen these findings by visualizing the sorting process using EM. We performed an in vitro budding experiment using sec18 mutant membranes and cytosol as described before and fixed the reactions directly after a 10-min incubation with cytosol and energy. The fixed material was embedded, and thin sections were prepared as described (Prescianotto-Baschong and Riezman, 2002). We performed a double immunolabeling on these sections with antibodies against Gas1p and Gap1p followed by IgG colloidal gold detection. The large gold particles were used to localize Gas1p, and the small gold particles were used to localize Gap1p. In a first set of experiments, we used 10- and 5-nm gold particles for Gas1p and Gap1p detection, respectively. Then, we used 15- and 10-nm gold particles for Gas1p and Gap1p, respectively. Since we wanted to analyze protein sorting upon ER exit, we focused on vesicles in the process of budding from the ER membranes. ER membranes were easily distinguished from other membranes by the high density of attached ribosomes. We could clearly observe vesicles budding from these ER membranes. The majority of these vesicles contained gold particles of only one size, meaning one type of cargo (Fig. 4, A and B). Quantitative analysis revealed that only 14% of the labeled budding vesicles were labeled for both Gas1p and Gap1p. These results are consistent with the biochemical findings shown above and visualize the segregation of Gas1p from Gap1p upon ER exit.
Figure 4.
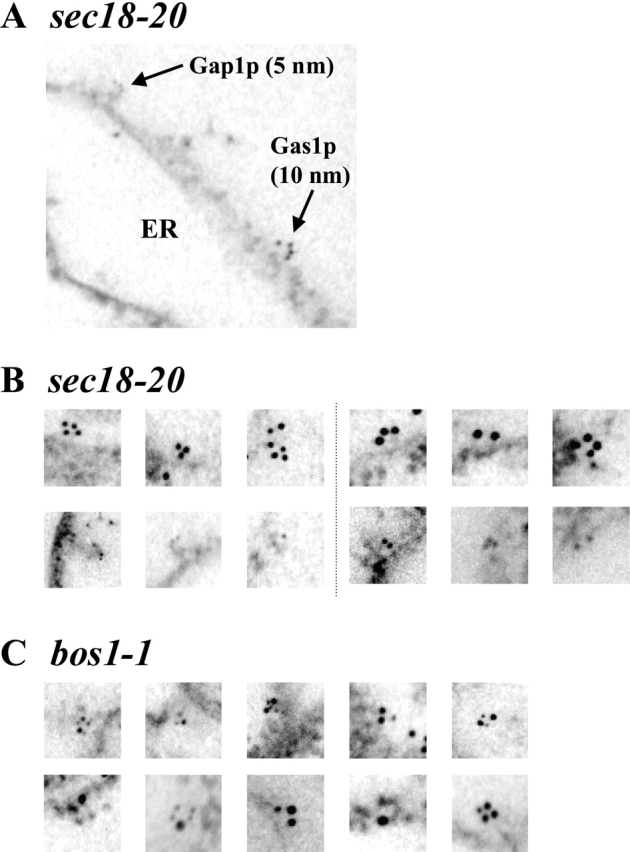
Immuno-EM of Gas1p and Gap1p upon exit from the ER in sec18–20 and bos1–1 mutants. In vitro budding reactions were performed using sec18–20 membranes and cytosol (A and B) or from bos1–1 membranes and wild-type cytosol (C). Extracts were fixed 10 min after addition of cytosol and incubation at 30°C. Immunodetection and quantification were performed as described in Materials and methods. For the quantification, >60 sections were counted for each mutant. In A, Gap1p and Gas1p were labeled with 5- and 10-nm gold particles, respectively. In B, Gas1p was labeled with either 10-nm (three left) or 15-nm (three right) gold particles (top); Gap1p was labeled with either 5-nm (three left) or 10-nm (three right) gold particles (bottom). In C, Gap1p and Gas1p were labeled with 5- and 10-nm gold particles, respectively (top) or with 10- and 15-nm gold particles, respectively (bottom).
When we analyzed the budding reaction in bos1–1 mutant extracts by the same methods, we found that 52% of the ER-forming vesicles contained a mixture of gold particles of different sizes, meaning that Gas1p and Gap1p were incorporated in the same type of vesicles (Fig. 4 B). Again, this observation is in agreement with the biochemical data showing that Bos1p is required for sorting upon ER exit and provides evidence that the inefficiency of codetection seen using sec18 extracts was not due to an inherent deficiency of the method.
Sorting of GPI-anchored proteins from other secretory proteins can be measured in vivo and requires functional Bos1p
Sorting of GPI-anchored proteins from other secretory proteins can also be measured in vivo (Morsomme and Riezman, 2002). The recovery of ER-derived vesicles generated in vivo was only possible in mutants defective in tethering of ER-derived vesicles to the Golgi compartment (Sütterlin et al., 1997; Morsomme and Riezman, 2002). As previously, ER-derived vesicles were immunoisolated from radiolabeled Bet3p-depleted cells. 60% of Gap1p signal present in the vesicle fraction was immunoisolated after incubation with anti-HA antibodies, and only 8% of Gas1p signal was coimmunoisolated showing efficient sorting in vivo (Fig. 5 A). Next, ER-derived vesicles were generated in vivo at 37°C, conditions in which ER to Golgi transport is blocked and purified from the bos1–1 mutant. 66% of the Gap1p signal was immunoisolated from bos1–1-generated vesicles, and 41% of Gas1p signal was coimmunoisolated, indicating a strong sorting defect in vivo (Fig. 5 A). The vesicle fractions isolated from the different strains did not contain Sec61p, showing that the signals were not generated as a result of ER fragmentation (unpublished data). Moreover, Gas1p was protected from protease digestion in absence of detergent but sensitive to degradation in presence of detergent (Fig. 5 B), demonstrating its presence in a closed membrane compartment.
Figure 5.
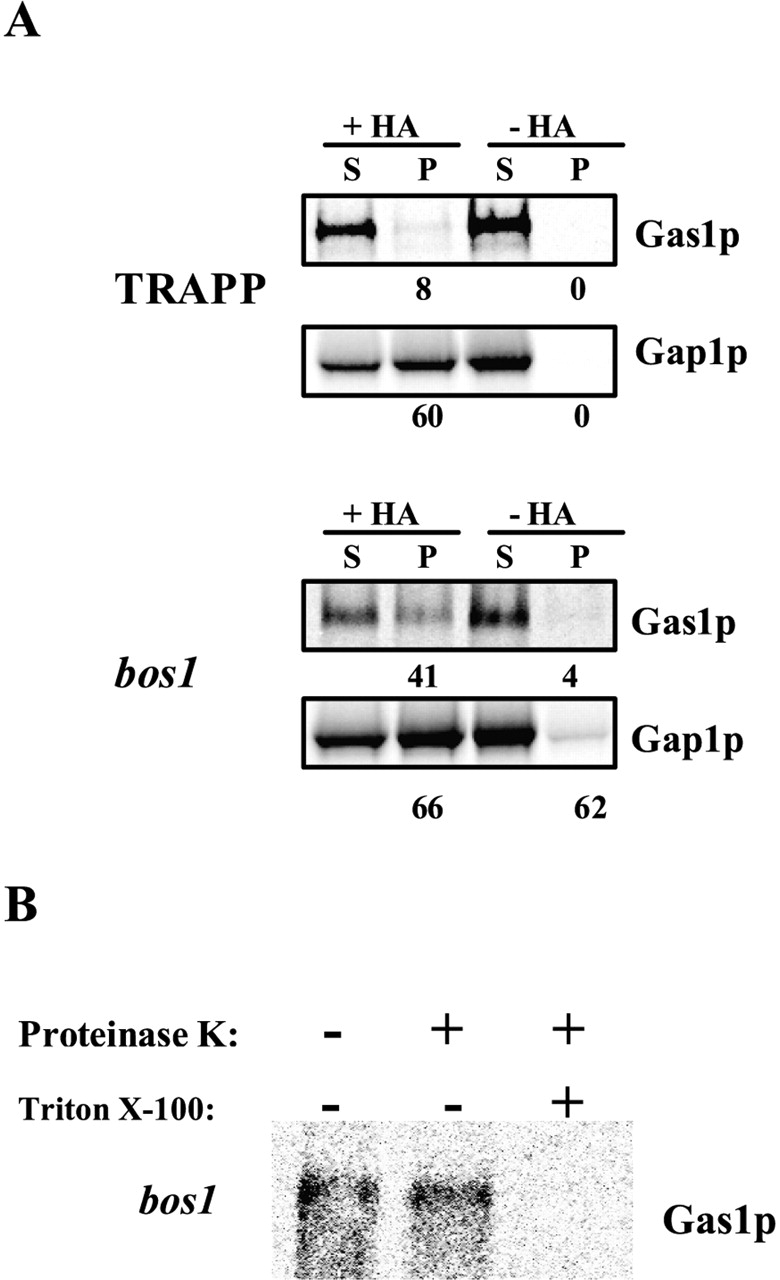
Bos1p is necessary for sorting in vivo. (A) After pulse–chase labeling, vesicles were purified through a Nycodenz® step gradient directly from Bet3p-depleted cells or bos1–1 mutant cells and then immunoisolated with or without monoclonal anti-HA antibody. The supernatants (S) and pellet (P) were processed for immunoprecipitation, and samples were analyzed as described above. The total recovery, S plus P, was set to 100%. Numbers represent the percentage recovery in the pellet. (B) The integrity of bos1–1-generated vesicles was tested as described above.
Gas1p maturation is specifically affected in bos1–1 mutant cells
The ER to Golgi transport of Gas1p can be followed in vivo by measuring the modification of the protein after its delivery to the Golgi complex (Doering and Schekman, 1996; Sütterlin et al., 1997). The ER form of Gas1p shows an apparent molecular weight of 105 kD. Gas1p is then modified in the Golgi compartment to its mature form of 125 kD. The other pathway of ER to Golgi transport can be followed by the maturation of carboxypeptidase Y (CPY), which is present in the ER in its p1 form, then modified in the Golgi complex to its p2 form, which is then cleaved into its mature form (m) in the vacuole. Transport of the two proteins to the Golgi compartment is reliably measured by analysis of the addition of α-1,6 mannose residues using antiserum against this carbohydrate modification. Gas1p transport to the Golgi compartment was shown previously to have specific requirements (Sütterlin et al., 1997). Therefore, we examined whether v-SNARE mutants could affect Gas1p transport in a specific manner. v-SNAREs apparently have at least two functions: a cargo-sorting function upon ER exit and a fusion function of ER-derived vesicles with the Golgi compartment. At 37°C, the transport of Gas1p and CPY was completely blocked in bos1–1, sec22–3, bet1–1, and sed5–1 mutant cells (unpublished data). This defect at restrictive temperature was probably due to the loss of fusion competence of ER-derived vesicles to the Golgi compartment. At 24°C, the transport of Gas1p and CPY from the ER to the Golgi compartment was similar in the bos1–1, sec22–3, and sed5–1 strains (Fig. 6 A). At 30°C, transport of both Gas1p and CPY was slightly delayed in bos1–1 and sec22–3 mutants and more severely affected in the sed5–1 mutant. However, at 30°C the maturation of Gas1p was affected in a particular manner in the bos1–1 strain compared with sec22–3 (Fig. 6 A). The molecular size of Gas1p increased continuously, but no clear shift from the 105- to 125-kD form was observed. The continuous increase of molecular shift was nevertheless due to addition of α-1,6 mannose residues because these forms were precipitated by anti–α1,6 mannose antibodies. Invertase was normally secreted and glycosylated at 24°C and at 30°C in the bos1–1 mutant (Fig. 6 B).
Figure 6.

In vivo Gas1p maturation is specifically affected in bos1–1 mutant at 30°C. Protein maturation in bos1–1, sec22–3, and sed5–1 mutants at 24 and 30°C. (A) Wild-type and mutant cells were pulse labeled and chased at the indicated temperatures for the indicated times. Gas1p and CPY were immunoprecipitated from cell lysates and separated by SDS-PAGE and quantified using a phosphorimager. (B) Analysis of invertase secretion in bos1–1 mutant. Cells were pulse–chase labeled, and then the cells and medium were separated and analyzed separately for invertase by immunoprecipitation, SDS-PAGE, and visualization using a phosphorimager.
The localization of radiolabeled Gas1p in the bos1–1 mutant after 60-min chase could be informative about the reasons for this defective glycosylation. To see if the radiolabeled Gas1p had arrived to the cell surface, we used a modification of a protease shaving technique (Nuoffer et al., 1991). We pulse labeled bos1–1 cells at 30°C, chased for 60 min, digested the cell wall, then incubated with or without proteinase K. At 24 and 30°C, 74 and 70% of Gas1p, respectively, was digested by proteinase K, indicating that the majority of the enzyme reached the plasma membrane (Fig. 7). In contrast, Gas1p was completely protected from protease digestion at 37°C, indicating that Gas1p did not reach the plasma membrane at restrictive temperature. As a control, the cytosolic glyceraldehyde-3-phosphate dehydrogenase 1, Tdh1p, was protected at the different temperatures (Fig. 7). These results show that, although Gas1p shows abnormal modification in bos1–1 at 30°C, it is transported to the cell surface.
Figure 7.

Gas1p reaches the cell surface in the bos1–1 mutant. Mutant bos1–1 cells were pulse labeled for 3 min and chased for 60 min. Cell walls were mildly digested and incubated with or without proteinase K as described. Proteinase K was inactivated and Gas1p and Tdh1p were immunoprecipitated from cell lysates and analyzed by SDS-PAGE and quantified using a phosphorimager.
Discussion
In this study, we have uncovered a novel function of vSNAREs on donor membranes, a cargo sorting function necessary for the segregation of GPI-anchored proteins from other secretory proteins upon ER exit. The in vitro sorting assay that we used in this study measures sorting of GPI-anchored proteins from other secretory proteins upon ER exit (Muniz et al., 2001). Using this assay with temperature-sensitive v-SNARE mutant extracts shows that these proteins are required for the sorting process. As shown previously for the sec18 mutant (Muniz et al., 2001), the fusion defect of Gas1p-containing vesicles generated from SNARE mutants with the Golgi membranes ensures that we analyze primary ER-derived vesicles. The sorting defect observed with wild-type extracts in the presence of specific antibodies directed against the v-SNAREs Bos1p and Sec22p suggests a direct involvement of these SNAREs in the sorting event. As observed previously in other studies (Allan et al., 2000; Liu and Barlowe, 2002), antibodies specifically inhibited the packaging of the corresponding SNARE into ER-derived vesicles. This observation suggests that the v-SNAREs Bos1p and Sec22p act as individual proteins rather than as cis–SNARE complexes for protein sorting and that their function in protein sorting is required before or during budding of the vesicles. In addition, the vesicles generated in the presence of anti-Bos1p, anti-Sec22p, or anti-Sed5p antibodies were not able to fuse with the Golgi membranes. Nevertheless, anti-Sed5p antibodies did not inhibit sorting of GPI-anchored proteins, showing again that the sorting defect is not due to a fusion defect as shown previously for vesicle-tethering mutants (Morsomme and Riezman, 2002).
In addition, we provide the first morphological evidence for sorting of cargo proteins upon ER exit. When we used sec18 membranes for the in vitro budding assay and fixed the membranes just after the budding reaction started, we could clearly discriminate between the two different pathways. To ensure that we analyzed the sorting event upon ER exit, we restricted our analysis to vesicles in the process of budding from the ER. The ER membranes were clearly identified by their electron-dense pattern due to the presence of ribosomes. Isolated vesicles containing separate cargoes were also detected, but since their origin was not certain, they were not included in our analysis. The role of Bos1p in sorting GPI-anchored proteins into distinct vesicles was also confirmed by EM. The analysis of budding profiles revealed that the proportion of ER-derived vesicles containing both Gap1p and Gas1p was 52% using bos1–1 mutant membranes and 14% using wild-type membranes. These numbers correlate well with the in vitro data and confirm the role of Bos1p in sorting of GPI-anchored proteins into distinct vesicles.
Importantly, sorting of GPI-anchored proteins from other secretory proteins upon exit of the ER can also be measured in vivo. The sorting defect observed in vitro for the bos1–1 mutant was reproduced in vivo. The physiological consequence of missorting can be illustrated by the maturation defect observed for Gas1p in the bos1–1 mutant at 30°C. Although transport of CPY was slightly delayed, Gas1p was slowly and continuously matured. In theory, this could be due to the slow transport kinetics. However, in the sec22–3 mutant the kinetics of CPY export was similar to that of bos1–1 mutant at 30°C, but the maturation of Gas1p was apparently normal albeit with a small delay. The maturation defect of Gas1p is thus specific for the bos1–1 mutant at 30°C, conditions where sorting is defective. This specific bos1–1 allele may be more appropriate to observe the predominance of the sorting defect on the fusion defect compared with other mutants such as sec22–3 or bet1–1. Unfortunately, we were not able to measure sorting in vivo for sec22–3 or bet1–1 mutants because recovery of Gas1p-containing vesicles was too low (unpublished data). One possible explanation for the low recovery could be that tethering of ER-derived vesicles is not defective in these mutants but is defective in the bos1–1 mutant used in this study. Previously, we have proposed that recovery of ER-derived vesicles from the in vivo sorting assay is efficient only when the vesicles are in an untethered state (Morsomme and Riezman, 2002). The maturation defect of Gas1p observed in the bos1–1 mutant at 30°C is not due to a general glycosylation defect since both CPY and invertase are glycosylated to the same extent at 24 and 30°C. Moreover, Gas1p travels through the Golgi apparatus because it receives α-1,6 mannose modification and reaches the plasma membrane where it can be digested by proteinase K. The fact that Gas1p-containing vesicles did not fuse in vitro at 30°C while Gas1p was transported to the cell surface in vivo at the same temperature indicates that the temperature sensitivity is more stringent in vitro than in vivo. Interestingly, it was shown recently that a soluble form of Gas1p is inefficiently matured in wild-type cells (Watanabe et al., 2002). Indeed, the mutant form of Gas1p (L526R), which is not GPI anchored, is released into the media and shows a slow maturation kinetic. Although there is no data concerning the sorting of the soluble form of Gas1p, we could speculate that the maturation defect reflects a sorting defect. Previous experiments suggested that the two ER-derived vesicle populations could reach two different Golgi domains or membranes (Muniz et al., 2001; Morsomme and Riezman, 2002). According to this hypothesis, the maturation defect of Gas1p could reflect the mistargeting of Gas1p to the wrong Golgi domain, leading to a specific glycosylation defect for Gas1p.
Although we identified several components necessary for protein sorting in the ER, the molecular mechanisms triggering this process are still unknown. Ypt1p was shown to activate v-SNARES on carrier vesicles, conferring them fusion competence (Lian and Ferro-Novick, 1993; Lian et al., 1994) and p115, the mammalian homologue of Uso1p, forms a cis-SNARE complex on the ER exit sites and is recruited to the membrane by the mammalian homologue of Ypt1p, Rab1 (Allan et al., 2000). It was reported recently that SNARE proteins can be concentrated into lipid microdomains (Lafont et al., 1999; Chamberlain et al., 2001; Lang et al., 2001), which might be relevant to GPI-anchored protein transport (Sütterlin et al., 1997; Bagnat et al., 2000) . However, if the formation of lipid rafts in the ER could possibly contribute to the sorting mechanism of GPI-anchored proteins in the ER, proteins are also certainly involved in this process. The SNARE proteins may constitute the link between the cytosolic factors involved in sorting (Ypt1p, Uso1p, and the COG complex) (Morsomme and Riezman, 2002) and the GPI-anchored proteins that are exclusively lumenal. On the other hand, v-SNAREs were shown to interact with COPII coat proteins (Matsuoka et al., 1998; Springer and Schekman, 1998), which are themselves responsible for efficient packaging of secreted proteins into the vesicles (Kuehn et al., 1998). It is thus possible that Ypt1p recruits the tethering factors onto the ER membrane where they would interact with specific SNAREs and coat proteins ensuring specific packaging and thus sorting of cargo molecules into ER-derived vesicles. These findings suggest that Rab-mediated recruitment of tethering factors to membranes and interaction of these proteins with v-SNAREs may be a general mechanism to couple protein sorting and packaging into vesicles to targeting, docking, and fusion.
Materials and methods
Strains and plasmids
The Saccharomyces cerevisiae strains used in this work are described in Table I. The different cytosols were prepared from the strains RH732 (WT) and RH2043 (sec18) as described previously (Salama et al., 1993). All the strains used for in vitro and in vivo assays were transformed with the multicopy vector pCNYG1 (GAS1, URA3) (Nuoffer et al., 1991) or Yeplac112-GAS1 (GAS1, TRP1). When indicated, the strains were also transformed with the pPL269 plasmid (CEN, LYS2) (Kuehn et al., 1996) or with PCM189-GAP1HA expressing the HA-tagged version of GAP1 plasmid (CEN, URA3).
Table I. Yeast strains used in this study.
| Strain
|
Genotype
|
Source
|
|---|---|---|
| RH256 | Mata ura3 his4 leu2 lys2 trp1 | This laboratory |
| RH732 | Mata pep4Δ::URA3 his4 leu2 lys2 bar1 | This laboratory |
| PLY129 | Mata gap1Δ::LEU2 lys2Δ201 leu2 ura3 ade2 | P.O. Ljungdahla |
| RH2043 | Mata sec18-20 pep4Δ::URA3 his4 leu2 ura3 bar1 | This laboratory |
| RH3780 | Matα sec18-20 gap1Δ::LEU2 lys2Δ201 leu2 ura3 ade2 | This laboratory |
| RH2222 | Mata sec22-3 lys2Δ201 leu2 ura3 his4 bar1 | This laboratory |
| RH2782 | Matα bet1-1 his4 | S. Ferro-Novickb |
| RH5535 | Matα bet1-1 ura3 leu2 trp1 | This study |
| RH3339 | Mata gos1_::LEU2 lys2Δ201 leu2 ura3 his4 | This laboratory |
| RH3470 | Mata bos1-1 lys2Δ201 leu2 | R. Schekmanc |
| RH5543 | Matα bos1-1 ura3 leu2 trp1 his4 lys2 | This study |
| RH5541 | Matα sed5-1 ura3 leu2 trp1 his3 lys2 suc2 | D. Banfieldd |
| SFNY431 | Matα Gal+ bet3Δ::URA3 ura3 leu2 pGR10 (LEU2, BET3, GAL1) | S. Ferro-Novick |
| RH5502 | Matα Gal+ bet3Δ::URA3 ura3 leu2 lys2 trp1 his4 pGR10 (LEU2, BET3, GAL1) | This laboratory |
Ludwig Institute for Cancer Research, Stockholm, Sweden.
Yale Medical School, New Haven, CN.
University of California, Berkeley, CA.
Hong Kong University of Science and Technology, Hong Kong.
In vitro sorting assay
The in vitro sorting assay was performed as described previously (Muniz et al., 2001). The strains PLY129, RH3780, RH2222, RH5535, RH3339, RH5543, RH5541, and RH5502 were all transformed with a GAS1 multicopy plasmid and the HA-tagged version of GAP1. The different strains were first grown in a SUD medium (0.16% yeast nitrogen base without amino acids and without [NH4]2SO4, 2% glucose, 0.1% urea) supplemented with the required amino acids at 24°C and then grown overnight in 200 ml SDYE medium (0.67% yeast nitrogen base without amino acids, 2% glucose, 0.2% yeast extract) supplemented with the required amino acids and nutrients at 24°C. Cells were harvested at 5 × 106 cells/ml, washed twice with SD medium supplemented with the required amino acids, resuspended in 4 ml, and incubated 15 min at 24°C. Cells were then incubated at 32°C for 5 min and pulse labeled for 3 min with Trans-label mix (NEN). The labeling reaction was stopped by the addition of 20 mM NaN3 and incubation on ice for 15 min. Permeabilized cells were prepared as described previously (Muniz et al., 2000). To deplete the cells of Bet3p, SFNY431 and RH5502 were grown to stationary phase in SG medium (0.67% yeast nitrogen base without amino acids, 2% raffinose, 0.5% galactose) supplemented with the required amino acids and nutrients, then diluted to an OD600 = 0.01 into SDYE medium, grown for 14 h at 24°C, and processed as above to obtain cytosol or permeabilized cells.
The in vitro ER-budding assay was performed in a total volume of 300 μl in the presence of 25 × 107 permeabilized cells, 600 μg cytosol, 1 mM ATP, 1 mM creatine phosphate, 1 mM phosphocreatine kinase, 0.2 mM GTP, 0.1 mM GDP-mannose, 6 μg Sar1p, 1 mM protease inhibitor mix (leupeptin, pepstatin, and antipain) for 1 h at the indicated temperature. Vesicle immunoisolation was performed as described previously (Muniz et al., 2001).
In vivo sorting assay
Yeast cells were grown, harvested, and washed as described for in vitro assays. For the labeling step, cells were preincubated at 37°C for 5 min, pulse labeled for 6 min at 37°C with Trans-label mix (NEN), and chased for 10 min at 37°C by adding 1/100 volume of chase cocktail (0.3% methionine, 0.3% cysteine, 0.3 M [NH4]2SO4). The labeling reaction was stopped by the addition of 20 mM NaN3 and incubation on ice for 15 min. Permeabilized spheroplasts were prepared as described previously (Kuehn et al., 1996) and directly sedimented (14000 rpm for 10 min at 4°C). The supernatant was subjected to flotation on a Nycodenz® step gradient, and the ER-derived vesicle-containing fractions were processed as described (Muniz et al., 2001).
EM
The permeabilized spheroplasts were washed and incubated with cytosol and energy exactly as described for the in vitro budding/sorting assay. After 10-min incubation at 30°C, the spheroplasts were fixed overnight at 4°C by addition of glutaraldehyde (0.2% final concentration) and formaldehyde (3% final concentration), washed in 50 mM Hepes, pH 7.0, 3 mM KCl, and free aldehyde groups were quenched for 30 min as described (van Tuinen and Riezman, 1987). Dehydration, infiltration, and polymerization in LRGold resin (London Resin Company, Ltd.) were done according to the supplier's instructions. 50- to 60-nm sections were cut and mounted on 200-mesh nickel grids. Gas1p and Gap1p antibodies were raised in rabbits and used for immunolabeling at dilutions determined empirically. Grids were placed upside down on 50-μl droplets of blocking solution (150 mM NaCl, 10 mM potassium phosphate, pH 7.5, 0.1% Tween 20, 2% fatty acid–free BSA [Sigma-Aldrich]) for 20 min. The grids were then transferred to droplets containing appropriate dilutions of the primary antibodies in blocking buffer and incubated for 4 h at room temperature. The grids were then washed three times for 5 min with PBS solution and 0.2% BSA and three times for 5 min in PBS solution. The grids were incubated for 10 min in blocking buffer before transferring to droplets with secondary antibody gold conjugates and incubation for 2 h. After washing as above, they were fixed for 5 min in 1% glutaraldehyde in PBS solution to preserve the immunolabeling and then washed (by dipping 10 times in a 100-ml beaker) in distilled water. Free aldehydes were again quenched with 50 mM NH4Cl and washed with distilled water. Labeling with the second primary antibodies was done essentially as described for the first primary antibodies except the blocking solution contained 0.5% Tween 20 in PBS solution. After washing in water, the grids were stained in 6% uranyl acetate for 10 min and in Reynold's lead citrate for 30–60 s.
For quantification, immunolabeled vesicles were counted when (a) they were labeled with more than one IgG colloidal gold particle and (b) they were still attached to the ER membrane. More than 60 vesicles were counted per experiment. ER membranes were identified by their electron-dense pattern due to the presence of ribosomes.
Pulse–chase experiments
Pulse–chase experiments were performed as described previously (Sütterlin et al., 1997). Invertase was induced by preincubating the cells at 24°C for 30 min in a medium containing 0.1% glucose.
Plasma membrane proteinase K shaving
The cells were grown, pulsed labeled, and chased for 60 min as described for the pulse–chase experiments. Cell walls were mildly digested by a 10-min incubation in 10 mM Mesna followed by a 20-min incubation with lyticase at 24°C. Spheroplasts were incubated for 30 min with 200 μg/ml proteinase K. Digestion was stopped by addition of 1 mM PMSF and 0.5 N NaOH followed by incubation on ice for 10 min. Proteins were TCA precipitated and analyzed by immunoprecipitation, SDS-PAGE, and quantitation using a phosphorimager.
Acknowledgments
We are grateful to R. Schekman (University of California, Berkeley, CA), S. Ferro-Novick (Yale Medical School, New Haven, CN), C. Barlowe (Darmouth College, Hanover, NH), H. Pelham (Laboratory of Molecular Biology-MRC, Cambridge, UK), T. Söllner (Memorial Sloan-Kettering Cancer Center, New York, NY), and R. Watanabe (University of Geneva, Geneva, Switzerland) for strains, plasmids, and antibodies and to members of the Riezman lab for comments and support. We particularly thank Reika and Sabrina for stimulating and helpful discussions and critical reading of the manuscript.
This work was supported by a grant from the Swiss National Science Foundation (to H. Riezman), the Fonds National de la Recherche Scientifique (Belgium) and a Human Frontier Science Program long-term fellowship (to P. Morsomme).
Pierre Morsomme's current address is Unité de Biochimie Physiologique, Institut des Sciences de la Vie, University of Louvain, Croix du Sud 2-20, B-1348 Louvain-la-Neuve, Belgium.
Abbreviations used in this paper: COG, conserved oligomeric Golgi; CPY, carboxypeptidase Y; GPI, glycosylphosphatidylinositol.
References
- Allan, B.B., B.D. Moyer, and W.E. Balch. 2000. Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science. 289:444–448. [DOI] [PubMed] [Google Scholar]
- Bagnat, M., S. Keranen, A. Shevchenko, and K. Simons. 2000. Lipid rafts function in biosynthetic delivery of proteins to the cell surface in yeast. Proc. Natl. Acad. Sci. USA. 97:3254–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, X., N. Ballew, and C. Barlowe. 1998. Initial docking of ER-derived vesicles requires Uso1p and Ypt1p but is independent of SNARE proteins. EMBO J. 17:2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, K.S., J. Hanna, I. Simon, J. Krise, P. Barbero, and S.R. Pfeffer. 2001. Role of rab9 GTPase in facilitating receptor recruitment by tip47. Science. 292:1373–1376. [DOI] [PubMed] [Google Scholar]
- Chamberlain, L.H., R.D. Burgoyne, and G.W. Gould. 2001. SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc. Natl. Acad. Sci. USA. 98:5619–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis, S., H.M. McBride, R.D. Burgoyne, and M. Zerial. 1999. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 397:621–625. [DOI] [PubMed] [Google Scholar]
- Doering, T.L., and R. Schekman. 1996. GPI anchor attachment is required for Gas1p transport from the endoplasmic reticulum in COP II vesicles. EMBO J. 15:182–191. [PMC free article] [PubMed] [Google Scholar]
- Kuehn, M.J., R. Schekman, and P.O. Ljungdahl. 1996. Amino acid permeases require COPII components and the ER resident membrane protein Shr3p for packaging into transport vesicles in vitro. J. Cell Biol. 135:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn, M.J., J.M. Herrmann, and R. Schekman. 1998. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature. 391:187–190. [DOI] [PubMed] [Google Scholar]
- Lafont, F., P. Verkade, T. Galli, C. Wimmer, D. Louvard, and K. Simons. 1999. Raft association of SNAP receptors acting in apical trafficking in Madin-Darby canine kidney cells. Proc. Natl. Acad. Sci. USA. 96:3734–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, T., D. Bruns, D. Wenzel, D. Riedel, P. Holroyd, C. Thiele, and R. Jahn. 2001. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 20:2202–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian, J.P., and S. Ferro-Novick. 1993. Bos1p, an integral membrane protein of the endoplasmic reticulum to Golgi transport vesicles, is required for their fusion competence. Cell. 73:735–745. [DOI] [PubMed] [Google Scholar]
- Lian, J.P., S. Stone, Y. Jiang, P. Lyons, and S. Ferro-Novick. 1994. Ypt1p implicated in v-SNARE activation. Nature. 372:698–701. [DOI] [PubMed] [Google Scholar]
- Liu, Y., and C. Barlowe. 2002. Analysis of Sec22p in endoplasmic reticulum/Golgi transport reveals cellular redundancy in SNARE protein function. Mol. Biol. Cell. 13:3314–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, K., Y. Morimitsu, K. Uchida, and R. Schekman. 1998. Coat assembly directs v-SNARE concentration into synthetic COPII vesicles. Mol. Cell. 2:703–708. [DOI] [PubMed] [Google Scholar]
- McBride, H.M., V. Rybin, C. Murphy, A. Giner, R. Teasdale, and M. Zerial. 1999. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell. 98:377–386. [DOI] [PubMed] [Google Scholar]
- McLauchlan, H., J. Newell, N. Morrice, A. Osborne, M. West, and E. Smythe. 1998. A novel role for Rab5-GDI in ligand sequestration into clathrin-coated pits. Curr. Biol. 8:34–45. [DOI] [PubMed] [Google Scholar]
- Morsomme, P., and H. Riezman. 2002. The Rab GTPase Ypt1p and tethering factors couple protein sorting at the ER to vesicle targeting to the Golgi apparatus. Dev. Cell. 2:307–317. [DOI] [PubMed] [Google Scholar]
- Muniz, M., C. Nuoffer, H.P. Hauri, and H. Riezman. 2000. The Emp24 complex recruits a specific cargo molecule into endoplasmic reticulum–derived vesicles. J. Cell Biol. 148:925–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz, M., P. Morsomme, and H. Riezman. 2001. Protein sorting upon exit from the endoplasmic reticulum. Cell. 104:313–320. [DOI] [PubMed] [Google Scholar]
- Nuoffer, C., P. Jeno, A. Conzelmann, and H. Riezman. 1991. Determinants for glycophospholipid anchoring of the Saccharomyces cerevisiae GAS1 protein to the plasma membrane. Mol. Cell. Biol. 11:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescianotto-Baschong, C., and H. Riezman. 2002. Ordering of compartments in the yeast endocytic pathway. Traffic. 3:37–49. [DOI] [PubMed] [Google Scholar]
- Salama, N.R., T. Yeung, and R.W. Schekman. 1993. The Sec13p complex and reconstitution of vesicle budding from the ER with purified cytosolic proteins. EMBO J. 12:4073–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schekman, R., and L. Orci. 1996. Coat proteins and vesicle budding. Science. 271:1526–1533. [DOI] [PubMed] [Google Scholar]
- Seachrist, J.L., S.A. Laporte, L.B. Dale, A.V. Babwah, M.G. Caron, P.H. Anborgh, and S.S. Ferguson. 2002. Rab5 association with the angiotensin II type 1A receptor promotes Rab5 GTP binding and vesicular fusion. J. Biol. Chem. 277:679–685. [DOI] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.M. Gaullier, A. Brech, J. Callaghan, B.H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Springer, S., and R. Schekman. 1998. Nucleation of COPII vesicular coat complex by endoplasmic reticulum to Golgi vesicle SNAREs. Science. 281:698–700. [DOI] [PubMed] [Google Scholar]
- Sütterlin, C., T.L. Doering, F. Schimmoller, S. Schroder, and H. Riezman. 1997. Specific requirements for the ER to Golgi transport of GPI-anchored proteins in yeast. J. Cell Sci. 110:2703–2714. [DOI] [PubMed] [Google Scholar]
- van IJzendoorn, S.C., M.J. Tuvim, T. Weimbs, B.F. Dickey, and K.E. Mostov. 2002. Direct interaction between Rab3b and the polymeric immunoglobulin receptor controls ligand-stimulated transcytosis in epithelial cells. Dev Cell. 2:219–228. [DOI] [PubMed] [Google Scholar]
- VanRheenen, S.M., X. Cao, V.V. Lupashin, C. Barlowe, and M.G. Waters. 1998. Sec35p, a novel peripheral membrane protein, is required for ER to Golgi vesicle docking. J. Cell Biol. 141:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen, S.M., X. Cao, S.K. Sapperstein, E.C. Chiang, V.V. Lupashin, C. Barlowe, and M.G. Waters. 1999. Sec34p, a protein required for vesicle tethering to the yeast Golgi apparatus, is in a complex with Sec35p. J. Cell Biol. 147:729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tuinen, E., and H. Riezman. 1987. Immunolocalization of glyceraldehyde-3-phosphate dehydrogenase, hexokinase, and carboxypeptidase Y in yeast cells at the ultrastructural level. J. Histochem. Cytochem. 35:327–333. [DOI] [PubMed] [Google Scholar]
- Watanabe, R., K. Funato, K. Venkataraman, A.H. Futerman, and H. Riezman. 2002. Sphingolipids are required for stable membrane association of glycosylphosphatidylinositol-anchored proteins in yeast. J. Biol. Chem. 277:49538–49544. [DOI] [PubMed] [Google Scholar]