

Abstract
Formation of senile plaques containing the β-amyloid peptide (Aβ) derived from the amyloid precursor protein (APP) is an invariant feature of Alzheimer's disease (AD). APP is cleaved either by β-secretase or by α-secretase to initiate amyloidogenic (release of Aβ) or nonamyloidogenic processing of APP, respectively. A key to understanding AD is to unravel how access of these enzymes to APP is regulated. Here, we demonstrate that lipid rafts are critically involved in regulating Aβ generation. Reducing cholesterol levels in N2a cells decreased Aβ production. APP and the β-site APP cleavage enzyme (BACE1) could be induced to copatch at the plasma membrane upon cross-linking with antibodies and to segregate away from nonraft markers. Antibody cross-linking dramatically increased production of Aβ in a cholesterol-dependent manner. Aβ generation was dependent on endocytosis and was reduced after expression of the dynamin mutant K44A and the Rab5 GTPase-activating protein, RN-tre. This inhibition could be overcome by antibody cross-linking. These observations suggest the existence of two APP pools. Although APP inside raft clusters seems to be cleaved by β-secretase, APP outside rafts undergoes cleavage by α-secretase. Thus, access of α- and β-secretase to APP, and therefore Aβ generation, may be determined by dynamic interactions of APP with lipid rafts.
Keywords: lipid rafts; β-amyloid; BACE; Alzheimer's disease; endocytosis
Introduction
Formation of senile plaques composed of a 4-kD small peptide, the amyloid β-peptide (Aβ)* is one of the hallmarks of Alzheimer's disease (AD). Aβ derives from a large type I transmembrane protein, the amyloid precursor protein (APP) (for review see Selkoe, 2001). It is cleaved out sequentially by enzymes termed β- and γ-secretase. The β-site APP cleavage enzyme (BACE1) has been identified recently as a novel membrane-bound aspartyl-protease (De Strooper and Annaert, 2000; Esler and Wolfe, 2001) and cleaves APP in its luminal domain to generate a secreted ectodomain (βAPP). The remaining 10-kD β-cleaved COOH-terminal stub of APP (βCTF) fragment is subsequently the substrate for γ-secretase, which cleaves the transmembrane domain of APP to release Aβ. γ-Secretase is a multiprotein complex, requiring presenilins-1 and -2 for activity (De Strooper and Annaert, 2000; Weihofen et al., 2002). A third enzyme, the α-secretase, cleaves APP in the middle of the Aβ region to generate a secreted ectodomain (αAPP) and a shorter α-cleaved COOH-terminal stub of APP (αCTF) that is also cleaved by γ-secretase. α-secretase activity was shown to be associated with members of the ADAM (a disintegrin and metalloprotease) family (ADAM 9, 10, and 17) (Buxbaum et al., 1998; Koike et al., 1999; Lammich et al., 1999). α-Cleavage is the dominant processing step, and since it cuts APP within the Aβ region it prevents Aβ formation. Since α- and β-cleavages directly compete for their substrate APP, the key in understanding Aβ generation is to find out how access of these enzymes to APP is regulated.
There is growing evidence that cholesterol is of particular importance in regulating α- and β-cleavage (Simons et al., 2001). The E4 allele of apolipoprotein E has been shown to be a major risk factor for AD (Corder et al., 1993; Strittmatter et al., 1993), levels of total cholesterol and LDL in serum were reported to correlate with the amount of Aβ in AD brains (Kuo et al., 1998), and there is epidemiological evidence that elevated cholesterol levels during mid-life increase the risk of developing AD (Kivipelto et al., 2001). Elevated dietary cholesterol uptake increased amyloid plaque formation in rabbits and transgenic mice (Sparks et al., 1994; Refolo et al., 2000), and cholesterol loading and depletion affected Aβ generation in cultured cells and in an animal model (Simons et al., 1998; Fassbender et al., 2001). There is also a correlation between cellular cholesteryl-ester levels and Aβ production (Puglielli et al., 2001), and it was demonstrated that aggregated Aβ binds cholesterol in vitro (Avdulov et al., 1997). Interestingly, two independent retrospective studies reported a strong decrease in the incidence of AD and dementia in patients treated with 3-hydroxy-3-methylglutaryl–coenzyme A reductase inhibitors (Jick et al., 2000; Wolozin et al., 2000).
All of these studies point out that cholesterol is critically involved in Aβ generation. However, little is known about the mechanisms by which cholesterol affects this process. We previously hypothesized that the association of APP with lipid rafts determines Aβ production (Simons et al., 1998). Rafts are lateral assemblies of sphingolipids and cholesterol within the membrane (for review see Simons and Toomre, 2000). They are thought to form ordered platforms, which float around in the liquid-disordered matrix of the cellular membrane and represent versatile devices to compartmentalize membrane processes. Biochemically, the components of lipid rafts are characterized by their insolubility in detergents such as Triton X-100 or CHAPS at 4°C (Fiedler et al., 1993; Brown and London, 1997). A fraction of APP and BACE1 were shown to be associated with detergent-resistant membranes (DRMs) in a cholesterol-dependent manner (Bouillot et al., 1996; Simons et al., 1998; Riddell et al., 2001). α-Secretase cleavage, on the other hand, was elevated after inhibition of β-secretase activity by cholesterol depletion, and ADAM 10, a putative α-secretase, was soluble after detergent extraction (Kojro et al., 2001). The most straightforward interpretation of these data is that APP is present in two cellular pools, one associated with lipid rafts where Aβ is generated and another outside of rafts where α-cleavage takes place. This model of membrane compartmentalization would explain how the same protein could be processed in two different mutually exclusive ways.
If cleavage of APP by BACE1 occurred in rafts, it would be important to know how and where this interaction is regulated. Therefore, in this paper we have studied these relationships and provide evidence that Aβ generation critically depends on lipid rafts for enzyme activation to occur.
Results
Cholesterol depletion inhibits β-cleavage in N2a cells
To study the significance of lipid rafts for APP processing, we have used a neuronal cell line, mouse neuroblastoma N2a. We first analyzed the effect of cholesterol depletion on APP processing, which was done by a combination of lovastatin treatment and methyl-β-cyclodextrin (MβCD) extraction. Lovastatin in the presence of small amounts of mevalonate decreases de novo synthesis of cholesterol by inhibiting 3-hydroxy-3-methylglutaryl–coenzyme A reductase and MβCD extracts plasma membrane cholesterol. N2a cells were grown for 36 h in lipid-deficient FCS in the presence of lovastatin, and immediately before metabolic labeling they were treated with 10 mM MβCD for 5–30 min. Depending on the time of extraction, total cellular cholesterol levels could be reduced to 15% of control cells. Increasing time of exposure and concentrations of MβCD or prolonged treatment with lovastatin started to affect cell viability.
To easily monitor the influence of cholesterol depletion on APP processing, N2a cells were infected with adenoviruses to express either human wild-type APP (wtAPP) or the Swedish mutant of APP (swAPP). swAPP is dominantly β cleaved, resulting in a several-fold higher production of Aβ than for wtAPP. After MβCD extraction, the cells were metabolically labeled with [35S]methionine and chased for up to 2 h. Immunoprecipitations from conditioned medium revealed that Aβ production was dependent on cellular cholesterol levels (Fig. 1 A). Decreasing cellular cholesterol by 85% totally abolished Aβ secretion. Remarkably, already relatively small changes in total cellular cholesterol levels were found to have strong effects. A 20–30% decrease showed a 50–60% reduction in Aβ secretion. βCTF, which is generated by β-cleavage, was also clearly reduced. On the other hand, the production of αCTF by α-cleavage was increased (Fig. 1 B). As expected from results of Kojro et al. (2001), the soluble ectodomain generated by α-cleavage was also strongly increased in cholesterol-depleted cells. In general, processing of wtAPP and swAPP were similarly affected by cholesterol depletion.
Figure 1.

Cholesterol depletion inhibits β-cleavage. (A) N2a cells were grown in the presence (+) or absence (−) of lovastatin/ mevalonate/delipidated FCS and 10 h after infection with adenoviruses to express wtAPP treated with 10 mM MβCD for the indicated times. Cells were then labeled with [35S]methionine for 40 min and chased for 2 h. Immunoprecipitations of extracellular medium (Aβ; antibody 70JE) and cell lysate (total wtAPP; antibody IP60) revealed that Aβ secretion decreased after cholesterol depletion. The extent of cholesterol depletion was determined as described in Materials and methods. (B) 10 h after infection with adenoviruses to express swAPP, N2a cells were labeled for 30 min and chased for 30 min in the presence of 10 mM MβCD (leading to an ∼40–50% decrease in total cellular cholesterol). Immunoprecipitations from extracellular medium (antibody 6E10) and cell lysate (antibody IP60) showed a decreased production of Aβ and the COOH-terminal fragment generated by β-cleavage (βCTF). At the same time, the COOH-terminal fragment generated by α-cleavage (αCTF) and the soluble ectodomain generated by α-cleavage (αAPPs) were increased.
These results show that cholesterol depletion of N2a cells inhibits β-cleavage, whereas α-cleavage is increased. They suggest that cholesterol is critically involved in regulating the access of α- and β-secretase to APP.
APP and BACE copatch with placental alkaline phosphatase but segregate from transferrin receptor
Lipid rafts are most abundant in the plasma membrane. In fibroblasts, individual rafts have a size of ∼50 nm, corresponding to ∼3,500 sphingolipid molecules and probably not more than 10–30 proteins (Pralle et al., 2000). This means that two different species of raft proteins would rarely be in the same raft. However, it was shown previously that raft and nonraft markers could be cross-linked with antibodies into distinct patches (Harder et al., 1998; Janes et al., 1999; Prior et al., 2001). Raft markers copatch and segregate away from nonraft markers. Cross-linking with antibodies can thus be used as an assay for raft association. We tested whether antibody cross-linking induced copatching of APP and BACE1 with a raft marker, the glycosyl phosphatidylinositol (GPI)-anchored protein placental alkaline phosphatase (PLAP). As a nonraft marker, we used a mutant human transferrin receptor (TfR), where the cytosolic aa 5–41 (TfR del 5–41) have been removed. This mutant is defective in endocytosis due to the deletion of the sorting signal. Patches of TfR del 5–41 were shown to be segregated from components found in lipid rafts (Harder et al., 1998).
Our experiments were performed with BACE1A-CFP and YFP-wtAPP. CFP and YFP are the cyan and yellow color variants of the green fluorescent protein, respectively. Control experiments demonstrated that these fluorescent protein (FP)-containing constructs showed the same proteolytic processing and immunofluorescence behavior as the corresponding untagged proteins (unpublished data). BACE1A-CFP was cross-linked with the polyclonal antibody 7523 recognizing the NH2-terminal part of BACE1 (Capell et al., 2002). YFP-wtAPP was cross-linked with antiserum KG77 or mouse monoclonal antibody 3E6, both directed against the FP. Control experiments with wtAPP or BACE1A-VSVG and anti-APP antibody 5313 or anti-BACE1 antibody 7523 showed essentially the same patching (unpublished data). Also, we did not see significant differences in staining of swAPP and wtAPP.
Both BACE1 and wtAPP colocalized with PLAP at the plasma membrane in the majority of cells, but they clearly segregated from TfR del 5–41 (Fig. 2). BACE1 and wtAPP could also be localized to the same patches upon cross-linking (Fig. 3). For quantitative analyses of the extent of copatching, images of 10 randomly selected cells on one coverslip were taken and assigned into four categories: (1) coclustering (>80% overlap); (2) partial coclustering (clearly overlapping spots 30–80%); (3) random distribution, and (4) segregation. The data from five independent experiments (Fig. 4) indicate that cross-linked wtAPP and BACE1 copatched with the raft protein PLAP and segregated from the nonraft protein TfR del 5–41.
Figure 2.

Copatching of PLAP and TfR del 5–41 with YFP-wtAPP and BACE1A-CFP. 10 h after transient transfection, the cells were incubated for 45 min at 10°C with the primary antibodies. Patching of YFP-wtAPP and BACE1A-CFP was achieved with polyclonal antibodies KG77 and 7523, respectively. PLAP and TfR del 5–41 were patched with mouse monoclonal antibodies from Dako and Roche, respectively. Thereafter, the cells were washed and incubated for 45 min with mixed Cy5- and Cy3-labeled secondary antibodies. (A) YFP-wtAPP and BACE1A-CFP segregate from TfR del 5–41. (B) Colocalization of cross-linked YFP-wtAPP and BACE1A-CFP with PLAP. Bar, 10 μm.
Figure 3.

Copatching of YFP-wtAPP and BACE1A-VSVG at the cell surface. (A) Immunofluorescence of BACE1A-VSVG using polyclonal antiserum 7523 against the NH2-terminal part of BACE1 (green) and YFP-wtAPP using mAb 3E6 against the FP (red). Both proteins were randomly distributed at the cell surface when the antibody was added after fixation. (B) Colocalization of YFP-wtAPP (red) and BACE1A-VSVG (green) after antibody induced patching with the same antibodies as above. Bar, 10 μm.
Figure 4.

Quantification of copatching of PLAP, TfR del 5–41, BACE1A-CFP, and YFP-wtAPP. Patches of different proteins were scored into four groups: (1) copatching (>80% overlap), (2) partial copatching (30–80%, clearly overlapping patches), (3) random distribution, and (4) segregation. The percentages of cells belonging to each group are expressed as mean ± SD (n = 5).
Antibody induced cross-linking increases association to detergent-resistant membranes
Different proteins associate with rafts with different kinetics and partition coefficients. Antibody-induced patching may stabilize association of raft proteins with DRMs (Harder et al., 1998; Janes et al., 1999). Thus, oligomerization, i.e., by antibody cross-linking, can be used to monitor specific raft lipid–protein interactions. Therefore, we investigated the association of APP and BACE1 with DRMs under cross-linking conditions. Association of a protein with DRMs is shown by its insolubility in detergents such as Triton X-100 or CHAPS at 4°C (Fiedler et al., 1993; Brown and London, 1997), which leads to flotation to low densities in sucrose or OptiPrep gradients.
Initial experiments revealed that in N2a cells only a minor amount (<5%) of both APP and BACE1 were resistant to extraction with 1% Triton X-100. However, when the cells were extracted with 20 mM CHAPS a significantly higher amount of BACE1 and APP floated to the low density membrane fraction in a cholesterol-dependent manner (unpublished data). Therefore, we used CHAPS-extracted membranes to examine the effect of antibody-induced patching on DRM association. N2a cells were infected with adenoviruses to express YFP-swAPP or BACE1A-CFP, metabolically labeled for 2 h with [35S]methionine, and chased for 2 h in the absence of antibody or in the presence of anti-FP (KG77) or anti-BACE1 (7523) antibodies, respectively. Cells were then extracted with 20 mM CHAPS, and the detergent extracts were subjected to OptiPrep step gradient centrifugation. A significantly higher fraction of APP and BACE1 floated with DRMs after antibody-induced patching (Fig. 5). Quantification revealed that without cross-linking ∼18.0 ± 2.6% of APP (n = 3) and 24.6 ± 2.3% of BACE1 (n = 4) were found in the upper two fractions (DRMs). Antibody cross-linking increased the DRM-associated fraction to 25.1 ± 1.2% (n = 3) and 32.3 ± 0.7% (n = 4) of APP and BACE1, respectively. Thus, both APP and BACE1 increased their detergent resistance upon cross-linking, probably reflecting increased raft affinity caused by oligomerization. Similar results have been obtained for other raft proteins, which increase their raft association by forming oligomers (Simons and Toomre, 2000; Cheng et al., 2001).
Figure 5.
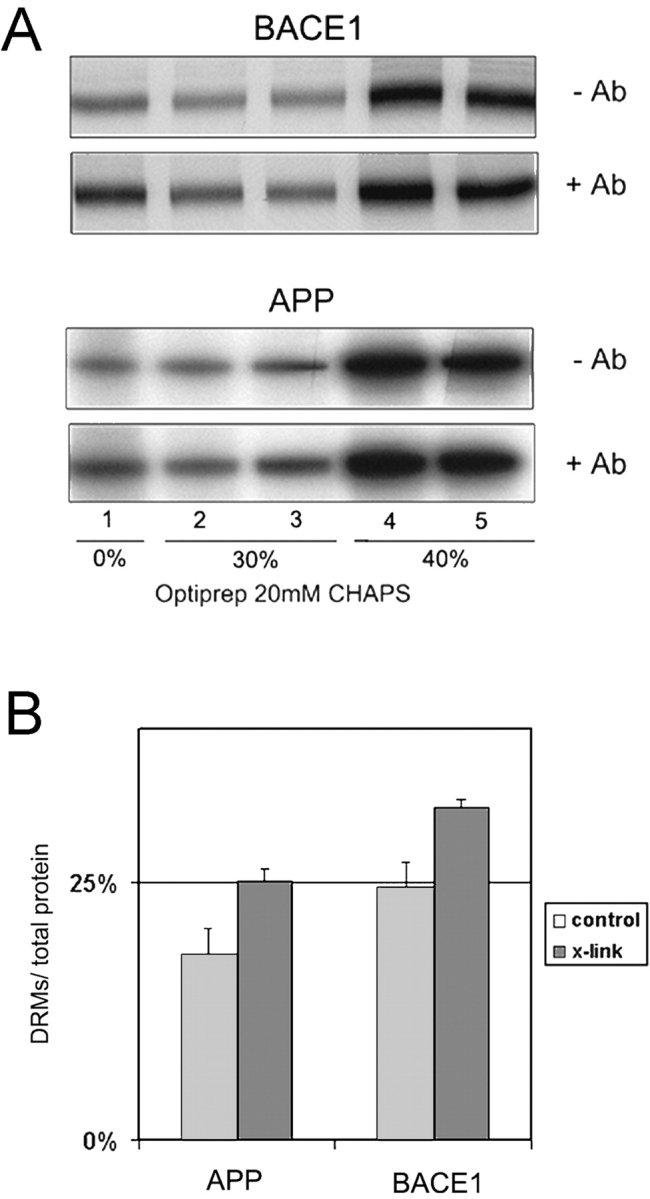
Effect of antibody cross-linking on association of BACE1A-CFP and YFP-swAPP to DRMs. 10 h after adenovirus infection to express BACE1A-CFP or YFP-swAPP, the cells were labeled for 2 h with [35S]methionine and chased for 2 h in the presence of antibody KG77 (anti-FP) or antibody 7523 (anti-BACE1). The cells were subsequently lysed in 20 mM CHAPS/TNE at 4°C. (A) After flotation in an OptiPrep step gradient, BACE1A-CFP and YFP-swAPP were immunoprecipitated with antibody KG77 from the collected fractions. (B) Quantification; antibody-induced patching significantly increased the amount of APP (n = 3) and of BACE1 (n = 4) in the top two fractions (DRM associated). The amount in the top two fractions was correlated to the total amount of protein in all fractions.
Cross-linking with antibodies increases Aβ formation
If β-cleavage were to take place in cholesterol/sphingolipid-enriched microdomains, then antibody cross-linking should not only increase the association of APP and BACE1 with DRMs and induce their copatching at the surface of living cells, but cross-linking should also increase Aβ production. To find out whether this is the case, we analyzed the effect of antibody cross-linking on Aβ secretion. Cells were infected with adenoviruses to express YFP-wtAPP and BACE-VSVG. They were metabolically labeled for 40 min with [35S]-methionine and chased for 2 h in the presence of antibodies KG77 (anti-FP), 7523 (anti-BACE1), or both. Antibody cross-linking increased Aβ secretion significantly (Fig. 6, A and B).
Figure 6.

Effect of antibody cross-linking and cholesterol depletion on Aβ secretion. (A) Cells were infected with adenoviruses to express YFP-wtAPP and BACE1-VSVG, metabolically labeled for 40 min with [35S]methionine, and chased for 2 h in the presence of the indicated antibodies. YFP-wtAPP was cross-linked with anti-FP antibody KG77, and BACE1 was cross-linked with antibody 7523. (B) Quantification of the two independent experiments of A. The ratio was arbitrarily set to 100% in cells not cross-linked with antibodies. Secreted Aβ was normalized to the total amount of APP found in the cell lysate. (C) Aβ secreted from cross-linked/cholesterol-depleted cells. N2a cells were grown in the presence (+depletion) or absence (−depletion) of lovastatin/mevalonate/lipid-deficient FCS. 10 h after infection with adenoviruses to express YFP-wtAPP, the cells were treated for 5 min with 10 mM MβCD, labeled for 40 min with [35S]methionine, and chased for 2 h in the presence (+Ab) or absence (−Ab) of anti-FP antibody KG77. (D) Quantification of five independent experiments. The amount of secreted Aβ was normalized to the total amount of APP present in the cell lysate. The ratio was arbitrarily set to 100% in cells neither cross-linked nor cholesterol depleted.
We next examined the effect of cholesterol depletion on antibody-induced Aβ production. Cells were grown in the presence of lovastatin as before and treated immediately before labeling for 5 min with 10 mM MβCD. This procedure depleted total cellular cholesterol by 50–60%. Antibody cross-linking did not stimulate Aβ secretion in cholesterol-depleted cells (Fig. 6, C and D). Thus, under conditions where rafts were disrupted increased amyloidogenic processing of APP was no longer detectable.
Endocytosis is essential for β-cleavage
Antibody cross-linking might not only lead to copatching of raft components, it could also alter endocytosis of cross-linked proteins. Previous studies suggest that endocytosis is required for Aβ generation (Koo and Squazzo, 1994; Perez et al., 1999; Huse et al., 2000; Daugherty and Green, 2001). Therefore, we decided to inhibit endocytosis by transiently expressing RN-tre or the dynamin II mutant K44A in N2a cells. RN-tre is a Rab5-specific GTPase-activating protein and inhibits clathrin-dependent endocytosis (Lanzetti et al., 2000). Dynamin is involved in fission of vesicles from the plasma membrane. It was shown that expression of the mutant K44A inhibits both clathrin-dependent and some clathrin-independent endocytotic pathways (Damke et al., 1994; Henley et al., 1998).
N2a cells were transiently transfected with equal amounts of plasmids encoding for swAPP, RN-tre, or dynamin K44A and labeled for 1 h with [35S]methionine. Immunoprecipitations from cell lysates with antibody IP60 (raised against the COOH terminus of APP) and from media with antibody 70JE (Aβ) were performed (Fig. 7 A). APP biosynthesis was unchanged after expression of RN-tre or dynamin K44A; however, the COOH-terminal fragment generated by β-cleavage (βCTF) and secretion of Aβ were significantly reduced. Expression of dynamin K44A inhibited Aβ secretion by 80–90% (Fig. 7 A). Remarkably, the membrane-bound fragment generated by α-cleavage (αCTF) was only slightly increased (correlated to total APP). Thus, under our experimental conditions endocytosis was essential for β-cleavage to occur, whereas α-cleavage was not appreciably stimulated by inhibiting endocytosis.
Figure 7.
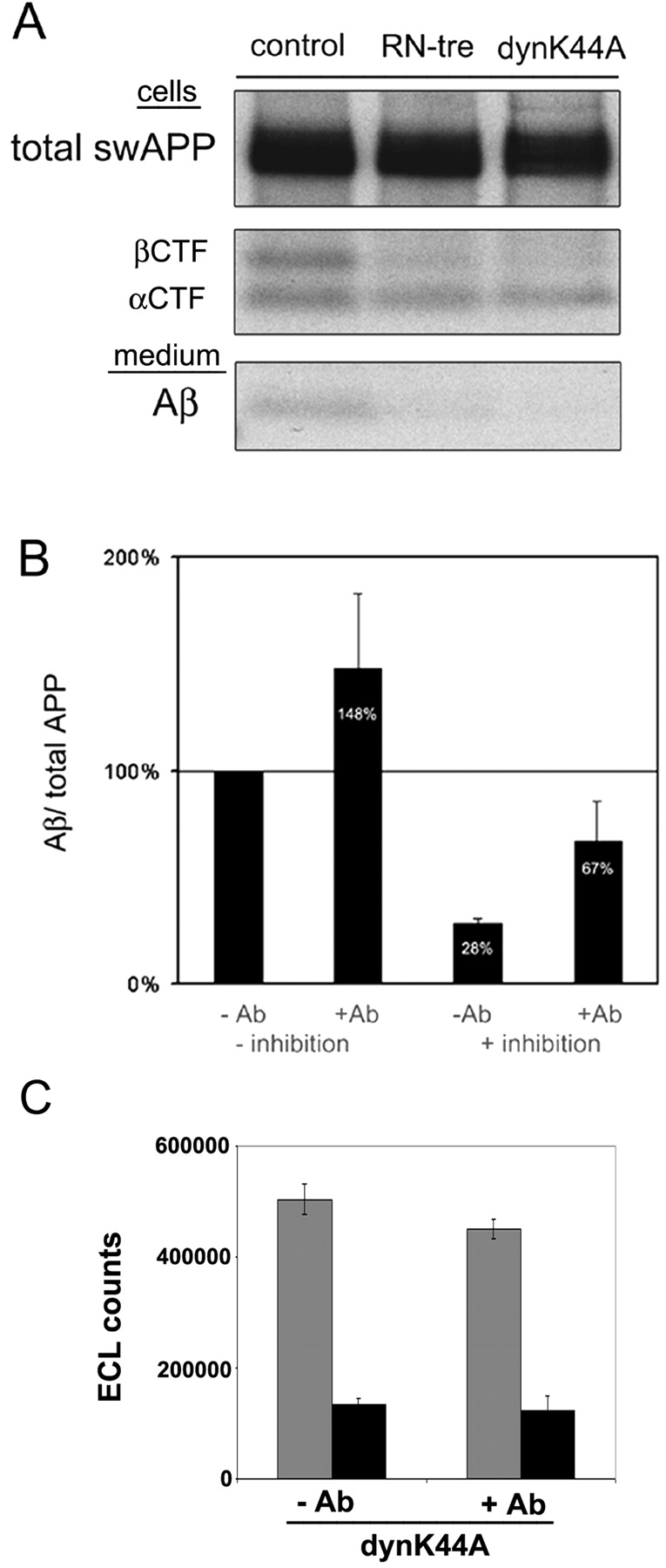
Endocytosis is essential for the generation of Aβ, and antibody cross-linking overcomes the block caused by inhibition of endocytosis. (A) Inhibition of endocytosis and APP processing. After transient transfection with YFP-swAPP and RN-tre or dynamin K44A, N2a cells were metabolically labeled for 1 h with [35S]methionine. Immunoprecipitations from cell lysate (swAPP, αCTF, βCTF; antibody IP60) and medium (Aβ; antibody 70JE) show a decrease of fragments generated by β-cleavage (βCTF and Aβ) upon expression of endocytosis inhibitors. (B) The effect of copatching of APP and BACE1 on Aβ secretion under conditions where endocytosis is inhibited. Cells were transfected with YFP-swAPP/BACE1A-VSVG/empty vector (−inhibition) or YFP-swAPP/BACE1A-VSVG/dynamin K44A (+inhibition), labeled with [35S]methionine for 40 min, and chased for 2 h in the presence (+Ab) or absence (−Ab) of antibodies KG77 (anti-FP) and 7523 (anti-BACE1). Quantification of three independent experiments showed a significant increase in Aβ generation upon copatching under both conditions. The values are normalized to the total amount of APP present in the cell lysate. The ratio has been arbitrarily set to 100% in nonpatched cells transfected with YFP-swAPP/BACE1A VSVG/empty vector. (C) Effect of antibody cross-linking on internalization of biotin transferrin under conditions where endocytosis is inhibited by expression of dynamin K44A. Cells were transfected with human transferrin receptor (TfR) and dynK44A-GFP, incubated on ice with biotin transferrin, and chased for 10 min at 37°C in the presence (+Ab) or absence (−Ab) of anti–human TfR antibodies. Biotin transferrin was quantified on an Origen M8 analyzer in cells where remaining surface transferrin was removed by acid wash (black columns) or controls (no acid wash; grey columns). Cross-linking did not increase internalization of transferrin.
Antibody-induced cross-linking stimulates formation of Aβ at the cell surface
If endocytosis is required for β-cleavage to occur and β-cleavage critically depends on lipid rafts, one could perhaps stimulate the cleavage already at the cell surface by cross-linking APP and BACE1-containing rafts and thus overcome the block of β-cleavage caused by inhibition of endocytosis. N2a cells were transfected with constructs to express dynamin K44A, YFP-swAPP, and BACE1A-VSVG, respectively, and pulse–chase experiments were performed as before. We blocked endocytosis and cross-linked APP and BACE1 by applying the respective ectodomain antibodies to living cells. Copatching of APP and BACE1 resulted in a significant increase in Aβ secretion under conditions of strict inhibition of endocytosis by dynamin K44A (Fig. 7 B). Cross-linking in cells expressing RN-tre revealed similar results (unpublished data). To control whether antibody cross-linking increased endocytosis after inhibiting internalization with dynamin K44A, we performed a control experiment using human transferrin receptor as our probe. Indeed cross-linking did not increase internalization of the transferrin receptor (Fig. 7 C), supporting our conclusion that we can stimulate Aβ production at the surface by raft patching.
Discussion
The data presented here strengthen the evidence that both APP and BACE1 partition into lipid rafts in cellular membranes and that amyloidogenic processing seems to occur raft associated. First of all, we could demonstrate that Aβ production was critically dependent on the integrity of lipid rafts. Lipid rafts are cholesterol- and sphingolipid-enriched microdomains within cellular membranes, and removal of raft lipids from cells leads to disruption of raft functions (Simons and Toomre, 2000). Consistent with previous results, we could demonstrate that a decrease of cellular cholesterol levels inhibited Aβ generation in N2a cells. The involvement of rafts in Aβ production is further supported by previous work showing that cholesterol and GM1 both bind to Aβ and facilitate amyloid fibril formation (Yanagisawa and Ihara, 1998; Ariga et al., 2001; Kakio et al., 2001). Like many other raft-associated proteins, BACE1 is also palmitoylated at three cysteine residues within its transmembrane/cytosolic tail (Benjannet et al., 2001) and is mainly apically sorted in epithelial cells (Capell et al., 2002).
Another important result in support of raft association was that after antibody-induced cross-linking both APP and BACE1 copatched on the surface of living cells with each other and with PLAP, a GPI-anchored raft-associated protein. All three proteins segregated from patches containing cross-linked transferrin receptor, which served as a nonraft marker. We and others have previously used this assay to monitor how proteins associate with rafts at the cell surface (Harder et al., 1998; Janes et al., 1999; Prior et al., 2001). Antibody cross-linking of raft surface antigens leads to the formation of large clusters, which are easily observable in the light microscope. Copatching is dependent on cholesterol, since patching is inhibited by cholesterol removal.
Importantly, antibody cross-linking increased the DRM association of both APP and BACE1, as was previously shown for other raft-associated proteins (Harder et al., 1998). Because only a small fraction of APP and BACE1 associated with DRMs, these proteins are probably found (at steady state) in two membrane pools, one raft associated and another localized outside of rafts. How partitioning between these two pools is regulated is not clear. It has been reported that both APP and BACE1 can dimerize and that homodimerization of APP increases Aβ production (unpublished data; Scheuermann et al., 2001). Oligomerization of raft components can lead to increased raft affinity. Many surface receptors, such as Fc(ɛ) receptors and T and B cell receptors, dimerize or oligomerize after ligand binding, and this has been shown to increase association with DRMs (Janes et al., 2000; Langlet et al., 2000; Cheng et al., 2001). Therefore, dimerization might be important for regulating raft association of APP and BACE1.
Partitioning of APP and BACE1 into rafts alone seems not to be sufficient to induce β-cleavage. Cleavage normally also depends on endocytosis. This was demonstrated by our experiments designed to inhibit endocytosis. In two approaches, we either expressed RN-tre, a Rab5 GTPase-activating protein (Lanzetti et al., 2000), or the dynamin mutant K44A (Damke et al., 1994). The former perturbs clathrin-dependent endocytosis (Lanzetti et al., 2000) and the latter both clathrin-dependent and some clathrin-independent endocytic pathways (Damke et al., 1994; Henley et al., 1998). The results were clear cut: Aβ generation was strongly inhibited, whereas APP was still α cleaved. The latter was expected from reports demonstrating that α-cleavage occurs at the cell surface (Haass et al., 1992; Parvathy et al., 1999). Previous work has reported that β-cleavage may happen already late in the secretory pathway, or after delivery to the cell surface, and during endocytosis (Koo and Squazzo, 1994; Perez et al., 1999; Huse et al., 2000; Daugherty and Green, 2001; Kamal et al., 2001). Inhibition of endocytosis by our approaches suggests that, at least in N2a cells, most of this cleavage occurs after internalization. Since cholesterol depletion is also known to decrease the rate of endocytosis (Rodal et al., 1999), this is also likely to contribute to the decreased β-cleavage. However, the inhibitory effect on Aβ production by inhibition of endocytosis could be overcome by cross-linking surface APP and/or BACE1 with antibodies.
To account for these results, we envisage that β-cleavage would normally not take place at the cell surface because surface APP and BACE1 are most likely present in separate rafts (Fig. 8). Rafts are small and highly dispersed at the cell surface and are suggested to contain only a subset of ∼10–30 protein molecules (Pralle et al., 2000). Therefore, the likelihood is low that APP and BACE1 are in the same individual raft. For β-cleavage to occur rafts would have to be clustered to get APP and BACE1 into the same raft platform. Thus, we hypothesize that APP and BACE1 meet after endocytosis by clustering and coalescence of APP- or BACE1-containing rafts within endosomes (Fig. 8 A). How and where clustering is accomplished during internalization from the plasma membrane is not known. However, raft clustering can be artificially induced at the cell surface by cross-linking with antibodies (Fig. 8 B). This could lead to the increased β-cleavage in clusters/patches containing both APP and BACE1 that we had observed. Remarkably, we did not detect a dramatic increase in α-secretase processing of APP after inhibition of endocytosis. We assume that this is due to a continued raft association of a fraction of cell surface APP, which would not be accessible to α-cleavage. On the other hand, cholesterol depletion would shift the partitioning of APP from lipid rafts to the surrounding lipid bilayer and lead to the observed increase of α-cleavage.
Figure 8.
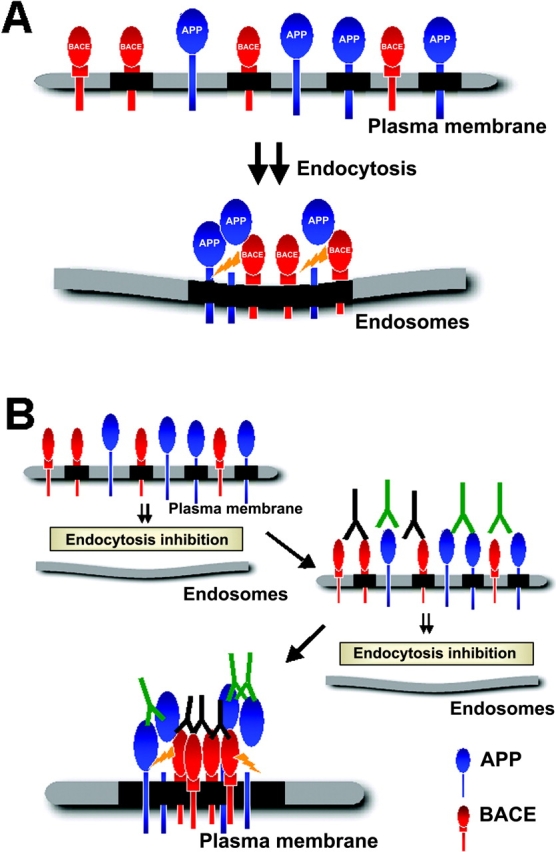
Model for raft clustering and β-cleavage of APP. (A) Under normal (steady state) conditions, two cellular pools of APP and presumably also of BACE1 exist at the plasma membrane. One is associated with rafts (black), and another is outside of rafts. Rafts are small and highly dispersed at the cell surface. Because they contain only a few proteins, APP and BACE1 are mainly localized in separate rafts at the plasma membrane. Endocytosis is necessary for APP- and BACE1-containing rafts to cluster for consecutive β-cleavage. We assume that raft APP is cleaved by raft BACE1. (B) If endocytic clustering is inhibited, e.g., by the expression of dynamin K44A, the block can be relieved by copatching with antibodies against BACE1 and APP. This leads to coalescence of rafts at the plasma membrane and results in β-cleavage. These conditions are postulated to mimic the process normally taking place after internalization.
The inhibition of β-cleavage by cholesterol depletion suggests that BACE1 processing of APP critically depends on the lipid raft environment. In living cells, BACE1 seems to require intact rafts for activity, and BACE1 outside of rafts appears to be inactive. Other such examples of raft-dependent processes are known. The conformational change of the cellular prion protein (PrPc) to its pathogenic scrapie form critically depends on the integrity of rafts (Taraboulos et al., 1995; Baron et al., 2002). Membrane vesicle transport (Klopfenstein et al., 2002), many signal transduction pathways, like Ras and GDNF signaling (Roy et al., 1999; Tansey et al., 2000), or T and B cell activation and allergic response mechanisms are raft dependent (Janes et al., 1999; Langlet et al., 2000; Cheng et al., 2001). However, clearly more work will be necessary to demonstrate whether β-cleavage of APP is indeed directly dependent on the raft lipid environment as postulated in the hypothetical scheme in Fig. 8. Also, the mechanisms regulating the trafficking and clustering of APP and BACE1 need to be identified. Nevertheless, our data support a crucial role for lipid rafts in APP processing and Aβ generation. Compartmentalization by lipid rafts seems to be important in regulating the access of APP to α- and β-secretases.
Also the γ-secretase complex was shown to be raft associated (Li et al., 2000; Wahrle et al., 2002). Moreover, the β-cleaved COOH-terminal fragment, the substrate for γ-cleavage, is found in DRMs and so is the product Aβ (Lee et al., 1998; Riddell et al., 2001). How and where γ-secretase acts to cleave out Aβ is not known. Interestingly, Yanagisawa and coworkers have in a series of publications demonstrated that cholesterol-dependent sequestration of Aβ promotes fibrillogenesis of soluble Aβ and suggested that Aβ associated with rafts undergoes a conformational change, which promotes amyloid plaque formation (Yanagisawa et al., 1995; Mizuno et al., 1999; Kakio et al., 2001). Thus, the stage is set for a molecular dissection of how cholesterol and lipid rafts contribute to amyloid plaque formation in the pathogenesis of AD.
Materials and methods
Cells
Mouse neuroblastoma N2a cells were cultured at 37°C in DME supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (Invitrogen). Cells used for microscopy were grown on 11-mm diameter collagen-coated coverslips in complete medium without phenol red.
Reagents and antibodies
MβCD, mevalonate, and cycloheximide were from Sigma-Aldrich, and lovastatin was from Calbiochem. The following antibodies were used against APP: rabbit polyclonal antibody IP60 (Ehehalt et al., 2002) directed against the very COOH terminus of APP, mouse monoclonal antibody 6E10 (Senetek) that detects αAPP and Aβ, rabbit polyclonal antibody 70JE (Ehehalt et al., 2002) against aa 1–11 of Aβ, and the rabbit polyclonal antibody 5313 (Steiner et al., 1999) directed against the NH2-terminal part of the APP ectodomain. Antibodies against FP were rabbit polyclonal antibody KG77 raised against recombinant GFP expressed in bacteria and mouse monoclonal antibody 3E6 (Molecular Probes). Rabbit polyclonal antibody 7523 (Capell et al., 2002) was against the NH2-terminal end of BACE1. The anti–human transferrin receptor monoclonal antibody was from Roche. Mouse monoclonal and rabbit polyclonal anti-PLAP antibodies were from Dako.
Constructs and generation of recombinant adenoviruses
The BACE1A-YFP/CFP constructs were described previously (Ehehalt et al., 2002); BACE1A containing a VSVG tag (BACE1A-VSVG) was constructed as follows. BACE1A-YFP in pGEMT (Promega) was digested with AflII and NotI to release the YFP moiety. The VSVG epitope (MYTDIEMNRLGK) was then added using two complementary oligonucleotides to reconstitute the AflII and NotI restriction sites. The oligonucleotides used were 5′-TTAAGGGTATGTATACTGATATCGAAATGAATCGATTGGGTAAGTGAGC-3′ and 5′-GGCCGCTCACTTACCCAATCGATTCATTTCGATATCAGTATACATACCC-3′. BACE1A-VSVG was subsequently transferred as a SalI-NotI fragment into the mammalian expression vector pShuttle–cytomegalovirus (CMV) (He et al., 1998).
The YFP-APP construct was obtained as follows. APP was tagged in the ectodomain by replacing the naturally occurring Kunitz-type protease inhibitor domain with the FP. This was necessary because insertion of the FP at the very NH2 terminus of APP resulted in a misfolded chimeric protein, incapable of leaving the ER. YFP was amplified by PCR from pEYFP-N1 (CLONTECH Laboratories, Inc.) to add an XcmI site at the 5′ end, an XhoI site at the 3′ end, and spacers on either side of YFP. The oligonucleotides used were 5′-ACCACAGAGTCTGTGGAAGAGGTGGTTCGAGGCGGCGGATCTACCGTGGGCAGCGCACCGGTCGCCACCATG-3′ (the XcmI site is underlined and the bolded sequence matches plasmid pEYFP-N1) and 5′-TCTCGAGATACTTGTCAACGGCATCAGGGGTACTGGCTGCTGTTGTAFGAACTCCGCCGCCGGTAGATGCGGTCACGCTGCCGGTG-CCCTTGTACAGCTCGTCCATG-3′ (the XhoI site is underlined, and the bolded sequence matches plasmid pEYFP-N1). This PCR product was ligated as an XcmI-XhoI fragment into pGEMT-APP695 digested with XcmI and XhoI. YFP-APP was subsequently transferred as a SalI-NotI fragment into the mammalian expression vector pShuttle-CMV. Adenoviruses were prepared as described (He et al., 1998).
PLAP under control of the Rous sarcoma virus promoter and the TfR del 5–41 expression construct in pCMV5 were described previously (Harder et al., 1998). Dynamin II K44A in pCMV5 was provided by S. Schmid (Scripps Research Institute, La Jolla, CA) (Damke et al., 1994; Fish et al., 2000), and the cDNA of RN-tre (Lanzetti et al., 2000) was provided by M. Zerial (Max Planck Institute of Molecular Cell Biology and Genetics).
Transfection, viral infection, and cholesterol depletion
24 h after seeding of N2a cells into 3.5-cm dishes, they were infected with recombinant adenoviruses for 0.5 h at 37°C in complete medium. After a change of medium, the cells were incubated for 10–16 h at 37°C and then used for biochemical assays.
Transient transfections using calcium phosphate precipitation were performed with 1–3 μg of each expression plasmid as described by Chen and Okayama (1988). For cholesterol depletion, the cells were grown for 1 d in normal medium and then for 1 d in either DME supplemented with 2 mM l-glutamine, 10% lipid-deficient FCS, 2 μM lovastatin, and 0.25 mM mevalonate, or in complete medium. They were then infected and grown for a further 12–16 h in the same medium. The cells were treated for 5–30 min with 10 mM MβCD in methionine-free medium (labeling medium) and thereafter metabolically labeled with 100 μCi/dish of [35S]methionine (NEN). Depending on the experiment, the cells were chased for 0.5–2 h in labeling medium containing an excess of methionine (150 μg/ml) and 20 μg/ml cycloheximide to inhibit protein synthesis.
Cholesterol determinations were done with the Amplex Red Cholesterol Assay kit (Molecular Probes), which revealed a depletion of up to 80% of total cellular cholesterol after treatment with a combination of lovastatin and MβCD.
Immunoprecipitation and quantification
After metabolic labeling, the cell culture medium was collected and cell extracts were prepared using PBS containing 2% NP-40, 0.2% SDS, and 25 μg/ml each of chymostatin, leupeptin, antipain, and pepstatin A. Immunoprecipitates were recovered on protein A–Sepharose CL4B beads (Amersham Biosciences) and analyzed either on 10% polyacrylamide (Laemmli, 1970) or 10–20% Tris-Tricine (Invitrogen) gels. Individual bands were quantified using the Fujifilm BAS 1800II image plate reader and Science Lab 99 Image Gauge v3.3 software (Raytest Isotopenmessgeraete).
Immunofluorescence and antibody-induced patching
For immunofluorescence microscopy, the cells were fixed for 4 min at 8°C with 4% paraformaldehyde in PBS followed by an incubation in methanol for 4 min at –20°C. Fixed cells were incubated for 1 h at RT with a proper dilution of antibodies in PBS/0.2% gelatin. After three washes with PBS/0.2% gelatine, they were incubated with the respective secondary antibodies in PBS/0.2% gelatin for 1 h at RT.
To cluster raft proteins, the respective antibodies were diluted in CO2-independent medium (Invitrogen) containing 20 mg/ml BSA. Antibodies against PLAP were diluted 1:35, the anti–human transferrin receptor monoclonal antibody 1:100, the polyclonal anti-FP (KG77) and polyclonal anti-BACE1 (7523) antibodies 1:100, and the monoclonal anti-FP (3E6) 1:50. The cells were incubated for 45 min with the respective combination of antibodies at 10°C, briefly washed, and further incubated for 45 min at 10°C with mixed fluorescently labeled secondary antibodies. Cy3-labeled secondary antibodies were diluted 1:500, and the Cy5-labeled secondary antibodies were diluted 1:100. The cells were fixed as described above. Fluorescent images were acquired on an Olympus BX61 microscope.
Quantification of copatching
Because of variation in expression levels of transiently expressed proteins and differences in cell shape, quantification of copatching was done as described before (Harder et al., 1998). Briefly, images of 10 randomly selected cells expressing the two marker proteins were taken from one coverslip. An individual not involved in recording scored the images in a blind fashion into four categories: (1) coclustering (>80% overlap), (2) partial coclustering (clearly overlapping patches; 30–80%), (3) random distribution, and (4) segregation. The percentage of cells in each class from five independent experiments were expressed as mean ± SD.
Preparation of DRMs
Detergent extraction with CHAPS was performed as described (Fiedler et al., 1993). N2a cells were grown in 3.5-cm dishes, infected with adenoviruses to express BACE1A-YFP or YFP-APP, labeled for 2 h with [35S]methionine, and chased for 2 h in labeling medium containing an excess of methionine (150 μg/ml), 20 μg/ml cycloheximide, and for some samples antibodies 7523 or KG77 (1:100). The cells were washed once with PBS and scraped on ice into 300 μl 25 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 3 mM EDTA (TNE) buffer containing 25 μg/ml each of chymostatin, leupeptin, antipain, and pepstatin A. The cells were homogenized through a 25 G needle and centrifuged for 5 min at 3,000 rpm. The postnuclear supernatant was subjected to extraction for 30 min at 4°C in 20 mM CHAPS/TNE. The extracts were adjusted to 40% OptiPrep (Nycomed) and overlaid in a TLS 55 centrifugation tube with 1.4 ml of 30% OptiPrep/TNE and 200 μl TNE. After centrifugation for 2 h at 55,000 rpm, five fractions were collected from the top, and BACE1A-YFP or YFP-APP were recovered by immunoprecipitation with antibody KG77.
Uptake of biotin transferrin
N2a cells seeded on coverslips were transfected with either human transferrin receptor (TfR) and dynK44A-GFP or with TfR and pEGFP-N1 (CLONTECH Laboratories, Inc.). On the next day, the coverslips were washed by dipping in ice-cold PBS and then incubated for 20 min on ice with human biotin transferrin (50 μg/ml; Sigma-Aldrich) in 30 μl of low carbonate MEM supplemented with 2 mg/ml BSA (medium/BSA). After another wash with ice-cold PBS, the coverslips were incubated for 20 min on ice with a 1:40 dilution of the monoclonal anti–human TfR antibody (Roche) in 30 μl of medium/BSA or with medium/BSA alone. The cells were washed again in ice-cold PBS and subsequently incubated for 10 min at 37°C in 50 μl of medium/BSA to allow internalization of biotin transferrin. Biotin transferrin that remained on the surface was removed by three washes (2 min each) with ice-cold 0.5 M acetic acid and 0.5 M NaCl. Control cells used to determine the total amount of biotin transferrin present were incubated with ice-cold PBS for the same time.
The cells were lysed in 200 μl of PBS, 2% NP-40, and 0.2% SDS. Part of the lysate was incubated with streptavidin-coated magnetic beads (Dynal), a sheep antitransferrin antibody (Scottish Antibody Production Unit), and a rabbit anti–sheep secondary antibody (Dianova) coupled to a ruthenium trisbipyridine chelate (IGEN International, Inc.). These reagents have been described previously by Horiuchi et al. (1997). Biotin transferrin was then quantified on an Origen M8 analyzer (IGEN International, Inc.).
Acknowledgments
We thank Marino Zerial, Michel Bagnat, and Mikael Simons for critically reading the manuscript. We would like to thank Marta Miaczynska for help with the biotin transferrin uptake experiments.
R. Ehehalt was supported by grant EH196/1-1 from the Deutsche Forschungsgemeinschaft (DFG), P. Keller was supported by a grant from the Max Planck Gesellschaft, and K. Simons and P. Keller were supported by the DFG Schwerpunktprogramm SPP 1085 “Zellulaere Mechanismen der Alzheimer Erkrankung.”
Footnotes
Abbreviations used in this paper: Aβ, amyloid β-peptide; AD, Alzheimer's disease; APP, amyloid precursor protein; αAPP, α-cleaved ectodomain of APP; BACE1, β-site APP cleavage enzyme; CMV, cytomegalovirus; αCTF, α-cleaved COOH-terminal stub of APP; βCTF, β-cleaved COOH-terminal stub of APP; DRM, detergent-resistant membrane; FP, fluorescent protein; MβCD, methyl-β-cyclodextrin; PLAP, placental alkaline phosphatase; swAPP, Swedish mutant of APP; TfR, human transferrin receptor; wtAPP, wild-type APP.
References
- Ariga, T., K. Kobayashi, A. Hasegawa, M. Kiso, H. Ishida, and T. Miyatake. 2001. Characterization of high-affinity binding between gangliosides and amyloid beta-protein. Arch. Biochem. Biophys. 388:225–230. [DOI] [PubMed] [Google Scholar]
- Avdulov, N.A., S.V. Chochina, U. Igbavboa, C.S. Warden, A.V. Vassiliev, and W.G. Wood. 1997. Lipid binding to amyloid beta-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J. Neurochem. 69:1746–1752. [DOI] [PubMed] [Google Scholar]
- Baron, G.S., K. Wehrly, D.W. Dorward, B. Chesebro, and B. Caughey. 2002. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J. 21:1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjannet, S., A. Elagoz, L. Wickham, M. Mamarbachi, J.S. Munzer, A. Basak, C. Lazure, J.A. Cromlish, S. Sisodia, F. Checler, et al. 2001. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J. Biol. Chem. 276:10879–10887. [DOI] [PubMed] [Google Scholar]
- Bouillot, C., A. Prochiantz, G. Rougon, and B. Allinquant. 1996. Axonal amyloid precursor protein expressed by neurons in vitro is present in a membrane fraction with caveolae-like properties. J. Biol. Chem. 271:7640–7644. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and E. London. 1997. Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes? Biochem. Biophys. Res. Commun. 240:1–7. [DOI] [PubMed] [Google Scholar]
- Buxbaum, J.D., K.N. Liu, Y. Luo, J.L. Slack, K.L. Stocking, J.J. Peschon, R.S. Johnson, B.J. Castner, D.P. Cerretti, and R.A. Black. 1998. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273:27765–27767. [DOI] [PubMed] [Google Scholar]
- Capell, A., L. Meyn, R. Fluhrer, D.B. Teplow, J. Walter, and C. Haass. 2002. Apical sorting of beta-secretase limits amyloid beta-peptide production. J. Biol. Chem. 277:5637–5643. [DOI] [PubMed] [Google Scholar]
- Chen, C.A., and H. Okayama. 1988. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 6:632–638. [PubMed] [Google Scholar]
- Cheng, P.C., A. Cherukuri, M. Dykstra, S. Malapati, T. Sproul, M.R. Chen, and S.K. Pierce. 2001. Floating the raft hypothesis: the roles of lipid rafts in B cell antigen receptor function. Semin. Immunol. 13:107–114. [DOI] [PubMed] [Google Scholar]
- Corder, E.H., A.M. Saunders, W.J. Strittmatter, D.E. Schmechel, P.C. Gaskell, G.W. Small, A.D. Roses, J.L. Haines, and M.A. Pericak-Vance. 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 261:921–923. [DOI] [PubMed] [Google Scholar]
- Damke, H., T. Baba, D.E. Warnock, and S.L. Schmid. 1994. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127:915–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty, B.L., and S.A. Green. 2001. Endosomal sorting of amyloid precursor protein-P-selectin chimeras influences secretase processing. Traffic. 2:908–916. [DOI] [PubMed] [Google Scholar]
- De Strooper, B., and W. Annaert. 2000. Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 113:1857–1870. [DOI] [PubMed] [Google Scholar]
- Ehehalt, R., B. Michel, D. De Pietri Tonelli, D. Zacchetti, K. Simons, and P. Keller. 2002. Splice variants of the beta-site APP-cleaving enzyme BACE1 in human brain and pancreas. Biochem. Biophys. Res. Commun. 293:30–37. [DOI] [PubMed] [Google Scholar]
- Esler, W.P., and M.S. Wolfe. 2001. A portrait of Alzheimer secretases—new features and familiar faces. Science. 293:1449–1454. [DOI] [PubMed] [Google Scholar]
- Fassbender, K., M. Simons, C. Bergmann, M. Stroick, D. Lutjohann, P. Keller, H. Runz, S. Kuhl, T. Bertsch, K. von Bergmann, et al. 2001. Simvastatin strongly reduces levels of Alzheimer's disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 98:5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler, K., T. Kobayashi, T.V. Kurzchalia, and K. Simons. 1993. Glycosphingolipid-enriched, detergent-insoluble complexes in protein sorting in epithelial cells. Biochemistry. 32:6365–6373. [DOI] [PubMed] [Google Scholar]
- Fish, K.N., S.L. Schmid, and H. Damke. 2000. Evidence that dynamin-2 functions as a signal-transducing GTPase. J. Cell Biol. 150:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass, C., E.H. Koo, A. Mellon, A.Y. Hung, and D.J. Selkoe. 1992. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 357:500–503. [DOI] [PubMed] [Google Scholar]
- Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, T.C., S. Zhou, L.T. da Costa, J. Yu, K.W. Kinzler, and B. Vogelstein. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 95:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley, J.R., E.W. Krueger, B.J. Oswald, and M.A. McNiven. 1998. Dynamin-mediated internalization of caveolae. J. Cell Biol. 141:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi, H., R. Lippe, H.M. McBride, M. Rubino, P. Woodman, H. Stenmark, V. Rybin, M. Wilm, K. Ashman, M. Mann, and M. Zerial. 1997. A novel rab5 GDP/GTP exchange factor complexed to rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell. 90:1149–1159. [DOI] [PubMed] [Google Scholar]
- Huse, J.T., D.S. Pijak, G.J. Leslie, V.M. Lee, and R.W. Doms. 2000. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J. Biol. Chem. 275:33729–33737. [DOI] [PubMed] [Google Scholar]
- Janes, P.W., S.C. Ley, and A.I. Magee. 1999. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 147:447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes, P.W., S.C. Ley, A.I. Magee, and P.S. Kabouridis. 2000. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin. Immunol. 12:23–34. [DOI] [PubMed] [Google Scholar]
- Jick, H., G.L. Zornberg, S.S. Jick, S. Seshadri, and D.A. Drachman. 2000. Statins and the risk of dementia. Lancet. 356:1627–1631. [DOI] [PubMed] [Google Scholar]
- Kakio, A., S.I. Nishimoto, K. Yanagisawa, Y. Kozutsumi, and K. Matsuzaki. 2001. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 276:24985–24990. [DOI] [PubMed] [Google Scholar]
- Kamal, A., A. Almenar-Queralt, J.F. LeBlanc, E.A. Roberts, and L.S. Goldstein. 2001. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 414:643–648. [DOI] [PubMed] [Google Scholar]
- Kivipelto, M., E.L. Helkala, M.P. Laakso, T. Hanninen, M. Hallikainen, K. Alhainen, H. Soininen, J. Tuomilehto, and A. Nissinen. 2001. Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. BMJ. 322:1447–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein, D.R., M. Tomishige, N. Stuurman, and R.D. Vale. 2002. Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell. 109:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike, H., S. Tomioka, H. Sorimachi, T.C. Saido, K. Maruyama, A. Okuyama, A. Fujisawa-Sehara, S. Ohno, K. Suzuki, and S. Ishiura. 1999. Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem. J. 343:371–375. [PMC free article] [PubMed] [Google Scholar]
- Kojro, E., G. Gimpl, S. Lammich, W. Marz, and F. Fahrenholz. 2001. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-ecretase ADAM 10. Proc. Natl. Acad. Sci. USA. 98:5815–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo, E.H., and S.L. Squazzo. 1994. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269:17386–17389. [PubMed] [Google Scholar]
- Kuo, Y.M., M.R. Emmerling, C.L. Bisgaier, A.D. Essenburg, H.C. Lampert, D. Drumm, and A.E. Roher. 1998. Elevated low-density lipoprotein in Alzheimer's disease correlates with brain abeta 1-42 levels. Biochem. Biophys. Res. Commun. 252:711–715. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Lammich, S., E. Kojro, R. Postina, S. Gilbert, R. Pfeiffer, M. Jasionowski, C. Haass, and F. Fahrenholz. 1999. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA. 96:3922–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlet, C., A.M. Bernard, P. Drevot, and H.T. He. 2000. Membrane rafts and signaling by the multichain immune recognition receptors. Curr. Opin. Immunol. 12:250–255. [DOI] [PubMed] [Google Scholar]
- Lanzetti, L., V. Rybin, M.G. Malabarba, S. Christoforidis, G. Scita, M. Zerial, and P.P. Di Fiore. 2000. The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature. 408:374–377. [DOI] [PubMed] [Google Scholar]
- Lee, S.J., U. Liyanage, P.E. Bickel, W. Xia, P.T. Lansbury, Jr., and K.S. Kosik. 1998. A detergent-insoluble membrane compartment contains A beta in vivo. Nat. Med. 4:730–734. [DOI] [PubMed] [Google Scholar]
- Li, Y.M., M. Xu, M.T. Lai, Q. Huang, J.L. Castro, J. DiMuzio-Mower, T. Harrison, C. Lellis, A. Nadin, J.G. Neduvelil, et al. 2000. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 405:689–694. [DOI] [PubMed] [Google Scholar]
- Mizuno, T., M. Nakata, H. Naiki, M. Michikawa, R. Wang, C. Haass, and K. Yanagisawa. 1999. Cholesterol-dependent generation of a seeding amyloid beta-protein in cell culture. J. Biol. Chem. 274:15110–15114. [DOI] [PubMed] [Google Scholar]
- Parvathy, S., I. Hussain, E.H. Karran, A.J. Turner, and N.M. Hooper. 1999. Cleavage of Alzheimer's amyloid precursor protein by alpha-secretase occurs at the surface of neuronal cells. Biochemistry. 38:9728–9734. [DOI] [PubMed] [Google Scholar]
- Perez, R.G., S. Soriano, J.D. Hayes, B. Ostaszewski, W. Xia, D.J. Selkoe, X. Chen, G.B. Stokin, and E.H. Koo. 1999. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Biol. Chem. 274:18851–18856. [DOI] [PubMed] [Google Scholar]
- Pralle, A., P. Keller, E.L. Florin, K. Simons, and J.K. Horber. 2000. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J. Cell Biol. 148:997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior, I.A., A. Harding, J. Yan, J. Sluimer, R.G. Parton, and J.F. Hancock. 2001. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 3:368–375. [DOI] [PubMed] [Google Scholar]
- Puglielli, L., G. Konopka, E. Pack-Chung, L.A. Ingano, O. Berezovska, B.T. Hyman, T.Y. Chang, R.E. Tanzi, and D.M. Kovacs. 2001. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 3:905–912. [DOI] [PubMed] [Google Scholar]
- Refolo, L.M., B. Malester, J. LaFrancois, T. Bryant-Thomas, R. Wang, G.S. Tint, K. Sambamurti, K. Duff, and M.A. Pappolla. 2000. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 7:321–331. [DOI] [PubMed] [Google Scholar]
- Riddell, D.R., G. Christie, I. Hussain, and C. Dingwall. 2001. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 11:1288–1293. [DOI] [PubMed] [Google Scholar]
- Rodal, S.K., G. Skretting, O. Garred, F. Vilhardt, B. van Deurs, and K. Sandvig. 1999. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell. 10:961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S., R. Luetterforst, A. Harding, A. Apolloni, M. Etheridge, E. Stang, B. Rolls, J.F. Hancock, and R.G. Parton. 1999. Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nat. Cell Biol. 1:98–105. [DOI] [PubMed] [Google Scholar]
- Scheuermann, S., B. Hambsch, L. Hesse, J. Stumm, C. Schmidt, D. Beher, T.A. Bayer, K. Beyreuther, and G. Multhaup. 2001. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J. Biol. Chem. 276:33923–33929. [DOI] [PubMed] [Google Scholar]
- Selkoe, D.J. 2001. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81:741–766. [DOI] [PubMed] [Google Scholar]
- Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- Simons, M., P. Keller, B. De Strooper, K. Beyreuther, C.G. Dotti, and K. Simons. 1998. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA. 95:6460–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, M., P. Keller, J. Dichgans, and J.B. Schulz. 2001. Cholesterol and Alzheimer's disease: is there a link? Neurology. 57:1089–1093. [DOI] [PubMed] [Google Scholar]
- Sparks, D.L., S.W. Scheff, J.C. Hunsaker, III, H. Liu, T. Landers, and D.R. Gross. 1994. Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126:88–94. [DOI] [PubMed] [Google Scholar]
- Steiner, H., K. Duff, A. Capell, H. Romig, M.G. Grim, S. Lincoln, J. Hardy, X. Yu, M. Picciano, K. Fechteler, et al. 1999. A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and notch signaling. J. Biol. Chem. 274:28669–28673. [DOI] [PubMed] [Google Scholar]
- Strittmatter, W.J., A.M. Saunders, D. Schmechel, M. Pericak-Vance, J. Enghild, G.S. Salvesen, and A.D. Roses. 1993. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA. 90:1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey, M.G., R.H. Baloh, J. Milbrandt, and E.M. Johnson, Jr. 2000. GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron. 25:611–623. [DOI] [PubMed] [Google Scholar]
- Taraboulos, A., M. Scott, A. Semenov, D. Avrahami, L. Laszlo, S.B. Prusiner, and D. Avraham. 1995. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 129:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle, S., P. Das, A.C. Nyborg, C. McLendon, M. Shoji, T. Kawarabayashi, L.H. Younkin, S.G. Younkin, and T.E. Golde. 2002. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol- rich membrane microdomains. Neurobiol. Dis. 9:11–23. [DOI] [PubMed] [Google Scholar]
- Weihofen, A., K. Binns, M.K. Lemberg, K. Ashman, and B. Martoglio. 2002. Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science. 296:2215–2218. [DOI] [PubMed] [Google Scholar]
- Wolozin, B., W. Kellman, P. Ruosseau, G.G. Celesia, and G. Siegel. 2000. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 57:1439–1443. [DOI] [PubMed] [Google Scholar]
- Yanagisawa, K., and Y. Ihara. 1998. GM1 ganglioside-bound amyloid beta-protein in Alzheimer's disease brain. Neurobiol. Aging. 19:S65–S67. [DOI] [PubMed] [Google Scholar]
- Yanagisawa, K., A. Odaka, N. Suzuki, and Y. Ihara. 1995. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer's disease. Nat. Med. 1:1062–1066. [DOI] [PubMed] [Google Scholar]