

Abstract
Stimulation of cell surface death receptors activates caspase-8, which targets a limited number of substrates including BAP31, an integral membrane protein of the endoplasmic reticulum (ER). Recently, we reported that a caspase-resistant BAP31 mutant inhibited several features of Fas-induced apoptosis, including the release of cytochrome c (cyt.c) from mitochondria (Nguyen, M., D.G. Breckenridge, A. Ducret, and G.C. Shore. 2000. Mol. Cell. Biol. 20:6731–6740), implicating ER-mitochondria crosstalk in this pathway. Here, we report that the p20 caspase cleavage fragment of BAP31 can direct pro-apoptotic signals between the ER and mitochondria. Adenoviral expression of p20 caused an early release of Ca2+ from the ER, concomitant uptake of Ca2+ into mitochondria, and mitochondrial recruitment of Drp1, a dynamin-related protein that mediates scission of the outer mitochondrial membrane, resulting in dramatic fragmentation and fission of the mitochondrial network. Inhibition of Drp1 or ER-mitochondrial Ca2+ signaling prevented p20-induced fission of mitochondria. p20 strongly sensitized mitochondria to caspase-8–induced cyt.c release, whereas prolonged expression of p20 on its own ultimately induced caspase activation and apoptosis through the mitochondrial apoptosome stress pathway. Therefore, caspase-8 cleavage of BAP31 at the ER stimulates Ca2+-dependent mitochondrial fission, enhancing the release of cyt.c in response to this initiator caspase.
Keywords: apoptosis; caspase-8; BID; BAX; Drp1
Introduction
Mitochondria are key regulators of apoptosis that integrate diverse apoptotic stimuli into a core death pathway (Green and Reed, 1998). Mitochondrial control of apoptosis is governed by the BCL-2 family of proteins, which include anti-apoptotic BCL-2 and BCL-xL and pro-apoptotic BAX and BAK; the balance between these opposing members is regulated by a third subgroup called the “BH3-only” proteins (Cory and Adams, 2002). Current models hold that certain BH3-only proteins invoke a mitochondrial phase of apoptosis by directing the insertion of BAX into mitochondria and inducing oligomerization of BAX and BAK in the outer mitochondrial membrane (OMM),* causing an efflux of intermembrane space proteins, including cytochrome c (cyt.c; Korsmeyer et al., 2000). Once in the cytosol, cyt.c complexes with Apaf-1 and procaspase-9 forming the apoptosome, a direct activator of downstream effector caspases 3 and 7 (Budihardjo et al., 1999). Activation of the TNF family of cell surface death receptors is coupled to the mitochondrial phase of apoptosis by the BH3-only protein BID. Binding of Fas to its ligand or agonistic antibody induces the recruitment and autoactivation of initiator procaspase-8 (Krammer, 2000). In turn, caspase-8 cleaves BID, generating tBID, which translocates from the cytosol to mitochondria and induces organelle dysfunction and cyt.c release (Li et al., 1998; Luo et al., 1998). BID plays an obligate role in transducing signals from death receptors to mitochondria in at least some cell types because hepatocytes from BID−/− mice do not release cyt.c in response to Fas, despite normal activation of caspase-8 (Yin et al., 1999). In some contexts, caspase-8 can bypass mitochondria and directly cleave downstream caspases. However, in many cell types, the BID-dependent mitochondrial loop is required to amplify weak death receptor signals and relieve the inhibitory effect of IAP proteins on caspase activity (Scaffidi et al., 1998; Yin et al., 1999; Deng et al., 2002; Fulda et al., 2002).
Although it is clear that BCL-2 family members govern mitochondrial dysfunction, it remains unclear at what point the functions of these proteins intercede with gross alterations in mitochondrial morphology that occur during apoptosis. Normal mitochondrial morphology can vary dramatically between cell types, but in most cases mitochondria form long “wormlike” tubules that may (Rizzuto et al., 1998) or may not (Collins et al., 2002) make up interconnected networks. The distribution of mitochondria depends on interactions with microtubules whereas mitochondrial size and shape is the result of constant fusion and fission processes (Bereiter-Hahn and Voth, 1994). Little is known about the mechanism of mitochondrial fission and fusion except that it is regulated by a group of evolutionary conserved GTPases; fusion is dependent on Fzo/Mfn, whereas fission relies on a dynamin related protein, Drp1 (Osteryoung, 2001; Shaw and Nunnari, 2002). During apoptosis mitochondria remodel inner membrane cristae (Scorrano et al., 2002), fragment into small punctiform organelles that sometimes cluster in the perinuclear region (Desagher and Martinou, 2000; Frank et al., 2001; Pinton et al., 2001), and eventually undergo matrix swelling leading to OMM rupture (Petit et al., 1998; Mootha et al., 2001). Recently, Frank et al. (2001) demonstrated that fragmentation of the mitochondrial network during apoptosis is caused by large-scale activation of Drp1-dependent mitochondrial fission, and that this event is requisite for the mitochondrial phase of apoptosis. How apoptotic signals converge on the fission machinery, however, is unclear.
In the current paper, we present evidence that caspase cleavage of BAP31 at the ER can trigger the onset of mitochondrial fission. BAP31 is a polytopic integral protein of the ER membrane that forms a large hetero-oligomeric complex with the related BAP29 protein and components of the actomyosin network (Adachi et al., 1996; Ng et al., 1997; Nguyen et al., 2000). After activation of cell surface death receptors, human BAP31 is cleaved at two identical caspase recognition sites in its cytosolic tail, generating a membrane-embedded fragment, called p20, which induces apoptosis when expressed ectopically (Ng et al., 1997; Nguyen et al., 2000). Cleavage of BAP31 seems to be an important event in the Fas pathway because cells expressing a caspase-resistant BAP31 (crBAP31) mutant retain a near normal morphology after stimulation and resist apoptotic membrane blebbing/fragmentation, disruption of the actin network, and irreversible loss of cell growth potential after removal of the Fas stimulus (Nguyen et al., 2000). In addition, crBAP31 prevents mitochondrial remodeling and the release of cyt.c in the face of activated caspases, suggesting that events at the ER can modulate mitochondrial dysfunction in intact cells (Nguyen et al., 2000). To better understand this communication between ER and mitochondria, and how BAP31 contributes to Fas signaling in general, we investigated the role of p20 in apoptotic progression. We find that p20 stimulates ER Ca2+ release, resulting in the activation of Drp1-dependent fission of mitochondria, which ultimately sensitizes this organelle to caspase-8–induced cyt.c release.
Results
Caspase cleavage of BAP31 during Fas-mediated apoptosis generates p20
BAP31 and its cellular homologue and heterodimerizing partner, BAP29 (Adachi et al., 1996), are structurally conserved proteins sharing identical topology in the ER membrane and 47% sequence identity in man. Both proteins initiate with the NH2 terminus facing the lumen, followed by three transmembrane regions and a cytosolic tail containing a long coiled coil domain ending in a canonical KKXX ER retrieval sequence (Fig. 1 A). Human BAP31 contains two identical caspase cleavage sites (AAVD.G) at D164 and D238 that are preferentially cleaved by caspase-8 (Ng et al., 1997; Wang et al., 2003). Fig. 1 B shows that in human KB cells stimulated with agonistic anti-Fas antibody BAP31 was cleaved generating the p27 and p20 membrane-embedded fragments. Only the former cleavage site is conserved in mouse Bap31, suggesting that cleavage at D164 is critical. The two caspase cleavage sequences are not conserved in BAP29, which remained structurally intact during apoptosis (Fig. 1 B).
Figure 1.


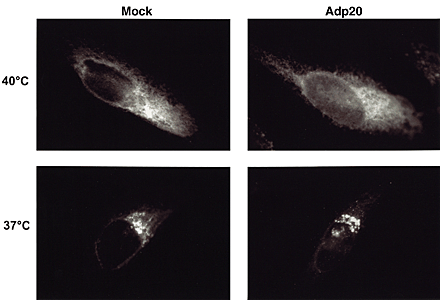
BAP31, but not BAP29, is cleaved during Fas-mediated apoptosis. (A) Schematic representation of human BAP31, the p20 caspase cleavage product, and BAP29 in the ER membrane. Both BAP31 and BAP29 contain three transmembrane domains, a cytosolic tail containing a coiled coil domain (boxed region), and terminate with a canonical KKXX ER retrieval sequence. The caspase-8 recognition sites in BAP31 are shown. (B) KB cells were untreated or stimulated with 500 ng/ml anti-Fas activating antibody (CH11) and 10 μg/ml cycloheximide (CHX) for 7 h, and cell lysates were analyzed by SDS-PAGE and immunoblotting with anti-BAP31 (left) or anti-BAP29 (right) pAbs. The positions of BAP31, its p27 and p20 cleavage products, and BAP29 are indicated. (C) Adenoviral-expressed p20-HA (Adp20) localizes to the ER. H1299 cells were infected with Adp20 for 20 h, then fixed and double stained with anti-HA and anti-calreticulin antibodies or anti-HA and anti-TOM20 antibodies.
Previously, we observed that the p20 caspase cleavage fragment of BAP31 is cytotoxic when expressed ectopically (Ng et al., 1997), indicating that caspase cleavage of BAP31 might generate a pro-apoptotic gain of function. To study the mechanism of action of p20, we created an adenoviral vector (Adp20) expressing this fragment (aa 1–164 of human BAP31) with a COOH-terminal hemagglutinin (HA) tag. The endogenous p20 protein generated during Fas-mediated apoptosis was associated with microsomes and remained resistant to alkali extraction (pH 11.5), indicative of a membrane-integrated protein (unpublished data). Immunofluorescence microscopic analysis of Adp20-infected human H1299 cells revealed that exogenous p20 strongly colocalized with endogenous calreticulin, a resident ER lumen protein, but p20 did not colocalize with TOM20, a marker of the OMM. Therefore, caspase cleavage of BAP31 generates a pro-apoptotic p20 fragment that remains at the ER.
Prolonged expression of p20 induces apoptosis
Expression of p20 was observed by 10 h post-infection of KB cells with Adp20, and remained stable for over 50 h (Fig. 2 A). 30–40 h after infection, Adp20 induced the activation of caspases, measured by the hydrolysis of the caspase substrate DEVD-amc and by processing of procaspase-3, in many cell types including KB, H1299, HeLa, and Rat1 cells (Fig. 2 B; unpublished data). The mechanism of this caspase activation seemed to occur via the classical mitochondrial apoptosome stress pathway. For example, p20 expression resulted in the insertion of BAX into the OMM, homo-oligomerization of BAK, and release of cyt.c from mitochondria in the presence of the pan-caspase inhibitor, zVAD-fmk (Fig. 2 C; unpublished data). In contrast, p20-induced caspase activation was abrogated in APAF-1–null cells (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200212059/DC1). Control adenovectors expressing either LacZ or the reverse tet transactivating protein (RTA) did not cause any of the aforementioned apoptotic changes (unpublished data). Inhibition of caspases using zVAD-fmk, or overexpression of BCL-2 or BCL-xL, blocked downstream morphological features of apoptosis including loss of plasma membrane integrity as assessed by trypan blue uptake (Fig. 2 D). In the absence of these inhibitors, cells showed typical signs of apoptosis, including nuclear condensation and fragmentation, membrane blebbing and cell surface exposure of phosphatidylserine (unpublished data).
Figure 2.
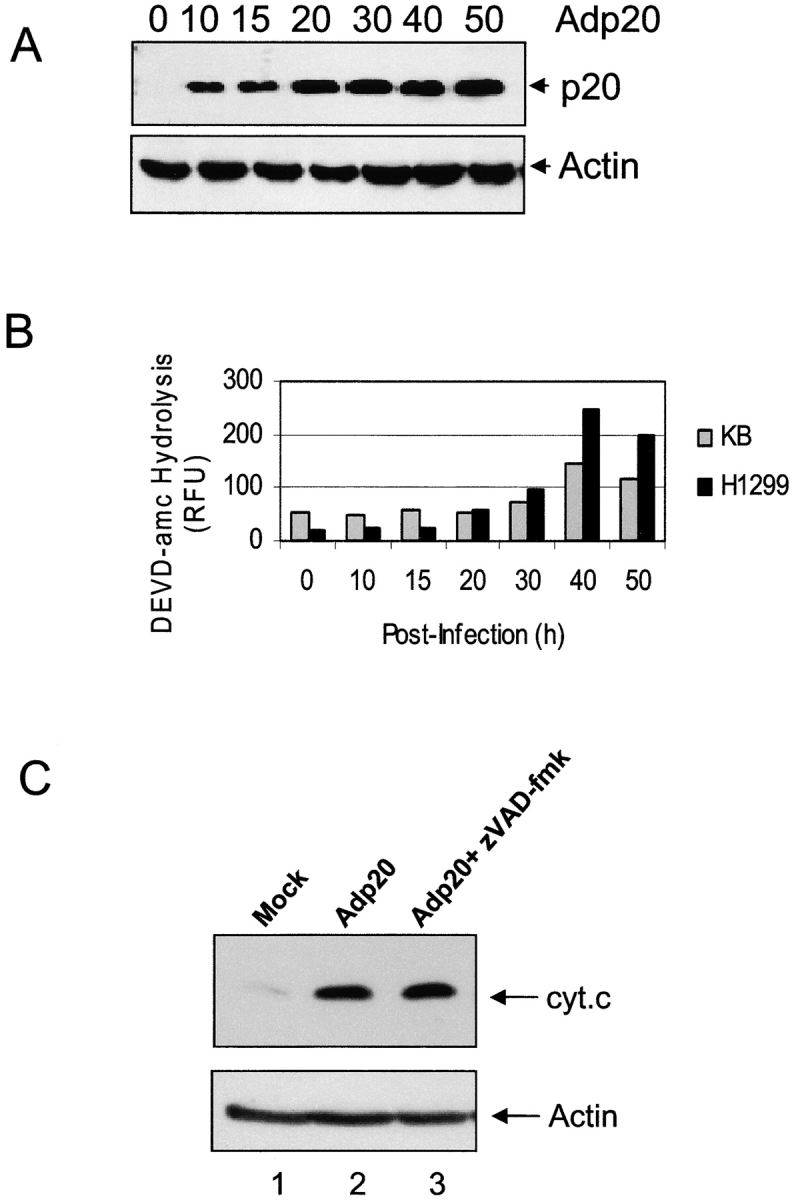


Prolonged expression of p20 induces mitochondrial apoptosis. (A) Expression of p20 in KB cells. Cells were infected with Adp20 and cell lysates were collected and analyzed by immunoblotting at the times indicated post-infection. (B) KB and H1299 cells were infected with Adp20, and effector caspase (DEVDase) activity was measured at the indicated times post-infection by the ability of cell lysates to hydrolyze the fluorogenic caspase substrate DEVD-amc. Shown is a representative experiment. (C) KB cells were mock infected or infected with Adp20 for 35–40 h in the absence or presence of 50 μM zVAD-fmk, and equivalent amounts of post-mitochondrial supernatants were analyzed for the presence of cyt.c by SDS-PAGE and immunoblotting. The membrane was reprobed with anti-actin antibody to confirm equal loading. (D) Parental KB cells, or KB cells stably overexpressing BCL-2 or BCL-xL, were mock infected or infected with Adp20 in the absence or presence of 50 μM zVAD-fmk, and at 45 h post infection, cell death was assessed by trypan blue staining. Shown is mean ± SD of three independent experiments. (E) Wt, Bap31-null, and Bap29,31-null mouse ES cells were treated and analyzed as in D.
p20 strongly heterodimerizes with full-length BAP31 (Nguyen et al., 2000) and therefore, might cause apoptosis by exerting a dominant-negative influence on endogenous BAP31 or BAP29. However, as shown in Fig. 2 E, cell death was observed in Bap31 or Bap29,31 double-deleted mouse cells (Breckenridge et al., 2002) infected with Adp20, demonstrating that p20 has an intrinsic pro-apoptotic activity at the ER that is separate from the functions of BAP31 and BAP29. Moreover, export of ectopic VSV G protein from the ER occurred at 22 h after Adp20 infection, suggesting that p20 does not exert a gross influence on ER-Golgi trafficking at this early time (Fig. S2). Collectively, these results indicate that p20 can activate mitochondrial apoptosis. However, it is noteworthy that this pathway did not culminate until at least 30–40 h after infection (Fig. 2), whereas ectopic tBID induces cyt.c release within several hours of its expression (Li et al., 1998). Therefore, a more relevant function for p20 in physiological cell death might relate to an early sensitization of mitochondria to a costimulus.
p20 sensitizes mitochondria to caspase-8–induced cyt.c release
Given that BAP31 is a caspase-8 substrate, p20 might cooperate with other products generated by caspase-8, such as tBID, to enhance mitochondrial dysfunction. According to this model, immediately after its expression, p20 should activate a signal that is slow to induce cyt.c release on its own, but able to synergize with other apoptotic signals during Fas-mediated apoptosis. Therefore, we investigated whether p20 could enhance caspase-8–driven cyt.c release. Death receptor–dependent caspase-8 activation was mimicked by infecting H1299 cells with adenovector-expressing triplicate copies of Fpk (a mutant of FKBP) fused to the catalytic subunits of caspase-8 (AdMFpk3FLICE; Muzio et al., 1998). After its expression in cells, oligomerization and autoactivation of the caspase-8 fusion protein was induced with the Fpk-dimerizing compound, FK1012Z. This approach has the benefit of delivering synchronized caspase-8 signals to cells without stimulating caspase-8–independent pathways activated by death receptors (Schulze-Osthoff et al., 1998; Wang et al., 2001). In Fig. 3, H1299 cells were co-infected with AdRTA (control adenovector) and AdMFpk3FLICE, or with Adp20 and AdMFpk3FLICE. 16 h after infection, a time when Adp20 alone did not induce cyt.c release or caspase activation (Figs. 2 and 3), the cells were exposed to a short treatment (45 or 90 min) with FK1012Z or vehicle (DMSO) alone, and the mitochondrial and post-mitochondrial fractions were isolated. Compared with caspase-8 activation in the presence of the control protein RTA, caspase-8 activation in the presence of ectopic p20 strongly induced release of cyt.c to the cytosol (Fig. 3 A), but it did not affect the amount of caspase-8–generated tBID that was recovered in the mitochondrial fraction (Fig. 3 B). In all cases, equivalent amounts of MFpk3FLICE were produced (unpublished data). Therefore, these results suggest that p20-mediated signals from the ER might cooperate with other caspase-8–generated signals to increase cyt.c release from mitochondria.
Figure 3.

p20 sensitizes mitochondria to caspase-8–induced cyt.c release. H1299 cells were mock infected, or co-infected with AdRTA (control) and AdMFpk3FLICE or with Adp20 and AdMFpk3FLICE. 16 h post-infection, FK1012Z or vehicle alone (DMSO; Wang et al., 2003) were added for 45 or 90 min, and the amount of cyt.c in the post-mitochondrial supernatant and tBID in the mitochondrial fraction were assessed by SDS-PAGE and Western blot. The intensity of the cyt.c (A) and tBID (B) signals, relative to loading controls, was determined using ImageQuantTM software (Amersham Biosciences) and is expressed in arbitrary units. Shown is a representative of three independent experiments.
p20 mediates its effect through an early release of Ca2+ from the ER
Next, we sought to identify the early ER signaling events after p20 expression. Release of Ca2+ from the ER occurs as an early event during many forms of apoptosis, including the Fas pathway, and Ca2+ has been implicated as a second messenger between ER and mitochondria during apoptosis (Hajnoczky et al., 2000; Breckenridge and Shore, 2002). We tested whether p20 expression altered ER Ca2+ homeostasis by loading Adp20-infected cells with the Ca2+-sensitive fluorescent indicator Fura2-AM and measuring the increase in cytosolic Ca2+ that results from thapsigargin (TG)-induced depletion of ER stores. TG invokes a rapid emptying of ER Ca2+ stores to the cytosol by irreversibly inhibiting SERCA pumps that normally maintain the concentration of ER Ca2+ ([Ca2+]ER) several orders of magnitude above that of the cytosol ([Ca2+]c). Fig. 4 A reveals that expression of p20 in H1299 cells in the presence of zVAD-fmk caused an early, time-dependent decrease in ER Ca2+ stores. The kinetics of ER Ca2+ release was concomitant with an increase in the concentration of mitochondrial Ca2+ ([Ca2+]m), measured by Rhod2 fluorescence. These changes in ER and mitochondrial Ca2+ levels could be measured as early as 12–14 h post-infection (i.e., 2–4 h after p20 protein appears; Fig. 2 A), making them the earliest events we observed in the p20 pathway.
Figure 4.



p20-induced mitochondrial fragmentation is mediated by an ER-mitochondria Ca2 + signal. (A) Top, p20 induces a time dependent release of Ca2+ from the ER. H1299 cells were infected with Adp20 in the presence of 50 μM zVAD-fmk and at the indicated times post-infection, cells were loaded with Fura2-AM in Ca2+-free buffer and ER calcium stores were measured as the sudden difference in Fura2 fluorescence recorded after the addition of TG (see Materials and methods). Shown is the mean and SD of five independent experiments. Bottom, elevated [Ca2+]m after Adp20 infection. HeLa cells were treated as in A, except cells were loaded with Rhod2-AM and the [Ca2+]m was estimated as described in the Materials and methods. (B) p20 induces dramatic fragmentation of mitochondria, which is inhibited by predepletion of ER Ca2+ stores with TG. H1299 cells were infected with 50 μM Adp20 + zVAD-fmk in the absence or presence of 50 nm TG for 24 h, and mitochondria were visualized by anti-cyt.c staining. Representative images are shown. (C) Reducing ER Ca2+ stores, chelating cytosolic Ca2+, or preventing mitochondrial Ca2+ uptake inhibits p20-induced fragmentation of mitochondria. As in B, but H1229 cells, or H1299 cells pretreated with 50 nm TG, 2 μM BAPTA-AM, or 20 μM Ru360, or H1299 b5-BCL-2 cells were infected with Adp20 + zVAD-fmk for 24 h and the number of cells showing signs of mitochondrial fragmentation was quantified. Shown is the mean ± SD of five independent experiments.
To determine whether the release of ER Ca2+ affected early responses of mitochondria to p20, we examined the consequence of inhibiting Ca2+ signaling between the ER and mitochondria. We began by adopting two experimental conditions that reduce the amount of Ca2+ that could be released from the ER by p20 (Pinton et al., 2001). In the first case, H1299 cells were incubated with a low concentration of TG (50 nM). Addition of TG resulted in an immediate emptying of ER Ca2+ stores, which remained depleted for over 24 h (Fig. S3 A). In a second approach, we took advantage of the ability of BCL-2 to lower the [Ca2+]ER by increasing the passive leak of Ca2+ from the organelle (Foyouzi-Youssefi et al., 2000; Pinton et al., 2000). However, to minimize the antiapoptotic activity of BCL-2 at the mitochondria, we created H1229 cells stably overexpressing BCL-2 selectively targeted to the ER with the membrane insertion sequence of cytochrome b5 (H1299 b5-BCL-2; Zhu et al., 1996). H1299 b5-BCL-2 cells had an ∼40% reduction in resting ER Ca2+ levels, resulting in a substantial decrease in the total amount of Ca2+ released in response to Adp20 (Fig. S3 B). We also tested the effect of two other agents: BAPTA-AM, a cytosolic Ca2+ chelator that can inhibit Ca2+ transmission between the ER and mitochondria (Byrne et al., 1999; Sharma et al., 2000); and Ru360, an inhibitor of mitochondrial Ca2+ uptake (Matlib et al., 1998).
H1299 cells treated with Adp20 + zVAD-fmk for 24 h and stained with cyt.c displayed dramatically fragmented mitochondria compared with mock-infected cells (Fig. 4 B). Remarkably, in cells pre-treated with TG or expressing b5-BCL-2, the mitochondrial network remained intact and highly interconnected with no signs of mitochondrial fragmentation (Fig. 4 B; unpublished data). Quantification of the two mitochondrial phenotypes revealed that TG and b5-BCL-2 reduced the number of cells showing signs of mitochondrial fragmentation from 52% to 10 and 13%, respectively (Fig. 4 C). Pretreatment of cells with BAPTA or Ru360 also reduced the number of cells manifesting fragmented mitochondria in response to p20 (Fig. 4 C). The expression of p20 was not affected by any of the treatments (unpublished data). TG, b5-BCL-2, BAPTA, and Ru360 also inhibited p20-induced release of cyt.c from mitochondria, which occurred subsequent to fragmentation (unpublished data; see following section). Therefore, inhibition of Ca2+ transport between the ER and mitochondria inhibits the effect of p20 on mitochondrial morphology and redistribution of cyt.c.
p20-induced fragmentation of the mitochondrial network
The observation that p20 caused mitochondrial fragmentation was extended in Fig. 5. p20 induced an early fragmentation of the mitochondrial network into small punctiform organelles in all cell types tested, including H1299, Rat1, and HeLa cells (Figs. 4 and 5; unpublished data). The gross morphological changes in the mitochondrial network could be observed 15–16 h after Adp20 infection (i.e., 2–3 h after the onset of Ca2+ release), a time when p20 sensitized mitochondria to caspase-8–induced cyt.c release (Fig. 3). Induction of mitochondrial fragmentation by p20 occurred in the absence of zVAD-fmk–sensitive caspase activation and cell shrinkage or disruption of microtubules. For example, Fig. 5 A shows that Rat1 fibroblasts expressing p20 + zVAD-fmk for 20 h and costained with anti-tubulin and anti-TOM20 antibodies displayed a normal microtubule distribution despite having fragmented mitochondria. The punctiform mitochondria could be observed in living cells stained with MitoTracker® Red (unpublished data), indicating that fragmented mitochondria maintain membrane potential and were not an artifact of fixation. Co-staining of Rat1 fibroblasts expressing p20 + zVAD-fmk with antibody to cyt.c and antibody selective for the active conformation of BAX (Desagher et al., 1999) revealed that the transition of mitochondria into punctiform organelles preceded cyt.c release and activation of BAX. As exemplified in Fig. 5 B, most cells expressing p20 + zVAD-fmk for 25 h displayed fragmented mitochondria but showed no signs of cyt.c release or BAX conformation-specific immunoreactivity. BAX immunoreactivity could only be observed in apoptotic cells that had released cyt.c from mitochondria, and all cells that had undergone cyt.c release stained positive for BAX. These results suggest that p20 induces early fragmentation of mitochondria, which precedes BAX activation and cyt.c release. Given that disintegration of the mitochondrial network has been demonstrated to contribute to apoptotic progression (Desagher and Martinou, 2000; Frank et al., 2001) but that BAX/BAK activation and cyt.c release are normally stimulated by BH3-only molecules (Korsmeyer et al., 2000), it is likely that p20 mediates its sensitizing effect by inducing early fragmentation of mitochondria.
Figure 5.

p20 induces fragmentation of the mitochondrial network as an early event. (A) Mitochondrial restructuring and fragmentation occur in the absence of cell shrinkage. Rat1 fibroblasts were infected with Adp20 in the presence of 50 μM zVAD-fmk (to prevent caspase activation and cell detachment) for 20 h, fixed, and double stained with anti-tubulin and anti-TOM20 antibodies. (B) Mitochondrial fragmentation occurs before activation of BAX and cyt.c release. As in A, except cells were infected for 25 h and double stained with anti-cyt.c antibody (arrows, lower left) and the active conformation-specific anti-BAX-NT antibody (aa 1–21, Upstate Biotechnology; arrows, lower right).
p20 induces Drp1 translocation to mitochondria
Recently, fragmentation of mitochondria during staurosporin-induced apoptosis was demonstrated to be dependent on mitochondrial fission mediated by Drp1 (Frank et al., 2001). Therefore, we investigated whether Drp1-dependent fission played a role in the p20 pathway. We began by examining the subcellular localization of Drp1 because GFP-tagged Drp1 was shown to redistribute from a predominately cytosolic location to predicted sites of division along mitochondrial tubules after treatment with staurosporin (Frank et al., 2001). Fig. 6 A documents that endogenous Drp1 was recruited to mitochondria before the onset of mitochondrial fragmentation in HeLa cells treated with Adp20. HeLa cells were used for this experiment because their mitochondria form long, clearly defined tubules ideal for colocalization studies; however, the results were also confirmed in Rat1 and H1299 cells. In mock-infected HeLa cells, Drp1 was distributed throughout the cytosol and showed only minor colocalization with mitochondria stained with TOM20 (Fig. 6 A), likely because Drp1 normally cycles on and off mitochondria continuously (Frank et al., 2001; Smirnova et al., 2001). In contrast, Drp1 showed a strong colocalization with mitochondria in HeLa cells infected with Adp20 + zVAD-fmk for 17 h (Fig. 6 A, bottom panels). Enlargement of the merged image revealed that Drp1 formed clusters along the surface of mitochondrial tubules before the onset of fragmentation. Interestingly, in Caenorhabditis elegans, similar clusters of GFP-Drp1 on mitochondrial tubules were shown to coincide with future sites of membrane scission (Labrousse et al., 1999). Pretreatment of H1299 cells with TG or expression of b5-BCL-2 reduced the amount of endogenous Drp1 recovered in the mitochondrial fraction after p20 expression (Fig. 6 B, compare lane 2 with lanes 3 and 4) and inhibited mitochondrial fission (Fig. 4 C).
Figure 6.

Drp1 mediates p20-induced mitochondrial fission. (A) Recruitment of endogenous Drp1 to mitochondria. HeLa cells were mock infected (top) or infected with Adp20 (bottom) in the presence of zVAD-fmk, and 17 h post-infection, cells were fixed, double stained with anti-Drp1 (green) and anti-TOM20 (red) antibodies, and imaged by confocal immunofluorescence microscopy. Enlargement of the merged overlay revealed that clusters of Drp1 relocate along mitochondrial filaments before the onset of fission. (B) H1299 cells, H1299 cells treated with TG, or H1299 b5-BCL-2 cells were infected with Adp20+zVAD for 18 h and the mitochondrial fraction was isolated and analyzed for the presence of Drp1 by SDS-PAGE and immunoblotting. The blot was reprobed with anti-TOM20 antibody to demonstrate equal protein loading.
Dominant-negative Drp1 K38E prevents p20-induced mitochondrial changes
To confirm that p20 mediates its sensitizing effect on mitochondria through Drp1, we examined the effect of a dominant-negative Drp1 mutant on p20-induced mitochondrial changes. Mutation of a conserved lysine (K38) in the GTP binding domain of Drp1 is predicted to reduce GTPase activity (Smirnova et al., 1998; Bleazard et al., 1999), and expression of such a mutant inhibits OMM scission (Labrousse et al., 1999). Ectopic expression of CFP-Drp1K38E in Rat1 cells offset the normal balance between mitochondrial fission and fusion and increased the connectivity of mitochondria compared with untransfected cells or cells transfected with wild-type CFP-Drp1 (Fig. 7 A, top panels; transfected [CFP-positive] cells are indicated by arrows). Overexpression of wild-type Drp1 does not induce fission in mammalian cells (Smirnova et al., 1998; Frank et al., 2001), and accordingly, Rat1 cells transiently transfected with CFP-Drp1 exhibited a normal mitochondrial phenotype and underwent fragmentation in response to Adp20 (Fig. 7 A). Cells transfected with CFP-Drp1K38E, on the other hand, resisted Adp20-induced mitochondrial fission and the highly interconnected network remained intact. As shown in Fig. 7 (B and C), CFP-Drp1K38E also inhibited p20-induced cyt.c release and caspase activation. Based on morphological criteria, as well as the recruitment of endogenous Drp-1 to mitochondria and dominant interference by the Drp1K38E mutant, we conclude that p20 activates Drp1-dependent mitochondrial fission, sensitizing this organelle for cyt.c release.
Figure 7.

Expression of a Drp1K38E dominant-negative mutant inhibits p20 induced disruption of the mitochondrial network. (A) Rat1 fibroblasts were transiently transfected with CFP-Drp1 or CFP- Drp1K38E, then either mock infected or infected with Adp20 in the presence of zVAD-fmk. 24 h post-infection cells were fixed, stained with anti-TOM20, and analyzed by fluorescence microscopy. Cells expressing CFP-Drp1 or CFP- Drp1K38E were identified under the cyan filter and are indicated with an arrow. (B) CFP- Drp1K38E inhibits cyt.c release. H1299 cells were treated as in B for 36 h, and immunofluorescence microscopy was used to assess the distribution of cyt.c in cells positive for CFP fluorescence. Shown is the mean ± SD of four independent experiments. (C) H1299 cells were transiently cotransfected with the indicated constructs and 36 h post-transfection cell lysates were collected and processed for DEVDase activity, shown is the mean ± SD of three independent experiments.
Discussion
Engagement of the TNF receptor family of death receptors, including TNF-R1, Fas, Trail-R1, and Trail-R2, with their cognate ligands leads to the recruitment and autoactivation of initiator procaspase-8 (Krammer, 2000). Recent studies implicate that caspase-8 substrates located at distinct cellular loci play key roles in mediating death receptor–induced apoptosis. For example, caspase-8 cleavage of the BH3-only molecule BID promotes mitochondrial release of cyt.c and Smac/Diablo (Yin et al., 1999; Li et al., 2002); cleavage of RIP prevents the activation of NF-κB survival responses (Lin et al., 1999); and cleavage of the cytolinker plectin is important for disassembly of microfilaments (Stegh et al., 2000). In this work, we investigated the consequence of caspase-8 cleavage of BAP31 at the ER by expressing the pro-apoptotic p20 cleavage fragment in cells using an adenovirus vector. This approach allowed us to isolate and delineate a predicted branch of the death receptor signaling cascade. Specifically, we found that p20 could mediate Ca2+-dependent apoptotic crosstalk between the ER and mitochondria, stimulating mitochondrial fission and sensitization of this organelle to caspase-8–induced cyt.c release.
The importance of BAP31 cleavage during Fas-mediated apoptosis was first highlighted by the observation that expression of crBAP31 strongly inhibited apoptotic membrane blebbing and release of cyt.c from mitochondria (Nguyen et al., 2000), suggesting that ER-mitochondrial signaling played a role in this pathway. When we reexamined photographs of mitochondria in crBAP31 cells undergoing Fas-induced apoptosis, it was apparent that mitochondrial fragmentation was also strongly inhibited (Nguyen et al., 2000). Thus, full-length BAP31 and p20 have opposing functions during Fas-mediated apoptosis, the former inhibiting mitochondrial fission and egress of cyt.c from mitochondria, and the latter stimulating these events. Importantly, however, p20 operates independently of BAP31 and BAP29 because p20 caused apoptosis in Bap31- and Bap29,31-null cells (Fig. 2 E). Therefore, caspase-8 cleavage of BAP31 converts it from an inhibitor to an activator of cell death; a paradigm that has been ascribed to other caspase targets such as BCL-2 (Cheng et al., 1997), BCL-xL (Clem et al., 1998), and RIP (Lin et al., 1999).
Cleavage of BAP31 may contribute to other cell death pathways that signal through caspase-8. For example, we recently reported that BAP31 and BAP29 play a role in the recruitment and activation of procaspase-8L at the ER during E1A-induced apoptosis (Breckenridge et al., 2002). The kinetics of procaspase-8L processing strongly correlated with BAP31 cleavage in response to E1A, suggesting that activated procaspase-8L may hydrolyze BAP31. The ensuing p20-induced Ca2+ release and mitochondrial fission might then enhance cyt.c release by other pro-apoptotic regulators that are activated by E1A, including BIK (Breckenridge and Shore, 2000; Mathai et al., 2002).
Based on studies using pharmacological modulators of Ca2+ signaling and inhibitors of apoptosis and mitochondrial fission, our results suggest that p20 induces an apoptotic pathway between the ER and mitochondria (Fig. 8 A). This is initiated by ER Ca2+ release coupled to mitochondrial Ca2+ uptake. An important caveat, of course, is that such conclusions rely on the specificity of the inhibitors that are widely used to interfere with Ca2+ signaling. Moreover, it cannot be ruled out that additional mechanisms are also involved. Importantly, however, it has been demonstrated that Drp1 recruitment to mitochondria initiates fission (Labrousse et al., 1999; Smirnova et al., 2001). Because either the lowering of ER Ca2+ stores, or chelating cytosolic Ca2+, or preventing mitochondrial Ca2+ uptake all prevented p20-induced fission of mitochondria, it is likely that ER-mitochondrial Ca2+ transmission acts upstream of Drp1 translocation in this context. Drp1 recruitment is likely mediated by an OMM receptor protein(s), and this complex likely cooperates with an inner mitochondrial membrane reorganizing enzyme(s) to mediate organelle fission (Shaw and Nunnari, 2002). Mitochondrial membranes are often in close proximity and privileged Ca2+ exchange between the two organelles has previously been implicated during apoptosis. For example, IP3 receptor– and ryanodine receptor–mediated Ca2+ spikes that modulate mitochondrial metabolism in healthy cells also sensitize mitochondria to pro-apoptotic stimuli during cell death (Szalai et al., 1999; Hajnoczky et al., 2000). Moreover, manipulations that increase [Ca2+]ER also increase agonist-induced Ca2+ spikes and enhance mitochondrial cyt.c release and apoptosis, whereas a lowering of ER Ca2+ stores has the opposite effect (Nakamura et al., 2000; Pinton et al., 2001). Modulation of the frequency, amplitude and spatio-temporal pattern of ER Ca2+ release during apoptosis may determine how mitochondria respond to Ca2+ signals (Berridge et al., 2000; Pacher and Hajnoczky, 2001). Our results suggest that caspase cleavage of BAP31 may be one mechanism to generate such pro-apoptotic ER-mitochondrial Ca2+-dependent crosstalk in the Fas pathway.
Figure 8.

A proposed mechanism of p20-induced mitochondrial fission. (A) p20 triggers a specific Ca2+ signal from the ER that is decoded by mitochondria. Mitochondria, in turn, recruit Drp1, which initiates organelle fission. Lowering ER Ca2+ stores by pretreatment with TG or expression of b5-BCL-2, chelating the Ca2+ released to the cytosol with BAPTA, blocking mitochondrial uptake of Ca2+ with Ru360, or inhibition of Drp1 by expression of Drp1K38E all prevent p20-induced mitochondrial fission. (B) A model depicting how in intact cells, cleavage of BAP31 at the ER sensitizes mitochondria to caspase-8–driven cyt.c release. Stimulation of Fas leads to caspase-8–dependent processing of BAP31 and BID, generating p20 and tBID. tBID translocates to mitochondria, where it induces the oligomerization of BAX/BAK into pores in the OMM. Simultaneously, p20 triggers ER Ca2+ release, causing Drp-1 translocation to mitochondria and subsequent organelle fission, enhancing the release of cyt.c to the cytosol.
In isolation, p20 caused ER Ca2+ release soon after its expression and Drp1 redistribution and mitochondrial fission were apparent within several hours of this event, but BAX activation, cyt.c release, and caspase activation were significantly delayed. Therefore, in the absence of a parallel BH3-dependent hit, mitochondria undergo fission in response to p20 and probably remain in a fragmented state (without releasing cyt.c) until a second signal responds and activates BAX/BAK. However, in a normal death receptor signaling context, simultaneous processing of BAP31 and BID by caspase-8 would be predicted to mount a dual attack on mitochondria, with p20 causing mitochondrial fission and tBID inducing cristae remodeling and activation of BAX and BAK (Scorrano et al., 2002; Fig. 8 B). Apoptotic cristae remodeling and mitochondrial fission may be intimately linked because cristae reorganization occurs during normal fission and fusion events in healthy cells (Bereiter-Hahn and Voth, 1994; Shaw and Nunnari, 2002) and mitochondrial fission is a requisite for cyt.c release (Frank et al., 2001). A “two hit” model in which an ER–mitochondrial Ca2+ signal and a direct mitochondrial insult synergize to promote the mitochondrial phase of apoptosis likely functions in other apoptosis pathways (Szalai et al., 1999; Pinton et al., 2001). Of note, tBID was reported to induce caspase-independent mitochondrial fragmentation on its own (Li et al., 1998), and therefore, p20 signaling may not be an obligate requirement for cyt.c release on the death receptor pathway but rather a sensitizer of this event. Indeed, the combined actions of p20 and tBID could cooperate in vivo because p20 strongly enhanced the ability of caspase-8 to promote cyt.c release without affecting the extent of BID cleavage (Fig. 3). This duality in signaling may be particularly relevant in physiological situations where apoptotic stimuli are sub-optimal or are countered by opposing survival signals, and the fate of the cell hinges on the balance of pro-apoptotic and anti-apoptotic signals received by mitochondria.
Materials and methods
Antibodies, plasmids, and reagents
The following antibodies were used in this work: chicken anti–human BAP31 (Ng et al., 1997), rabbit pAbs raised against the recombinant human BAP29, human TOM-20 (Goping et al., 1995), γ-actin (a gift from P. Braun, McGill University, Montreal, PQ), and human BAX aa 1–21 (Upstate Biotechnology); Rabbit pAb raised against the p15 caspase cleavage product of BID and purified by affinity selection; and mouse mAbs to pigeon cyt.c (BD Biosciences), chicken α-tubulin (clone DM1A; Sigma-Aldrich), rodent Drp1 (BD Biosciences), and HA (clone 16B12; BAbCO). Goat anti-calreticulin was provided by M. Michalak (University of Alberta, Edmonton, AB). Anti-human Fas activating antibody (Clone CH-11) was from Upstate Biotechnology. Standard PCR techniques were used to generate cDNA encoding p20 (aa 1–164 of human BAP31) with a COOH-terminal HA tag that was cloned in to pcDNA3. Plasmids encoding Drp1 and Drp1K38E fused to CFP at the NH2 terminus were gifts from H. McBride (Ottawa Heart institute, Ottawa, ON). Carbobenzoxy-valvy-alanyl-aspartyl-methyl ester-fluormethyl ketone (zVAD-fmk) was purchased from Enzyme System Products. Fura2-AM, Rhod2-AM, and BAPTA-AM were from Molecular Probes, Inc., and Ru360 was from Calbiochem. All other chemicals were purchased from Sigma-Aldrich, unless otherwise noted.
Cell culture, virus infection, and transfection
KB epithelial cells and H1299 lung carcinoma cells were maintained in MEM-α supplemented with 10% FBS and 100 U/ml streptomycin and penicillin. Rat1 fibroblast, CHO, and HeLa cells were grown in DME supplemented as above. KB cells stably expressing HA-Bcl-2 and HA-Bcl-XL have been described previously (Nguyen et al., 1998; Ruffolo et al., 2000). H1299 b5-BCL-2 cells were created by transfecting H1299 cells with pcDNA3 vector encoding human HA-Bcl-2 with amino acids 215–239 swapped with the transmembrane sequence of human cytochrome B5 (amino acids 107–134) and selecting for resistance to geneticin. Bap31- and Bap29-null mouse embryonic stem cells were maintained as described previously (Breckenridge et al., 2002).
For the construction of Adp20, AdMFpk3FLICE, and AdRTA, cDNAs encoding p20-HA, MFpk3FLICE (Muzio et al., 1998) and RTA were subcloned into a variant of pCA14 containing the T-REx™ promoter (Invitrogen), which functioned as a shuttle to produce the adenoviral vectors as described previously (Bett et al., 1994; Mathai et al., 2002). All adenoviral infections were conducted at a multiplicity of infection of 100 pfu/cell as described previously (Ng et al., 1997), except for in Fig. 3, where the viruses were mixed to generate a total of 100 pfu/cell. LipofectAMINE™ Plus (Invitrogen) was used for all transfections using the manufacturer's protocols. In experiments where Adp20 infection followed transfection of CFP-Drp1 or CFP-Drp1K38E, the transfection medium was removed 3 h after transfection and medium containing serum and Adp20 virus was added back.
Apoptotic assays
DEVDase activity was measured from 25 μg of cell lysate protein according to the manufacturer's protocol (Upstate Biotechnology). For statistical analysis of mitochondrial fission and cyt.c release, cells were stained for TOM20 or cyt.c, and the distribution of cyt.c and the morphology of mitochondria were analyzed by conventional immunofluorescence microscopy. In all cases, at least five independent experiments were conducted, where three counts of 150 randomly selected cells was done per experiment. In Fig. 7 B, only cells showing CFP expression were assessed for cyt.c release. Biochemical isolation of the heavy membrane fraction enriched in mitochondria or post-mitochondrial supernatant for measurement of cyt.c release and Drp1 recruitment was done as described previously (Ruffolo et al., 2000). In Fig. 3, the intensity of each cyt.c and tBID Western blot signal was quantified using ImageQuantTM (Amersham Biosciences) software and compared with the intensity of a loading control signal in the same lane (actin or TOM20, respectively) after the subtraction of background. The relative values were expressed as arbitrary units.
Fluorescence microscopy
Cells were typically seeded at 50% confluency on glass coverslips and mock infected or infected with Adp20, always in the presence of zVAD-fmk to prevent apoptosis and cell detachment. At the indicated times after infection, cells were washed in PBS, and were then fixed in PFA solution (4% PFA, 23 mM NaH2PO4, and 77 mM Na2HPO4, pH 7.3). Cells were briefly permeabilized in PBS/0.2% Triton X-100, then blocked in blocking solution (PBS containing 10% FCS and 0.1% Triton X-100). Primary and secondary antibody incubations were done in blocking solution for 1 h at RT using the indicated antibodies and goat anti–mouse IgG or goat anti–rabbit IgG secondary antibody coupled to Alexa® 488 (green) or Alexa® 594 (red; Molecular Probes, Inc.). Cells were visualized by confocal microscopy or by conventional fluorescence microscopy on an inverted microscope (TE-FM Epi-fl; Nikon).
Measurement of ER Ca2+ content
The ER Ca2+ store was measured as the sudden increase in [Ca2+]c on addition of TG. [Ca2+]c was measured by the cell permeable fluorescent indicator Fura2-AM. In brief, 2 × 106 cells were washed in Ca2+-free buffer (20 mM Hepes, pH 7.4, 143 mM NaCl, 6 mM KCL, 1 mM MgSO4, 0.1% glucose, and 250 μM sulfinpyrazone), then loaded with 3 mM Fura2-AM for 30 min at 37°C in Ca2+-free buffer containing 0.02% pluronic acid and 0.1% BSA. After a final wash, cells were resuspended in Ca2+-free buffer and [Ca2+]c was measured as 340/380 nm excitation wavelength ratio at 510 nm wavelength emission (340/380 ratio) in a luminescence spectrophotometer (model LS 50B; PerkinElmer). The ER calcium content was measured as the difference between the baseline 340/380 ratio before TG addition and the peak 340/380 ratio after TG addition. This value was arbitrarily set at 100% for untreated cells.
Measurement of [Ca2+]m
5 × 105 cells were collected, washed once in PBS, then resuspended in 1 ml Earl's balanced salt solution and loaded with 2 μM Rhod2-AM in the presence of 0.02% pluronic acid for 20 min at RT. Cells were washed twice in the same buffer and Rhod2 fluorescence (F) was measured as above at 550/580 excitation/emission wavelengths. Minimum and maximum fluorescence values (Fmax and Fmin, respectively) were then obtained on the sequential addition of EGTA and saturating amounts of CaCl2 in the presence of detergent. [Ca2+]m was determined by the equation
[Ca2+] = Kd (F − Fmin)/(Fmax − F) where Kd is the dissociation constant of Rhod2. Rhod2 was judged to be localized to mitochondria based on analysis by immunofluorescence microscopy and by the fact that fluorescence was reduced to basal levels on the addition of the mitochondrial uncoupler, CCCP.
Online supplemental material
Fig. S1 documents that Adp20-induced release of cyt.c from mitochondria, procaspase-3 processing, and caspase activation are blocked in APAF-1–null MEFs (Yoshida et al., 1998; a gift from T. Mak, Ontario Cancer Institute, Toronto, Canada). Fig. S2 shows that Adp20 infection does not affect ER-Golgi trafficking of temperature-sensitive VSV-G-EGFP. Fig. S3 documents that TG and b5-BCL-2 effectively lowered resting ER Ca2+ stores. Immunofluorescence microscopy confirmed that b5-BCL-2 was located exclusively at the ER. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200212059/DC1.
Supplemental Material
Acknowledgments
We are very grateful to J. Lynch and M. Michalak for Fura2 protocols and anti-calreticulin antibody.
D.G. Breckenridge is a recipient of a Canadian Institutes of Health Research Doctoral award. This work was supported by grants to G.C. Shore from the National Cancer Institute of Canada, and the Canadian Institutes of Health Research.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: crBAP31, caspase-resistant BAP31; cyt.c, cytochrome c; HA, hemagglutinin; OMM, outer mitochondrial membrane; RTA, reverse tet transactivating protein; TG, thapsigargin.
References
- Adachi, T., W.W. Schamel, K.M. Kim, T. Watanabe, B. Becker, P.J. Nielsen, and M. Reth. 1996. The specificity of association of the IgD molecule with the accessory proteins BAP31/BAP29 lies in the IgD transmembrane sequence. EMBO J. 15:1534–1541. [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn, J., and M. Voth. 1994. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc. Res. Tech. 27:198–219. [DOI] [PubMed] [Google Scholar]
- Berridge, M.J., P. Lipp, and M.D. Bootman. 2000. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1:11–21. [DOI] [PubMed] [Google Scholar]
- Bett, A.J., W. Haddara, L. Prevec, and F.L. Graham. 1994. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA. 91:8802–8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard, W., J.M. McCaffery, E.J. King, S. Bale, A. Mozdy, Q. Tieu, J. Nunnari, and J.M. Shaw. 1999. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge, D.G., and G.C. Shore. 2000. Regulation of apoptosis by E1A and Myc oncoproteins. Crit. Rev. Eukaryot. Gene Exp. 10:273–280. [DOI] [PubMed] [Google Scholar]
- Breckenridge, D.G., and G.C. Shore. 2002. The endoplasmic reticulum and apoptosis. Genetics of Apoptosis. S. Grimm, editor. BIOS Scientific Publishers Inc., Oxford. 95–113.
- Breckenridge, D.G., M. Nguyen, S. Kuppig, M. Reth, and G.C. Shore. 2002. The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 99:4331–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budihardjo, I., H. Oliver, M. Lutter, X. Luo, and X. Wang. 1999. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15:269–290. [DOI] [PubMed] [Google Scholar]
- Byrne, A.M., J.J. Lemasters, and A.L. Nieminen. 1999. Contribution of increased mitochondrial free Ca2+ to the mitochondrial permeability transition induced by tert-butylhydroperoxide in rat hepatocytes. Hepatology. 29:1523–1531. [DOI] [PubMed] [Google Scholar]
- Cheng, E.H., D.G. Kirsch, R.J. Clem, R. Ravi, M.B. Kastan, A. Bedi, K. Ueno, and J.M. Hardwick. 1997. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 278:1966–1968. [DOI] [PubMed] [Google Scholar]
- Clem, R.J., E.H. Cheng, C.L. Karp, D.G. Kirsch, K. Ueno, A. Takahashi, M.B. Kastan, D.E. Griffin, W.C. Earnshaw, M.A. Veliuona, and J.M. Hardwick. 1998. Modulation of cell death by Bcl-XL through caspase interaction. Proc. Natl. Acad. Sci. USA. 95:554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, T.J., M.J. Berridge, P. Lipp, and M.D. Bootman. 2002. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21:1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory, S., and J.M. Adams. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2:647–656. [DOI] [PubMed] [Google Scholar]
- Deng, Y., Y. Lin, and X. Wu. 2002. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 16:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher, S., and J.C. Martinou. 2000. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 10:369–377. [DOI] [PubMed] [Google Scholar]
- Desagher, S., A. Osen-Sand, A. Nichols, R. Eskes, S. Montessuit, S. Lauper, K. Maundrell, B. Antonsson, and J.C. Martinou. 1999. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyouzi-Youssefi, R., S. Arnaudeau, C. Borner, W.L. Kelley, J. Tschopp, D.P. Lew, N. Demaurex, and K.H. Krause. 2000. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 97:5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, S., B. Gaume, E.S. Bergmann-Leitner, W.W. Leitner, E.G. Robert, F. Catez, C.L. Smith, and R.J. Youle. 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell. 1:515–525. [DOI] [PubMed] [Google Scholar]
- Fulda, S., W. Wick, M. Weller, and K.M. Debatin. 2002. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 8:808–815. [DOI] [PubMed] [Google Scholar]
- Goping, I.S., D.G. Millar, and G.C. Shore. 1995. Identification of the human mitochondrial protein import receptor, huMas20p. Complementation of delta mas20 in yeast. FEBS Lett. 373:45–50. [DOI] [PubMed] [Google Scholar]
- Green, D.R., and J.C. Reed. 1998. Mitochondria and apoptosis. Science. 281:1309–1312. [DOI] [PubMed] [Google Scholar]
- Hajnoczky, G., G. Csordas, M. Madesh, and P. Pacher. 2000. Control of apoptosis by IP(3) and ryanodine receptor driven calcium signals. Cell Calcium. 28:349–363. [DOI] [PubMed] [Google Scholar]
- Korsmeyer, S.J., M.C. Wei, M. Saito, S. Weiler, K.J. Oh, and P.H. Schlesinger. 2000. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 7:1166–1173. [DOI] [PubMed] [Google Scholar]
- Krammer, P.H. 2000. CD95's deadly mission in the immune system. Nature. 407:789–795. [DOI] [PubMed] [Google Scholar]
- Labrousse, A.M., M.D. Zappaterra, D.A. Rube, and A.M. van der Bliek. 1999. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell. 4:815–826. [DOI] [PubMed] [Google Scholar]
- Li, H., H. Zhu, C.J. Xu, and J. Yuan. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 94:491–501. [DOI] [PubMed] [Google Scholar]
- Li, S., Y. Zhao, X. He, T.H. Kim, D.K. Kuharsky, H. Rabinowich, J. Chen, C. Du, and X.M. Yin. 2002. Relief of extrinsic pathway inhibition by the Bid-dependent mitochondrial release of Smac in Fas-mediated hepatocyte apoptosis. J. Biol. Chem. 277:26912–26920. [DOI] [PubMed] [Google Scholar]
- Lin, Y., A. Devin, Y. Rodriguez, and Z.G. Liu. 1999. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13:2514–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, X., I. Budihardjo, H. Zou, C. Slaughter, and X. Wang. 1998. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 94:481–490. [DOI] [PubMed] [Google Scholar]
- Mathai, J.P., M. Germain, R.C. Marcellus, and G.C. Shore. 2002. Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene. 21:2534–2544. [DOI] [PubMed] [Google Scholar]
- Matlib, M.A., Z. Zhou, S. Knight, S. Ahmed, K.M. Choi, J. Krause-Bauer, R. Phillips, R. Altschuld, Y. Katsube, N. Sperelakis, and D.M. Bers. 1998. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J. Biol. Chem. 273:10223–10231. [DOI] [PubMed] [Google Scholar]
- Mootha, V.K., M.C. Wei, K.F. Buttle, L. Scorrano, V. Panoutsakopoulou, C.A. Mannella, and S.J. Korsmeyer. 2001. A reversible component of mitochondrial respiratory dysfunction in apoptosis can be rescued by exogenous cytochrome c. EMBO J. 20:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio, M., B.R. Stockwell, H.R. Stennicke, G.S. Salvesen, and V.M. Dixit. 1998. An induced proximity model for caspase-8 activation. J. Biol. Chem. 273:2926–2930. [DOI] [PubMed] [Google Scholar]
- Nakamura, K., E. Bossy-Wetzel, K. Burns, M.P. Fadel, M. Lozyk, I.S. Goping, M. Opas, R.C. Bleackley, D.R. Green, and M. Michalak. 2000. Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol. 150:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, F.W., M. Nguyen, T. Kwan, P.E. Branton, D.W. Nicholson, J.A. Cromlish, and G.C. Shore. 1997. p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8-associated protein in the endoplasmic reticulum. J. Cell Biol. 139:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, M., P.E. Branton, S. Roy, D.W. Nicholson, E.S. Alnemri, W.C. Yeh, T.W. Mak, and G.C. Shore. 1998. E1A-induced processing of procaspase-8 can occur independently of FADD and is inhibited by Bcl-2. J. Biol. Chem. 273:33099–33102. [DOI] [PubMed] [Google Scholar]
- Nguyen, M., D.G. Breckenridge, A. Ducret, and G.C. Shore. 2000. Caspase-resistant BAP31 inhibits fas-mediated apoptotic membrane fragmentation and release of cytochrome c from mitochondria. Mol. Cell. Biol. 20:6731–6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteryoung, K.W. 2001. Organelle fission in eukaryotes. Curr. Opin. Microbiol. 4:639–646. [DOI] [PubMed] [Google Scholar]
- Pacher, P., and G. Hajnoczky. 2001. Propagation of the apoptotic signal by mitochondrial waves. EMBO J. 20:4107–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit, P.X., M. Goubern, P. Diolez, S.A. Susin, N. Zamzami, and G. Kroemer. 1998. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett. 426:111–116. [DOI] [PubMed] [Google Scholar]
- Pinton, P., D. Ferrari, P. Magalhaes, K. Schulze-Osthoff, F. Di Virgilio, T. Pozzan, and R. Rizzuto. 2000. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J. Cell Biol. 148:857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton, P., D. Ferrari, E. Rapizzi, F.D. Di Virgilio, T. Pozzan, and R. Rizzuto. 2001. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2. EMBO J. 20:2690–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto, R., P. Pinton, W. Carrington, F.S. Fay, K.E. Fogarty, L.M. Lifshitz, R.A. Tuft, and T. Pozzan. 1998. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 280:1763–1766. [DOI] [PubMed] [Google Scholar]
- Ruffolo, S.C., D.G. Breckenridge, M. Nguyen, I.S. Goping, A. Gross, S.J. Korsmeyer, H. Li, J. Yuan, and G.C. Shore. 2000. BID-dependent and BID-independent pathways for BAX insertion into mitochondria. Cell Death Differ. 7:1101–1108. [DOI] [PubMed] [Google Scholar]
- Scaffidi, C., S. Fulda, A. Srinivasan, C. Friesen, F. Li, K.J. Tomaselli, K.M. Debatin, P.H. Krammer, and M.E. Peter. 1998. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17:1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Osthoff, K., D. Ferrari, M. Los, S. Wesselborg, and M.E. Peter. 1998. Apoptosis signaling by death receptors. Eur. J. Biochem. 254:439–459. [DOI] [PubMed] [Google Scholar]
- Scorrano, L., M. Ashiya, K. Buttle, S. Weiler, S.A. Oakes, C.A. Mannella, and S.J. Korsmeyer. 2002. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell. 2:55–67. [DOI] [PubMed] [Google Scholar]
- Sharma, V.K., V. Ramesh, C. Franzini-Armstrong, and S.S. Sheu. 2000. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenerg. Biomembr. 32:97–104. [DOI] [PubMed] [Google Scholar]
- Shaw, J.M., and J. Nunnari. 2002. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 12:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova, E., L. Griparic, D.L. Shurland, and A.M. van der Bliek. 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 12:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova, E., D.L. Shurland, S.N. Ryazantsev, and A.M. van der Bliek. 1998. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 143:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegh, A.H., H. Herrmann, S. Lampel, D. Weisenberger, K. Andra, M. Seper, G. Wiche, P.H. Krammer, and M.E. Peter. 2000. Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol. Cell. Biol. 20:5665–5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai, G., R. Krishnamurthy, and G. Hajnoczky. 1999. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 18:6349–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B., M. Nguyen, D.G. Breckenridge, M. Stojanovic, P.A. Clemons, S. Kuppig, and G.C. Shore. 2003. Uncleaved BAP31 in association with A4 protein at the endoplasmic reticulum is an inhibitor of Fas-initiated release of cytochrome c from mitochondria. J. Biol. Chem. In press. [DOI] [PubMed] [Google Scholar]
- Wang, J., H.J. Chun, W. Wong, D.M. Spencer, and M.J. Lenardo. 2001. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA. 98:13884–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, X.M., K. Wang, A. Gross, Y. Zhao, S. Zinkel, B. Klocke, K.A. Roth, and S.J. Korsmeyer. 1999. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 400:886–891. [DOI] [PubMed] [Google Scholar]
- Yoshida, H., Y.Y. Kong, R. Yoshida, A.J. Elia, A. Hakem, R. Hakem, J.M. Penninger, and T.W. Mak. 1998. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 94:739–750. [DOI] [PubMed] [Google Scholar]
- Zhu, W., A. Cowie, G.W. Wasfy, L.Z. Penn, B. Leber, and D.W. Andrews. 1996. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}