

Abstract
Mitotic chromosome structure and DNA sequence requirements for normal chromosomal condensation remain unknown. We engineered labeled chromosome regions with altered scaffold-associated region (SAR) sequence composition as a formal test of the radial loop and other chromosome models. Chinese hamster ovary cells were isolated containing high density insertions of a transgene containing lac operator repeats and a dihydrofolate reductase gene, with or without flanking SAR sequences. Lac repressor staining provided high resolution labeling with good preservation of chromosome ultrastructure. No evidence emerged for differential targeting of SAR sequences to a chromosome axis within native chromosomes. SAR sequences distributed uniformly throughout the native chromosome cross section and chromosome regions containing a high density of SAR transgene insertions showed normal diameter and folding. Ultrastructural analysis of two different transgene insertion sites, both spanning less than the full chromatin width, clearly contradicted predictions of simple radial loop models while providing strong support for hierarchical models of chromosome architecture. Specifically, an ∼250-nm-diam folding subunit was visualized directly within fully condensed metaphase chromosomes. Our results contradict predictions of simple radial loop models and provide the first unambiguous demonstration of a hierarchical folding subunit above the level of the 30-nm fiber within normally condensed metaphase chromosomes.
Keywords: mitotic chromosome; large-scale structure of chromatin; scaffold-associated regions; transgenes; chromosome scaffold
Introduction
How chromatin folds into chromosomes remains a fundamental question in cell biology today. What DNA sequences, if any, are necessary and sufficient for reproducible packaging of DNA into chromosomes is currently unknown. Chromatin condensation observed using in vitro chromosome assembly systems with prokaryotic DNA has led to suggestions of minimal sequence requirements for chromosome architecture; however, the degree to which normal chromosome architecture was reconstituted in those works is unclear. “Successive coiling” models do not require any particular cis DNA sequences for proper maintenance of the structure. In contrast, scaffold-associated region/matrix-associated region (SAR/MAR)* sequences, identified experimentally as sequences remaining in “scaffold” preparations (SAR) or as sequences binding to scaffold preparations (MAR), have been proposed as the anchors of DNA loops to the chromosome scaffold predicted in radial loop models (Razin, 1996).
Experiments aimed at identifying the role of specific sequences have been constrained by experimental limitations associated with FISH for mapping the locations of specific DNA sequences within chromosomes. The harsh DNA denaturation conditions required for FISH lead to loss of the traditional chromosome scaffold. However, chromosomes extracted with 0.5–1.0 M univalent salt, producing a “partial halo/scaffold” preparation, show a relative enrichment of nontranscribed spacer sequences from the human ribosomal repeats within the scaffold fraction relative to transcribed sequences (Bickmore and Oghene, 1996). A more significant, preferential positioning of the Pax6 gene region to the edge of the chromosome scaffold with flanking genomic regions oriented into the “halo” fraction also was observed (Bickmore and Oghene, 1996).
Investigation of several chromosome loci by FISH at best have shown only a slight deviation from a random, lateral distribution within unextracted chromosomes (Baumgartner et al., 1991). Reduction in chromatid diameters in mammalian mitotic chromosome in regions containing integrated yeast chromosomes have been explained as the result of the increased SAR frequency in yeast DNA (McManus et al., 1994). Alternatively, a higher density of transcriptionally active regions with a resulting impairment in chromosome condensation has been proposed (McManus et al., 1994).
Here, we have attempted an approach, based on engineering chromosome regions with altered sequence content, which is motivated by two long-term goals. First, by labeling chromosome regions of variable size, we should be able to discern folding motifs within native chromosomes. Second, by experimentally manipulating the sequence composition of these engineered chromosome regions, we should be able to directly test the targeting of specific sequences and sequence requirements for normal chromosome condensation. In this paper, we describe the folding of mitotic chromosome regions, hundreds to thousands of kb in size, engineered to contain a large fraction of foreign DNA containing lac operator repeats and varying SAR density.
Results
Experimental system
For improved visualization of specific chromosome regions within intact, native chromosomes, we isolated cell clones containing high copy number insertions of vector sequences tagged with lac operator sequences. These lac operator sequences can be identified by light or electron microscopy through combining lac repressor staining with either immunofluorescence or immunogold detection (Robinett et al., 1996). To test the possible role of SAR sequences in maintenance of the structure of mitotic chromosomes, we created regions in CHO chromosomes that have an altered density of SAR sequences as compared with native CHO chromosomes. The average size of a chromatin loop in the radial loop model is ∼50 kbp (Razin, 1996). Predictions of the radial loop model are that these SAR sequences should be targeted to the chromosome scaffold axis. Moreover, significant variations in the mean spacing of SAR sequences should lead to changes in mean loop sizes, leading to a predicted change in chromosome diameter and/or packing density within the chromosome. In contrast, the successive coiling model (Sedat and Manuelidis, 1978) and “chromonema” model (Belmont and Bruce, 1994), not involving the concepts of loops and anchoring sequences, do not predict any change in the appearance of mitotic chromosomes in response to a change in spacing between SARs.
Fig. 1 outlines our experimental protocol. Two flanking SAR sequences from the human β–interferon gene were used to flank the dihydrofolate reductase (DHFR)–selectable marker. Both the “control” vector and the “double SAR” (dSAR) vector were transfected into CHO DG44 cells, containing a double deletion of the endogenous DHFR gene (Urlaub et al., 1986). After lac repressor immunostaining, the number of cells showing large spots was below 1% of the total cell pool of stable transformants. To enrich for cell clones with large inserts, fluorescence-activated cell sorting was used to select cells with high DHFR expression, using a cell permeable, fluorescein-labeled methotrexate (MTX) conjugate. MTX binds to DHFR, and binding is proportional to DHFR expression (Kaufman et al., 1978). The cell population corresponding to the highest 1% fluorescence intensity was sorted and subcloned by serial dilution.
Figure 1.

Experimental approach. (A) A pSVII-dhfr vector derivative was used to create control and dSAR constructs with 32 copies of a lac operator 8-mer repeat. (B) DHFR (−) CHO cells (DG44) were stably transfected with either control or dSAR supercoiled DHFR expression vectors. fluorescein-labeled MTX–stained cells with larger inserts were selected with FACS® and cloned. (C) Mitotic chromosomes were isolated and used for extraction and staining by different methods; lac repressor immunostaining or in vivo expression of an EGFP–lac repressor–NLS fusion protein and FISH. (D) Cell flow cytometry (abscissa, logarithm of relative intensity; ordinate, cell number) after staining with fluorescein-labeled MTX. Average DHFR expression is ∼20× higher in cells transformed with the dSAR versus control vector.
Characterization of cell clones
Previous work showed that expression of the human β–interferon gene was increased 20–30-fold by flanking SAR sequences (Klehr et al., 1991). Consistent with these observations, flow cytometry analysis demonstrated that pools of cells carrying the stably integrated dSAR construct express on average ∼20 times as much DHFR per cell as compared with the pools of cells with the control construct (Fig. 1 D).
Two clones from the control vector (Con-1, Con-610) and two clones from the dSAR vector (dSAR-d11, dSAR-g12) were selected for detailed characterization and analysis. Southern blot analysis was used to estimate vector copy number in these stable cell clones. Different dilutions of DNA samples from these cell lines were digested with EcoRI, which cuts within the lac operator array, producing a 292-bp DNA band corresponding to a lac operator 8-mer (Robinett et al., 1996). As standards, varying amounts of the dSAR vector were digested as well, and were run on the gel. After blotting and hybridization to a lac operator DNA probe, integrated intensities of signals from each lane were plotted for different exposures. This analysis produced estimates of 1,000, 10, 1,000, and 300 copies for the Con-1, Con-610, dSAR-d11, and dSAR-g12 clones, respectively.
A rough estimate of the ratio of vector sequence to total DNA was calculated for two of the cell lines, dSAR-d11 and Con-1, with the largest labeled chromosome regions. Measurements of longitudinal lengths of inserts of 20 isolated chromosomes of the dSAR-d11 clone immunostained with lac repressor gave an average of 0.5 μm. Metaphase chromosomes from CHO DG44 cells have a DNA content of ∼70 Mbp per 1 μm length of chromosome arm (Li et al., 1998). Application of this to our cell lines yielded an estimate of 70 and 60% vector DNA content for Con-1 and dSAR-d11, respectively.
The Mbp size range of integration regions and high number of vector copies make it difficult to construct a physical map of the insertion sites. To probe the length of the uninterrupted vector arrays inside insert regions, we cut genomic DNA with restriction enzymes with no sites within either of the plasmid vectors, using pulse-field gel electrophoresis and Southern blotting for analysis. Assuming normal, nonrepetitive DNA flanking the vector insertion sites, use of a frequent cutter restriction enzyme should produce flanking, genomic sequences only several kb in size, negligible relative to a vector concatemer hundreds of kb in size. If an insert region consisted of multiple copies of vector DNA only, we would predict single bands hundreds of kb to tens of Mbp in size.
One cell line, Con-610, produced clear evidence for large, contiguous vector concatemers (Fig. 2). Two distinct bands, the first >150 kbp and the second >200 kbp in size were observed after both NheI and EcoRV digests, and two closely sized bands between 200 and 250 kb in size were observed after MluI digestion. The remaining clones showed mixtures of high mol wt fragments with smaller fragments. The MluI digest produced fragments hundreds of kb to several Mbp in size from the dSAR-g12, dSAR-d11, and Con-1 clones (Fig. 2 C). However, this finding was not confirmed by NheI and EcoRV digests. Even though high molecular DNA vector fragments were observed for these three clones, their fraction in the NheI digest is less compared with the MluI digest. For the EcoRV digest, most of the insert DNA can be cut into fragments of ∼50 kbp, corresponding to only two or three copies of the vector. Vector fragment lengths of several hundred kbp and longer represented only a small fraction of the hybridization signal.
Figure 2.

Insert DNA is noncontiguous in clones with large inserts. Genomic DNA of clones Con-610, Con-1, dSAR-g12, and dSAR-d11 was cut with MluI, NheI, and EcoRV, endonucleases not cutting the vectors. (A) Hybridization pattern of the EcoRV digest. (B) Hybridization pattern of the MluI digest. (C) Hybridization pattern of the NheI digest. For all panels, lane M is phage λ DNA 48.5-kbp ladder.
Previous work with CHO cells had shown that stable transgene arrays contained up to hundreds of vector copies (Chen and Okayama, 1988). Based on restriction digest analysis of the integrated vectors in these cell lines, these transgene arrays were thought to consist of contiguous concatemers of vector copies (Barsoum, 1990). Our Southern analysis clearly excludes a contiguous organization for vector sequences arranged into large, Mbp scale arrays. Most inserts appear to consist of short vector arrays separated by genomic DNA. Presumably, each insertion that appears as a single spot by light microscopy is the result of a complex recombination event.
DNA halo analysis of vector sequences
To characterize our cell lines further, we mapped the location of vector sequences within mitotic halos. Previously, biochemical assays were used to demonstrate association of SAR/MAR sequences with nuclear scaffolds or matrices, or, in a small number of cases, with mitotic scaffolds (Mirkovitch et al., 1988; Laemmli et al., 1992; Saitoh and Laemmli, 1994). In these experiments, no data addressed how SAR sequences were distributed within undigested halos, and localization to scaffold fractions was based on biochemical fractionation rather than microscopic visualization of spatial localization. Moreover, because the proportion of SAR sequences in the matrix versus supernatant (halo) fraction has frequently been normalized by DNA concentration rather than cell number, scaffold sequences that show substantial enrichment in the scaffold fraction may partition in absolute terms predominately to the halo fraction (Bode and Maass, 1988). Attempts to locate endogenous DNA sequences within mitotic halos have been hindered by disruption of the mitotic halo during the FISH DNA denaturation step. Here, we have used lac repressor staining, thus allowing examination of intact halos. No consistent trend was observed. Vector sequences from large chromosome transgene insertion sites all showed scaffold enrichment, independent of SAR content. Smaller transgene insertion sites showed modest or no scaffold enrichment, for the vector with SARs, with the small Con-610 insert region from a vector without SARs showing depletion from the central “scaffold region.”
Mitotic halos were prepared from isolated mitotic chromosomes using either high salt or LIS extraction. The scaffold fraction of vector DNA was defined as the high intensity DAPI staining located in the central halo area, which retained the general shape of the unextracted chromosome. The measured width of this DAPI-rich core is approximately one half to two thirds that expected for the native chromatid, and compares well with the observed width of axial condensin immunofluorescence staining observed within native chromosomes and mitotic halos (Maeshima and Laemmli, 2003). Consistent with previous papers (Bickmore and Oghene, 1996), traditional FISH conditions led to the loss of the central scaffold structure (Fig. 3 , A–C). In contrast, staining with purified lac repressor, followed by antibody detection, or direct labeling with purified GFP–dimer lac repressor fusion protein did not change the appearance of scaffold and halos. Similar results were obtained with both methods of halo production. The large insert from the dSAR-d11 cell line consistently showed the strongest concentration of lac operator staining over the chromosome scaffold (Fig. 3, J–L). The large insert in the Con-1 cell line showed strong staining appearing qualitatively uniform across the halo and scaffold regions (Fig. 3, P–R), and the large insert in the dSAR-g12 cell line showed an increased association at the scaffold edges (Fig. 3, G–I). In contrast, the insert region from Con-610 (Fig. 3, M–O) and the small inserts regions for the dSAR-d11 and dSAR-g12 cell lines (Fig. 3, D–F) instead showed a large fraction of staining extending as fibers or loops well into the halo region, without significant accumulation near the chromosome scaffold.
Figure 3.
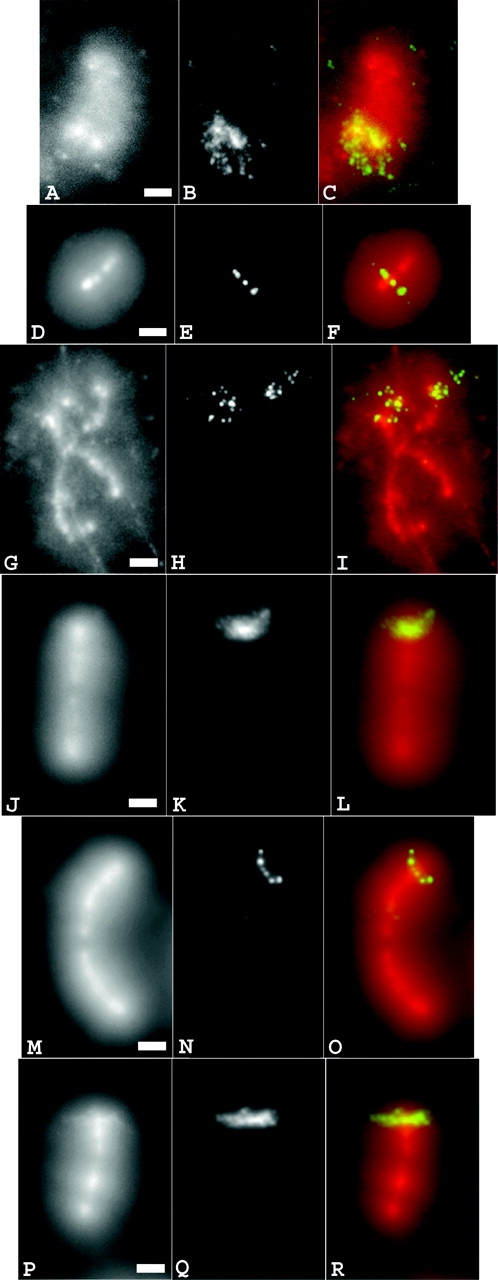
Visualization of vector sequence distribution within chromosome halos. (A–C) FISH on smaller insert of clone dSAR-g12, isolated, fixed, and extracted with 2 M salt buffer. (D–F) Isolated smaller insert of dSAR-g12 extracted with LIS buffer and stained with GFP–lacI fusion protein. (G–R) Isolated chromosomes extracted with 2 M salt buffer and stained with lac repressor. (G–I) dSAR-g12, larger insert; (J–L) dSAR-d11, larger insert; (M–O) Con-610; (P-R) Con-1. (A, D, G, J, M, and P) total DNA staining (DAPI); (B) FISH signal; (C) combined DAPI and FISH signal; (E) GFP signal; (F) combined DAPI and GFP signal; (H, K, N, and Q) lac repressor immunostaining; (I, L, O, and R) combined DAPI and immunofluorescence signal. Bars, 2 μm.
The real question in analyzing these images is whether the apparent association (or lack of association) with the scaffold region is meaningful; ideally, we need to normalize the observed DNA distribution to test whether the vector sequence is distributed between halo and scaffold quantitatively different than the total DNA. We wish to measure the fold enrichment of vector sequence relative to total DNA present in the scaffold fraction. This parallels most closely typical biochemical analysis of sequence fractionation in the scaffold fraction, where relative distribution of putative SAR sequences are measured between equal amounts of scaffold versus halo (supernatant) DNA loaded per sample. This “scaffold enrichment ratio” was measured by normalizing the operator signal by the DNA signal present in the same compartment. Specifically, we computed the ratio of the fraction of total operator signal localizing to the scaffold region divided by the fraction of DAPI staining localizing to the same scaffold region. We defined the scaffold region as corresponding to the collection of all pixels whose DAPI intensity exceeded a given threshold. Because the scaffold perceived visually did not have an exact boundary (Fig. 4, A and B), we performed these calculations for a range of DAPI intensity thresholds, plotting the ratio of fractional operator signal over fractional DAPI signal as a function of the DAPI scaffold threshold used (Fig. 4 C). These results were averaged over a number of different chromosome halos for each insert from each cell line (Fig. 4 D). Peak DAPI staining varied no more than 10–20% between different chromosomes, and a simple linear normalization of DAPI intensity was used before averaging data from different chromosome halos.
Figure 4.

Quantitation of scaffold association of vector sequence. Large inserts show enrichment of vector sequences over scaffold. (A) Extracted chromosome of clone dSAR-g12. Green, total DNA signal; red, vector DNA. (B) The size and shape of the scaffold depend on the threshold level. Colors are coordinated with the colored bar and threshold levels of C (arrowheads and arrows point to boundaries of two scaffolds defined by two intensity threshold levels shown in C). Higher threshold levels result in smaller scaffold regions. In C and D, the ordinate “scaffold enrichment ratio” represents the fraction of total operator signal localizing to the scaffold region divided by the fraction of total DNA localizing to the same region. Here, the scaffold region refers to the entire image region with intensity greater than the specific intensity threshold. Thus, the scaffold defined by arrows includes (but is larger than) the region defined by arrowheads. (C) Scaffold enrichment ratio versus threshold for the halo image shown in A. Intensity thresholds (vertical gray lines, marked by arrow or arrowhead) correspond to the actual scaffold regions (marked by arrows or arrowheads) shown in B. (D) Graphs represent the scaffold enrichment ratio versus threshold averaged over several halos; seven halos for Con-1 (con1), five halos for Con-610 (610), five and seven halos for smaller (d11-s) and larger (d11-l) inserts of clone dSAR-d11, respectively, and seven and four for smaller (g12-s) and larger (g12-l) inserts of clone dSAR-g12, respectively. Threshold values to the right of the vertical line correspond to defined scaffold regions that visually correspond to the DAPI core staining. Bar, 1 μm.
Our quantitative analysis matched very well with our qualitative assessment. The dSAR-d11 large insert, which showed the strongest scaffold association in the original images, gave an approximately ninefold ratio for the fraction of operator DNA relative to DNA associating with the scaffold region, defined by high DAPI threshold. Large inserts from the Con-1 and dSAR-g12 cell lines gave ratios of 4 and 2–4, respectively. In contrast, small inserts showed little enrichment (dSAR-d11, small insert, ratio 1–3 depending on threshold), no enrichment (dSAR-g12, small insert), or actual depletion (Con-610) at the scaffold, and appeared in the images rather uniformly distributed through the halo in a series of dots often in linear patterns. No obvious trend was apparent as a function of SAR content of the inserts.
Chromosomes have normal diameter at the insertion site with SAR sequences distributed throughout the chromosome width
At light microscopy resolution, metaphase chromosomes within intact metaphase plates had a normal chromosomal width at the insert sites (Fig. 5). Staining was performed on fixed cells not expressing lac repressor by using purified lac repressor, followed by immunostaining with anti-lac repressor antibodies (Fig. 5, columns 1–4). For cells expressing GFP–dimer lac repressor, cells were fixed and stained with DAPI (Fig. 5, column 5). In both cases, repressor staining was uniform across the entire chromosome width for large insert sites. The chromosome width at these insertion sites, as defined by repressor staining, was the same as the width throughout the entire chromosome visualized by DAPI staining. In chromosomes with small insert sites, repressor-labeled spots were smaller than a full chromosome width; however, spots in different labeled chromosomes appeared at all positions, peripheral as well as interior, across the entire chromosome width.
Figure 5.

Normal chromosome morphology over insert regions by light microscopy. In native chromosomes, the vector insert appears as a band going over the entire width of the chromosome, or as a pair of spots within normal diameter chromosome. (A) Con-1 clone showing insert across entire chromosome width. (B) Con-610 clone with single insert site. (C) dSAR-d11 clone showing two insert sites. (D) dSAR-g12 clone showing two insert sites. (column 1) Lac repressor immunostaining (green) and DAPI staining (red) of metaphase cells; (columns 2–4) metaphase chromosomes at vector integration sites: lac repressor immunostaining (green), DAPI staining (red, or grayscale). (column 5) Chromosome structure at the regions of vector insertion for cells expressing GFP–lacI–NLS fusion protein. Bars, 1 μm (white, column 1; black, columns 2–5).
The only example of altered chromosome diameter was observed for the large insert in the dSAR-d11 cell line. In a small number of anaphase cells, this large insert region showed a slight stretching, with thinning of the chromosome. As described below, metaphase chromosome diameter within intact cells was normal for this region in all cells, and none of the other insert regions in any of the cell lines showed this behavior. We interpret this phenomenon as due to a decreased stability of this chromosome region in response to tension induced during anaphase. This interpretation is strengthened by observations of similar stretching, for this one region only, in a small number of traditional mitotic spreads produced by hypotonic swelling and alcohol and acetic acid fixation. The basis for this phenomenon is now being explored in a separate work.
The above observations were based on light microscopy. To examine the labeled chromosome regions at higher resolution, cell lines not expressing lac repressor were studied with EM. Mitotic cells were collected by shakeoff after a 3-h nocodazole block and were deposited on coverslips before fixation with formaldehyde. Precise localization of the inserts was provided by lac repressor staining followed by pre-embedding immunogold staining and silver amplification of the gold-labeled secondary antibody. For all insert regions in all cell clones, chromosome diameters were similar to surrounding chromosome regions (Fig. 6). Moreover, large insertion sites showed uniform gold staining across the cross-sectional width of the chromosome.
Figure 6.

Mitotic chromosomes have normal large-scale structure at the resolution of EM. Mitotic chromosomes of all four clones showed no change in the chromosome diameter at the sites of vector insert. Arrows indicate immunogold-stained insert sites. (A) Con-610 clone. (B) Con-1 clone. (C and D) dSAR-d11 clone. C shows smaller insert, D shows larger insert. (E and F) dSAR-g12 clone. E shows the smaller insert and F shows the larger insert. Bars: 1 μm for images, 0.5 μm for inserts.
These EM results unambiguously demonstrate normal chromosome width at the chromosome insert sites. By light microscopy, though, some insert regions appear as light staining DAPI or propidium iodide bands (Fig. 5). These are comparable to other light bands found elsewhere in the mitotic chromosomes. Despite these variations in staining density, wild-type chromosomes show uniform density by EM, leading to the conclusion that these staining variations over the wild-type chromosomes reflect variations in sequence content as opposed to variations in chromosome compaction. The heavy immunogold staining over insert regions (Fig. 6) obscures visualization of the uranyl and lead chromatin staining. To verify that the lighter staining over our insert regions is also a result of differential staining, as opposed to a lower local chromosome density, we used correlative light and electron microscopy. This approach showed that by EM, the insert regions were indistinguishable from the surrounding chromosomes (Fig. 7).
Figure 7.
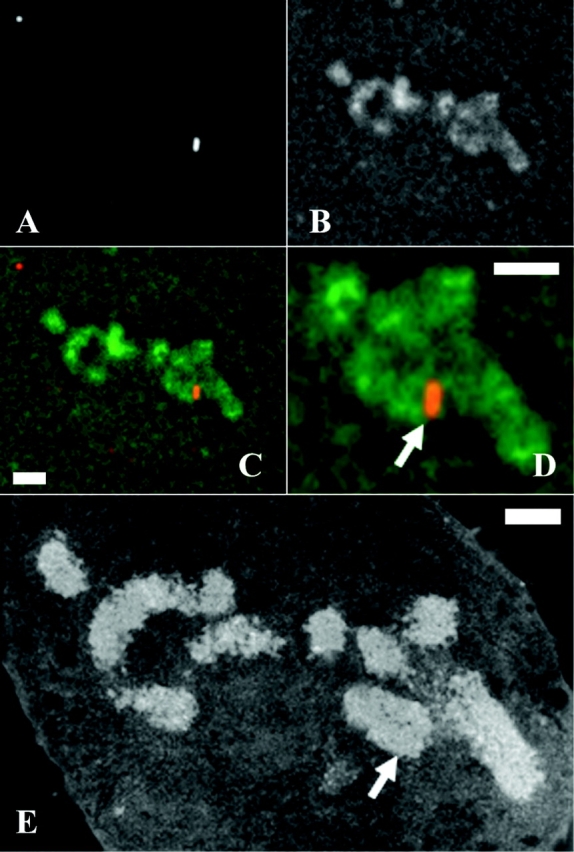
Normal chromosome morphology over insert regions—correlative light and electron microscopy. In native chromosomes of clone Con1, vector inserts appear as a band going over the entire width of the chromosome. Sections are 0.2 μm thick. (A–C) Fluorescent light microscopy of a single section; lac repressor immunostaining signal, staining for total DNA with DAPI, combined A and B, respectively. (D) Two-fold expanded image from C. (E) An EM image of the same section. Arrows indicate insert region labeled with immunofluorescence probes (D) and corresponding regions on EM sections (E). Bars, 1 μm.
Demonstration of a distinct, ∼250-nm-diam subunit within fully condensed metaphase chromosomes
Two independent cell lines have chromosome insertions that appear either as a band extending across the entire chromosome width (Fig. 5 C, columns 2 and 3; Fig. 5 D, columns 2–4) or instead, as a pair of spots, with each spot on an individual daughter chromatid spanning only a portion of the chromatid width (Fig. 5 C, columns 1 and 5; Fig. 5 D, column 1). Because of the high degree of stability of the insert number and size in these cell lines, we interpret the difference in appearance as due to a conformational difference rather than a difference in insert size. More specifically, we hypothesize that these inserts correspond in size to a coiling subunit that spans roughly one half the chromatid width. When the labeled region appears as a full band across the chromosome, we hypothesize that this is due to an orientation perpendicular to the chromosome axis but running across the two daughter chromatids. Instead, where the labeled region appears as an individual spot within each daughter chromatid, we hypothesize this is due to an orientation parallel to the optical axis.
Our lac repressor immunogold staining allows ultrastructural analysis of specific chromosome regions within unextracted, metaphase chromosomes. Serial thin sections from dSAR-d11 cells support this “half band, variable orientation” hypothesis (Fig. 8). Both types of orientations, inferred from the light microscopy results, were observed. In cases where a full-width immunogold-labeled band was observed spanning the entire chromatid width, the major volume of stained region was only observed in several (4–5) of the nominally 40-nm thick serial sections. The remaining serial sections demonstrated that the stained region only occupied approximately one half the chromatid diameter. In cases where a single spot per chromatid was observed, the labeled region occupied only approximately one half the chromatid width, but appeared in roughly twice the number of adjacent sections (7–10). Fig. 8 (I–L) shows an example with these two different orientations on the two sister chromatids, where the band-like staining (Fig. 8 I) appears in four sections and the spot-like staining (Fig. 8, J and K) appears in 10 sections. Note that the chromatid diameter is ∼500 nm. The length of the labeled band, corresponding to tangential sections, is slightly less than the chromatid diameter, typically in the 400-nm range. The width is in the 200–300-nm range. Therefore, the band corresponds to a feature with a roughly 2:1 aspect ratio. Therefore, these section numbers agree well with the expected size of these labeled regions, taking into account as well that the chromosomes are always going to be at some angle to the section so that the sections are never exactly transverse or perpendicular to the band or spot.
Figure 8.
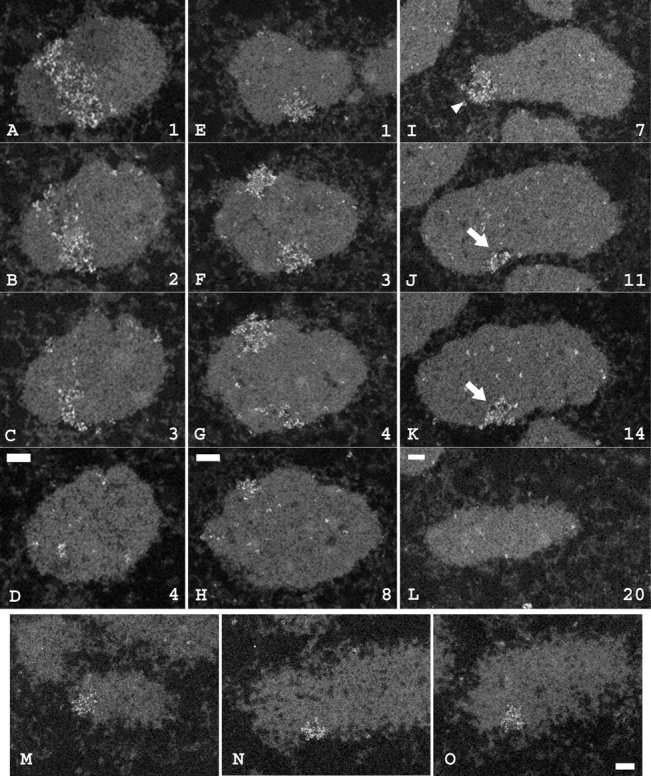
A distinct, ∼250-nm-diam coiling subunit within fully condensed metaphase chromosomes. A–D, E–H, and I–L show regions from individual serial sections from three different clone dSAR-d11 mitotic cells collected after nocodazole treatment, fixation, and immunogold labeling. Numbers represent positions of 40-nm thick sections in the original serial section stacks. Arrows and arrowhead label the two different labeled areas on the chromosome shown in I–L and displayed in three dimensions in Fig. 9, B and C. M–O show three independent examples of the small insert region from clone dSAR-g12. Bars: 0.2 μm for A–L, 0.3 μm for M–O.
More detailed ultrastructural analysis further supports this interpretation. Three-dimensional (3-D) analysis of this reconstruction, showing either different cross sections through the reconstruction (Fig. 9 B) or solid model displays from different chromosome orientations (Fig. 9 C), demonstrates that both the original band and spot orientations actually have similar conformations. In all examples, the width of the labeled region ranges from 200–300 nm, with the average width roughly 250 nm. Significantly, the comparable small insert from the dSAR-g12 cell line (Fig. 5 D and Fig. 6 F), also spanned approximately one half the chromatid width, and serial thin sections show the same ∼250-nm width (Fig. 8, M–O) for the insert region. Previous work showed a similar diameter for early prophase chromatids in CHO cells (unpublished data).
Figure 9.

3-D visualization of an ∼250-nm wide subunit spanning a fraction of the chromatid width supports a hierarchical folding model. (A) Dependence of labeled band width versus insert size reveals folding subunits. In a simple radial loop model (top), as the insert size increases, the labeled region spans an increasing fraction of the chromatid cross section, with the minimal width of a labeled band corresponding roughly to the diameter of a 30-nm chromatin fiber loop. With a successive coiling model, as the insert region increases in size, the labeled region spans an increasing fraction of the chromatid cross section, but the width of this labeled region does not increase until it spans a full chromatid cross section (bottom). The width of the minimal labeled region corresponds to the diameter of the folding subunit, significantly larger than a 30-nm chromatin loop. (B) Reconstructed orthogonal cross sections of EM serial section data built with NewVision. Arrows and arrowhead show the same nanogold-labeled areas as in Fig. 8, I–L. The spot-like labeled region appears as a labeled band extending across the chromatid in the orthogonal view. Thick red, green, and blue lines in images a, b, and c are, respectively, X, Y, and Z axes of a left orthogonal system with origin inside the nanogold-labeled insert. (C) Solid model display for same chromosome region shown in Fig. 8, I–L and Fig. 9 B reveals similar appearance of labeled regions on both chromatids. In the original serial sections, one region appeared as a band (Fig. 8 I, arrowhead) and one as a spot (Fig. 8, J and K, arrows). In the orthogonal views in B and in the solid model, both regions appear as ∼250–300-nm wide segments spanning only a fraction of the chromatid cross section.
As diagrammed in Fig. 9 A, the size of labeled chromosome regions smaller than a full chromatid cross section provides a simple yet powerful probe of mitotic chromosome architecture. For instance, in a simple radial loop model with 30-nm chromatin loops arranged radially around the chromatid axis (Pienta and Coffey, 1984), the minimal width of a labeled band corresponds to the 30-nm chromatin fiber diameter (Fig. 9 A, top row). This model assumes a helical arrangement of loops. One could imagine that these loops of 30-nm fibers might not be so precisely arranged, with loops forming clumps before extending further across the chromatid, but this would not produce bands of defined width with sharply demarcated borders. In contrast, in a helical hierarchical model the minimal width of a labeled band in a chromatid will be determined by the diameter of the fiber that, through an extra level of folding, gives rise to the metaphase chromatid (Fig. 9 A, bottom row). As our labeled chromosome regions increase in size, we expect to see the labeled region change progressively from a small spot, to larger domains within the chromatid, to a labeled region that spans the full chromatid width. In any hierarchical folding model of chromosome architecture, the characteristic width of the minimal size band that spans the full chromatid should correspond to a fundamental large-scale chromatin-folding subunit. Moreover, if the size of the insert running through the entire chromatid width is reduced twofold, one should expect to see a “half-band” of the same diameter (Fig. 9 A, bottom row).
Significantly, both half-band inserts from the independent dSAR-d11 and g12 cell lines show the same characteristic ∼250-nm width, with examples of banded staining with sharply defined, parallel edges (Fig. 8 A), providing strong evidence for folding of a 250-nm-diam chromatid subunit to form the metaphase chromatid.
Discussion
Here, we describe our initial results aimed at addressing models of chromosome structure by manipulating sequence composition of chromosome regions. We demonstrate that chromosome regions containing ∼60–70% vector sequence content condense into metaphase chromosome regions of normal diameter and apparently normal higher order structure. This was true even when the density of SAR sequences within these regions was increased by adding two SAR sequences from the human β–interferon gene to the vector transgene. No differential targeting of these SAR sequences with respect to the chromatid axis was observed. Selective visualization of labeled chromosome regions spanning approximately one half the chromatid width demonstrated the existence of an ∼250-nm-diam coiling subunit within native metaphase chromosomes.
Normal chromosome condensation with abnormal DNA sequence composition
Despite estimated vector composition ranging from 60–70%, we observed normal chromosome diameters for all of our clones. This argues for a relative independence of mitotic chromosome condensation to underlying sequence. In one of our clones, we obtained vector concatemers exceeding 200 kb in length; larger than the average loop size in radial loop models, yet normal chromosome diameter was again observed. Our results parallel similar findings in the literature(Monroe et al., 1992; McNally et al., 2000). In insect cells, large integrated arrays of vector sequence are observed after transfection, with stable inserts approaching the size of chromosome arms (Monroe et al., 1992), yet normal chromosome widths are observed in mitotic spreads. In mammalian cells, integration sites containing ∼2 Mbp vector concatemers again showed normal chromosome diameters in mitotic spreads when hybridized to vector sequence (McNally et al., 2000). We cannot exclude that in all of these examples, nonvector DNA integrated between the vector repeats is present, which maintains normal chromosome architecture. However our results imply that there is certainly a high degree of tolerance for large variations in DNA sequence and spacing.
In contrast, we have observed highly reproducible positioning of specific sequences with respect to the chromosome axis in a gene-amplified chromosome region (Dietzel and Belmont, 2001). Our observation of normal chromosome condensation with abnormal sequence composition does not imply that wild-type chromosomes necessarily show sequence-independent folding and positioning. Wild-type chromosomes might show highly reproducible folding and positioning of DNA sequences with this reproducibility serving other or additional purposes than chromosome condensation.
Hierarchical versus radial loop models of mitotic chromosome structure
Our work demonstrating the distribution of the 5′ and 3′ human β–interferon gene SARs throughout the normal chromatid diameter is inconsistent with simple radial loop models in which most SAR sequences are targeted to an axial chromosome structure. Moreover, our results raise serious questions concerning the original experimental basis for radial loop models. Specifically, our results show that the disposition of vector sequences within classic chromosome halo preparations do not necessarily parallel their distribution within unextracted chromosomes. For instance, the large insert within the dSAR-d11 cell line shows significant enrichment of vector sequences in halo preparations, yet no axial vector distributional bias is observed within native chromosomes. Given these observations, one necessarily has to reexamine the functional significance that has been assigned to the observed preferred distribution of certain sequences within mitotic halos versus scaffolds with respect to the structure of native chromosomes.
If chromosomes compact through a radial loop organization, then the concept of a strict, one to one anchoring of SAR sequences to a well-defined, central axis needs modification based on our results. One might imagine, for example, that only a subset of specific SAR sequences functions as mitotic loop attachment sites. SAR sequences might also be facultative in the sense that only particular copies of SAR sequences would function as loop attachment sites, depending on spacing of nearby SAR sequences, or even that non-SAR sequences could function similarly to SAR sequences when no nearby SARs are present. In fact, biochemical approaches showing enrichment of SAR sequences in scaffold fractions typically express the relative enrichment based on the enrichment of SAR sequence per DNA concentration. This is similar to the normalization we used for our cytological approach in Fig. 4 D. However, if the absolute amount of scaffold DNA is small, even in cases where significant SAR enrichment in the scaffold fraction is present, the absolute amount of SAR sequence might still be higher in the halo fraction. Along these lines, SAR sequences embedded within repetitive vector transgene copies might not function appropriately as SAR sequences, perhaps failing to bind properly to chromosomal proteins required for normal chromosomal positioning and function. In large repetitive arrays lacking SAR sequences, one would have to imagine that other sequences assume the function of SARs.
Helical coil/radial loop models, in which a 200–300-nm prophase chromatid, itself organized as a radial loop structure, is coiled into a metaphase chromatid are derived from observations of human chromosomes isolated from special cell lines or after prolonged inhibition of microtubule polymerization (Rattner and Lin, 1985; Boy de la Tour and Laemmli, 1988). Our results suggesting hierarchical folding involving an ∼250-nm subunit are consistent with either such a helical/radial loop or other hierarchical models such as successive helical folding or folded chromonema models.
However, a recent paper suggested that the small percentage of chromosomes showing an obvious helical organization by light microscopy (∼1%) in these previous works may be related to overcondensation of chromosomes observed after a prolonged colcemid-induced mitotic block (Maeshima and Laemmli, 2003). Metaphase chromosomes from cells not exposed to colcemid showed a smaller chromosome diameter, the absence of obvious daughter chromatid coiling, and a centralized core of condensin and topoisomerase II staining, roughly 0.2–0.3 μm in diameter. This observation of an axial condensin core appears to contradict the helical coil/radial loop model, which would instead have predicted a helical coil to the scaffold proteins with a radius equal to one half the chromatid radius and equal to the predicted radial loop size.
Implications for coiling models of mitotic chromosome structure
A presumably helical folding of an ∼200–300-nm structure to form the final metaphase chromosome has been proposed in several models of chromosomes. Support for this model has come from visualization of surface features of metaphase chromosomes by scanning EM, visualization of apparent gyres by light microscopy of isolated metaphase chromosomes after treatment with specific buffers, isolation of chromosomes from cell lines apparently defective in normal chromosome condensation, or by banding patterns seen in TEM thin sections or suggestions of gyres in whole-mount metaphase chromosomes visualized as stereo-pairs (Belmont, 1997). However, experimental support for this model has been inconclusive, either because of the harsh treatments to which the chromosomes had been exposed to in these experiments and/or due to limitations in the visualization methods used. For example, scanning EM of metaphase chromosomes prepared using normal isolation buffers reveals a smooth surface. Instead, evidence for coiling of 200–300-nm gyres comes from a segmented chromosomal appearance observed after G-banding procedures, involving trypsinization and methanol/acetic acid fixation. Similarly, light microscopy methods have relied on harsh buffer conditions that alter chromosome structure to produce obvious zigzag or coiled structures (Ohnuki, 1965), or else treatment with polyglutamic acid that swells chromosomes through extraction of histone H1, producing coiled appearances in ∼1% of isolated chromosomes from cells blocked in mitosis and possibly overcondensed (see above).
Here, we have demonstrated the existence of an ∼250-nm-diam substructure within metaphase chromosomes visualized directly within mitotic cells exposed for several seconds to a chromosome isolation buffer, chosen for its preservation of chromosome morphology close to that observed within living cells (Belmont et al., 1989), followed by formaldehyde fixation. The diameter of this substructural unit is similar to the diameter of early prophase CHO chromosomes observed previously in serial section reconstructions (unpublished data). These observations are consistent with a coiling model in which early prophase chromatids coil to form thicker prometaphase and metaphase chromosomes.
Reproducibility of daughter chromatid folding
Reproducible chromosome banding patterns indicate folding reproducibility between daughter chromatids, at least at the level of resolution probed by these works. A fundamental question with regard to models of chromosome compaction is to what level of resolution is this reproducibility between daughter chromatid folding maintained. Our ability to use immunogold staining on relatively intact chromosomes to examine specific chromosome regions smaller than chromosome bands sheds some light on this question of daughter chromatid reproducibility. EM immunostaining results specifically showing close longitudinal alignment of band-like staining across both daughter chromatids indicate a high degree of alignment between daughter chromatids. The boundaries between labeled and unlabeled regions are aligned to an estimated 20–40 nm between daughter chromatids. Yet, the examples of insertion sites spanning a “half chromosome band” are intriguing, in that some examples show a transverse band extending across both chromatids, whereas other examples show a band-like staining on one chromatid and a spot-like staining on the opposing daughter chromatid, or two spots on opposite sides of the daughter chromatids, away from their region of contact. These results not only show a variation in folding between different chromosomes, but even within daughter chromatids of the same chromosome. The implication of these observations is that either the packing of sequences within daughter chromatids is not reproducible at this sub-band level of resolution, which might be difficult to reconcile with the observed precision in longitudinal alignment of the labeled bands, or the packing is reproducible between daughter chromatids, but that the daughter chromatids are not rotationally fixed. More detailed analysis of the reproducibility of daughter chromatid alignment is now in progress.
Future directions
Previous approaches to understanding mitotic chromosome organization have been largely descriptive and correlative, examining the structure of normal wild-type chromosomes. Although models were presented that invoked specific looped domains organized by specific sequences, no direct experimental testing of these models was conducted. Our experiments establish an experimental paradigm in which sequence composition of chromosome regions are directly manipulated to test sequence requirements for chromosome compaction and stability, and in which specific chromosome sequences can be mapped at high spatial resolution within native mitotic chromosomes. More elaborate DNA construction methods exploiting BAC and/or YAC cloning vectors should allow further dissection of folding motifs underlying mitotic chromosome organization.
Materials and methods
Plasmids
Plasmid vectors with lac operator repeats were based on the modified pSV2-DHFR vector (Robinett et al., 1996). Two oligonucleotides with sites for rare cutting endonucleases were introduced at the SalI and PvuII sites to yield pSV2-dao2. 2.2-kb EcoRI-EcoRI and 4.3-kb EcoRI-HindIII fragments containing 5′ and 3′ SAR sequences from the human β–interferon gene locus (provided by Dr. Juergen Bode, German Research Center for Biotechnology, Braunschweig, Germany; Bode and Maass, 1988) were respectively cloned into the PvuII and SalI sites of pSVII-dao2 (Fig. 1 A). The 256-copy lac operator array was added at the last step of cloning using the SalI and KpnI sites (Robinett et al., 1996). The bacterial EGFP–lacI–NLS expression vector was created by ligating the XhoI-DraI restriction fragment from p3′SS–EGFP–dimer lacI (Tumbar et al., 1999) carrying the fusion protein sequence into the modified pET28b plasmid (New England Biolabs, Inc.). Modification of the original pET28b consisted of linearization with NotI, filling “sticky” ends with Klenow polymerase, and SalI digest.
Cell culture
CHO cells with a double deletion in the DHFR locus (DG44 CHO cell line; Urlaub et al., 1986) were grown in Ham's F12 medium (GIBCO BRL) supplemented with 10% defined FBS (HyClone). Ham's F12 medium, lacking thymidine and hypoxanthine, with 10% dialyzed FBS (HyClone) was used for selection and passaging of stably transfected cells. Calcium phosphate–mediated transfection of cells with plasmid DNA was based on protocols described elsewhere (Chen and Okayama, 1988). For in vivo expression of EGFP–lacI–NLS fusion protein, isolated clones were transformed with p3′SS-EGFP–dimer lacI–NLS (Tumbar et al., 1999) using FuGENE™ reagent (Boehringer). Stable transformants were selected with hygromycin. Cell sorting according to DHFR expression levels was done after growing cells for 8 h in regular Ham's F12 medium with 10% FBS and 20 μM fluorescein-labeled MTX (Molecular Probes, Inc.). 100 μM glycine, 30 μM hypoxanthine, and 30 μM thymine were added to relieve toxic effects of fluorescein-labeled MTX (Sherwood et al., 1990). The copy number was determined by quantitative Southern blot analysis of digested genomic DNA, with transfection plasmid as a control.
Southern blot analysis
High mol wt DNA was isolated from a 5 × 105 cells/ml cell suspension in agarose blocks by a conventional method described earlier (Riethman et al., 1997). DNA probe synthesis and detection were done with the Renaissance® random primer fluorescein labeling kit and CDP-Star kit, respectively (NEN Life Science Products).
In situ hybridization
Labeling of probe DNA was done with ChromaTide™ Alexa® 594–5-dUTP from Molecular Probes, Inc., using nick translation. In situ hybridization on halos was done as described elsewhere (Robinett et al., 1996) with minor changes. DNA was denatured at 70°C for 5 min. Hybridization was done in 60% formamide, 2× SSC, 5% dextran sulfate, pH 7.0, with 2 ng/μl of probe DNA and 1 μg/μl of salmon sperm DNA (Sigma-Aldrich) overnight at 37°C. Coverslips were washed 30 min in 60% formamide for 30 min in 2× SSC at 37°C, and finally for 5 min in PBS with extra 5 mM MgCl2 and 0.1 mM EDTA.
Immunostaining
Lac repressor staining of fixed cells was performed as described elsewhere (Li et al., 1998). For 1 l of PBS, 8.00 g NaCl, 2.16 g Na2HPO4(7H2O), 0.20 g KCl, and 0.20 g KH2PO4 are dissolved in diionized water, pH 7.4. DNA was stained either with 0.2 μg/ml DAPI or with 0.5 μg/ml propidium iodide in PBS with extra 5 mM MgCl2 and 0.1 mM EDTA.
Chromosome and nuclear halo preparation
Isolation of mitotic chromosomes was performed according to a previously published protocol (Bickmore and Oghene, 1996). Chromosomes were extracted with high salt buffer (10 mM Tris-HCl, 10 mM EDTA, 0.1% NP-40, 1 μg/ml PMSF, 1 μg/ml aprotonin [Sigma-Aldrich], and 2 M NaCl, pH 8.0) for 5 min at RT with one change of extraction buffer. Alternatively, chromosomes and nuclei were extracted with LIS-containing buffer (Craig et al., 1997; 25 mM LIS lithium salt, 5 mM Hepes/NaOH, 2 mM KCl, 2 mM EDTA/KOH, 0.25 mM spermidine, and 0.1% digitonin, pH 7.4) for 10 min at RT. After extraction, halos were fixed in 1.6% formaldehyde (Polyscience) in PBS with extra 5 mM MgCl2 and 0.1 mM EDTA for 10 min at RT for further staining.
Isolation of the EGFP–lacI–NLS fusion protein
GFP–lacI–NLS fusion protein was expressed in BL21 Escherichia coli cells of the BL21 transformed with the pET28b expression vector. Recombinant protein was isolated with TALON™ resin column (CLONTECH Laboratories, Inc.) as described earlier (Tumbar et al., 1999).
Microscopy and processing and analysis of images
3-D optical sections were collected with an inverted fluorescence microscope (IMT-2; Olympus) and deconvolved as described previously (Hiraoka et al., 1991). Analysis of images was done with NewVision (Pixton and Belmont, 1996), and quantitative analysis of images was done with MATLAB® 6 (The MathWorks, Inc.). EM images were normalized for intensity variation, aligned by cross-correlation, and interpolated to compensate for the larger pixel spacing in the z direction. The interpolated image stack was smoothed twice with a 3 × 3 × 3 box filter and reduced 4× in size in the x and y dimensions. 3-D models were built using MATLAB® 6.
Immunostaining for EM
Staining of mitotic cells for electron and correlative microscopy was done as described earlier (Li et al., 1998; Tumbar et al., 1999). Cells were silver enhanced (Burry et al., 1992) and prepared for EM by dehydration, embedding, and sectioning. Electron micrographs were digitized for analysis and storage, or images were collected with a CCD camera (model 2kx2k; Tietz).
Acknowledgments
We thank Dr. Juergen Bode for generous advice and for DNA constructs containing the 5′ and 3′ human β–interferon SAR DNA.
This work was supported by National Institutes of Health grants GM42516 and GM 58460 to A.S. Belmont.
Y. Wang's present address is Kraft Foods, Inc., 1 Kraft Court, Glenview, IL 60025.
Footnotes
Abbreviations used in this paper: DHFR, dihydrofolate reductase; dSAR, double SAR construct; MAR, matrix-associated region; MTX, methotrexate; SAR, scaffold-associated region.
References
- Barsoum, J. 1990. Introduction of stable high-copy-number DNA into Chinese hamster ovary cells by electroporation. DNA Cell Biol. 9:293–300. [DOI] [PubMed] [Google Scholar]
- Baumgartner, M., B. Dutrillaux, N. Lemieux, A. Lilienbaum, D. Paulin, and E. Viegas-Pequignot. 1991. Genes occupy a fixed and symmetrical position on sister chromatids. Cell. 64:761–766. [DOI] [PubMed] [Google Scholar]
- Belmont, A.S. 1997. Large-scale chromatin organization. Genome Structure and Function. C. Nicolini, editor. Kluwer Academic Publishers, Dordrecht, Netherlands. 261–276.
- Belmont, A.S., and K. Bruce. 1994. Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J. Cell Biol. 127:287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, A.S., M.B. Braunfeld, J.W. Sedat, and D.A. Agard. 1989. Large-scale chromatin structural domains within mitotic and interphase chromosomes in vivo and in vitro. Chromosoma. 98:129–143. [DOI] [PubMed] [Google Scholar]
- Bickmore, W.A., and K. Oghene. 1996. Visualizing the spatial relationships between defined DNA sequences and the axial region of extracted metaphase chromosomes. Cell. 84:95–104. [DOI] [PubMed] [Google Scholar]
- Bode, J., and K. Maass. 1988. Chromatin domain surrounding the human interferon-beta gene as defined by scaffold-attached regions. Biochemistry. 27:4706–4711. [DOI] [PubMed] [Google Scholar]
- Boy de la Tour, E., and U.K. Laemmli. 1988. The metaphase scaffold is helically folded: sister chromatids have predominantly opposite helical handedness. Cell. 55:937–944. [DOI] [PubMed] [Google Scholar]
- Burry, R.W., D.D. Vandre, and D.M. Hayes. 1992. Silver enhancement of gold antibody probes in pre-embedding electron microscopic immunocytochemistry. J. Histochem. Cytochem. 40:1849–1856. [DOI] [PubMed] [Google Scholar]
- Chen, C.A., and H. Okayama. 1988. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 6:632–638. [PubMed] [Google Scholar]
- Craig, J.M., S. Boyle, P. Perry, and W.A. Bickmore. 1997. Scaffold attachments within the human genome. J. Cell Sci. 110:2673–2682. [DOI] [PubMed] [Google Scholar]
- Dietzel, S., and A.S. Belmont. 2001. Reproducible but dynamic positioning of DNA in chromosomes during mitosis. Nat. Cell Biol. 3:767–770. [DOI] [PubMed] [Google Scholar]
- Hiraoka, Y., J.R. Swedlow, M.R. Paddy, D.A. Agard, and J.W. Sedat. 1991. Three-dimensional multiple-wavelength fluorescence microscopy for the structural analysis of biological phenomena. Semin. Cell Biol. 2:153–165. [PubMed] [Google Scholar]
- Kaufman, R.J., J.R. Bertino, and R.T. Schimke. 1978. Quantitation of dihydrofolate reductase in individual parental and methotrexate-resistant murine cells. Use of a fluorescence activated cell sorter. J. Biol. Chem. 253:5852–5860. [PubMed] [Google Scholar]
- Klehr, D., K. Maass, and J. Bode. 1991. Scaffold-attached regions from the human interferon beta domain can be used to enhance the stable expression of genes under the control of various promoters. Biochemistry. 30:1264–1270. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K., E. Kas, L. Poljak, and Y. Adachi. 1992. Scaffold-associated regions: cis-acting determinants of chromatin structural loops and functional domains. Curr. Opin. Genet. Dev. 2:275–285. [DOI] [PubMed] [Google Scholar]
- Li, G., G. Sudlow, and A.S. Belmont. 1998. Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: precise choreography of condensation/decondensation and nuclear positioning. J. Cell Biol. 140:975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima, K., and U.K. Laemmli. 2003. A two-step scaffolding model for mitotic chromosome assembly. Dev. Cell. 4:467–480. [DOI] [PubMed] [Google Scholar]
- McManus, J., P. Perry, A.T. Sumner, D.M. Wright, E.J. Thomson, R.C. Allshire, N.D. Hastie, and W.A. Bickmore. 1994. Unusual chromosome structure of fission yeast DNA in mouse cells. J. Cell Sci. 107:469–486. [DOI] [PubMed] [Google Scholar]
- McNally, J.G., W.G. Muller, D. Walker, R. Wolford, and G.L. Hager. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 287:1262–1265. [DOI] [PubMed] [Google Scholar]
- Mirkovitch, J., S.M. Gasser, and U.K. Laemmli. 1988. Scaffold attachment of DNA loops in metaphase chromosomes. J. Mol. Biol. 200:101–109. [DOI] [PubMed] [Google Scholar]
- Monroe, T.J., M.C. Muhlmann-Diaz, M.J. Kovach, J.O. Carlson, J.S. Bedford, and B.J. Beaty. 1992. Stable transformation of a mosquito cell line results in extraordinarily high copy numbers of the plasmid. Proc. Natl. Acad. Sci. USA. 89:5725–5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuki, Y. 1965. Demonstration of the spiral structure of human chromosomes. Nature. 208:916–917. [DOI] [PubMed] [Google Scholar]
- Pienta, K.J., and D.S. Coffey. 1984. A structural analysis of the role of the nuclear matrix and DNA loops in the organization of the nucleus and chromosome. J. Cell Sci. Suppl. 1:123–135. [DOI] [PubMed] [Google Scholar]
- Pixton, J.L., and A.S. Belmont. 1996. NewVision: a program for interactive navigation and analysis of multiple 3-D data sets using coordinated virtual cameras. J. Struct. Biol. 116:77–85. [DOI] [PubMed] [Google Scholar]
- Rattner, J.B., and C.C. Lin. 1985. Radial loops and helical coils coexist in metaphase chromosomes. Cell. 42:291–296. [DOI] [PubMed] [Google Scholar]
- Riethman, H., B. Birren, and A Gnirke. 1997. Preparation, manipulation and mapping of HMW DNA. Genome Analysis. Vol. 1. B. Birren, E.D. Green, S. Klapholz, R.M. Myers, and J. Roskams, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 103–108.
- Razin, S.V. 1996. Functional architecture of chromosomal DNA domains. Crit. Rev. Eukaryot. Gene Expr. 6:247–269. [DOI] [PubMed] [Google Scholar]
- Robinett, C.C., A. Straight, G. Li, C. Willhelm, G. Sudlow, A. Murray, and A.S. Belmont. 1996. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 135:1685–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh, Y., and U.K. Laemmli. 1994. Metaphase chromosome structure: bands arise from a differential folding path of the highly AT-rich scaffold. Cell. 76:609–622. [DOI] [PubMed] [Google Scholar]
- Sedat, J., and L. Manuelidis. 1978. A direct approach to the structure of eukaryotic chromosomes. Cold Spring Harb. Symp. Quant. Biol. 42:331–350. [DOI] [PubMed] [Google Scholar]
- Sherwood, S.W., Y.G. Assaraf, A. Molina, and R.T. Schimke. 1990. Flow cytometric characterization of antifolate resistance in cultured mammalian cells using fluoresceinated methotrexate and daunorubicin. Cancer Res. 50:4946–4950. [PubMed] [Google Scholar]
- Tumbar, T., G. Sudlow, and A.S. Belmont. 1999. Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J. Cell Biol. 145:1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlaub, G., P.J. Mitchell, E. Kas, L.A. Chasin, V.L. Funanage, T.T. Myoda, and J. Hamlin. 1986. Effect of gamma rays at the dihydrofolate reductase locus: deletions and inversions. Somat. Cell Mol. Genet. 12:555–566. [DOI] [PubMed] [Google Scholar]