

Abstract
Scaffold, anchoring, and adaptor proteins coordinate the assembly and localization of signaling complexes providing efficiency and specificity in signal transduction. The PKA, PKC, and protein phosphatase-2B/calcineurin (CaN) scaffold protein A–kinase anchoring protein (AKAP) 79 is localized to excitatory neuronal synapses where it is recruited to glutamate receptors by interactions with membrane-associated guanylate kinase (MAGUK) scaffold proteins. Anchored PKA and CaN in these complexes could have important functions in regulating glutamate receptors in synaptic plasticity. However, direct evidence for the assembly of complexes containing PKA, CaN, AKAP79, and MAGUKs in intact cells has not been available. In this report, we use immunofluorescence and fluorescence resonance energy transfer (FRET) microscopy to demonstrate membrane cytoskeleton–localized assembly of this complex. Using FRET, we directly observed binding of CaN catalytic A subunit (CaNA) and PKA-RII subunits to membrane-targeted AKAP79. We also detected FRET between CaNA and PKA-RII bound simultaneously to AKAP79 within 50 Å of each other, thus providing the first direct evidence of a ternary kinase–scaffold–phosphatase complex in living cells. This finding of AKAP-mediated PKA and CaN colocalization on a nanometer scale gives new appreciation to the level of compartmentalized signal transduction possible within scaffolds. Finally, we demonstrated AKAP79-regulated membrane localization of the MAGUK synapse-associated protein 97 (SAP97), suggesting that AKAP79 functions to organize even larger signaling complexes.
Keywords: calcineurin; cyclic AMP–dependent protein kinases; glutamate; microscopy; fluorescence
Introduction
Recent research advances have shown that molecular associations regulating subcellular targeting of signaling proteins are crucial for efficient and specific signal transduction (Smith and Scott, 2002). At the postsynaptic membrane of neuronal excitatory synapses, postsynaptic density-95, discs large, zona occludens-1 (PDZ)* domain–containing scaffold proteins combine with cytoskeletal proteins in the postsynaptic density (PSD) (Sheng and Scala, 2001). Organized around these PSD scaffolds are membrane receptors including N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) ionotropic glutamate receptors as well as intracellular signaling proteins. Importantly, it is believed that regulation of PSD scaffolding is interrelated with signaling by glutamate receptors in long-term potentiation (LTP) and long-term depression (LTD) plasticity linked to learning and memory (Sheng and Lee, 2001; Tomita et al., 2001).
One prototypic signaling scaffold that might have important functions in synaptic plasticity is A-kinase anchoring protein (AKAP) 79 (humanAKAP79/ratAKAP150) (Bregman et al., 1989; Carr et al., 1992). AKAP79 is a membrane/cortical cytoskeleton–targeted anchoring protein that binds the cAMP-dependent protein kinase (PKA) regulatory (R) subunits, PKC, and protein phosphatase 2B/calcineurin catalytic A subunit (CaNA) (Coghlan et al., 1995; Klauck et al., 1996; Dell'Acqua et al., 1998; Gomez et al., 2002). In hippocampal neurons, AKAP79, PKA, and CaN are localized on postsynaptic dendritic spines with F-actin and PSD-95 family membrane-associated guanylate kinase (MAGUK) PDZ scaffolds (Gomez et al., 2002). AKAP79 is recruited to NMDA and AMPA glutamate receptors at synapses through binding the MAGUKs PSD-95 and synapse associated protein 97 (SAP97), respectively, and anchored PKA and CaN in these complexes participate in the regulation of AMPA receptor phosphorylation (Rosenmund et al., 1994; Colledge et al., 2000; Dell'Acqua et al., 2002; Tavalin et al., 2002). Importantly, PKA, CaN, and SAP97 have also been implicated in the regulation of AMPA receptor activity in LTP and LTD (Hayashi et al., 2000; Lee et al., 2000; Carroll et al., 2001). AKAP79 is targeted to the membrane cytoskeleton through an NH2-terminal basic domain that binds phosphatidylinositol-4,5-bisphosphate and F-actin (Dell'Acqua et al., 1998; Gomez et al., 2002). We have recently shown that AKAP79–PKA synaptic targeting and association with MAGUKs depends on F-actin and is negatively regulated by NMDA receptor CaN signaling pathways that also control AMPA receptors in LTD (Beattie et al., 2000; Ehlers, 2000; Lin et al., 2000; Gomez et al., 2002). Thus, assembly and disassembly of AKAP79 complexes containing PKA, CaN, and MAGUKs may be central to synaptic plasticity.
Unfortunately, the biochemical precipitation techniques used to study the assembly of protein complexes are limited by steric hindrance from bulky fusion proteins or antibodies, use of detergents to solubilize complexes from cells, and poor yield of proteins precipitating through intermediate proteins. For instance, precipitation studies have shown limited amounts of CaN and PKA associated together with AKAP79 and have been unable to detect CaN and MAGUKs coprecipitating through the AKAP (Coghlan et al., 1995; Colledge et al., 2000). Even when biochemical methods work ideally, they still do not provide information about the existence or localization of the complex in intact living cells. Thus, precipitation methods cannot provide a complete, accurate depiction of the in vivo condition. As a first step in visualizing AKAP79 function in vivo, we have used microscopy techniques that allow observation of protein binding in living cells to provide direct evidence for membrane cytoskeleton–localized molecular assembly of the PKA–AKAP79–CaN ternary complex in association with SAP97. In this work, we have employed both immunofluorescence and fluorescence resonance energy transfer (FRET) microscopy using proteins tagged with CFP and YFP variants of GFP to study signaling complex reconstitution in a model cell line.
Results
Using GFP as an expression tag to study protein targeting has become an indispensable tool of modern cell biology (Tsien, 1998). CFP and YFP variants of GFP have been developed that can be imaged separately in the same cell. In addition, energy can be transferred from an excited CFP donor to YFP acceptor through a nonradiative dipole–dipole interaction called FRET. There is a strong inverse relationship between FRET and chromophore separation such that CFP–YFP FRET (CYFRET) occurs only if the two proteins are in very close proximity (<50 Å) (Fig. 1, A and B). Thus, CYFRET can be used to measure protein binding in living cells (Tsien, 1998; Miyawaki and Tsien, 2000). However, the majority of studies in living cells have focused on the regulation of intramolecular FRET for CFP–YFP indicator proteins reported as a ratio of YFP to CFP emission during CFP excitation. This ratio is a very sensitive indicator of changes in FRET because acceptor and donor emission have an inverse relationship due to CFP quenching upon FRET with YFP (Miyawaki and Tsien, 2000; Nagai et al., 2000; Zhang et al., 2001). However, CYFRET ratio images are not useful for protein–protein interactions in cells with uncertain stoichiometries due to variable CFP and YFP expression in transient transfections. Recently, a technique using image subtraction, called micro-FRET, that is not subject to these limitations has been shown to produce high-resolution sensitized FRET images showing protein complexes in live cells (Gordon et al., 1998; Sorkin et al., 2000). Thus, we have chosen to use micro-FRET to study PKA and CaN anchoring to AKAP79 in live cells. To independently confirm FRET measured by this method, we have also performed parallel YFP acceptor photobleaching measurement of FRET CFP donor quenching in fixed cells (Miyawaki and Tsien, 2000).
Figure 1.
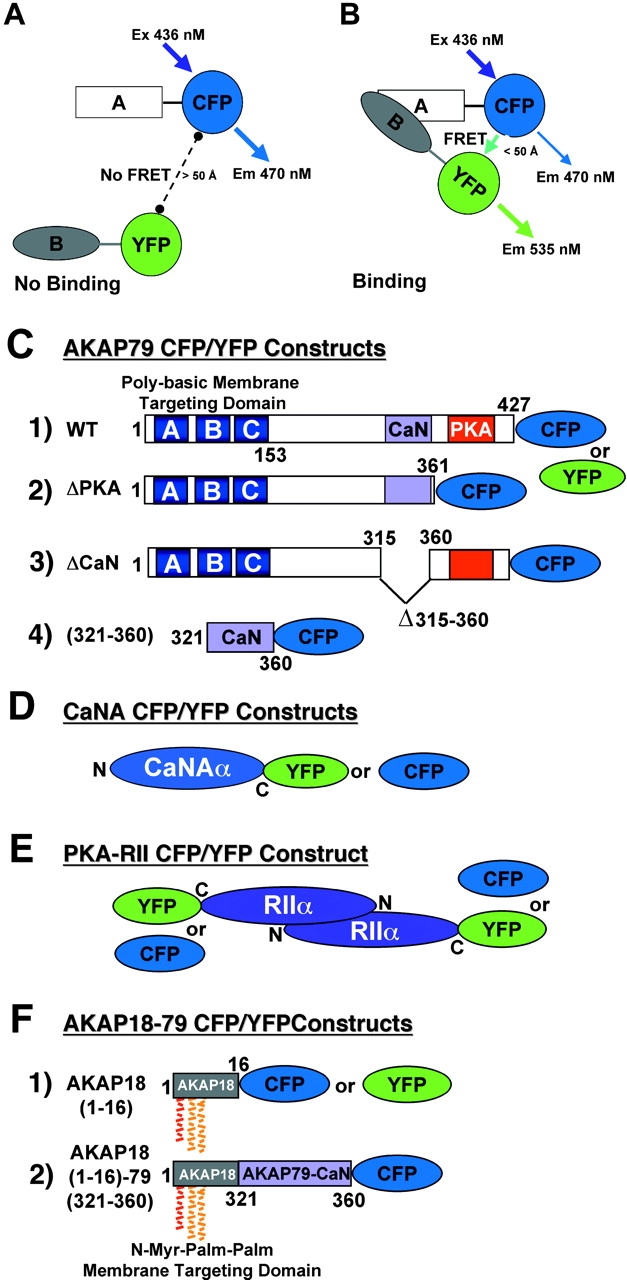
CFP/YFP FRET microscopy system for studying AKAP79 protein–protein interactions in living cells. (A) Model showing no FRET between A–CFP and B–YFP tagged proteins in the absence of binding. (B) CFP to YFP FRET (CYFRET) resulting in increased YFP acceptor emission and quenching of CFP donor emission seen when A–CFP and B–YFP tagged proteins are bound within 50 Å of each other. (C) CFP- and YFP-tagged (1) AKAP79 WT, (2) ΔPKA, (3) ΔCaN, and (4) (321–360) constructs used for CYFRET imaging of AKAP79 binding to CFP- and YFP-tagged (D) CaNAα and (E) PKA-RIIα. (F) (1) AKAP18(1–16) and (2) AKAP18(1–16)–AKAP79(321–360) hybrid CFP and YFP fusion proteins for CYFRET imaging of CaNA binding. AKAP79 anchors the CaNA,B holoenzyme through binding the catalytic CaNA subunit (Coghlan et al., 1995; Kashishian et al., 1998) and anchors the PKA heterotetrameric R2C2 holoenzyme through binding a surface created by dimerization of the of NH2 terminus of the regulatory subunit (RIIα/β) (Newlon et al., 2001). The NH2 and COOH termini of each protein are indicated by N and C or numbers starting with 1 at the NH2 terminus. The AKAP79 CaN (315–360, pink) and PKA (388–413, red) binding sites are indicated as are the AKAP79 (1–153, A, B, and C poly-basic, blue) and AKAP18 (1–16, NH2-terminal Gly-myristoylated, dual Cys-palmitoylated) membrane targeting domains.
Imaging AKAP79 binding to CaN and PKA in living cells with CFP/YFP micro-FRET microscopy
To facilitate FRET studies of the AKAP79 scaffold, we generated cDNA expression vectors for CFP and YFP COOH-terminal fusion proteins of AKAP79 (Fig. 1 C), CaNA catalytic subunit (Fig. 1 D), and PKA-RII regulatory subunit (Fig. 1 E) for expression in COS7 cells. COS7 cells do not express endogenous AKAP79 and have thus been used in transfection studies to reconstitute AKAP79, PKA, MAGUK, and glutamate receptor complexes, map the CaNA binding site on AKAP79, and study AKAP79 targeting (Colledge et al., 2000; Dell'Acqua et al., 2002; Gomez et al., 2002). Importantly, previous AKAP79 COS7 transfection studies are in good agreement with parallel experiments done in cultured neurons and brain extracts with endogenous proteins. Using micro-FRET subtraction to generate a corrected FRET (FRETC) image (see Materials and methods), we first set out to image binding between AKAP79 and CaNA. In agreement with earlier studies in fixed COS7 cells, expression of AKAP79WT–CFP (Fig. 1 C, 1) with CaNA–YFP (Fig. 1 D) resulted in targeting of CaN to plasma membrane ruffles and intracellular vesicles in living cells where extensive colocalization with the AKAP was seen (Fig. 2 A, CFP/YFP Overlay). In contrast, in cells coexpressing CaN–YFP with an AKAP79ΔCaN–CFP (Fig. 1 C, 3) mutant lacking the CaN binding site (Δ315–360) (Dell'Acqua et al., 2002), CaN was in the cytoplasm and not with membrane-targeted AKAP (Fig. 2 A). In cells expressing AKAP79WT–CFP and CaNA–YFP, FRETC (monochrome) was seen at membrane sites where the two proteins colocalized, but in cells expressing AKAP79ΔCaN, no FRET was detected.
Figure 2.
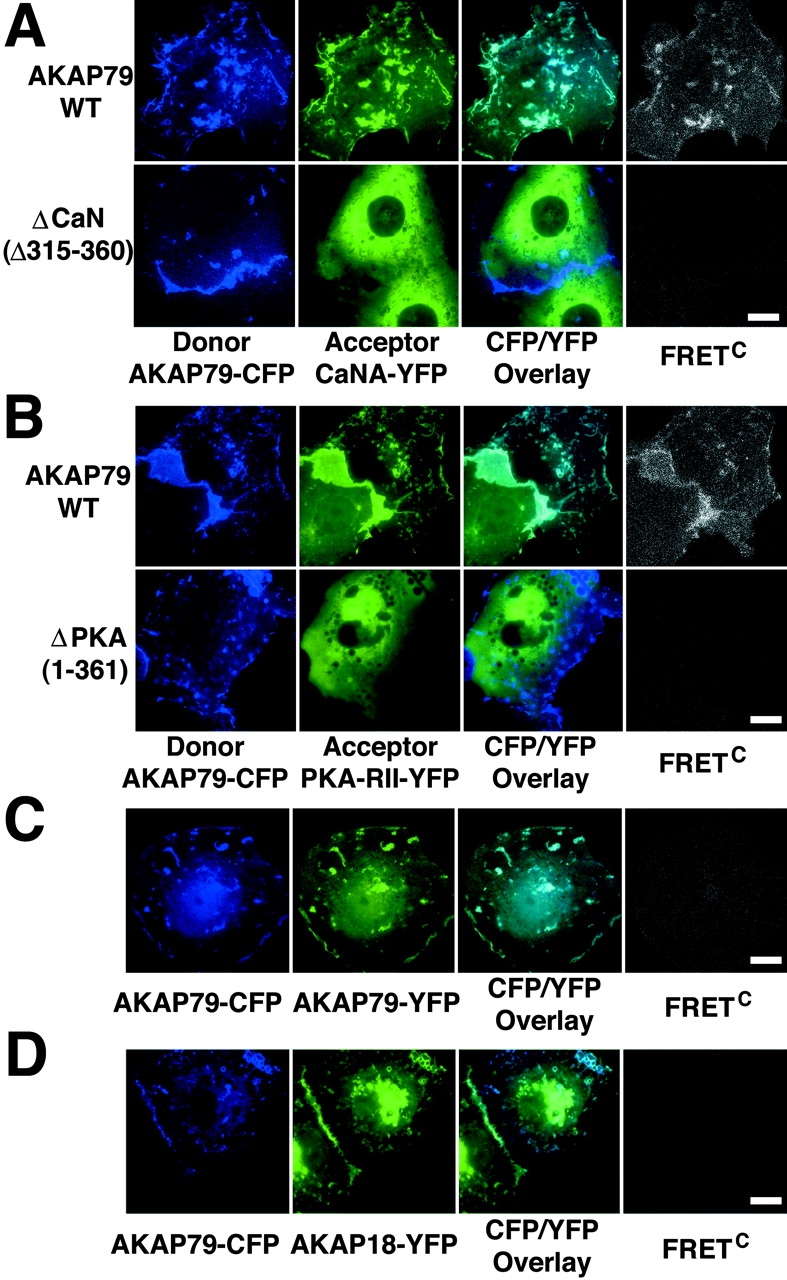
CFP/YFP micro-FRET microscopy imaging of AKAP79 binding to CaN and PKA in living cells. (A) Plasma membrane/cortical colocalization (CFP/YFP Overlay) and direct binding (FRETC) seen for CaNA–YFP (green) and AKAP79–CFP WT but not ΔCaN (Δ315–360) (blue) in live COS7 cells. (B) Plasma membrane/cortical colocalization (CFP/YFP Overlay) and direct binding (FRETC) seen for PKA-RIIα–YFP (green) and AKAP79–CFP WT but not ΔPKA (1–361) (blue) in live COS7 cells. (C) Negative control showing no FRETC for AKAP79–CFP (blue) and AKAP79–YFP (green) that colocalize at the plasma membrane (CFP/YFP Overlay) but do not bind to each other. (D) Negative control showing no FRETC for AKAP79–CFP (blue) and AKAP18(1–16)–YFP (green) that colocalize (CFP/YFP Overlay) but do not bind to each other. Bars, ∼15 μm.
Because the amounts of CFP donor and YFP acceptor will vary from cell to cell in transient transfections and the intensity of energy transfer is related to the number of YFP acceptor/CFP donor pairs, we also calculated FRET values from membrane ruffles and vesicles normalized (FRETNC) to the amounts of CFP and YFP. Thus, FRETNC allows comparison of FRET from multiple cells for a given acceptor–donor pairing to demonstrate reproducibility (Table I; see Materials and methods). We calculated a FRETNC value of 2.8 ± 0.8 (×10−5) for AKAP79WT–CFP and CaNA–YFP. A FRETNC value of 1.4 ± 0.4 was obtained (not depicted) when the chromophores were reversed with CaNA–CFP and AKAP79WT–YFP (Table I). For the AKAP79ΔCaN mutant and CaN, lack of FRET is reflected in a negative FRETNC value represented in Table I by <0. Negative FRETNC values are obtained when there is no FRET due to overcorrection for CFP and YFP cross-bleeding built into the micro-FRET method (Table I; Materials and methods).
Table I.
Normalized FRETN c for CFP donor and YFP acceptor pairs used to characterize AKAP79, CaNA, and PKA-RII binding interactions in live cells
| CFP donor | YFP acceptor | |||
|---|---|---|---|---|
| AKAP79–YFP | CaNA–YFP | PKA-RII–YFP | AKAP18(1–16)–YFP | |
| AKAP79–CFP | <0 | 2.8 ± 0.8 | 12.1 ± 1.1 | <0 |
| AKAP79ΔCaN–CFP | − | <0 | − | − |
| AKAP79ΔPKA–CFP | − | − | <0 | − |
| CaNA–CFP | 1.4 ± 0.4 | − | − | − |
| PKA-RII–CFP | 13.8 ± 3.0 | − | − | − |
| AKAP79WT + CaNA–CFP | − | − | 9.1 ± 1.6 | − |
| AKAP79ΔCaN + CaNA–CFP | − | − | <0 | − |
| AKAP79ΔPKA + CaN–CFP | − | − | <0 | − |
| AKAP79ΔCaN + ΔPKA + CaN–CFP | − | − | <0 | − |
| AKAP79(321–360)–CFP | − | 13.3 ± 1.0 | − | − |
| AKAP18(1–16)AKAP79(321–360)–CFP | − | 35.6 ± 10.0 | − | − |
| AKAP18(1–16)–CFP | − | <0 | − | − |
Mean fluorescence intensities in arbitrary linear units were measured (see Materials and methods) for CFP, YFP, and raw, uncorrected CYFRET for regions of CFP and YFP overlap in membrane ruffles and vesicles. For each image, corrected FRETc was calculated from raw CYFRET by fractional subtraction (see Materials and methods) of CFP and YFP intensity values. The FRETc intensity values were then normalized (FRETNc) to the amounts of CFP donor and YFP acceptor present by the product (CFP)(YFP). FRETNc is thus proportional to Keq = [donorCFP · acceptorYFP]/[donorCFP][acceptorYFP], the equilibrium constant for the binding interaction. FRETNc values are represented above as the mean ± SEM (×10−5) for multiple images (n = 5–8) for each donor–acceptor pairing. Negative FRETc values are seen for CFP–YFP pairs that fail to exhibit FRET due to bleedthrough overcorrection (see Materials and methods) that prevents false positive FRET measurements. Negative FRET pairs are thus represented by FRETNc values < 0 above. Thus, positive FRETNc values in this table and the FRETc images shown in the figures are actually slight underestimates of the actual FRET signals.
The PKA-RII binding site on AKAP79 (388–409) (Carr et al., 1992) is even closer to the COOH terminus of the AKAP than the CaNA binding site (Fig. 1 C, 1), so we next focused on FRET imaging of AKAP79–PKA binding. In cells expressing AKAP79WT–CFP (Fig. 1 C, 1), the PKA-RII–YFP protein (Fig. 1 E) was colocalized with the AKAP in membrane ruffles and vesicles where FRETC was also seen (Fig. 2 B). Accordingly, deletion of the PKA-RII binding site (ΔPKA[1–361]; Fig. 1 C, 2) prevented both membrane targeting of RII and FRET between AKAP79 and RII. Normalized FRETNC values for multiple cells confirmed reproducible FRET between AKAP79WT–CFP and RII–YFP (12.1 ± 1.1) and the absence of FRET between ΔPKA and RII (<0; Table I). Reversal of the chromophores with PKA-RII–CFP and AKAP79WT–YFP also produced membrane FRETC signals (not depicted) with a similar FRETNC value of 13.8 ± 3.0 (Table I).
The controls above demonstrating that FRET depends on CaN and PKA binding to AKAP79 use AKAP deletion mutants that also disrupt CFP/YFP colocalization. Thus, we performed controls confirming that image subtraction was measuring FRET and not colocalized CFP/YFP bleedthrough. We coexpressed AKAP79WT–CFP with AKAP79WT–YFP (Fig. 1 C, 1) or a construct fusing YFP onto the COOH terminus of the membrane-targeting domain of the unrelated AKAP18 (AKAP18[1–16]–YFP; Fig. 1 F, 1) (Trotter et al., 1999). The CFP/YFP overlay images for cells expressing these protein pairs revealed significant colocalization of AKAP79–CFP with AKAP79–YFP (Fig. 2 C) or AKAP18–YFP (Fig. 2 D) at the membrane. However, no FRETC was detected for these pairings despite membrane overlap (Fig. 2, C and D). Negative FRETNC values (<0; Table I) calculated from multiple images confirmed a lack of AKAP79 intermolecular FRET with itself or AKAP18. In contrast to AKAP79, AKAP18–YFP also showed very strong localization to intracellular perinuclear/Golgi membranes (Fig. 2 D). Importantly, this strong Golgi YFP signal did not bleed into the FRETC image, further confirming the accuracy of image subtraction.
Confirmation of AKAP79 binding and FRET with CaN and PKA in fixed cells using YFP acceptor photobleaching
The advantage of the micro-FRET method is that it generates high-resolution, real-time images of sensitized FRET emission even if the protein–protein interaction is unregulated. However, it is an indirect FRET measurement that relies on controls done in separate cells expressing donor or acceptor alone. FRET can also be directly measured within a single cell by determining the amount of CFP donor quenching that is relieved by YFP acceptor photobleaching (CFPpostbleach − CFPprebleach = ΔCFP) and can be expressed as an apparent donor FRET efficiency percentage (%ΔCFP/CFPpostbleach). Unfortunately, this photobleaching method is not suitable for use in our live cell system because membrane ruffles and vesicles change shape and location during the 2 min required to bleach YFP. Nonetheless, in fixed cells, we were able to do single cell comparisons of FRETC determined by micro-FRET with apparent FRET efficiency (%ΔCFP/CFPpostbleach) determined by acceptor photobleaching.
As in live cells, coexpression of AKAP79–CFP (Fig. 1 C, 1) with CaNA–YFP (Fig. 1 D) or PKA-RII–YFP (Fig. 1 E) resulted in membrane anchoring of CaN (Fig. 3 A) or RII (Fig. 3 B) to AKAP79, detected by the micro-FRET method in fixed cells. AKAP79–CaN and AKAP79–RII FRETC signals (pseudo-color) at specific membrane locations are shown in images gated to the presence of YFP acceptor (blue underlay) to represent relative FRET intensities on a scale of blue (no FRET) to green (low FRET) to red (high FRET) (Fig. 3, A and B, top, second panel from right). Sites of high relative FRETC intensity (orange-red) included vesicles and plasma membrane ruffles where the donor and acceptors were highly colocalized. Micro-FRET measurements of FRETNC in membrane structures for these fixed samples were very reproducible across multiple experiments (n = 8–12) for AKAP79–CFP paired with CaN–YFP (14.0 ± 1.2) or RII–YFP (13.0 ± 1.6). This FRETNC value for AKAP79–RII in fixed cells is very similar to that determined in live cells for RII (12.1 and 13.8; Table I), whereas the FRETNC value for AKAP79–CaNA in fixed cells is larger than that determined in live cells for CaN (1.4 and 2.8; Table I). Thus, quantitative comparison of FRET for the same protein complex in live cells versus fixed cells is difficult because fixation may trap the complex in a form with somewhat altered chromophore separation and orientations compared with in living cells. Nonetheless, we can measure FRETC in fixed cells for AKAP79 binding to PKA and CaN.
Figure 3.

Confirmation of micro-FRET detection of AKAP79 binding to CaN and PKA by YFP acceptor photobleaching imaging of FRET CFP donor quenching in fixed COS7 cells. (A) Membrane colocalization of AKAP79–CFP (blue) and CaNA–YFP (green) with micro-FRETC (pseudo-color/gated to YFP [blue underlay]), and relief of FRET CFP donor quenching by YFP acceptor photobleaching (ΔCFP pseudo-color/gated to CFPpost [blue underlay]) observed for AKAP79WT (top) but not ΔCaN (Δ315–360) (bottom). B) Membrane colocalization of AKAP79–CFP (blue) and PKA-RII–YFP (green) with micro-FRETC (pseudo-color/gated to YFP [blue underlay]) and relief of FRET CFP donor quenching by YFP photobleaching (ΔCFP pseudo-color/gated to CFPpost [blue underlay]) observed for AKAP79WT (top) but not ΔPKA (1–361) (bottom). Bars, ∼15 μm.
Importantly, for both CaN and PKA-RII, selective bleaching of YFP relieved quenching of AKAP79–CFP donor fluorescence, showing FRET in the same membrane structures where FRETC signals were also detected (Fig. 3, A and B, top, right panels). This dequenching of CFP (ΔCFP, pseudo-color) is represented in subtracted images gated to CFP fluorescence after bleaching (CFPpost, blue underlay) to once again show relative FRET intensity on a scale of blue (no FRET) to red (high FRET). Calculation of apparent FRET efficiency from multiple experiments (n = 8–12) showed that this photobleaching method was also very reliable in detecting FRET between AKAP79–CFP and CaN–YFP (8.4 ± 1.2%) or RII–YFP (5.9 ± 1.2%). Final confirmation of the abilities of both the micro-FRET and photobleaching methods to measure FRET produced by binding of CaN or PKA to the AKAP scaffold was provided by analysis of the ΔPKA (Fig. 1 C, 2) and ΔCaN (Fig. 1 C, 3) mutants (Fig. 3, A and B, bottom). The corresponding FRETC and ΔCFP images each showed a lack of FRET for these protein pairings that was confirmed by quantitative analysis (n = 5) showing negative FRETNC (<0) and FRET efficiency values (ΔPKA, −3.5 ± 1.3%; ΔCaN, −0.7 ± 0.9%).
Direct binding of CaN to AKAP79 residues 321–360 detected in living cells by CFP/YFP FRET
Our findings demonstrate the use of FRET to image binding between AKAP79 and CaN or PKA in both living and fixed cells, and from our controls with ΔCaN and ΔPKA mutants, we can infer that these interactions involve the appropriate AKAP binding sites. However, we wished to compliment these “loss of function” controls with a “gain of function” approach using the AKAP–CaN interaction as an example. Recently, we demonstrated that residues contained in 321–360 of AKAP79 can bind CaN in vitro and inhibit signaling in cells, suggesting that this region is sufficient for AKAP79–CaN anchoring (Dell'Acqua et al., 2002). To show that CYFRET observed between AKAP79 and CaN involves binding between AKAP79 residues 321–360 and the CaNA subunit, we constructed three additional CFP donors (AKAP79[321–360]–CFP, Fig. 1 C, 4; AKAP18[1–16]–CFP, Fig. 1 F, 1; and AKAP18[1–16]AKAP79[321–360]–CFP, Fig. 1 F, 2). In agreement with our previous studies, expression of the isolated CaN binding domain (321–360) fused to CFP (Fig. 1 C, 4) produced an untargeted protein found in both the cytoplasm and the nucleus (Fig. 4, A and B). This (321–360)–CFP construct bound to CaNA–YFP in the cytoplasm and prevented plasma membrane anchoring of CaNA–YFP to AKAP79WT (Texas red antibody staining) (Fig. 4 A). In parallel live cells, where untagged AKAP79 cannot be seen, colocalization of (321–360)–CFP and CaNA–YFP and FRETC were seen exclusively in the cytoplasm (Fig. 4 B). FRETNC measurements showed this FRET to be reproducible (13.3 ± 3.0; Table I).
Figure 4.

Direct observation of CaN binding to AKAP79 residues (321–360) in living cells using CYFRET microscopy. (A) Displacement of CaNA–YFP (green) from plasma membrane–targeted AKAP79 (anti-AKAP79, TxRd, red) by an untargeted AKAP79 CaN binding site peptide, AKAP79(321–360)–CFP (blue), seen in the RGB composite as red in membrane ruffles and CFP-blue/YFP-green overlap in the cytoplasm. (B) Colocalization (CFP/YFP Overlay) and direct binding (FRETC) seen for CaNA–YFP (green) and AKAP79(321–360)–CFP (blue) in the cytoplasm of COS7 cells. (C) The AKAP79(321–360) CaN binding site confers CaNA binding activity on AKAP18 in living cells. Plasma membrane and Golgi targeting (CFP/YFP overlay) and direct binding (FRETC) of CaNA–YFP (green) to AKAP18(1–16)–AKAP79(321–360)–CFP (blue, bottom) but not AKAP18(1–16)–CFP (blue, top) in live COS7 cells. Bars, ∼20 μm.
As shown above, the AKAP18 NH2-terminal targeting domain (residues 1–16; Fig. 1 F, 1) localizes YFP (Fig. 2 D) or CFP (Fig. 4 C) to both plasma membrane and, especially, intracellular Golgi/perinuclear membranes. However, the AKAP18(1–16) targeting domain does not bind CaN and thus cannot target cytoplasmic CaNA–YFP to these locations (Fig. 4 C, top). The lack of interaction between these two proteins was confirmed by no detection of FRETC in these cells (Fig. 4 C; Table I, FRETNC < 0). In contrast, fusion of the AKAP79 CaN binding region (321–360) onto AKAP18(1–16) (Fig. 1 F, 2) conferred CaN targeting activity on AKAP18. Importantly, both CFP/YFP colocalization and FRETC were observed for CaNA–YFP and the AKAP18(1–16)AKAP79(321–360)–CFP hybrid (Fig. 1 F, 2) in plasma membrane and especially Golgi membranes (Fig. 4 C, bottom). Measurement of FRETNC (Table I) revealed this FRET to be reproducible (35.6 ± 10.0). These findings of FRET with CaNA conferred by AKAP79 residues 321–360 confirm through a gain of function approach that our FRETC images and measurements are truly representative of protein binding in live cells.
FRET imaging of the CaN–AKAP79–PKA ternary complex in living cells
The identified CaN (321–360) and PKA (388–409) binding sites on AKAP79 are in very close proximity to each other in primary sequence (Fig. 1 C, 1). Our FRET measurements above indicate that both proteins bound to AKAP79 are <50 Å from C/YFP at the AKAP COOH terminus. These findings raise the issue of whether PKA and CaN are bound to the same AKAP molecule simultaneously or if there is competition due to steric hindrance between the closely opposed binding sites. To address this issue, we sought to reconstitute CaN–AKAP79–PKA ternary complexes in COS7 cells. As a first step, we coexpressed AKAP79WT–CFP (Fig. 1 C, 1) and PKA-RII–YFP (Fig. 1 E) with myc-tagged CaNA (Dell'Acqua et al., 2002). Cells were fixed and labeled with anti-myc antibodies to visualize CaNA along with AKAP79–CFP and RII–YFP. These studies revealed that AKAP79WT–CFP, PKA-RII–YFP, and myc–CaNA were all colocalized in plasma membrane ruffles (Fig. 5 A, top). To show that this colocalization in plasma membrane structures was due to independent binding of CaN and PKA to their respective AKAP79 binding sites, we analyzed the ΔPKA (Fig. 1 C, 2) and ΔCaN (Fig. 1 C, 3) mutants. Deletion of the PKA anchoring site on AKAP79 led to cytoplasmic localization of RII–YFP but retention of AKAP79–CFP and myc–CaNA at the membrane (Fig. 5 A, middle). Deletion of the CaN anchoring site on AKAP79 had the opposite effect, causing cytoplasmic localization of myc–CaNA but maintaining membrane overlap for AKAP79–CFP and RII–YFP (Fig. 5 A, bottom). These results do not support a model of competition between PKA and CaN binding to AKAP79 and suggest that these proteins can bind independently to closely spaced, but functionally separate, binding sites on the AKAP.
Figure 5.

Direct observation of a CaN–AKAP79–PKA ternary in living cells using CYFRET imaging. (A) Plasma membrane/cortical targeting of CaN and PKA mediated through binding to functionally separable sites on AKAP79. Colocalization of PKA-RII–YFP (green) and CaNA–myc with AKAP79WT–CFP (blue) is seen as white in the RGB composite (top). Selective loss of PKA-RII (green) but not CaNA–myc (red) from the plasma membrane by deletion of the AKAP79–RII binding site (ΔPKA), seen as pink in the RGB composite (middle). Selective loss of CaNA–myc (red) but not PKA-RII (green) from the plasma membrane by deletion of the AKAP79–CaNA binding site (ΔCaN), seen as blue-green in the RGB composite (bottom). (B) Plasma membrane colocalization of CaNA–CFP (blue), PKA-RII–YFP (green), and untagged AKAP79WT (anti-AKAP79, TxRd, red), seen as white in RGB composite. (C) FRETC measured between CaNA–CFP and PKA-RII–YFP bound to AKAP79WT in a ternary complex at the plasma membrane (top). Loss of CaNA–CFP from the AKAP79-RII–YFP complex and loss of FRETC upon disruption of the CaN binding site (ΔCaN) (second row). Loss of RII–YFP from the AKAP79–CaNA–CFP complex and loss of FRETC upon disruption of the PKA binding site (ΔPKA) (third row). Membrane colocalization but no FRETC for CaNA–CFP and RII–YFP targeted through binding to separate AKAP molecules (ΔPKA + ΔCaN) (bottom). Bars, ∼20 μm.
Because both proteins bind in close proximity to each other and exhibit FRET with AKAP COOH-terminal C/YFP, we reasoned that it might be possible to directly observe the CaN–AKAP–PKA ternary complex by measuring FRET from CaNA–CFP to PKA-RII–YFP bound to the same AKAP molecule. We cotransfected CaNA–CFP, PKA-RII–YFP, and untagged AKAP79WT into COS7 cells. In agreement with Fig. 5 A, in cells fixed and antibody stained to visualize AKAP79WT, colocalization of CaN–CFP, RII–YFP, and AKAP was seen in membrane ruffles and vesicles (Fig. 5 B). In parallel live cells (where AKAP cannot be visualized), membrane colocalization of CaN–CFP and RII–YFP was seen in CFP/YFP overlay images and FRETC signals were detected at these sites of overlap (Fig. 5 C, top). A FRETNC value of 9.1 ± 1.6 (Table I) showed the FRET to be reproducible. The lack of any significant direct interaction between CaN and RII that could produce FRET was confirmed by analysis of untagged AKAP79 ΔCaN and ΔPKA mutants. In ΔCaN-expressing cells, CaNA–CFP was cytoplasmic, RII–YFP targeted to cortical ruffles, and no FRETC was observed (Fig. 5 C, second row; Table I, FRETNC < 0). In ΔPKA-expressing cells, RII–YFP was cytoplasmic, CaNA–CFP targeted to cortical ruffles, and no FRETC was observed (Fig. 5 C, third row; Table I, FRETNC < 0). Thus, the membrane colocalization and FRET seen for CaNA–CFP and RII–YFP in the presence of AKAP79WT can be attributed to both proteins binding to AKAP79. This FRET between CaN and PKA-RII in the presence of AKAP79WT could be due to CaN and PKA binding separate AKAP molecules clustered in close proximity to one another. However, we thought that this scenario was unlikely because coclustered AKAP79–CFP and AKAP79–YFP molecules did not produce detectable FRET (Fig. 2 C; Table I). Nonetheless, to directly rule out this possibility, we coexpressed untagged ΔCaN and ΔPKA mutants with CaNA–CFP and RII–YFP in order to colocalize CaN and PKA in membrane structures through obligate binding to separate AKAP molecules. As expected, membrane colocalization of CaNA–CFP and RII–YFP could be seen in these ΔCaN + ΔPKA–expressing cells, but no FRET was observed (Fig. 5 C, bottom row; Table I, FRETNC < 0). Proper expression and membrane targeting of the untagged ΔPKA and ΔCaN mutants was confirmed (as in Fig. 5 B) in parallel fixed cells stained with anti-AKAP79 antibodies (unpublished data). Thus, detection of FRET between CaNA–CFP and PKA-RII–YFP in cells expressing AKAP79WT is most consistent with direct observation of the ternary kinase–AKAP–phosphatase complex in living cells.
Plasma membrane targeting and recruitment of SAP97 to the AKAP79 signaling scaffold
AKAP79 can bind MAGUK scaffold proteins including SAP97 and PSD-95 (Colledge et al., 2000). In particular, binding of AKAP79 to SAP97 is believed to recruit AKAP, PKA, and CaN to GluR1 subunits for the regulation of AMPA receptor phosphorylation (Colledge et al., 2000; Dell'Acqua et al., 2002; Tavalin et al., 2002). Using our transfection system, we set out to characterize the interaction between AKAP79 and SAP97 in living cells. In agreement with previous studies (Tiffany et al., 2000), SAP97–YFP expressed alone was predominantly cytoplasmic (Fig. 6 A, top). However, expression of SAP97–YFP with AKAP79–CFP resulted in substantial SAP97 targeting to membrane ruffles in live cells (Fig. 6 A, bottom). In fixed cells, these sites of membrane colocalization of SAP97 with AKAP79 were found to be enriched in cortical F-actin stained with phalloidin (Fig. 6 B, top). We have previously shown that the highly-basic NH2 terminus of AKAP79 is necessary and sufficient for binding to phosphatidylinositol-4,5-bisphosphate and F-actin in vitro and targeting to dendritic spines in neurons and membrane ruffles in COS7 cells (Fig. 6 E; Dell'Acqua et al., 1998; Gomez et al., 2002). AKAP79 binds to the SAP97 MAGUK SH3 and GK domains (Fig. 6 F); however, the MAGUK binding site of AKAP79 is less defined but is thought to be located COOH terminal to the targeting domain (1–153) and NH2 terminal to the CaN anchoring site (315–360) (Fig. 6 E; Colledge et al., 2000). Thus, we expressed SAP97–YFP with AKAP79(1–153)–CFP as an additional negative control to support the need for AKAP79–MAGUK binding in SAP97 cortical targeting. As seen previously (Gomez et al., 2002), AKAP79(1–153)–CFP targeted to membrane ruffles enriched with F-actin (Fig. 6 B, middle); however, SAP97–YFP remained cytoplasmic with little or no colocalization with cortical actin or the AKAP fragment (Fig. 6 B, bottom). Coexpression of SAP97–YFP with a COOH-terminal fragment of AKAP79 ([150–427]–CFP) that is unable to localize to membranes or bind actin (Fig. 6 E; Gomez et al., 2002) resulted in no targeting of either protein to cortical actin (Fig. 6 B, bottom). This confirmed that binding of SAP97 to determinants found COOH terminal to the AKAP targeting domain must be required for SAP97 targeting to membrane ruffles with AKAP79 (Fig. 6 E).
Figure 6.

Targeting of SAP97 to the CaN–AKAP79–PKA membrane cytoskeleton signaling complex. (A) AKAP79 regulates plasma membrane targeting of SAP97. SAP97–YFP (green) is primarily cytoplasmic when expressed alone (−AKAP79) but is targeted to the membrane ruffles in association with AKAP79–CFP (+AKAP79, blue) in live COS7 cells (CFP/YFP Overlay). (B) AKAP79 regulates targeting of SAP97 to the cortical membrane cytoskeleton through determinants that lie COOH terminal to the AKAP NH2-terminal targeting domain. SAP97–YFP (green) is targeted to the membrane ruffles enriched in F-actin (Texas red–phalloidin, red) in association with AKAP79–CFP (WT[1–427], top row, colocalization of SAP97, AKAP, and F-actin, seen as white in RGB composite panel). SAP97–YFP (green) is cytoplasmic when expressed with AKAP79(153)–CFP (blue) NH2-terminal domain, which targets to F-actin membrane ruffles (N[1–153], middle row, colocalization of (1–153) with F-actin, seen as pink in RGB composite panel) or an untargeted CFP–(150–427) AKAP79 fragment (C[150–427], bottom row). (C) AKAP79 regulates cortical colocalization of SAP97 with CaN in COS7 cells. CaNA–YFP (green) and myc–SAP97 (TxRd, red) are both cytoplasmic when expressed alone (−AKAP79) but are colocalized at the plasma membrane with each other and AKAP79–CFP (blue) in cells expressing all three proteins (+AKAP79), seen as white in the RGB composite. (D) AKAP79 coordinates targeting of CaN, PKA, and SAP97 to the same plasma membrane structures in COS7 cells. AKAP79 (anti-79, Cy5, monochrome) mediated membrane colocalization of CaNA–CFP (blue), PKA-RII–YFP (green), and myc–SAP97 (TxRd, red), seen as white in the RGB composite panel. Diagrams showing structures of the (E) AKAP79–CFP WT(1–427), N(1–153), and C(150–427) and (F) SAP97–YFP fusion proteins used above. Relevant cellular targeting, protein binding domains, and other structural motifs are indicated for both AKAP and SAP97 (see Results and Discussion for more details). Bars: (A and B) ∼15 μm; (C and D) ∼20 μm.
Unfortunately, our efforts to measure FRET between AKAP79–CFP and SAP97–YFP as well as a chromophore-reversed pair were unsuccessful (FRETC images not depicted). It is possible that the chromophore separation distances are too great (i.e., >50 Å) or chromophore orientations in the complex are not favorable for FRET. Despite this lack of FRET between AKAP79 and SAP97, we combined CFP/YFP imaging with immunofluorescence to examine membrane-localized assembly of higher-order signaling complexes. Under conditions where we can measure FRET between AKAP79–CFP and CaN–YFP (Fig. 2 A; Fig. 3 A), we also detected cortical colocalization of the AKAP and CaN with myc-tagged SAP97 (Fig. 6 C, bottom). In contrast, coexpression of CaNA–YFP with myc–SAP97 in the absence of AKAP79 resulted in cytoplasmic localization of both proteins (Fig. 6 C, top). Under conditions where we measured FRET between CaNA–CFP and PKA-RII–YFP bound to AKAP79 (Fig. 5 C), we observed distinct targeting of CaN, PKA, and SAP97 to overlapping plasma membrane sites (Fig. 6 D, AKAP79 detected with Cy5 antibodies, shown in monochrome). These data suggest that SAP97 can be recruited to membranes/cortical cytoskeleton in association with the CaN–AKAP79–PKA ternary complex. Thus, this macromolecular signaling complex is also likely to be assembled endogenously on neuronal dendritic spines where it could function in the regulation of glutamate receptor signaling pathways underlying synaptic plasticity.
Discussion
FRET ratio methods with GFP variants have been used to study cAMP-regulated binding between PKA catalytic (C) and R subunits, PKA R subunit binding to untargeted AKAP anchoring peptides, and phosphorylation of PKA substrate indicators in living cells (Ruehr et al., 1999; Nagai et al., 2000; Zaccolo et al., 2000; Zhang et al., 2001; Zaccolo and Pozzan, 2002). However, FRET techniques have not been used to image binding of PKA or other signaling proteins to intact AKAP scaffolds at relevant subcellular locations as we have done here. Furthermore, we have extended the use of FRET to observe, for the first time, a ternary protein complex in living cells. Our FRET measurements demonstrate that PKA-RII and CaNA are simultaneously bound within ∼50 Å of each other on a membrane-targeted AKAP79 molecule. This important finding significantly extends our understanding of the level of molecular compartmentalization of signaling proteins that is made possible by scaffolding proteins. Recent intramolecular FRET studies with PKA–substrate indicators have elegantly demonstrated that artificially tethering the substrate and kinase in the same molecular complex allows very rapid cAMP-stimulated phosphorylation (Zhang et al., 2001). Our FRET observations of PKA and CaN anchored together on a nanometer scale nicely support proposed models where AKAP79 organizes a signaling complex that facilitates not only cAMP-stimulated phosphorylation but also efficient Ca2+-stimulated dephosphorylation of associated target substrates (Tavalin et al., 2002).
Evidence for the existence of complexes containing AKAP79, PKA, CaN, and MAGUKs had previously been obtained using immunoprecipitation and GST precipitation with recombinant proteins and cell extracts (Coghlan et al., 1995; Colledge et al., 2000; Gomez et al., 2002). Due to the inherent limitations of precipitation reagents and the use of cell extracts in these biochemical approaches, results from such studies may not always be entirely representative of intact living cells. For instance, one previous in vitro precipitation study suggested competition between CaN and MAGUK binding to AKAP79 (Colledge et al., 2000), whereas a second electrophysiological study in live cells found no evidence for such competition (Tavalin et al., 2002). Earlier attempts to map the CaN binding site on AKAP79 using in vitro biochemical methods or the yeast two-hybrid system gave inconsistent results and were only able to narrow the binding site to the COOH-terminal two thirds of AKAP79 (Dell'Acqua et al., 1998; Kashishian et al., 1998). In contrast, our more recent studies combining cellular immunofluorescence with in vitro biochemical assays were successful in mapping the CaN anchoring site to a single 40-amino acid region in very close proximity to the PKA anchoring site (Dell'Acqua et al., 2002).
Thus, in order to get a more accurate view of AKAP79 scaffolding functions in vivo, we thought it was imperative to study the formation of AKAP79 complexes in intact cells using not only immunofluorescence, which shows protein colocalization on a scale of 200 nm, but also FRET microscopy, which detects true protein–protein proximity within 5 nm (50 Å). Our current results show that by using micro-FRET, we can directly visualize, on this 5-nm scale, binding of AKAP79 to CaNA and PKA-RII in a ternary complex at the plasma membrane/cortical cytoskeleton of living cells. These binding interactions were confirmed by measurement of FRET in fixed cells using both micro-FRET and acceptor photobleaching methods. Importantly, the CaN and PKA binding interactions were shown to be through noncompeting sites near the AKAP COOH terminus. FRET signal strength is a function of not only the distance between chromophores but also the binding affinity of the protein–protein interaction and chromophore orientations within the complex. Thus, differences in any of these parameters can explain the relative differences in FRET signals between acceptor–donor pairs. For example, the weaker FRETNC that we measured for AKAP79–CaN versus AKAP79–PKA binding could reflect both the 20–50-fold weaker reported affinity of AKAP79 for CaNA versus RII as well as the closer proximity of the PKA-RII binding site to the AKAP COOH terminus (Fig. 1 C, 1; Carr et al., 1992; Kashishian et al., 1998; Dell'Acqua et al., 2002). However, consistent with chromophore separation contributing to our observed differences in FRETNC, when the CaN binding site (321–360) is fused directly to CFP with no intervening sequence (Fig. 1 C, 4; Fig. 1 F, 2), larger CaNA–YFP FRETNC values (13.3 and 35.6) are measured than for full-length AKAP79–CFP (2.8) (Table I). Extending this reasoning to the CaN–AKAP–PKA complex is problematic because the formation of a ternary complex is affected by the concentration of all three proteins and multiple binding affinities. Nonetheless, stronger FRETNC is seen for CaNA–CFP with RII–YFP in the AKAP79 ternary complex (9.1) versus with AKAP79–YFP in a binary complex (1.4) (Table I). This result could be explained by the shorter distance between CaN and RII bound to the AKAP compared with the distance between CaN and the AKAP COOH terminus (Fig. 1 C, 1). It would also follow from similar FRETNC values (9.1 and 12.1) that the distances between RII and CaN bound to AKAP79 may be similar to the distance between bound RII and the AKAP79 COOH terminus. Interestingly, the spacings between the CaN and PKA binding sites and the PKA binding site and AKAP COOH terminus are in fact both 20–30 amino acids in primary sequence (Fig. 1 C, 1).
By combining CFP/YFP imaging with immunofluorescence, our current work also shows that AKAP79 can regulate the membrane cytoskeletal targeting of SAP97 in concert with PKA and CaN and is not consistent with competition between CaN and MAGUK binding. Thus, in neurons, AKAP79 is likely to coordinate the assembly and targeting of PKA–CaN–SAP97 complexes that are recruited to the COOH terminus of AMPA-GluR1 receptors through SAP97 PDZ domains (Fig. 6 F; Leonard et al., 1998). Recently a great deal of attention in the field of hippocampal synaptic plasticity has focused on regulation of the phosphorylation state and localization of AMPA receptors through PKA and CaN pathways that are likely to be organized by AKAP79–MAGUK scaffolding. NMDA receptor Ca2+ influx in LTP, a synaptic strengthening process, leads to increased AMPA receptor activity and phosphorylation (Lee et al., 2000). In LTP, interactions of AMPA-GluR1 with SAP97 PDZ domains (Fig. 6 F) and phosphorylation by PKA may participate in both recruiting new receptors to the postsynaptic membrane and increasing channel activity (Banke et al., 2000; Hayashi et al., 2000; Passafaro et al., 2001; Shi et al., 2001). In contrast, NMDA receptor activation in LTD, a weakening of synaptic strength, leads to dephosphorylation of the GluR1 PKA site and removal of synaptic AMPA receptors by endocytosis through CaN activation (Beattie et al., 2000; Ehlers, 2000; Lee et al., 2000; Lin et al., 2000; Carroll et al., 2001). AKAP79 promotes similar PKA and CaN regulation of SAP97-linked AMPA-GluR1 receptors in heterologous systems and thus could play an important role in organizing PKA and CaN signals at synapses (Colledge et al., 2000; Dell'Acqua et al., 2002; Tavalin et al., 2002). In support of this model, we have recently shown that NMDA–CaN signaling pathways controlling AMPA receptors also negatively regulate the localization and association of the endogenous AKAP79/150–PKA complex with MAGUKs in neurons (Gomez et al., 2002).
Localization of AKAP79/150 and AMPA receptors with MAGUKs at synapses is dependent on the actin cytoskeleton, and, interestingly, both SAP97 and AMPA-GluR1 can be linked to the cytoskeleton through 4.1 actin binding proteins (Lue et al., 1994; Shen et al., 2000; Zhou et al., 2001; Gomez et al., 2002). However, targeting of SAP97 to cortical actin in epithelial cells depends on an NH2-terminal domain (SAP97[1–65]) not present in other MAGUKs as well as secondarily on 4.1 binding (Fig. 6 E; Reuver and Garner, 1998; Wu et al., 1998). Thus, it has been proposed that common binding of SAP97 and GluR1 to 4.1 and actin may stabilize AMPA receptors at synapses. However, our findings show that AKAP79 can also regulate targeting of SAP97 to membrane cytoskeleton structures. Thus, the association of AKAP79 with SAP97 may be an additional targeting signal for both proteins that provides AKAP–SAP97–AMPA receptor complexes further attachment to the postsynaptic cytoskeleton.
As discussed above, the molecular compartmentalization of CaN and PKA in the AKAP79 complex and how they respond to cAMP and Ca2+ signals is likely to be very important for efficient bidirectional regulation of AMPA receptor phosphorylation and trafficking. AKAP79 binding to CaN inhibits phosphatase activity in vitro even with CaNB and Ca2+-calmodulin activators present (Coghlan et al., 1995; Dell'Acqua et al., 2002). In contrast, AKAP79 targets basally active PKA holoenzyme that can be further activated by elevated cAMP through increased C subunit release; however, it is unknown whether the inhibitory CaNA binding to AKAP79 is regulated by synaptic Ca2+ signals that activate CaN. Future applications of FRET imaging methods in transfected neurons will allow us to examine the regulation of CaN–AKAP79–PKA binding interactions and targeting by neuronal Ca2+ signaling pathways involved in synaptic plasticity.
Materials and methods
Construction of cDNA expression plasmids
The cDNAs for human AKAP79 wild type (WT), ΔPKA (1–361), and ΔCaN (Δ315–360) mutants were expressed from pCDNA3 (Invitrogen) as previously described (Dell'Acqua et al., 1998, 2002). The coding sequences for AKAP79WT, (1–153), ΔPKA(1–361), ΔCaN(Δ315–360), or the 321–360 CaN binding fragment were subcloned into pEGFPN1 (CLONTECH Laboratories, Inc.) as previously described (Dell'Acqua et al., 1998, 2002). CFP and YFP variants of these fusion proteins were generated by exchange of GFP using BamHI-–NotI fragments from pECFPN1 or pEYFPN1 (constructed by PCR with ECFP and EYFP from pRSET into pEGFPN1, 2, or 3). CFP–AKAP79(150–427) in pECFPC2 was constructed as previously described (Gomez et al., 2002). pEGFPN2-AKAP18(1–16) was constructed as previously described (Trotter et al., 1999), and ECFP and EYFP variants were generated by BamHI–NotI exchange. AKAP79(321–360)–CFP was fused onto the COOH terminus of the AKAP18(1–16) sequence as a BglII–NotI PCR product into BamHI–NotI-digested AKAP18(1–16) vector to generate the AKAP18(1–16)–AKAP79(321–360)–ECFP fusion. Construction of mouse CaNAα subunit tagged with myc (9E10) epitope on its COOH terminus in pCDNA3.1MycHis(+)A (Invitrogen) was previously described (Dell'Acqua et al., 2002). CaNA was transferred from this vector as a HindIII–EcoRI fragment into pECFPN2 or pEYFPN2. The mouse PKA-RIIα coding sequence was subcloned into pEGFPN3 as previously described (Gomez et al., 2002), and ECFP and EYFP versions were obtained by BamHI–NotI exchange. The myc–SAP97 expression plasmid was provided by Marcie Colledge (Oregon Health and Science University, Portland, OR) and Morgan Sheng (Massachusetts Institute of Technology, Cambridge, MA). The SAP97 coding sequence was amplified with a 5′ T7 promoter primer and a 3′ sequence-specific primer with an EcoRI restriction site. The SAP97 PCR product was cloned as a BglII–EcoRI fragment into pEYFPN2.
COS7 cell culture and transfection
COS7 cells at 20–50% confluency (24–48 h after plating on glass coverslips in six-well plates) were transfected by calcium phosphate precipitation with cDNA expression constructs (0.5–2 μg each plasmid) for 4–16 h at 5% CO2, 37°C. Cells were washed twice with PBS, fed with DME, 10% FBS, and 1% penicillin/streptomycin (Invitrogen; GIBCO BRL), and grown for 24–48 h before live cell imaging or fixation for immunocytochemistry.
Immunocytochemistry and digital fluorescence microscopy
COS7 cells were washed in PBS, fixed in 3.7% formaldehyde/PBS, and permeabilized in 0.2% Triton X-100/PBS. For staining of myc–CaNA, myc–SAP97, or AKAP79, cells were blocked in PBS + 3.0% BSA for 30 min, followed by incubation with primary antibodies (1:250 anti-myc [9E10] [Santa Cruz] or 1:1,000 anti-AKAP79 [918I] [Icos]) for 1 h, followed by washing in PBS. Next, coverslips were incubated for 1 h with secondary antibodies (goat anti-mouse–Texas red, goat anti-rabbit–Texas red, or goat anti-rabbit–Cy5 [Molecular Probes or Amersham Biosciences]) and, in some cases, 1:500 Texas red–phalloidin (Molecular Probes) followed by washing in PBS. Coverslips were mounted on glass slides using the Pro-long anti-fade (Molecular Probes). Specific indirect immunofluorescence and/or intrinsic CFP and YFP fluorescence was detected with chroma filter sets using a Nikon TE-300 inverted microscope (100×plan-apo, oil, 1.4 NA) with Micromax (Princeton Instruments) or Sensicam (Cooke) digital CCD cameras and Slidebook 3.0 software (Intelligent Imaging Innovations). All images (including FRET images, see below) were exported from Slidebook in Tiff RGB format and figures assembled using Adobe Photoshop 5.5®.
CFP/YFP FRET microscopy and image analysis
For CYFRET imaging experiments in living cells, COS7 cells were transfected on 25-mm round glass coverslips and, 24–48 h later, placed in an imaging chamber (Molecular Probes) at room temperature in media on the stage of the microscope described above. CYFRET image acquisition and analysis were done by the three-filter “micro-FRET” image subtraction method described by Sorkin et al. (2000). In brief, three images (100-ms or 250-ms exposure sets, 2 × 2 binning) were obtained: a YFP excitation/YFP emission image; a CFP excitation/CFP emission image; and a CFP excitation/YFP emission image (raw, uncorrected CYFRET). After imaging, background images were taken. Background-subtracted YFP and CFP images were then fractionally subtracted from raw CYFRET images based on measurements for CFP bleedthrough (0.50–0.56) and YFP cross-excitation (0.015–0.02). This fractional subtraction generated corrected FRETC images, represented in monochrome or pseudo-color (gated to YFP acceptor levels), showing sensitized FRET within cells. The subtraction coefficients are rounded up from average cross-bleed values determined in cells expressing CFP- or YFP-tagged constructs alone. Thus, these coefficients result in the underestimation of FRETC signals for true FRET partners but prevent false positive detection of FRET.
Independent YFP acceptor photobleaching FRET measurement of CFP donor quenching was performed in fixed cells mounted without anti-fade reagent. YFP was selectively bleached by excitation at 535 nM for 2 min. The prebleach CFP image was then subtracted from the post-bleach CFP image to generate a pseudo-color image showing CFP quenching (ΔCFP) due to FRET (gated to post-bleach CFP levels). Calculation of CFP quenching apparent FRET efficiency values from the photobleaching method and normalized FRETNC values from the micro-FRET method used Slidebook mask analysis. CFP quenching apparent FRET efficiency values were obtained by dividing the mean intensity for change in CFP fluorescence after YFP photobleaching by the post-bleach CFP mean intensity (CFPpostbleach − CFPprebleach = ΔCFP/CFPpostbleach) for masks covering entire cells. Values from multiple cells were averaged to give the mean apparent FRET efficiency expressed as a percentage ± SEM. FRETC intensity values were obtained by fractional subtraction (using coefficients described above) of mean intensities for CFP and YFP from raw CYFRET for masks covering membrane ruffles and vesicles. FRETC was converted to FRETNC by dividing with the product of CFP and YFP intensities (FRETNC = FRETC/CFP × YFP). FRETNC values were averaged for multiple cells to generate mean FRETNC values ± SEM (×10−5) (Table I). Negative FRETNC values are obtained when there is no FRET due to the overcorrection for CFP and YFP cross-bleeding. FRETNC is proportional to the equilibrium constant for the binding interaction (Gordon et al., 1998; Sorkin et al., 2000).
Acknowledgments
Thanks to Dr. Roger Tsien (University of California, San Diego, CA) for providing pRSET-EC(Y)FP and Greg Scott in the laboratory of Dr. John D. Scott (Oregon Health and Science University) for assistance in constructing pEC(Y)FPN1,2,3; Dr. Iain Fraser (California Institute of Technology, Pasadena, CA) for providing the AKAP18(1-16)–GFP vector; Drs. Marcie Colledge and Morgan Sheng for providing pCMVneo-myc–SAP97; and Dr. Yvonne Lai (Icos Corporation, Bothel, WA) for providing anti-AKAP79 (918I). Thanks also to Shuvo Alam, Dr. Jessica Gorski, and Eric Horne (University of Colorado Health Sciences Center) for technical assistance. Final thanks to Alexander Sorkin for providing advice regarding FRET imaging.
This work was supported by grants from the American Heart Association (0130228N) and the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS40701) to M.L. Dell'Acqua.
Footnotes
Abbreviations used in this paper: AKAP, A-kinase anchoring protein; AMPA, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; CaN, calcineurin; CaNA, calcineurin catalytic A subunit; FRET, fluorescence resonance energy transfer; LTD, long-term depression; LTP, long-term potentiation; MAGUK, membrane-associated guanylate kinase; NMD, N-methyl-d-aspartate; PDZ, postsynaptic density-95, discs large, zona occludens-1; PSD, postsynaptic density; R, regulatory; SAP97, synapse-associated protein 97.
References
- Banke, T.G., D. Bowie, H.K. Lee, R.L. Huganir, A. Schousboe, and S.F. Traynelis. 2000. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 20:89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie, E.C., R.C. Carroll, X. Yu, W. Morishita, H. Yasuda, M. von Zastrow, and R.C. Malenka. 2000. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci. 3:1291–1300. [DOI] [PubMed] [Google Scholar]
- Bregman, D.B., N. Bhattacharyya, and C.S. Rubin. 1989. High affinity binding protein for the regulatory subunit of cAMP-dependent kinase IIβ: cloning, characterization and expression of cDNAs for rat brain P150. J. Biol. Chem. 264:4648–4656. [PubMed] [Google Scholar]
- Carr, D.W., R.E. Stofko-Hahn, I.D.C. Fraser, R.D. Cone, and J.D. Scott. 1992. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins: characterization of AKAP79. J. Biol. Chem. 267:16816–16823. [PubMed] [Google Scholar]
- Carroll, R.C., E.C. Beattie, M. von Zastrow, and R.C. Malenka. 2001. Role of AMPA receptor endocytosis in synaptic plasticity. Nat. Rev. Neurosci. 2:315–324. [DOI] [PubMed] [Google Scholar]
- Coghlan, V.M., B.A. Perrino, M. Howard, L.K. Langeberg, J.B. Hicks, W.M. Gallatin, and J.D. Scott. 1995. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science. 267:108–112. [DOI] [PubMed] [Google Scholar]
- Colledge, M., R.A. Dean, G.K. Scott, L.K. Langeberg, R.L. Huganir, and J.D. Scott. 2000. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 27:107–119. [DOI] [PubMed] [Google Scholar]
- Dell'Acqua, M.L., M.C. Faux, J. Thorburn, A. Thorburn, and J.D. Scott. 1998. Membrane targeting sequences on AKAP79 bind phosphatidylinositol-4,5-bisphosphate. EMBO J. 17:2246–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua, M.L., K.L. Dodge, S.J. Tavalin, and J.D. Scott. 2002. Mapping the protein phosphatase-2B anchoring site on AKAP79: binding and inhibition of phosphatase activity are mediated through residues 315-360. J. Biol. Chem. 277:48796–48802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers, M.D. 2000. Reinsertion and degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 28:511–525. [DOI] [PubMed] [Google Scholar]
- Gomez, L.L., S. Alam, K.E. Smith, E.H. Horne, and M.L. Dell'Acqua. 2002. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J. Neurosci. 22:7027–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, G.W., G. Berry, X.H. Liang, B. Levine, and B. Herman. 1998. Quantitative fluorescence energy transfer measurements using fluorescence microscopy. Biophys. J. 74:2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, Y., S.-H. Shi, J.A. Esteban, A. Piccini, J.-C. Poncer, and R. Malinow. 2000. Driving AMPA receptors by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 287:2262–2267. [DOI] [PubMed] [Google Scholar]
- Kashishian, A., M. Howard, C. Loh, M. Gallatin, M.F. Hoekstra, and Y. Lai. 1998. AKAP79 inhibits calcineurin through a site distinct from the immunophilin-binding region. J. Biol. Chem. 273:27412–27419. [DOI] [PubMed] [Google Scholar]
- Klauck, T.M., M.C. Faux, K. Labudda, L.K. Langeberg, S. Jaken, and J.D. Scott. 1996. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 271:1589–1592. [DOI] [PubMed] [Google Scholar]
- Lee, H.-K., M. Barbarosie, K. Kameyama, M.F. Bear, and R.L. Huganir. 2000. Regulation of AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 405:955–959. [DOI] [PubMed] [Google Scholar]
- Leonard, A.S., M.A. Davare, M.C. Horne, C.C. Garner, and J.W. Hell. 1998. SAP97 is associated with the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J. Biol. Chem. 273:19518–19524. [DOI] [PubMed] [Google Scholar]
- Lin, J.W., W. Ju, K. Foster, S.H. Lee, G. Ahmadian, M. Wyszynski, Y.T. Wang, and M. Sheng. 2000. Distinct molecular mechanisms and divergent endocytic pathways of AMPA receptor internalization. Nat. Neurosci. 3:1282–1290. [DOI] [PubMed] [Google Scholar]
- Lue, R.A., S.M. Marfatia, D. Branton, and A.H. Chisti. 1994. Cloning and characterization of hldg: the human homologue of the Drosophila discs large tumor supressor binds to protein 4.1. Proc. Natl. Acad. Sci. USA. 91:9818–9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki, A., and R.Y. Tsien. 2000. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327:472–500. [DOI] [PubMed] [Google Scholar]
- Nagai, Y., R. Aoki, T. Zama, S. Inouye, K. Hirose, M. Iino, and M. Hagiwara. 2000. A fluorescent indicator for visualizing cAMP-induced phosphorylation in vivo. Nat. Biotechnol. 18:313–316. [DOI] [PubMed] [Google Scholar]
- Newlon, M.G., M. Roy, D. Morikis, D.W. Carr, R. Westphal, J.D. Scott, and P.A. Jennings. 2001. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 20:1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro, M., V. Piech, and M. Sheng. 2001. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat. Neurosci. 4:917–926. [DOI] [PubMed] [Google Scholar]
- Rosenmund, C., D.W. Carr, S.E. Bergeson, G. Nilaver, J.D. Scott, and G.L. Westbrook. 1994. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 368:853–856. [DOI] [PubMed] [Google Scholar]
- Ruehr, M.L., D.R. Zakhary, D.S. Damron, and M. Bond. 1999. Cyclic AMP-dependent protein kinase binding to A-kinase anchoring protein in living cells by fluorescence resonance energy transfer of green fluorescent protein fusions. J. Biol. Chem. 274:33092–33096. [DOI] [PubMed] [Google Scholar]
- Reuver, S.M., and C.C. Garner. 1998. E-cadherin mediated cell adhesion recruits SAP97 into the cortical cytoskeleton. J. Cell Sci. 111:1071–1080. [DOI] [PubMed] [Google Scholar]
- Shen, L., F. Liang, L.D. Walensky, and R.L. Huganir. 2000. Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J. Neurosci. 20:7932–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, M., and S.H. Lee. 2001. AMPA receptor trafficking and the control of synaptic transmission. Cell. 105:825–828. [DOI] [PubMed] [Google Scholar]
- Sheng, M., and C. Scala. 2001. PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 24:1–29. [DOI] [PubMed] [Google Scholar]
- Shi, S.-H., H. Hayashi, J.A. Esteban, and R. Malinow. 2001. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 105:331–343. [DOI] [PubMed] [Google Scholar]
- Smith, F.D., and J.D. Scott. 2002. Signaling complexes: junctions on the intracellular information super highway. Curr. Biol. 12:R32–R40. [DOI] [PubMed] [Google Scholar]
- Sorkin, A., M. McClure, F. Huang, and R. Carter. 2000. Interaction of EGF receptor and Grb2 in living cells visualized by fluorescence resonance energy transfer microscopy. Curr. Biol. 10:1395–1398. [DOI] [PubMed] [Google Scholar]
- Tavalin, S.J., M. Colledge, J.W. Hell, L.K. Langeberg, R.L. Huganir, and J.D. Scott. 2002. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J. Neurosci. 22:3044–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffany, A.M., L.N. Manganas, E. Kim, Y.-P. Hsueh, M. Sheng, and J.S. Trimmer. 2000. PSD-95 and SAP97 exhibit distinct mechanisms for regulating K+ channel surface expression and clustering. J. Cell Biol. 148:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita, S., R.A. Nicoll, and D.S. Bredt. 2001. PDZ protein interactions regulating glutamate receptor function and plasticity. J. Cell Biol. 153:F19–F23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter, K.W., I.D.C. Fraser, G.K. Scott, M.J. Stutts, J.D. Scott, and S.L. Milgram. 1999. Alternative splicing regulates the subcellular localization of A-kinase anchoring protein 18 isoforms. J. Cell Biol. 147:1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien, R.Y. 1998. Green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- Wu, H., S.M. Reuver, S. Kuhlendahl, W.J. Chung, and C.C. Garner. 1998. Subcellular targeting and cytoskeletal attachment of SAP97 to the epithelial lateral membrane. J. Cell Sci. 111:2365–2376. [DOI] [PubMed] [Google Scholar]
- Zaccolo, M., and T. Pozzan. 2002. Discrete microdomains with high concentrations of cAMP in stimulated rat neonatal cardiac myocytes. Science. 295:1711–1715. [DOI] [PubMed] [Google Scholar]
- Zaccolo, M., F. De Giorgi, C.Y. Cho, L. Feng, T. Knapp, P.A. Negulescu, S.S. Taylor, R.Y. Tsien, and T. Pozzan. 2000. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2:25–29. [DOI] [PubMed] [Google Scholar]
- Zhang, J., M. Yulinag, S.S. Taylor, and R.Y. Tsien. 2001. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA. 98:14997–15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Q., M.-Y. Xiao, and R.A. Nicoll. 2001. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA. 98:1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]